
Bidirectional Allosteric Coupling between PIP2 Binding and the Pore of the Oncochannel TRPV6
 , , ,
, , ,
Abstract
:1. Introduction
2. Results
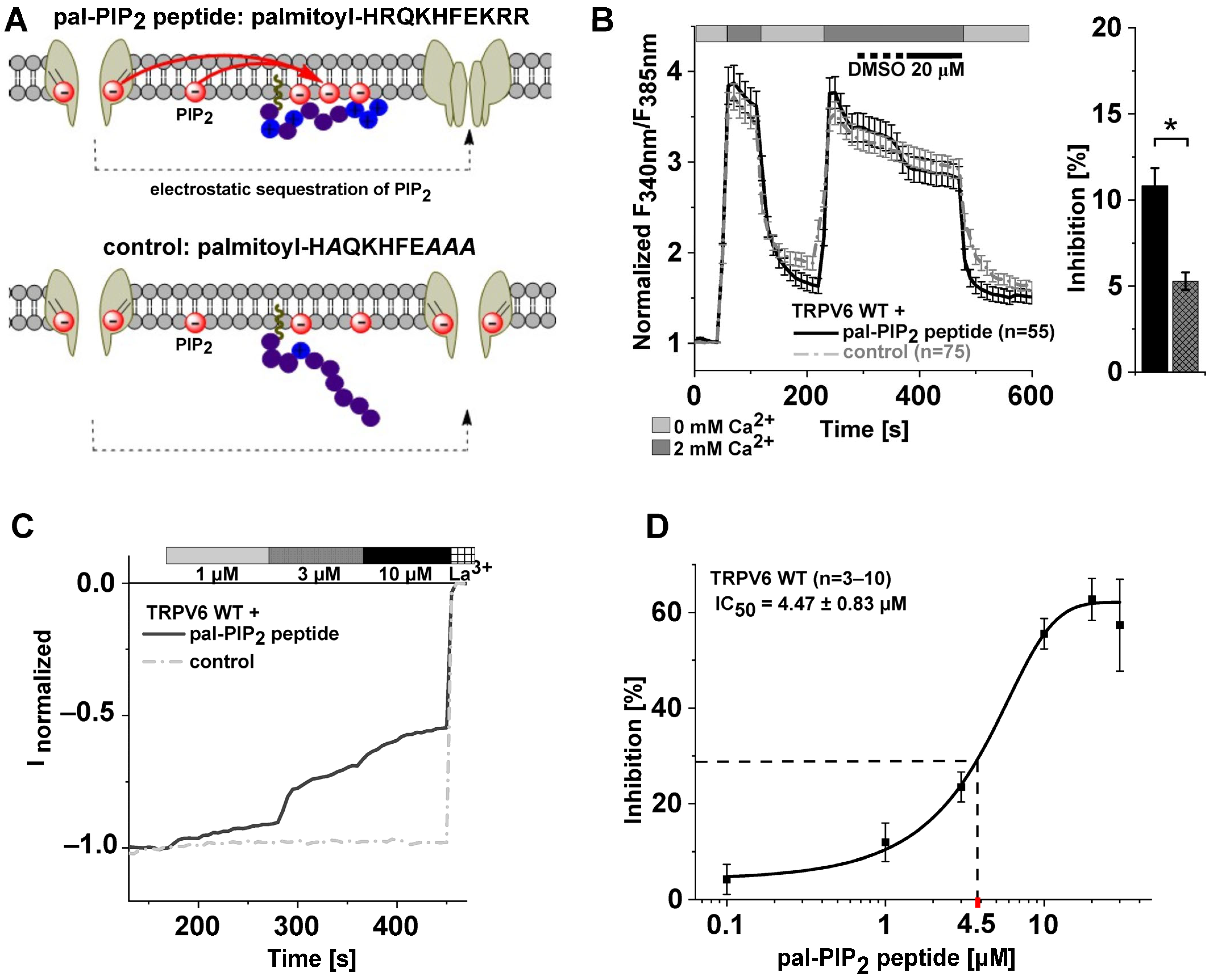
2.1. Electrostatic Sequestration of PIP2 Reduces TRPV6 Currents
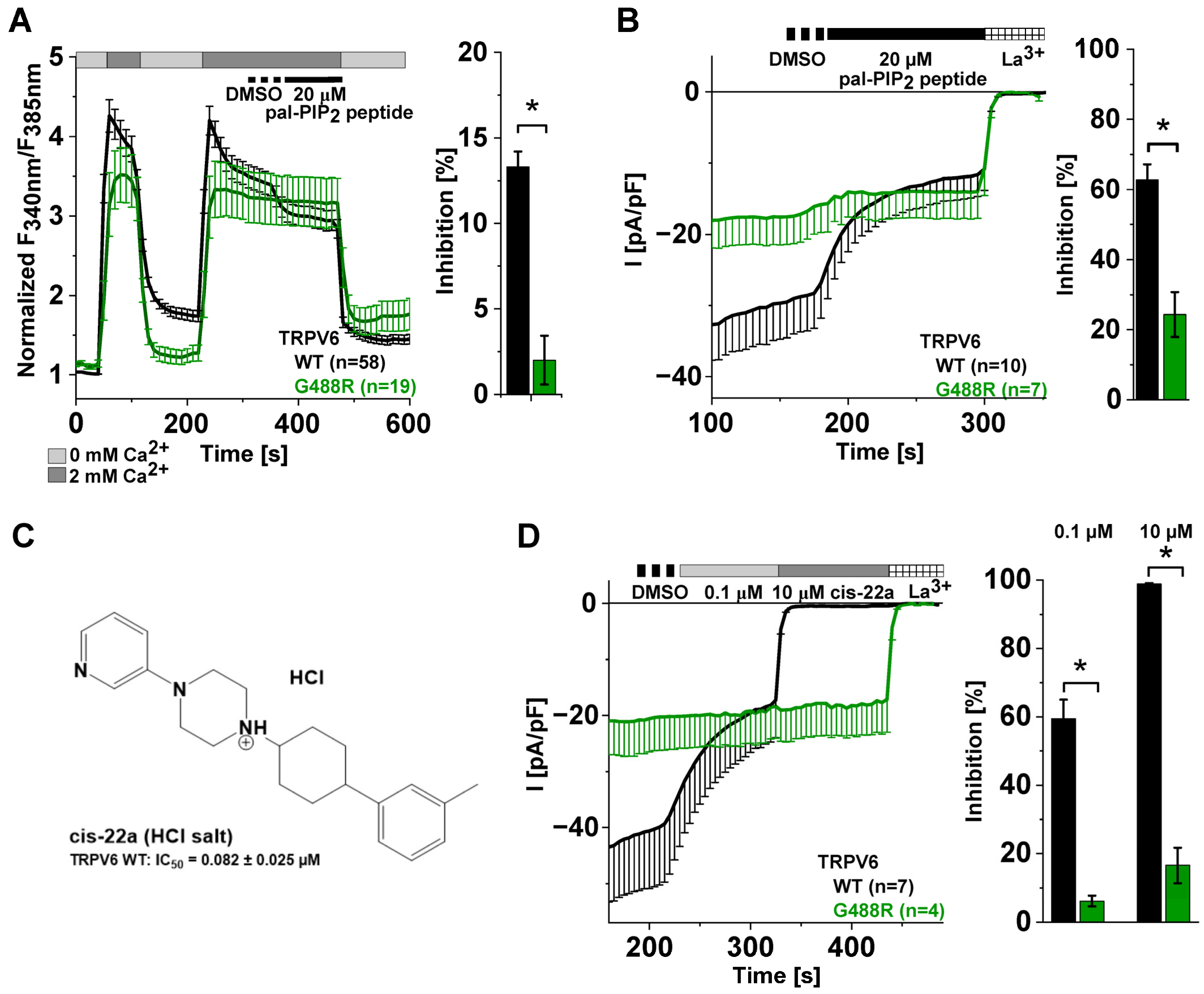
2.2. A Mutation-Based Increase in PIP2 Affinity Counteracts Inhibition by the Pal-PIP2 Peptide
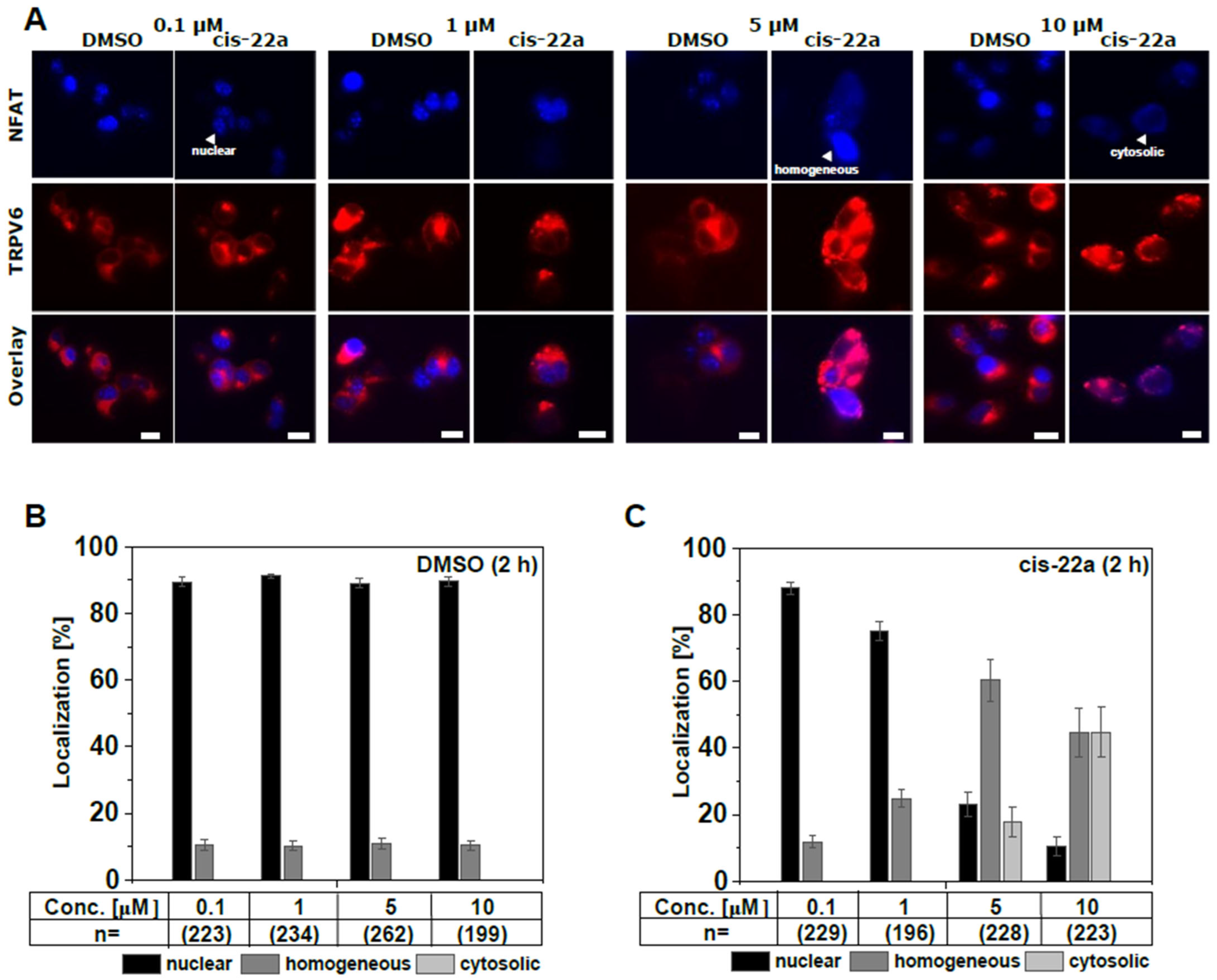
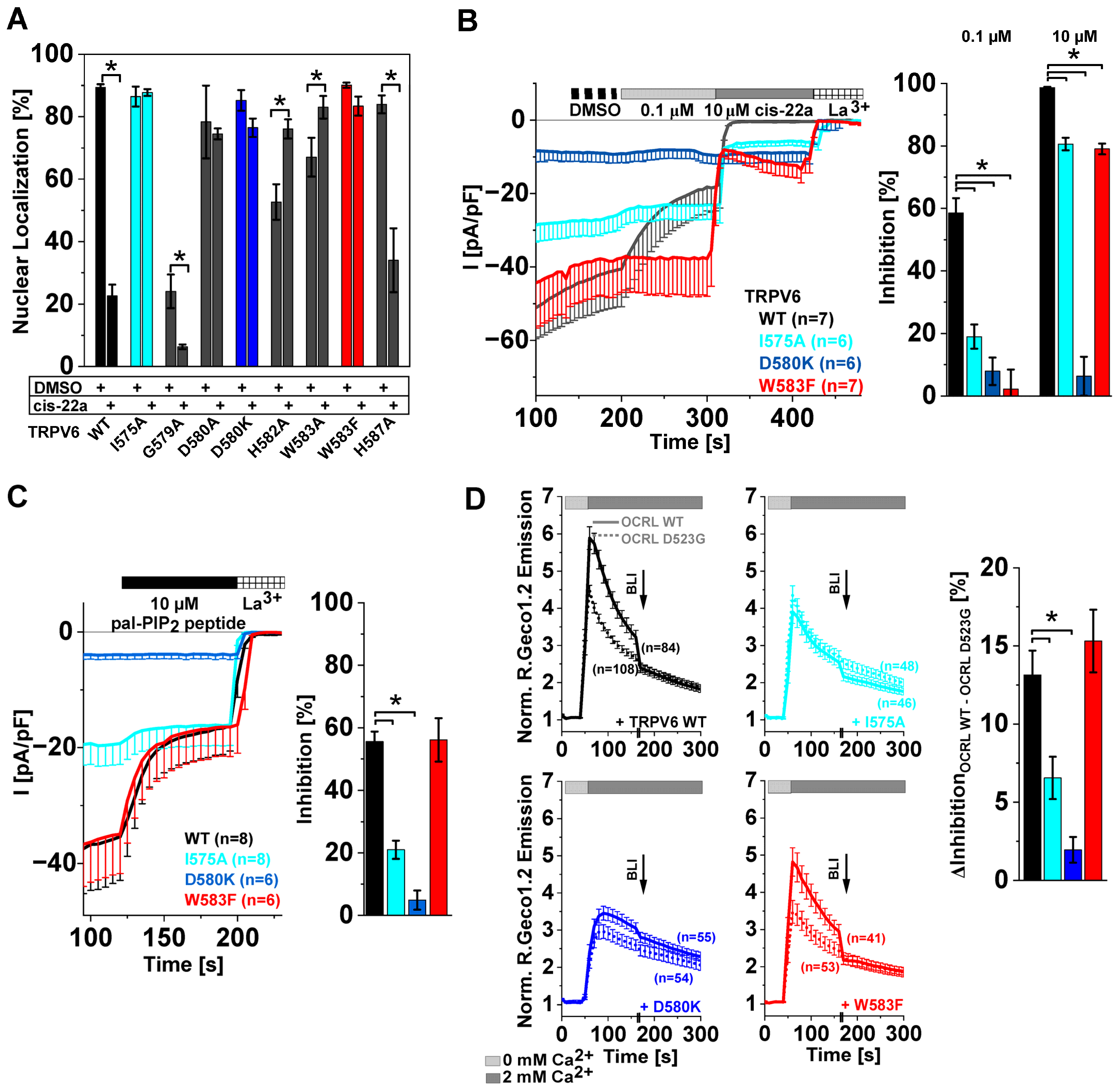
