
Antioxidant Efficacy of Hwangryunhaedok-tang through Nrf2 and AMPK Signaling Pathway against Neurological Disorders In Vivo and In Vitro

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. HHT Attenuated the Cognitive Impairments in Six-Month-Old 5xFAD Mice
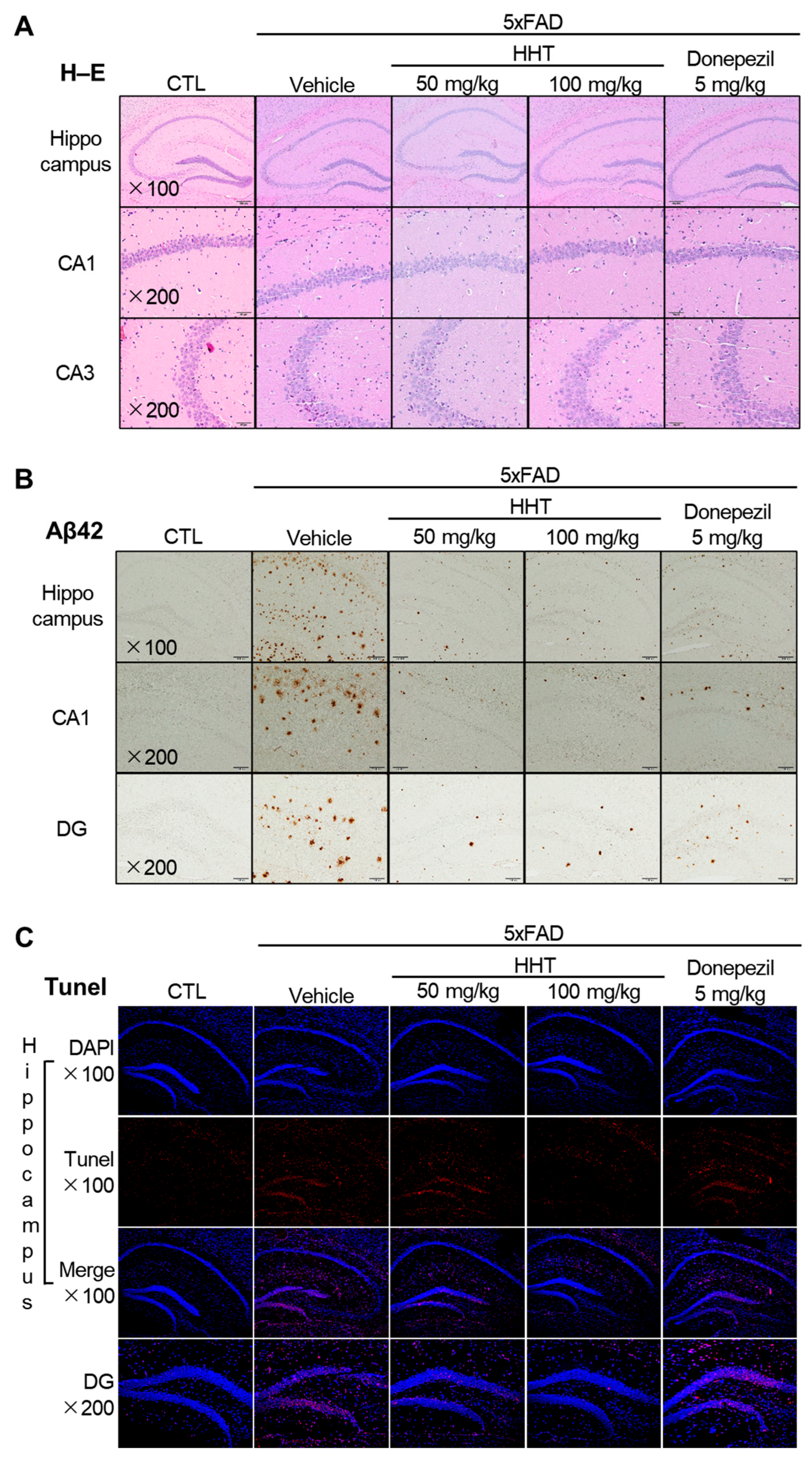
2.2. Protective Effects of HHT on Neuronal Damage in 5xFAD Mice
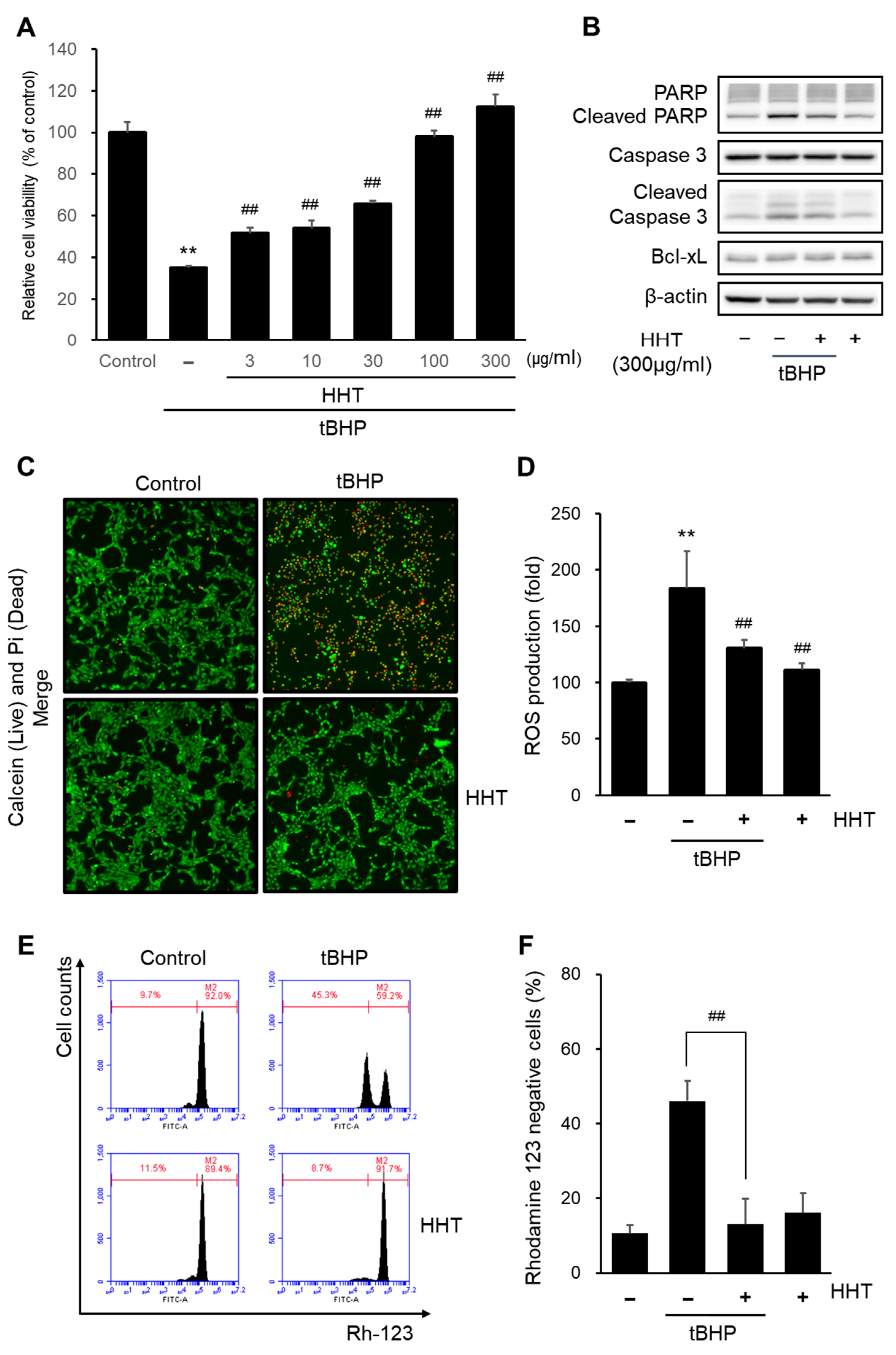
2.3. HHT Attenuates tBHP-Induced Cytotoxicity, ROS Generation, and Mitochondrial Dysfunction in HT-22 Cells
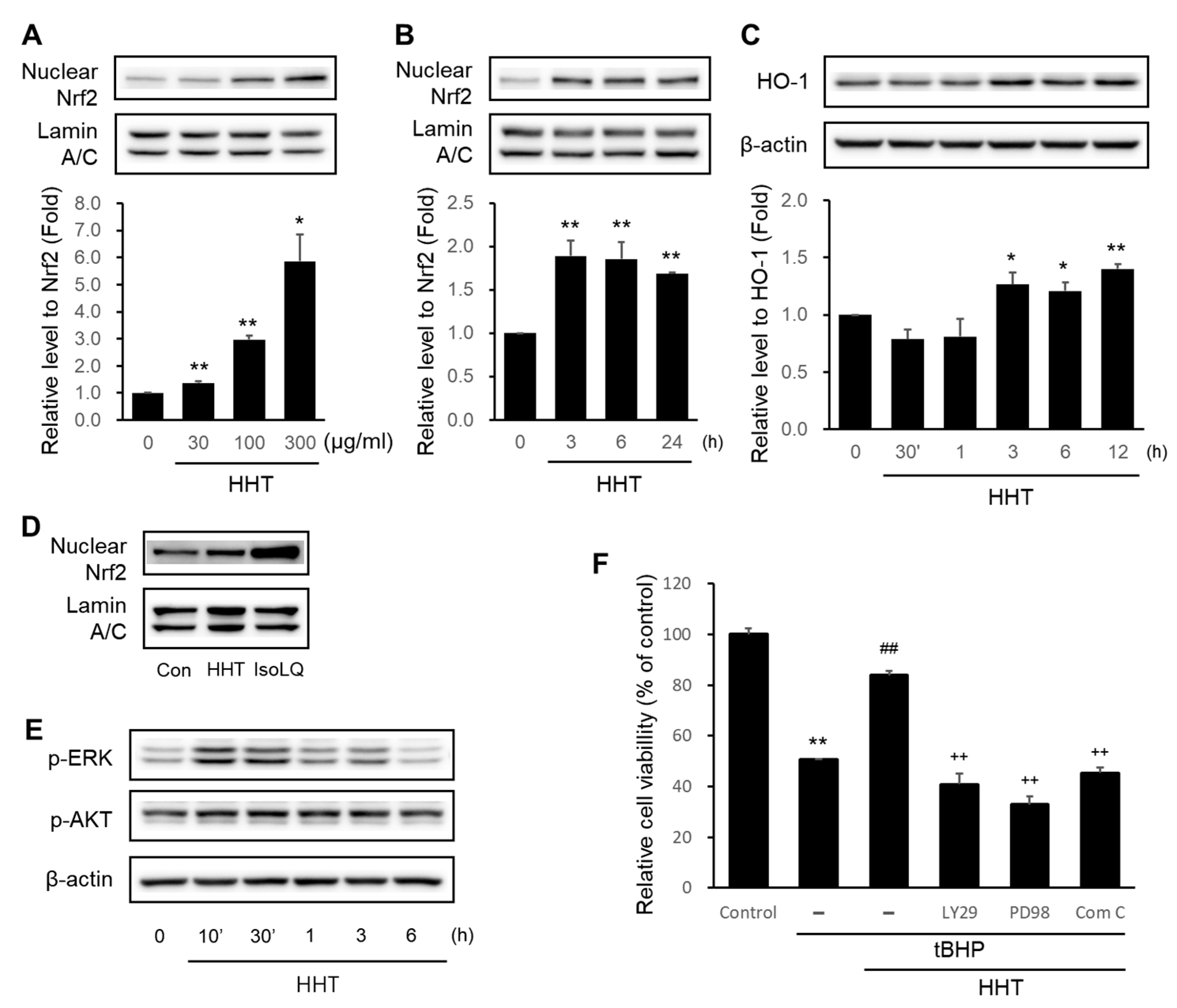
2.4. HHT Alleviates Cytotoxicity by Activating the Nrf2/HO-1 Signaling Pathway
2.5. HHT Attenuates Cytotoxicity through Activation of the AMPK Signaling Pathway
2.6. Baicalein in HHT Indicates Neuroprotective Effects
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Treatment of HHT in 5xFAD Mice
4.3. Morris Water Maze Test
4.4. Passive Avoidance Test
4.5. Tissue Processing
4.6. Hematoxylin and Eosin
4.7. Immunohistochemical Staining (IHC)
4.8. Terminal Deoxynucleotidyl-Transferase-Mediated dUTP Nick End Labeling (TUNEL) Staining
4.9. Cell Culture
4.10. MTT Assay
4.11. Measurement of Reactive Oxygen Species
4.12. Measurement of Mitochondrial Membrane Potential
4.13. Immunoblot Analysis
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid β-peptide (1-42)-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhao, B.; Ratka, A. Oxidative stress and β-amyloid protein in Alzheimer’s disease. Neuromolecular Med. 2011, 13, 223–250. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox. Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Selfridge, J.E.; Lezi, E.; Lu, J.; Swerdlow, R.H. Swerdlow RH. Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol. Dis. 2013, 51, 3–12. [Google Scholar] [CrossRef]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau with Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. 