
Single Nucleotide Polymorphism in Cell Adhesion Molecule L1 Affects Learning and Memory in a Mouse Model of Traumatic Brain Injury
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Following TBI, Injured L1-201 Mice Had Impaired Learning and Memory Relative to Injured WT Mice
2.2. There Is a Trend for Injured L1-201 to Have Impaired Gross Vestibular Motor Function Relative to Injured WT Mice; However, There Is no Significant Difference Seen
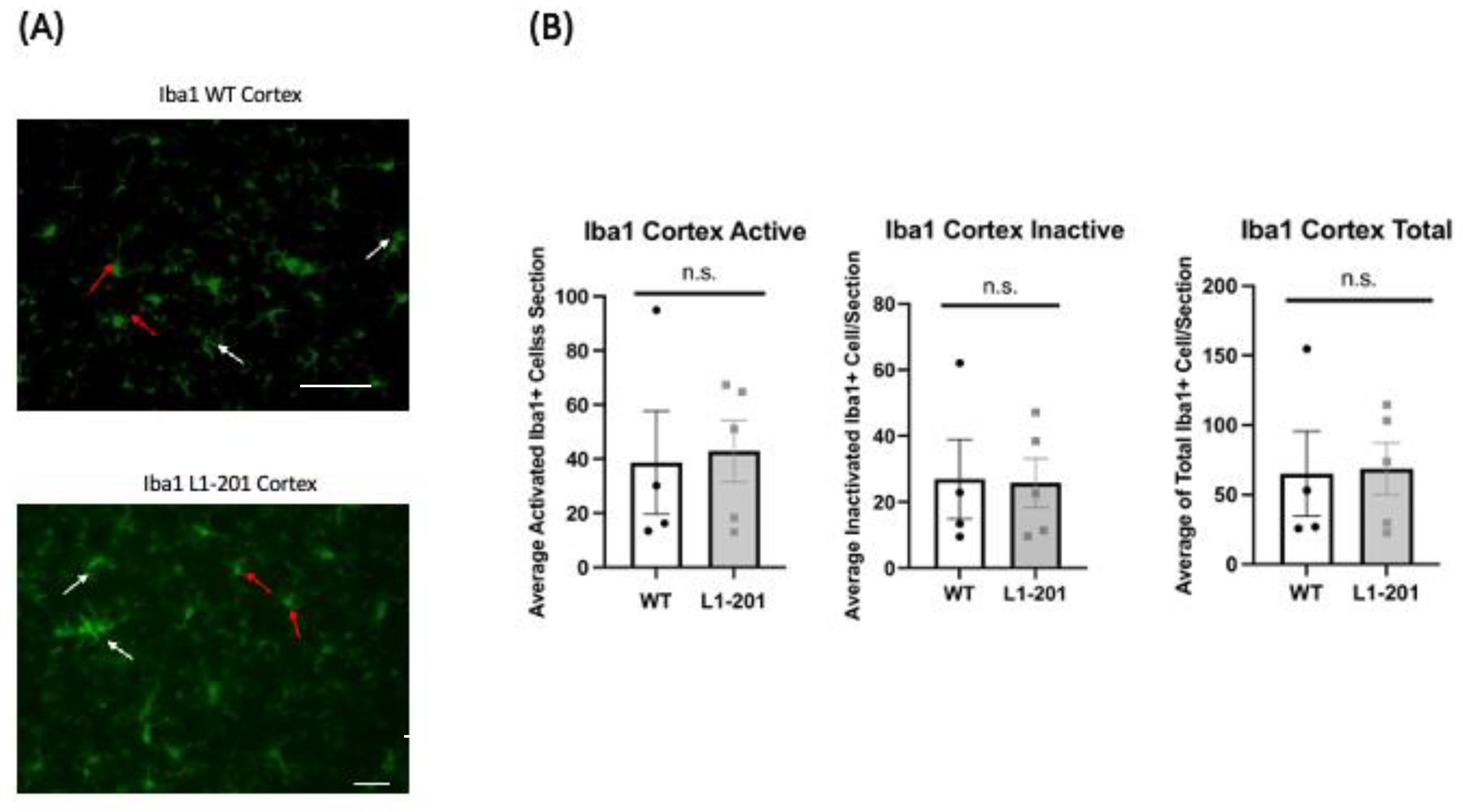
2.3. There Are no Significant Differences in IBA1 between the Injured L1-201 and Injured WT Mice in either the Cortex or Hippocampus at 21 DPI
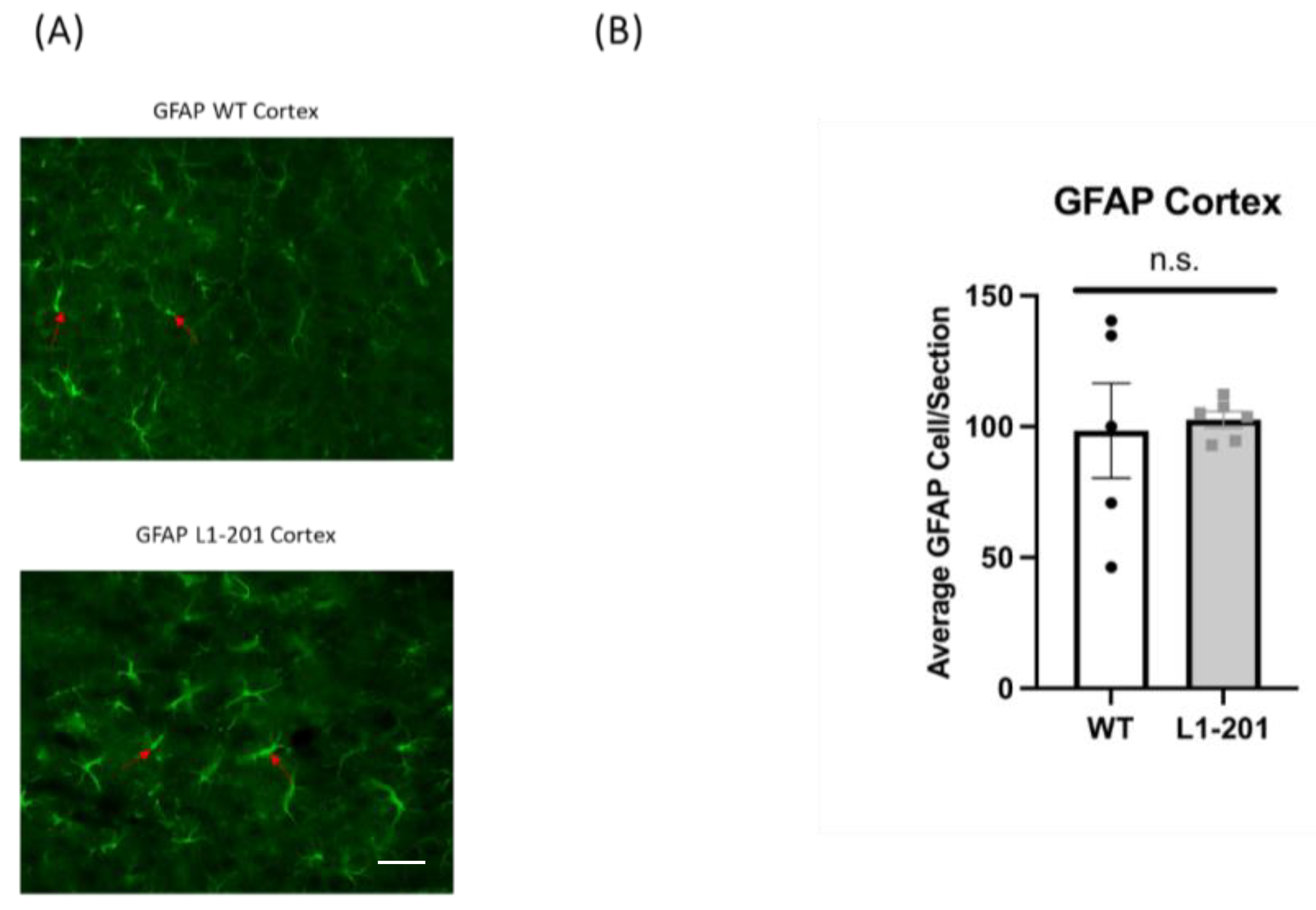
2.4. There Are no Significant Differences in GFAP between the Injured L1-201 and Injured WT Mice in either the Cortex or Hippocampus at 21 DPI
2.5. There Are no Significant Differences in Caspase between the Injured L1-201 and Injured WT Mice in either the Cortex or Hippocampus at 21 DPI
2.6. There Are no Significant Differences in the Number of Cells between the Injured L1-201 and Injured WT Mice in either the Cortex or Hippocampus at 21 DPI
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Lateral Fluid Percussion Injury
4.3. Vestibular Rotarod Test
4.4. Morris Water Maze Test
4.5. Immunohistochemistry
4.6. Quantification of Immunohistochemistry
4.7. Statistical Analysis
4.8. Animal Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, S.; Lee, D.W.; Schachner, M.; Park, H.C. Small compounds mimicking the adhesion molecule L1 improve recovery in a zebrafish demyelination model. Sci. Rep. 2021, 11, 5878. [Google Scholar] [CrossRef]
- Joseph, T.P.; Jagadeesan, N.; Sai, L.Y.; Lin, S.L.; Sahu, S.; Schachner, M. Adhesion Molecule L1 Agonist Mimetics Protect Against the Pesticide Paraquat-Induced Locomotor Deficits and Biochemical Alterations in Zebrafish. Front. Neurosci. 2020, 14, 458. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Lin, S.L.; Schachner, M. A fragment of cell adhesion molecule L1 reduces amyloid-β plaques in a mouse model of Alzheimer’s disease. Cell Death Dis. 2022, 13, 48. [Google Scholar] [CrossRef]
- Zhang, Y.; Yeh, J.; Richardson, P.M.; Bo, X. Cell adhesion molecules of the immunoglobulin superfamily in axonal regeneration and neural repair. Restor. Neurol. Neurosci. 2008, 26, 81–96. [Google Scholar]
