
KCNQ1 p.D446E Variant as a Risk Allele for Arrhythmogenic Phenotypes: Electrophysiological Characterization Reveals a Complex Phenotype Affecting the Slow Delayed Rectifier Potassium Current (IKs) Voltage Dependence by Causing a Hyperpolarizing Shift and a Lack of Response to Protein Kinase A Activation
 , , ,
, , ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
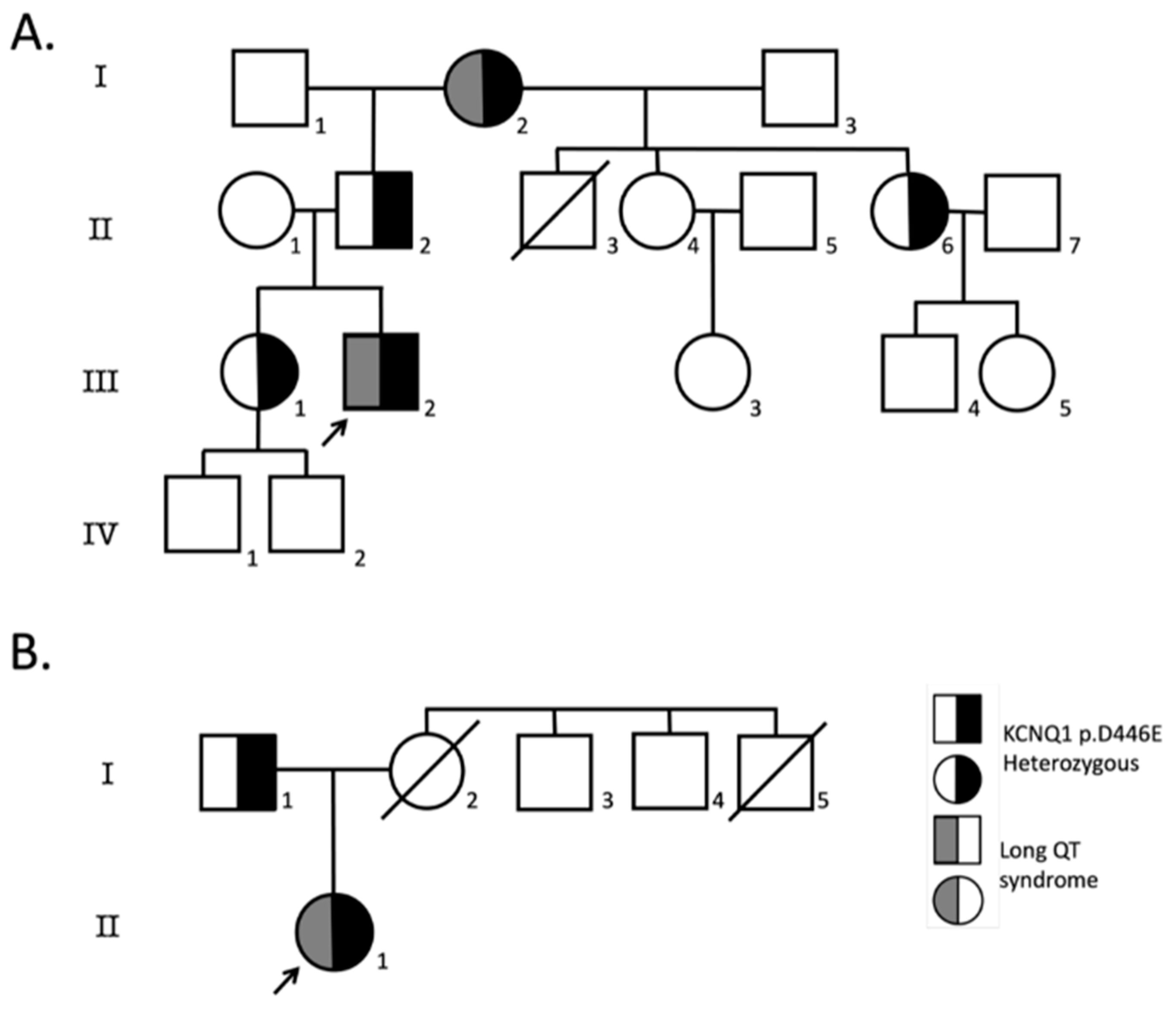
2.1. Patients
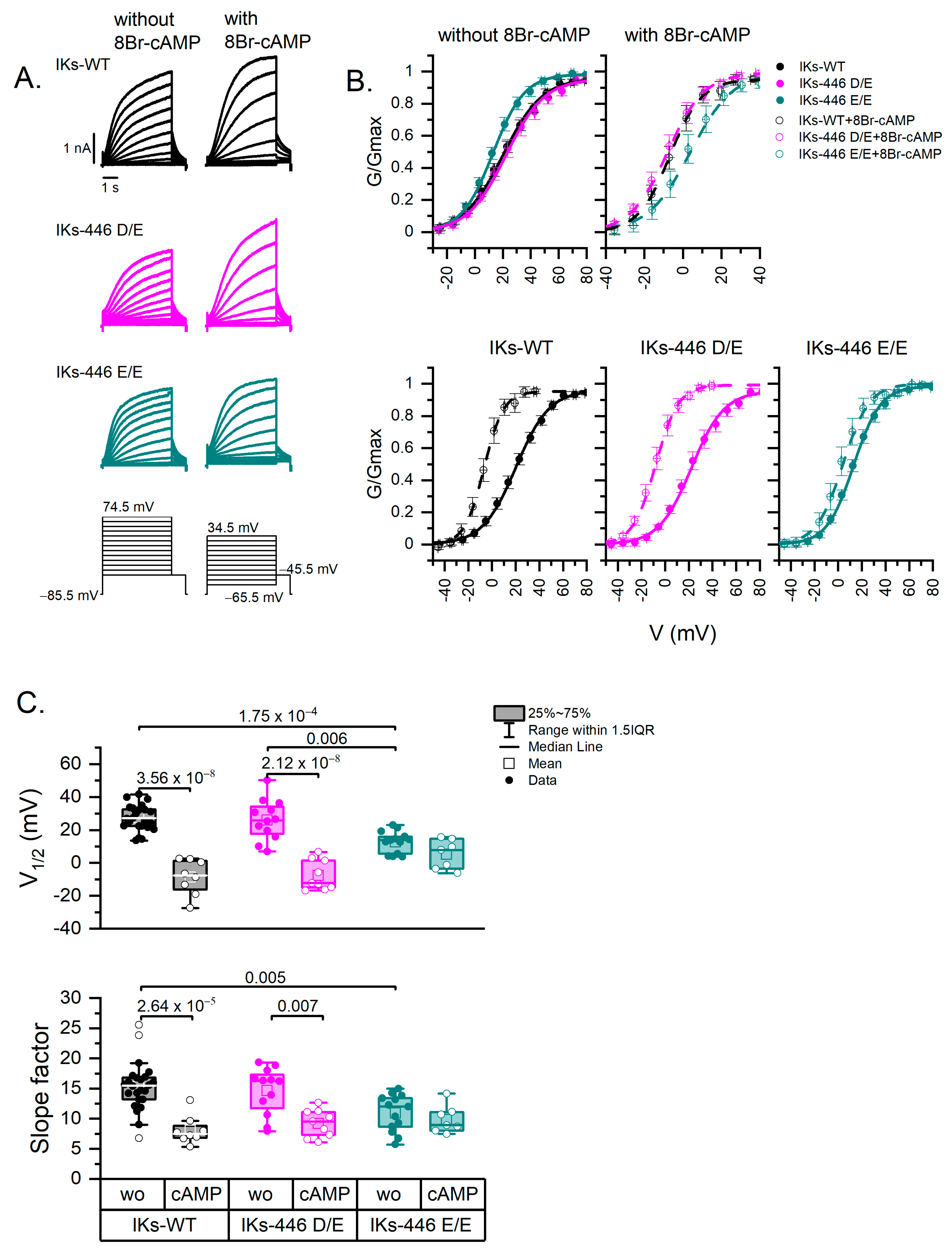
2.2. The Homozygous KCNQ1-446 E/E Channel Left-Shifts IKs Voltage Dependence and Impairs Its Response to PKA Activation
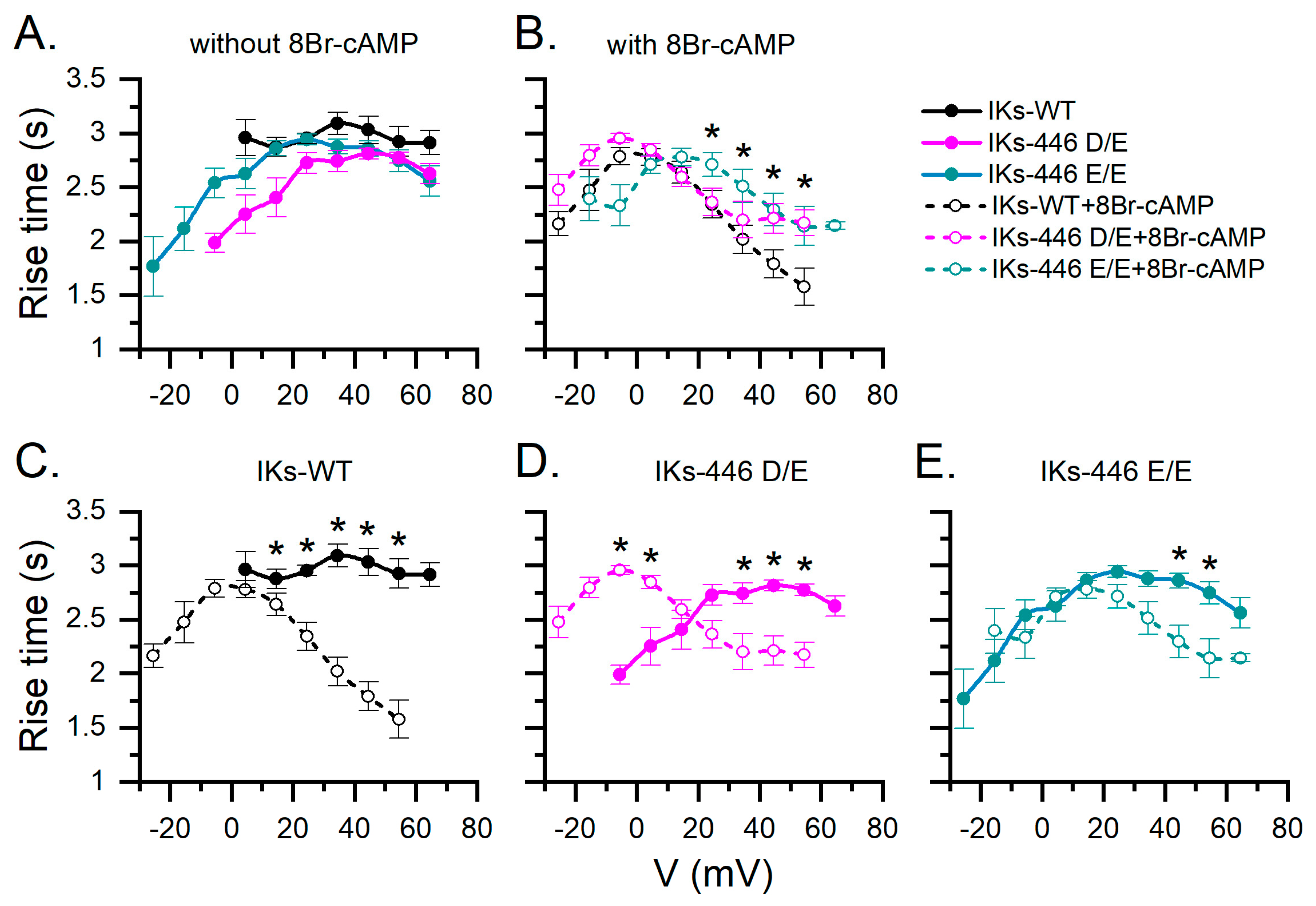
2.3. KCNQ1-446 E/E Slows Activation Time of IKs in Response to 8Br-cAMP and Extends Its Activation Voltage Range to Negative Values
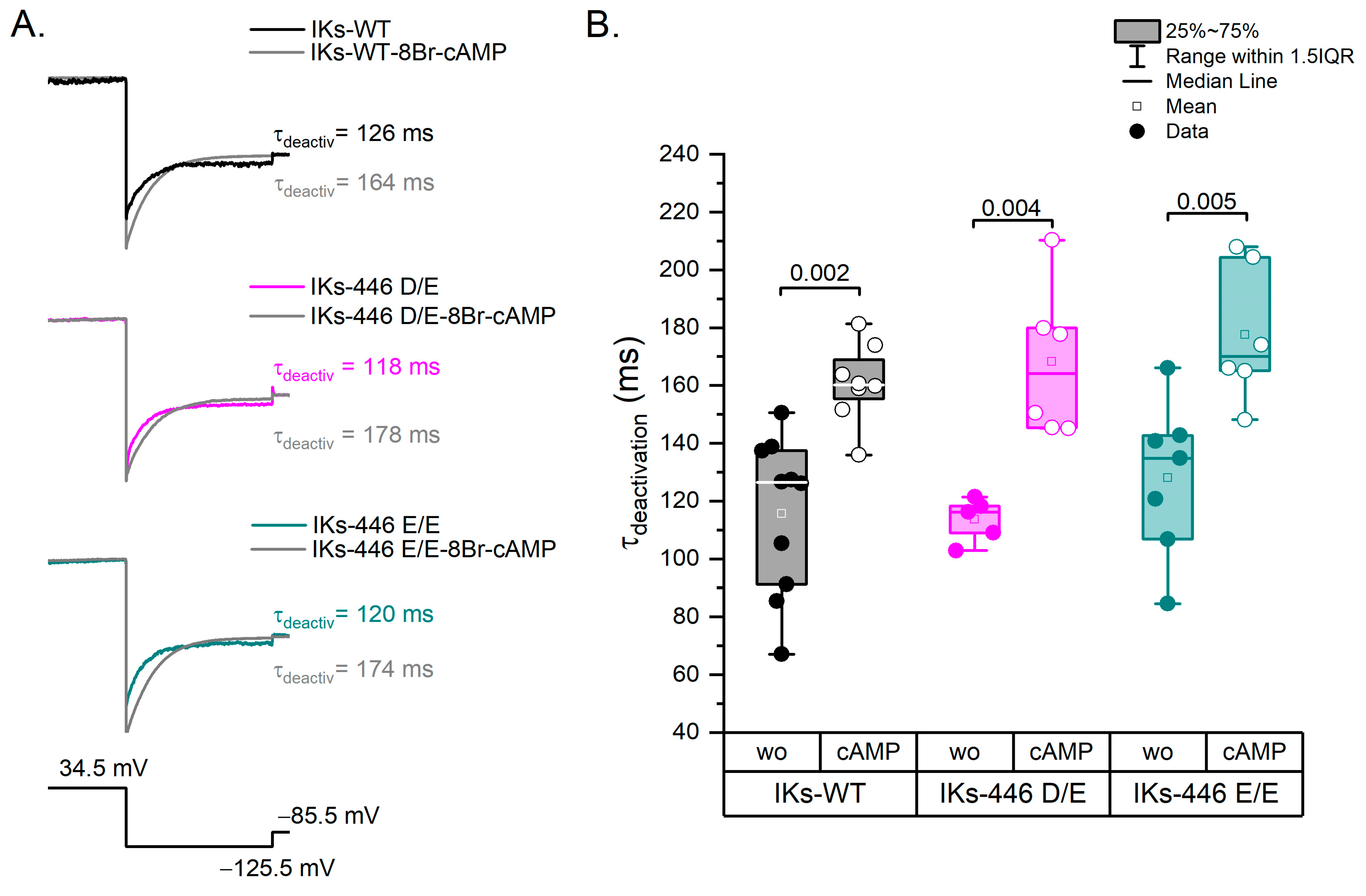
2.4. The KCNQ1 D446E Variant Does Not Affect IKs Deactivation Rate
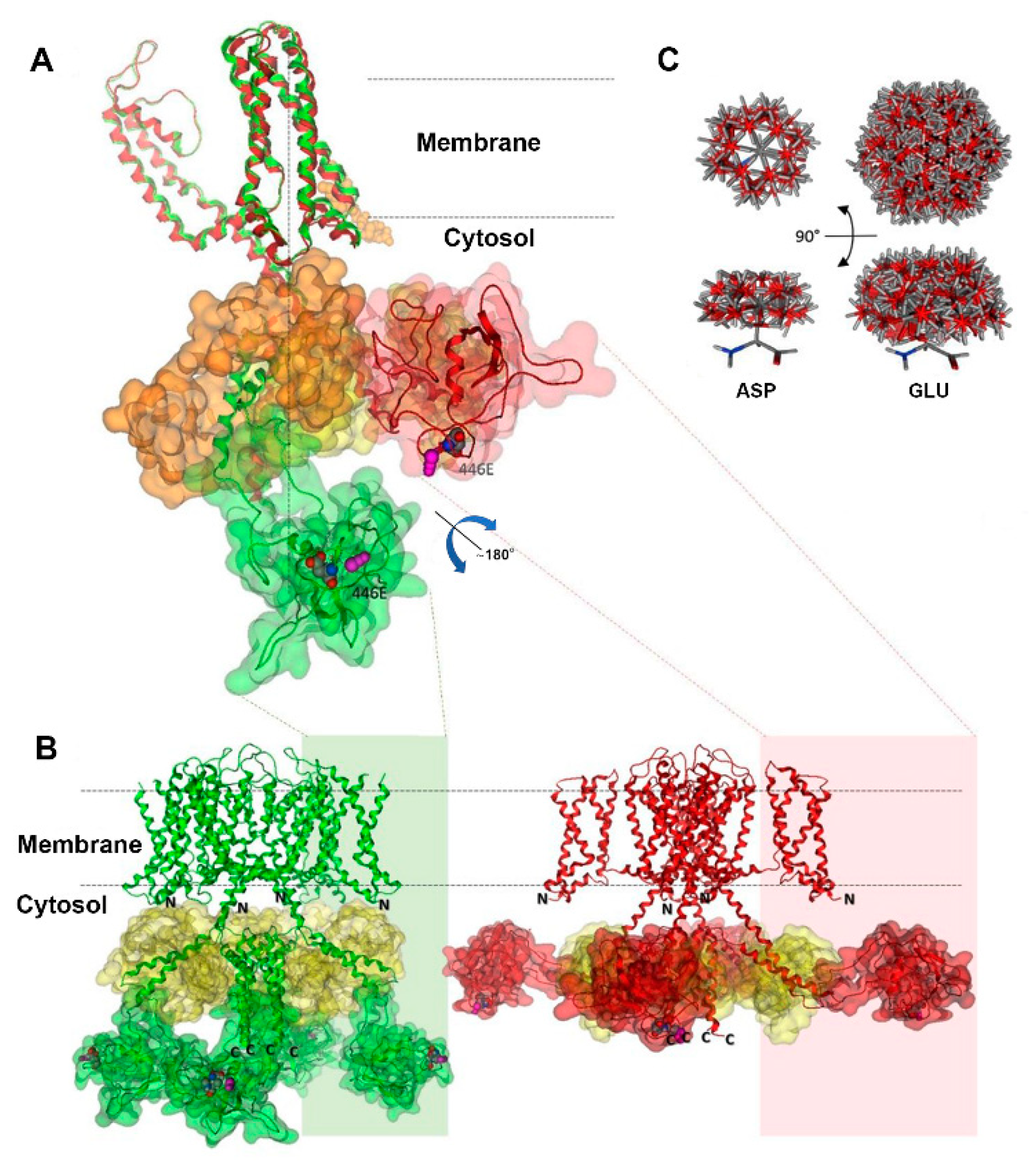
2.5. Kv7.1-D446E Modeling
3. Discussion
3.1. KCNQ1-p.D446E Left-Shifts the IKs Activation Curve Suggesting a Possible Role in Early Repolarization
3.2. KCNQ1-p.D446E Impairs the IKs Response to 8Br-cAMP (Loss of Function) Suggesting a Role in LQTS
3.3. Kv7.1-446E Modeling Predicts Slower Transition to the Stabilized Open State of the Channel
4. Materials and Methods
4.1. Subjects
4.2. Molecular Diagnosis
4.3. Site-Directed Mutagenesis
4.4. Cell Culture and Transfection
4.5. Electrophysiology
4.5.1. Genotypes
4.5.2. Recordings
4.5.3. Analysis
4.6. Statistical Analysis
4.7. Protein Modeling of KCNQ1 (Kv7.1)-D446E