2019, 76, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Poudel, A.; Kim, Y.-K.; Jo, H.-K.; Jung, H.-J. Development of simultaneous analysis for marker constituents in Hwangryunhaedok-tang and its application in commercial herbal formulas. J. Nat. Med. 2013, 67, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Ko, J.W.; Park, S.H.; Cho, Y.K.; Oh, S.R.; Ahn, K.S.; Ryu, J.M.; Kim, J.C.; Seo, C.S.; Shin, I.S. Protective effect of HwangRyunHaeDok-Tang water extract against chronic obstructive pulmonary disease induced by cigarette smoke and lipopolysaccharide in a mouse model. J. Ethnopharmacol. 2017, 200, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Seo, C.-S.; Kim, O.S.; Kim, J.-H.; Shin, H.-K. Simultaneous quantification and antiatherosclerosis effect of the traditional Korean medicine, Hwangryunhaedok-tang. BMC Complement. Altern. Med. 2015, 15, 108. [Google Scholar] [CrossRef]
- Hu, Y.; Hu, Z.; Wang, S.; Dong, X.; Xiao, C.; Jiang, M.; Lv, A.; Zhang, W.; Liu, R. Protective effects of Huang-Lian-Jie-Du-Tang and its component group on collagen-induced arthritis in rats. J. Ethnopharmacol. 2013, 150, 1137–1144. [Google Scholar] [CrossRef]
- Wu, W.; He, X.; Xie, S.; Li, B.; Chen, J.; Qu, Y.; Li, B.; Lei, M.; Liu, X. Protective effects of Huang-Lian-Jie-Du-Tang against Aβ25-35-induced memory deficits and oxidative stress in rats. J. Int. Med. Res. 2020, 48, 300060519893859. [Google Scholar] [CrossRef]
- Chen, M.; Wang, P.; Li, T.; Li, L.; Li, J.; Bai, H.; Lei, H.; Ma, Q. Comprehensive analysis of Huanglian Jiedu decoction: Revealing the presence of a self-assembled phytochemical complex in its naturally-occurring precipitate. J. Pharm. Biomed. Anal. 2021, 195, 113820. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lin, G.; Zuo, Z. Pharmacological effects and pharmacokinetics properties of Radix Scutellariae and its bioactive flavones. Biopharm. Drug Dispos. 2011, 32, 427–445. [Google Scholar] [CrossRef]