- Schäfer, M.K.; Frotscher, M. Role of L1CAM for axon sprouting and branching. Cell Tissue Res. 2012, 349, 39–48. [Google Scholar] [CrossRef]
- Budinich, C.S.; Chen, H.; Lowe, D.; Rosenberger, J.G.; Bernstock, J.D.; McCabe, J.T. Mouse brain PSA-NCAM levels are altered by graded-controlled cortical impact injury. Neural Plast. 2012, 2012, 378307. [Google Scholar] [CrossRef]
- Emery, D.L.; Fulp, C.T.; Saatman, K.E.; Schütz, C.; Neugebauer, E.; McIntosh, T.K. Newly born granule cells in the dentate gyrus rapidly extend axons into the hippocampal CA3 region following experimental brain injury. J. Neurotrauma 2005, 22, 978–988. [Google Scholar] [CrossRef]
- Wang, H.C.; Wang, P.M.; Lin, Y.T.; Tsai, N.W.; Lai, Y.R.; Kung, C.T.; Su, C.M.; Lu, C.H. Effects of Hyperbaric Oxygen Therapy on Serum Adhesion Molecules, and Serum Oxidative Stress in Patients with Acute Traumatic Brain Injury. J. Pers. Med. 2021, 11, 985. [Google Scholar] [CrossRef] [PubMed]
- Butenschön, J.; Zimmermann, T.; Schmarowski, N.; Nitsch, R.; Fackelmeier, B.; Friedemann, K.; Radyushkin, K.; Baumgart, J.; Lutz, B.; Leschik, J. PSA-NCAM positive neural progenitors stably expressing BDNF promote functional recovery in a mouse model of spinal cord injury. Stem Cell Res. Ther. 2016, 7, 11. [Google Scholar] [CrossRef]
- Yamanaka, H.; Kobayashi, K.; Okubo, M.; Fukuoka, T.; Noguchi, K. Increase of close homolog of cell adhesion molecule L1 in primary afferent by nerve injury and the contribution to neuropathic pain. J. Comp. Neurol. 2011, 519, 1597–1615. [Google Scholar] [CrossRef]
- Wu, J.; Leung, P.Y.; Sharp, A.; Lee, H.J.; Wrathall, J.R. Increased expression of the close homolog of the adhesion molecule L1 in different cell types over time after rat spinal cord contusion. J. Neurosci. Res. 2011, 89, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Chi, O.Z.; Theis, T.; Kumar, S.; Chiricolo, A.; Liu, X.; Farooq, S.; Trivedi, N.; Young, W.; Schachner, M.; Weiss, H.R. Adhesion molecule L1 inhibition increases infarct size in cerebral ischemia-reperfusion without change in blood-brain barrier disruption. Neurol. Res. 2021, 43, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Jakovcevski, I.; Djogo, N.; Hölters, L.S.; Szpotowicz, E.; Schachner, M. Transgenic overexpression of the cell adhesion molecule L1 in neurons facilitates recovery after mouse spinal cord injury. Neuroscience 2013, 252, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Roonprapunt, C.; Huang, W.; Grill, R.; Friedlander, D.; Grumet, M.; Chen, S.; Schachner, M.; Young, W. Soluble cell adhesion molecule L1-Fc promotes locomotor recovery in rats after spinal cord injury. J. Neurotrauma 2003, 20, 871–882. [Google Scholar] [CrossRef]
- Stumpel, C.; Vos, Y.J. L1 Syndrome. In GeneReviews(®); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Loers, G.; Appel, D.; Lutz, D.; Congiu, L.; Kleene, R.; Hermans-Borgmeyer, I.; Schäfer, M.K.E.; Schachner, M. Amelioration of the abnormal phenotype of a new L1 syndrome mouse mutation with L1 mimetics. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21329. [Google Scholar] [CrossRef] [PubMed]
- Hortsch, M.; Nagaraj, K.; Mualla, R. The L1 family of cell adhesion molecules: A sickening number of mutations and protein functions. Adv. Neurobiol. 2014, 8, 195–229. [Google Scholar] [CrossRef]
- Giarratana, A.O.; Teng, S.; Reddi, S.; Zheng, C.; Adler, D.; Thakker-Varia, S.; Alder, J. BDNF Val66Met Genetic Polymorphism Results in Poor Recovery Following Repeated Mild Traumatic Brain Injury in a Mouse Model and Treatment With AAV-BDNF Improves Outcomes. Front. Neurol. 2019, 10, 1175. [Google Scholar] [CrossRef]
- Giarratana, A.O.; Zheng, C.; Reddi, S.; Teng, S.L.; Berger, D.; Adler, D.; Sullivan, P.; Thakker-Varia, S.; Alder, J. APOE4 genetic polymorphism results in impaired recovery in a repeated mild traumatic brain injury model and treatment with Bryostatin-1 improves outcomes. Sci. Rep. 2020, 10, 19919. [Google Scholar] [CrossRef]
- Nagaraj, V.; Kim, R.; Martianou, T.; Kurian, S.; Nayak, A.; Patel, M.; Schachner, M.; Theis, T. Effects of L1 adhesion molecule agonistic mimetics on signal transduction in neuronal functions. Biochem. Biophys. Res. Commun. 