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adler, A.; Novelli, V.; Amin, A.S.; Abiusi, E.; Care, M.; Nannenberg, E.A.; Feilotter, H.; Amenta, S.; Mazza, D.; Bikker, H.; et al. An International, Multicentered, Evidence-Based Reappraisal of Genes Reported to Cause Congenital Long QT Syndrome. Circulation 2020, 141, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Policarová, M.; Novotný, T.; Bébarová, M. Impaired Adrenergic/Protein Kinase A Response of Slow Delayed Rectifier Potassium Channels as a Long QT Syndrome Motif: Importance and Unknowns. Can. J. Cardiol. 2019, 35, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, T.; Jian, Z.; Horvath, B.; Khabbaz, S.; Izu, L.T.; Chen-Izu, Y. Beta-Adrenergic Stimulation Reverses the IKr–IKs Dominant Pattern during Cardiac Action Potential. Pflug. Arch 2014, 466, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Schwartz, P.J.; Napolitano, C.; Bianchi, L.; Dennis, A.; De Fusco, M.; Brown, A.M.; Casari, G. A Recessive Variant of the Romano-Ward Long-QT Syndrome? Circulation 1998, 97, 2420–2425. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Nauffal, V.; Morrill, V.N.; Jurgens, S.J.; Choi, S.H.; Hall, A.W.; Weng, L.-C.; Halford, J.L.; Austin-Tse, C.; Haggerty, C.M.; Harris, S.L.; et al. Monogenic and Polygenic Contributions to QTc Prolongation in the Population. Circulation 2022, 145, 1524–1533. [Google Scholar] [CrossRef]
- Ingles, J.; Semsarian, C. Time to Rethink the Genetic Architecture of Long QT Syndrome. Circulation 2020, 141, 440–443. [Google Scholar] [CrossRef]