- He, X.; Wei, Z.; Zhou, E.; Chen, L.; Kou, J.; Wang, J.; Yang, Z. Baicalein attenuates inflammatory responses by suppressing TLR4 mediated NF-κB and MAPK signaling pathways in LPS-induced mastitis in mice. Int. Immunopharmacol. 2015, 28, 470–476. [Google Scholar] [CrossRef]
- Liang, W.; Huang, X.; Chen, W. The Effects of Baicalin and Baicalein on Cerebral Ischemia: A Review. Aging Dis. 2017, 8, 850–867. [Google Scholar] [CrossRef]
- Sowndhararajan, K.; Deepa, P.; Kim, M.; Park, S.J.; Kim, S. Baicalein as a potent neuroprotective agent: A review. Biomed. Pharmacother. 2017, 95, 1021–1032. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Liu, J.Q.; Zhao, X.T.; Qin, F.Y.; Zhou, J.W.; Ding, F.; Zhou, G.; Zhang, X.S.; Zhang, Z.H.; Li, Z.B. Isoliquiritigenin mitigates oxidative damage after subarachnoid hemorrhage in vivo and in vitro by regulating Nrf2-dependent Signaling Pathway via Targeting of SIRT1. Phytomedicine 2022, 105, 154262. [Google Scholar] [CrossRef]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Mo, C.; Wang, L.; Zhang, J.; Numazawa, S.; Tang, H.; Tang, X.; Han, X.J.; Li, J.; Yang, M.; Wang, Z.; et al. The crosstalk between Nrf2 and AMPK signal pathways is important for the anti-inflammatory effect of berberine in LPS-stimulated macrophages and endotoxin-shocked mice. Antioxid. Redox Signal. 2014, 20, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, X.; Jiang, A.; Li, X.; Chang, W.; Chen, J.; Ye, F. Metformin alleviates lead-induced mitochondrial fragmentation via AMPK/Nrf2 activation in SH-SY5Y cells. Redox Biol. 2020, 36, 101626. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Shen, J.; Xiao, J.; Chen, F.; Wang, M. Neuroprotective effect of cajaninstilbene acid against cerebral ischemia and reperfusion damages by activating AMPK/Nrf2 pathway. J. Adv. Res. 2020, 34, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.-C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging. 2012, 33, 196.e29–196.e40. [Google Scholar] [CrossRef]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Dejanovic, B.; Huntley, M.A.; De Mazière, A.; Meilandt, W.J.; Wu, T.; Srinivasan, K.; Jiang, Z.; Gandham, V.; Friedman, B.A.; Ngu, H.; et al. Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 2018, 100, 1322–1336.e7. [Google Scholar] [CrossRef]
- Sun, L.M.; Zhu, B.J.; Cao, H.T.; Zhang, X.Y.; Zhang, Q.C.; Xin, G.Z.; Pan, L.M.; Liu, L.F.; Zhu, H.X. Explore the effects of Huang-Lian-Jie-Du-Tang on Alzheimer’s disease by UPLC-QTOF/MS-based plasma metabolomics study. J. Pharm. Biomed. Anal. 2018, 151, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Durairajan, S.S.K.; Iyaswamy, A.; Shetty, S.G.; Kammella, A.K.; Malampati, S.; Shang, W.; Yang, C.; Song, J.; Chung, S.; Huang, J.; et al. A modified formulation of Huanglian-Jie-Du-Tang reduces memory impairments and β-amyloid plaques in a triple transgenic mouse model of Alzheimer’s disease. Sci. Rep. 2017, 7, 6238. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging. 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Wang, Z. Oxidative stress in carcinogenesis. Curr. Opin. Toxicol. 2018, 7, 116–121. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Arun, S.; Liu, L.; Donmez, G. Mitochondrial Biology and Neurological Diseases. Curr. Neuropharmacol. 2016, 14, 143–154. [Google Scholar] [CrossRef]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Elmlinger, M.W.; Kriebel, M.; Ziegler, D. Neuroprotective and anti-oxidative effects of the hemodialysate actovegin on primary rat neurons in vitro. Neuromolecular Med. 2011, 13, 266–274. [Google Scholar] [CrossRef]