2.3. Inhibition of TRPV6 WT by cis-22a Shifts Constitutive Nuclear NFAT Localization to a Mainly Homogeneous Distribution
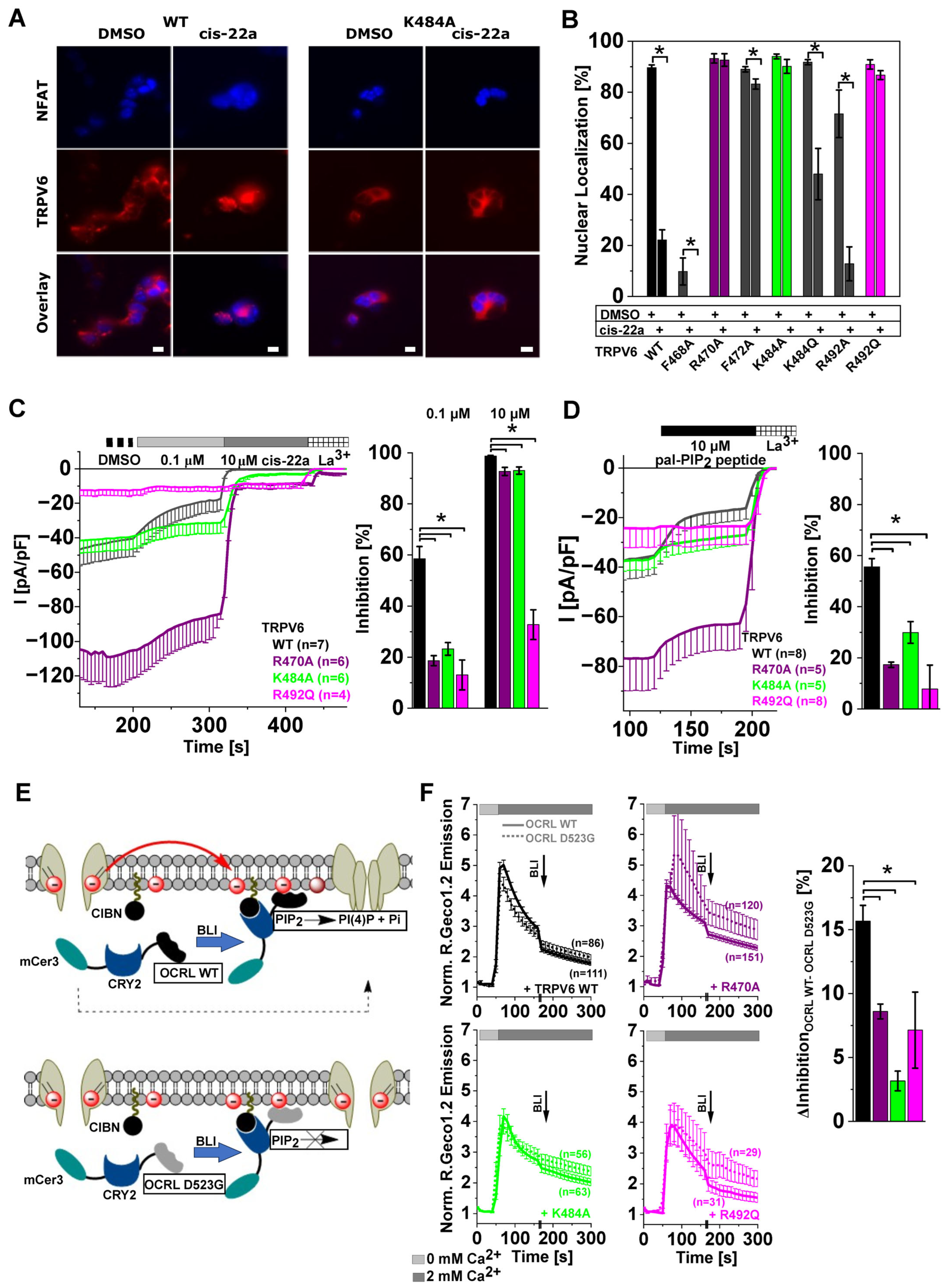
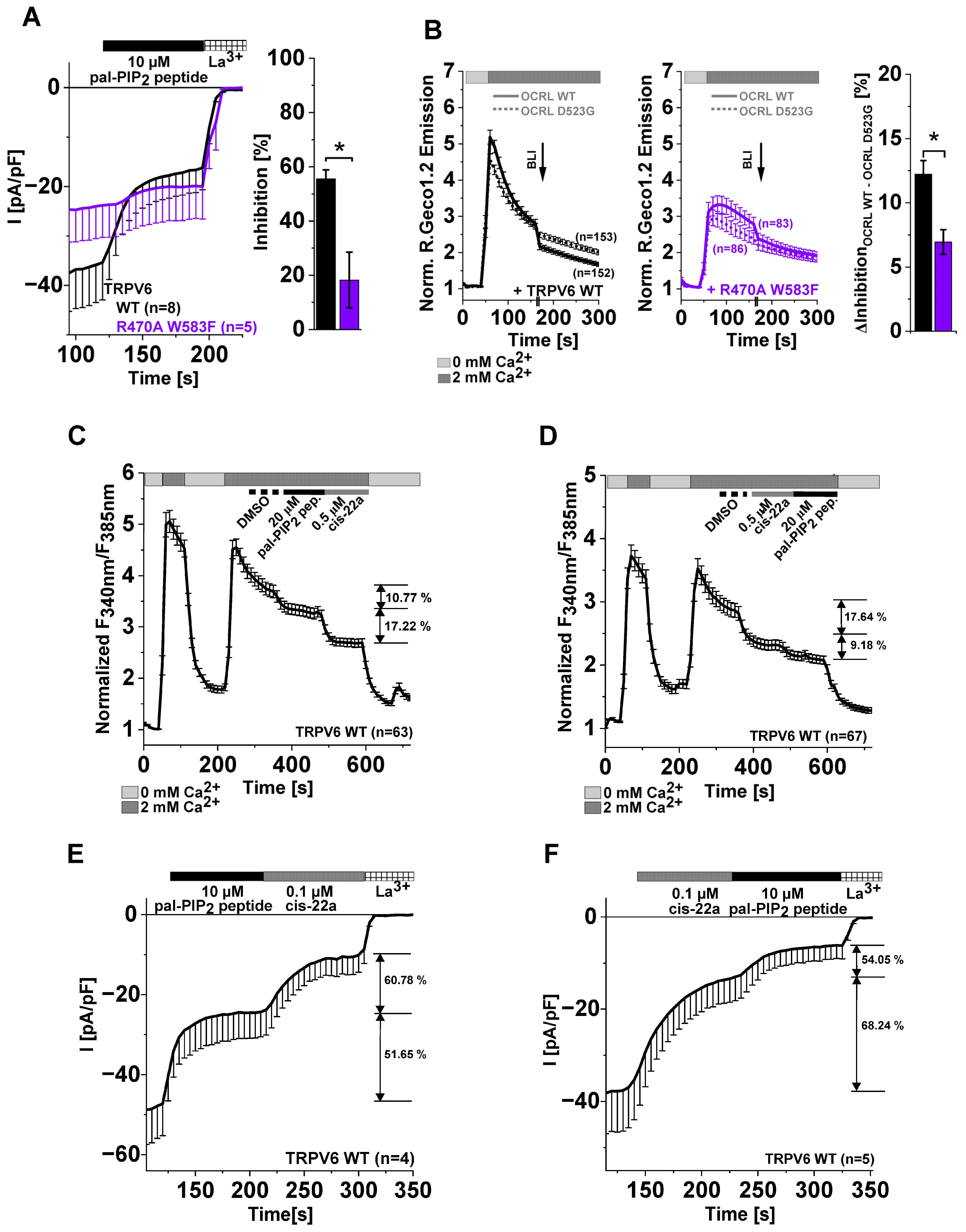
2.4. Site-Directed Mutagenesis in LBS-2 Alters Inhibition by cis-22a and Sensitivity to PIP2 Depletion
2.5. TRPV6 Current Inhibition by cis-22a and PIP2 Depletion Is Sensitive to Site-Directed Mutagenesis within the Ion Permeation Pathway
2.6. Differences in TRPV6 Channel Inhibition by cis-22a and PIP2 Depletion
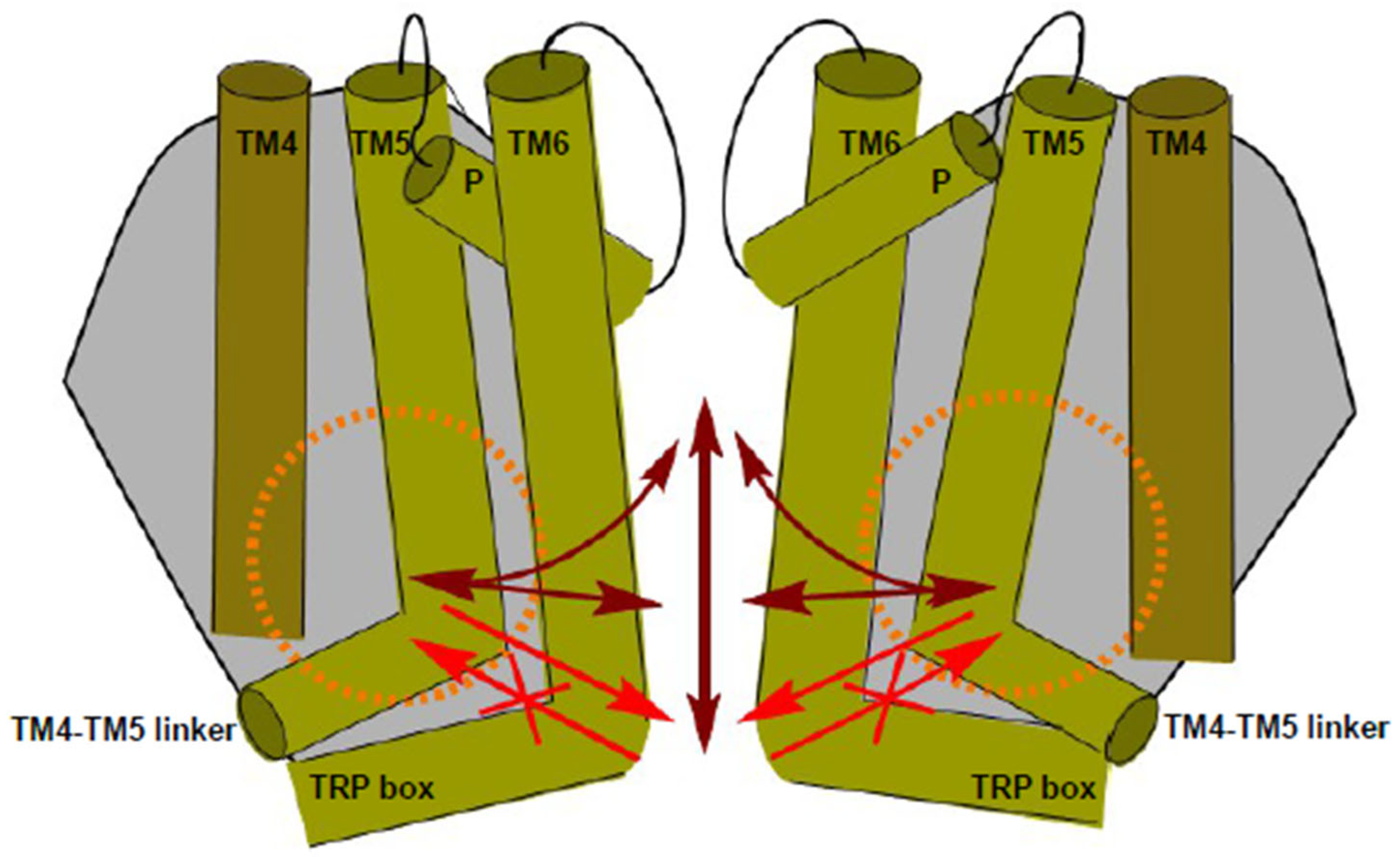