- Pandit, M.; Finn, C.; Tahir, U.; Frishman, W.H. Congenital Long QT Syndrome: A Review of Genetic and Pathophysiologic Etiologies, Phenotypic Subtypes and Clinical Management. Cardiol. Rev. 2022, 31, 318–324. [Google Scholar] [CrossRef]
- Steffensen, A.B.; Refsgaard, L.; Andersen, M.N.; Vallet, C.; Mujezinovic, A.; Haunsø, S.; Svendsen, J.H.; Olesen, S.-P.; Olesen, M.S.; Schmitt, N. IKs Gain- and Loss-of-Function in Early-Onset Lone Atrial Fibrillation. J. Cardiovasc. Electrophysiol. 2015, 26, 715–723. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Wilde, A.A.M.; Ackerman, M.J. The Genetic Architecture of Long QT Syndrome: A Critical Reappraisal. Trends Cardiovasc. Med. 2018, 28, 453–464. [Google Scholar] [CrossRef]
- Antúnez-Argüelles, E.; Rojo-Domínguez, A.; Arregui-Mena, A.L.; Jacobo-Albavera, L.; Márquez, M.F.; Iturralde-Torres, P.; Villarreal-Molina, M.T. Compound Heterozygous KCNQ1 Mutations (A300T/P535T) in a Child with Sudden Unexplained Death: Insights into Possible Molecular Mechanisms Based on Protein Modeling. Gene 2017, 627, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.; Priori, S.G.; Napolitano, C.; Surewicz, K.A.; Dennis, A.T.; Memmi, M.; Schwartz, P.J.; Brown, A.M. Mechanisms of I(Ks) Suppression in LQT1 Mutants. Am. J. Physiol.-Heart Circ. Physiol. 2000, 279, H3003–H3011. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Zhong, L.; Yan, Z.; Yao, J.; Zhang, Y.; Ye, F.; Huang, Y.; Lai, D.; Yang, W.; Hou, P.; et al. Structural Mechanisms for the Activation of Human Cardiac KCNQ1 Channel by Electro-Mechanical Coupling Enhancers. Proc. Natl. Acad. Sci. USA 2022, 119, e2207067119. [Google Scholar] [CrossRef] [PubMed]