2023, 642, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ashina, H.; Dodick, D.W.; Barber, J.; Temkin, N.R.; Chong, C.D.; Adler, J.S.; Stein, K.S.; Schwedt, T.J.; Manley, G.T. Prevalence of and Risk Factors for Post-traumatic Headache in Civilian Patients After Mild Traumatic Brain Injury: A TRACK-TBI Study. Mayo Clin. Proc. 2023, 98, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Stocchetti, N.; Zanier, E.R. Chronic impact of traumatic brain injury on outcome and quality of life: A narrative review. Crit. Care 2016, 20, 148. [Google Scholar] [CrossRef]
- Haarbauer-Krupa, J.; Pugh, M.J.; Prager, E.M.; Harmon, N.; Wolfe, J.; Yaffe, K. Epidemiology of Chronic Effects of Traumatic Brain Injury. J. Neurotrauma 2021, 38, 3235–3247. [Google Scholar] [CrossRef]
- Weaver, S.M.; Portelli, J.N.; Chau, A.; Cristofori, I.; Moretti, L.; Grafman, J. Genetic polymorphisms and traumatic brain injury: The contribution of individual differences to recovery. Brain Imaging Behav. 2014, 8, 420–434. [Google Scholar] [CrossRef]
- Kotloski, R.J.; Rutecki, P.A.; Sutula, T.P. Genetic Background Influences Acute Response to TBI in Kindling-Susceptible, Kindling-Resistant, and Outbred Rats. Front. Neurol. 2019, 10, 1286. [Google Scholar] [CrossRef]
- Alder, J.; Fujioka, W.; Giarratana, A.; Wissocki, J.; Thakkar, K.; Vuong, P.; Patel, B.; Chakraborty, T.; Elsabeh, R.; Parikh, A.; et al. Genetic and pharmacological intervention of the p75NTR pathway alters morphological and behavioural recovery following traumatic brain injury in mice. Brain Inj. 2016, 30, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Styren, S.D.; Miller, P.D.; Lagenaur, C.F.; DeKosky, S.T. Alternate strategies in lesion-induced reactive synaptogenesis: Differential expression of L1 in two populations of sprouting axons. Exp. Neurol. 1995, 131, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.D.; Chung, W.W.; Lagenaur, C.F.; DeKosky, S.T. Regional distribution of neural cell adhesion molecule (N-CAM) and L1 in human and rodent hippocampus. J. Comp. Neurol. 1993, 327, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Darwish, H.; Mahmood, A.; Schallert, T.; Chopp, M.; Therrien, B. Mild traumatic brain injury (MTBI) leads to spatial learning deficits. Brain Inj. 2012, 26, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Paterno, R.; Folweiler, K.A.; Cohen, A.S. Pathophysiology and Treatment of Memory Dysfunction After Traumatic Brain Injury. Curr. Neurol. Neurosci. Rep. 2017, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.W.; Lagenaur, C.F.; Yan, Y.M.; Lund, J.S. Developmental expression of neural cell adhesion molecules in the mouse neocortex and olfactory bulb. J. Comp. Neurol. 