- Zhao, K.; Luo, G.; Giannelli, S.; Szeto, H.H. Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochem. Pharmacol. 2005, 70, 1796–1806. [Google Scholar] [CrossRef]
- Liu, J.; Li, L.; Suo, W.Z. HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci. 2009, 84, 267–271. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, L.; Li, X.; Zhang, L.; Lv, J.; Guo, X.; Chen, H.; Zhao, T. Neuroprotective effects of an Nrf2 agonist on high glucose-induced damage in HT22 cells. Biol. Res. 2019, 52, 53. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Flohe, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef]
- Bahn, G.; Jo, D.G. Therapeutic Approaches to Alzheimer’s Disease Through Modulation of NRF2. Neuromolecular Med. 2019, 21, 1–11. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef]
- Choi, R.J.; Cheng, M.S.; Kim, Y.S. Desoxyrhapontigenin up-regulates Nrf2-mediated heme oxygenase-1 expression in macrophages and inflammatory lung injury. Redox Biol. 2014, 2, 504–512. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Petsouki, E.; Cabrera, S.N.S.; Heiss, E.H. AMPK and NRF2: Interactive players in the same team for cellular homeostasis? Free Radic. Biol. Med. 2022, 190, 75–93. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, J.; Hölscher, C. Therapeutic Potential of Baicalein in Alzheimer’s Disease and Parkinson’s Disease. CNS Drugs 2017, 31, 639–652. [Google Scholar] [CrossRef]
- Tsai, K.L.; Hung, C.H.; Chan, S.H.; Shih, J.Y.; Cheng, Y.H.; Tsai, Y.J.; Lin, H.C.; Chu, P.M. Baicalein protects against oxLDL-caused oxidative stress and inflammation by modulation of AMPK-alpha. Oncotarget 2016, 7, 72458–72468. [Google Scholar] [CrossRef]