- Rezus, C.; Floria, M.; Moga, V.D.; Sirbu, O.; Dima, N.; Ionescu, S.D.; Ambarus, V. Early Repolarization Syndrome: Electrocardiographic Signs and Clinical Implications. Ann. Noninvasive Electrocardiol. 2014, 19, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Haïssaguerre, M.; Derval, N.; Sacher, F.; Jesel, L.; Deisenhofer, I.; de Roy, L.; Pasquié, J.-L.; Nogami, A.; Babuty, D.; Yli-Mayry, S.; et al. Sudden Cardiac Arrest Associated with Early Repolarization. N. Engl. J. Med. 2008, 358, 2016–2023. [Google Scholar] [CrossRef] [PubMed]
- Tikkanen, J.T.; Anttonen, O.; Junttila, M.J.; Aro, A.L.; Kerola, T.; Rissanen, H.A.; Reunanen, A.; Huikuri, H. V Long-Term Outcome Associated with Early Repolarization on Electrocardiography. N. Engl. J. Med. 2009, 361, 2529–2537. [Google Scholar] [CrossRef] [PubMed]
- Siebermair, J.; Sinner, M.F.; Beckmann, B.-M.; Laubender, R.P.; Martens, E.; Sattler, S.; Fichtner, S.; Estner, H.L.; Kääb, S.; Wakili, R. Early Repolarization Pattern Is the Strongest Predictor of Arrhythmia Recurrence in Patients with Idiopathic Ventricular Fibrillation: Results from a Single Centre Long-Term Follow-up over 20 Years†. EP Eur. 2016, 18, 718–725. [Google Scholar] [CrossRef]