1991, 314, 290–305. [Google Scholar] [CrossRef] [PubMed]
- Flygt, J.; Ruscher, K.; Norberg, A.; Mir, A.; Gram, H.; Clausen, F.; Marklund, N. Neutralization of Interleukin-1beta following Diffuse Traumatic Brain Injury in the Mouse Attenuates the Loss of Mature Oligodendrocytes. J. Neurotrauma 2018, 35, 2837–2849. [Google Scholar] [CrossRef]
- Lim, S.W.; Shiue, Y.L.; Liao, J.C.; Wee, H.Y.; Wang, C.C.; Chio, C.C.; Chang, C.H.; Hu, C.Y.; Kuo, J.R. Simvastatin Therapy in the Acute Stage of Traumatic Brain Injury Attenuates Brain Trauma-Induced Depression-Like Behavior in Rats by Reducing Neuroinflammation in the Hippocampus. Neurocrit Care 2017, 26, 122–132. [Google Scholar] [CrossRef]
- Sun, G.W.; Ding, T.Y.; Wang, M.; Hu, C.L.; Gu, J.J.; Li, J.; Qiu, T. Honokiol Reduces Mitochondrial Dysfunction and Inhibits Apoptosis of Nerve Cells in Rats with Traumatic Brain Injury by Activating the Mitochondrial Unfolded Protein Response. J. Mol. Neurosci. 2022, 72, 2464–2472. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, G.; Xu, L.; Wen, X.; Cai, Y.; Fan, W.; Yao, X.; Huang, H.; Li, Q. Repair of Neurological Function in Response to FK506 Through CaN/NFATc1 Pathway Following Traumatic Brain Injury in Rats. Neurochem. Res. 2016, 41, 2810–2818. [Google Scholar] [CrossRef]
- Zhu, W.; Chi, N.; Zou, P.; Chen, H.; Tang, G.; Zhao, W. Effect of docosahexaenoic acid on traumatic brain injury in rats. Exp. Ther. Med. 2017, 14, 4411–4416. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, E.D.; Gao, Y.; Gavsyuk, A.F.; Maor, I.; DeWalt, G.J.; Eldred, W.D.; Mizrahi, A.; Davison, I.G. Rapid Changes in Synaptic Strength After Mild Traumatic Brain Injury. Front. Cell. Neurosci. 2019, 13, 166. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Chen, C.; Kang, S.S.; Liu, X.; Gu, X.; Yu, S.P.; Keene, C.D.; Cheng, L.; Ye, K. Neurotrophic signaling deficiency exacerbates environmental risks for Alzheimer’s disease pathogenesis. Proc. Natl. Acad. Sci. USA 2021, 118, 166. [Google Scholar] [CrossRef] [PubMed]