- de Oliveira, M.R.; Nabavi, S.F.; Habtemariam, S.; Erdogan Orhan, I.; Daglia, M.; Nabavi, S.M. The effects of baicalein and baicalin on mitochondrial function and dynamics: A review. Pharmacol. Res. 2015, 100, 296–308. [Google Scholar] [CrossRef]
- Fang, J.; Wang, H.; Zhou, J.; Dai, W.; Zhu, Y.; Zhou, Y.; Wang, X.; Zhou, M. Baicalin provides neuroprotection in traumatic brain injury mice model through Akt/Nrf2 pathway. Drug Des. Devel Ther. 2018, 12, 2497–2508. [Google Scholar] [CrossRef]
- Liu, M.; Chen, F.; Sha, L.; Wang, S.; Tao, L.; Yao, L.; He, M.; Yao, Z.; Liu, H.; Zhu, Z.; et al. (-)-Epigallocatechin-3-gallate ameliorates learning and memory deficits by adjusting the balance of TrkA/p75NTR signaling in APP/PS1 transgenic mice. Mol. Neurobiol. 2014, 49, 1350–1363. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, S.-J.; Lee, W.-Y.; Bak, S.B.; Lee, S.J.; Hwang, S.-J.; Kim, G.-W.; Koo, B.-S.; Park, S.-D.; Yoo, H.-H.; Kim, C.-O.; et al. Antioxidant Efficacy of Hwangryunhaedok-tang through Nrf2 and AMPK Signaling Pathway against Neurological Disorders In Vivo and In Vitro. Int. J. Mol. Sci. 2024, 25, 2313. https://doi.org/10.3390/ijms25042313
Bae S-J, Lee W-Y, Bak SB, Lee SJ, Hwang S-J, Kim G-W, Koo B-S, Park S-D, Yoo H-H, Kim C-O, et al. Antioxidant Efficacy of Hwangryunhaedok-tang through Nrf2 and AMPK Signaling Pathway against Neurological Disorders In Vivo and In Vitro. International Journal of Molecular Sciences. 2024; 25(4):2313. https://doi.org/10.3390/ijms25042313
Chicago/Turabian StyleBae, Su-Jin, Won-Yung Lee, Seon Been Bak, Seung Jin Lee, Su-Jin Hwang, Geun-Woo Kim, Byung-Soo Koo, Sun-Dong Park, Hye-Hyun Yoo, Choon-Ok Kim, and et al. 2024. "Antioxidant Efficacy of Hwangryunhaedok-tang through Nrf2 and AMPK Signaling Pathway against Neurological Disorders In Vivo and In Vitro" International Journal of Molecular Sciences 25, no. 4: 2313. https://doi.org/10.3390/ijms25042313