- Bourier, F.; Denis, A.; Cheniti, G.; Lam, A.; Vlachos, K.; Takigawa, M.; Kitamura, T.; Frontera, A.; Duchateau, J.; Pambrun, T.; et al. Early Repolarization Syndrome: Diagnostic and Therapeutic Approach. Front. Cardiovasc. Med. 2018, 5, 169. [Google Scholar] [CrossRef]
- Hancox, J.C.; Du, C.Y.; Butler, A.; Zhang, Y.; Dempsey, C.E.; Harmer, S.C.; Zhang, H. Pro-Arrhythmic Effects of Gain-of-Function Potassium Channel Mutations in the Short QT Syndrome. Philos. Trans. R. Soc. B Biol. Sci. 2023, 378, 20220165. [Google Scholar] [CrossRef]
- Patel, A.; Oommen, T.; Docekal, J.; Harris, D. Early Repolarization Syndrome Leading to Recurrent Cardiac Arrest in a Young Active Duty Patient. Mil. Med. 2023, usad229. [Google Scholar] [CrossRef]
- Kang, P.W.; Westerlund, A.M.; Shi, J.; White, K.M.; Dou, A.K.; Cui, A.H.; Silva, J.R.; Delemotte, L.; Cui, J. Calmodulin Acts as a State-Dependent Switch to Control a Cardiac Potassium Channel Opening. Sci. Adv. 2020, 6, eabd6798. [Google Scholar] [CrossRef]
- Terrenoire, C.; Clancy, C.E.; Cormier, J.W.; Sampson, K.J.; Kass, R.S. Autonomic Control of Cardiac Action Potentials. Circ. Res. 2005, 96, e25–e34. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, J.; Chen, L.; Kass, R.S. Requirement of Subunit Expression for CAMP-Mediated Regulation of a Heart Potassium Channel. Proc. Natl. Acad. Sci. USA 2003, 100, 2122–2127. [Google Scholar] [CrossRef] [PubMed]