- Loers, G.; Schachner, M. Recognition molecules and neural repair. J. Neurochem. 2007, 101, 865–882. [Google Scholar] [CrossRef] [PubMed]
- Bechara, A.; Nawabi, H.; Moret, F.; Yaron, A.; Weaver, E.; Bozon, M.; Abouzid, K.; Guan, J.L.; Tessier-Lavigne, M.; Lemmon, V.; et al. FAK-MAPK-dependent adhesion disassembly downstream of L1 contributes to semaphorin3A-induced collapse. EMBO J. 2023, 42, e113962. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Duncan, B.; Sperringer, J.E.; Zhang, E.; Haberman, V.; Wyatt, E.V.; Maness, P. Ankyrin B promotes developmental spine regulation in the mouse prefrontal cortex. Cereb. Cortex 2023, 33, 10634–10648. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, I.I.; Lutz, D. Functional Diversity of Neuronal Cell Adhesion and Recognition Molecule L1CAM through Proteolytic Cleavage. Cells 2022, 11, 3085. [Google Scholar] [CrossRef] [PubMed]
- Witheford, M.; Westendorf, K.; Roskams, A.J. Olfactory ensheathing cells promote corticospinal axonal outgrowth by a L1 CAM-dependent mechanism. Glia 2013, 61, 1873–1889. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, H.; Giarratana, A.O.; Theis, T.; Nagaraj, V.; Zhou, X.; Thakker-Varia, S.; Schachner, M.; Alder, J. Single Nucleotide Polymorphism in Cell Adhesion Molecule L1 Affects Learning and Memory in a Mouse Model of Traumatic Brain Injury. Int. J. Mol. Sci. 2024, 25, 3043. https://doi.org/10.3390/ijms25053043
Jiang H, Giarratana AO, Theis T, Nagaraj V, Zhou X, Thakker-Varia S, Schachner M, Alder J. Single Nucleotide Polymorphism in Cell Adhesion Molecule L1 Affects Learning and Memory in a Mouse Model of Traumatic Brain Injury. International Journal of Molecular Sciences. 2024; 25(5):3043. https://doi.org/10.3390/ijms25053043
Chicago/Turabian StyleJiang, Haoyu, Anna O. Giarratana, Thomas Theis, Vini Nagaraj, Xiaofeng Zhou, Smita Thakker-Varia, Melitta Schachner, and Janet Alder. 2024. "Single Nucleotide Polymorphism in Cell Adhesion Molecule L1 Affects Learning and Memory in a Mouse Model of Traumatic Brain Injury" International Journal of Molecular Sciences 25, no. 5: 3043. https://doi.org/10.3390/ijms25053043