- Spätjens, R.L.H.M.G.; Bébarová, M.; Seyen, S.R.M.; Lentink, V.; Jongbloed, R.J.; Arens, Y.H.J.M.; Heijman, J.; Volders, P.G.A. Long-QT Mutation p.K557E-Kv7.1: Dominant-Negative Suppression of IKs, but Preserved CAMP-Dependent up-Regulation. Cardiovasc. Res. 2014, 104, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.; Eldstrom, J.; Westhoff, M.; McAfee, D.; Balse, E.; Fedida, D. CAMP-Dependent Regulation of IKs Single-Channel Kinetics. J. Gen. Physiol. 2017, 149, 781–798. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Moreno, C.; Kotta, M.-C.; Pedrazzini, M.; Crotti, L.; Dagradi, F.; Castelletti, S.; Haugaa, K.H.; Denjoy, I.; Shkolnikova, M.A.; et al. Mutation Location and I Ks Regulation in the Arrhythmic Risk of Long QT Syndrome Type 1: The Importance of the KCNQ1 S6 Region. Eur. Heart J. 2021, 42, 4743–4755. [Google Scholar] [CrossRef]
- Asada, K.; Kurokawa, J.; Furukawa, T. Redox- and Calmodulin-Dependent S-Nitrosylation of the KCNQ1 Channel*. J. Biol. Chem. 2009, 284, 6014–6020. [Google Scholar] [CrossRef]
- Shugg, T.; Johnson, D.E.; Shao, M.; Lai, X.; Witzmann, F.; Cummins, T.R.; Rubart-Von-der Lohe, M.; Hudmon, A.; Overholser, B.R. Calcium/Calmodulin-Dependent Protein Kinase II Regulation of IKs during Sustained β-Adrenergic Receptor Stimulation. Heart Rhythm 2018, 15, 895–904. [Google Scholar] [CrossRef]
- Sun, J.; MacKinnon, R. Structural Basis of Human KCNQ1 Modulation and Gating. Cell 2020, 180, 340–347.e9. [Google Scholar] [CrossRef]
- Lopes, C.M.B.; Remon, J.I.; Matavel, A.; Sui, J.L.; Keselman, I.; Medei, E.; Shen, Y.; Rosenhouse-Dantsker, A.; Logothetis, D.E. Protein Kinase A Modulates PLC-Dependent Regulation and PIP2-Sensitivity of K+ Channels. Channels 2007, 1, 124–134. [Google Scholar] [CrossRef]
- Lundby, A.; Andersen, M.N.; Steffensen, A.B.; Horn, H.; Kelstrup, C.D.; Francavilla, C.; Jensen, L.J.; Schmitt, N.; Thomsen, M.B.; Olsen, J. V In Vivo Phosphoproteomics Analysis Reveals the Cardiac Targets of β-Adrenergic Receptor Signaling. Sci. Signal 2013, 6, rs11. [Google Scholar] [CrossRef] [PubMed]
- González-Garrido, A.; Domínguez-Pérez, M.; Jacobo-Albavera, L.; López-Ramírez, O.; Guevara-Chávez, J.G.; Zepeda-García, O.; Iturralde, P.; Carnevale, A.; Villarreal-Molina, T. Compound Heterozygous KCNQ1 Mutations Causing Recessive Romano–Ward Syndrome: Functional Characterization by Mutant Co-Expression. Front. Cardiovasc. Med. 2021, 8, 625449. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.H. JPCalc, a Software Package for Calculating Liquid Junction Potential Corrections in Patch-Clamp, Intracellular, Epithelial and Bilayer Measurements and for Correcting Junction Potential Measurements. J. Neurosci. Methods 1994, 51, 107–116. [Google Scholar] [CrossRef] [PubMed]
- UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Mackerell, A.D., Jr.; Feig, M.; Brooks, C.L., III. Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Garrido, A.; López-Ramírez, O.; Cerda-Mireles, A.; Navarrete-Miranda, T.; Flores-Arenas, A.I.; Rojo-Domínguez, A.; Arregui, L.; Iturralde, P.; Antúnez-Argüelles, E.; Domínguez-Pérez, M.; et al. KCNQ1 p.D446E Variant as a Risk Allele for Arrhythmogenic Phenotypes: Electrophysiological Characterization Reveals a Complex Phenotype Affecting the Slow Delayed Rectifier Potassium Current (IKs) Voltage Dependence by Causing a Hyperpolarizing Shift and a Lack of Response to Protein Kinase A Activation. Int. J. Mol. Sci. 2024, 25, 953. https://doi.org/10.3390/ijms25020953
González-Garrido A, López-Ramírez O, Cerda-Mireles A, Navarrete-Miranda T, Flores-Arenas AI, Rojo-Domínguez A, Arregui L, Iturralde P, Antúnez-Argüelles E, Domínguez-Pérez M, et al. KCNQ1 p.D446E Variant as a Risk Allele for Arrhythmogenic Phenotypes: Electrophysiological Characterization Reveals a Complex Phenotype Affecting the Slow Delayed Rectifier Potassium Current (IKs) Voltage Dependence by Causing a Hyperpolarizing Shift and a Lack of Response to Protein Kinase A Activation. International Journal of Molecular Sciences. 2024; 25(2):953. https://doi.org/10.3390/ijms25020953
Chicago/Turabian StyleGonzález-Garrido, Antonia, Omar López-Ramírez, Abel Cerda-Mireles, Thania Navarrete-Miranda, Aranza Iztanami Flores-Arenas, Arturo Rojo-Domínguez, Leticia Arregui, Pedro Iturralde, Erika Antúnez-Argüelles, Mayra Domínguez-Pérez, and et al. 2024. "KCNQ1 p.D446E Variant as a Risk Allele for Arrhythmogenic Phenotypes: Electrophysiological Characterization Reveals a Complex Phenotype Affecting the Slow Delayed Rectifier Potassium Current (IKs) Voltage Dependence by Causing a Hyperpolarizing Shift and a Lack of Response to Protein Kinase A Activation" International Journal of Molecular Sciences 25, no. 2: 953. https://doi.org/10.3390/ijms25020953