3. Discussion
4. Materials and Methods
4.1. DNA-Constructs and Reagents
4.2. Peptide Synthesis, Purification, and Characterization
4.3. Cell Culture and Transfection
4.4. Electrophysiology
4.5. Fura-2 Measurements
4.6. R.Geco1.2 Based Ca2+ Imaging
4.7. Subcellular Translocation
4.8. NFAT Screen
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Idevall-Hagren, O.; Dickson, E.J.; Hille, B.; Toomre, D.K.; De Camilli, P. Optogenetic control of phosphoinositide metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, E2316–E2323. [Google Scholar] [CrossRef]
- Katan, M.; Cockcroft, S. Phosphatidylinositol(4,5)bisphosphate: Diverse functions at the plasma membrane. Essays Biochem. 2020, 64, 513–531. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ghosh, C.; Xing, Y.; Sun, Y. Phosphatidylinositol 4,5-bisphosphate in the Control of Membrane Trafficking. Int. J. Biol. Sci. 2020, 16, 2761–2774. [Google Scholar] [CrossRef] [PubMed]
- Suh, B.-C.; Hille, B. PIP2 Is a Necessary Cofactor for Ion Channel Function: How and Why? Annu. Rev. Biophys. 2008, 37, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Huang, Y.; Lu, Y.; Xiong, J.; Wong, C.-O.; Yang, P.; Xia, J.; Chen, D.; Du, G.; Venkatachalam, K.; et al. Drosophila TRPML Forms PI(3,5)P2-activated Cation Channels in Both Endolysosomes and Plasma Membrane. J. Biol. Chem. 2014, 289, 4262–4272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; Xu, H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc. Natl. Acad. Sci. USA 2012, 109, 11384–11389. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-P.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; MacKinnon, R. Structural Basis of Human KCNQ1 Modulation and Gating. Cell 2020, 180, 340–347.e349. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Q.; Qiu, Y.; Li, Z.; Chen, Z.; Jiang, H.; Li, Y.; Yang, H. Migration of PIP2 lipids on voltage-gated potassium channel surface influences channel deactivation. Sci. Rep. 2015, 5, 15079. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, H.; Chen, Y.; Dong, S.; Wang, F.; Wang, S.; Li, G.-L.; Shu, Y.; Xu, F. Structural insights into the lipid and ligand regulation of a human neuronal KCNQ channel. Neuron 2022, 110, 237–247.e234. [Google Scholar] [CrossRef]
- Rohács, T.; Lopes, C.M.B.; Jin, T.; Ramdya, P.P.; Molnár, Z.; Logothetis, D.E. Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc. Natl. Acad. Sci. USA 2003, 100, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.-Z.; Liou, H.-H.; Krishna, U.M.; Falck, J.R.; Huang, C.-L. Structural determinants and specificities for ROMK1-phosphoinositide interaction. Am. J. Physiol. -Ren. Physiol. 2002, 282, F826–F834. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.B.; Tao, X.; MacKinnon, R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 2011, 477, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Ren, F.; Zangerl-Plessl, E.-M.; Heyman, S.; Stary-Weinzinger, A.; Yuan, P.; Nichols, C.G. Structural basis of control of inward rectifier Kir2 channel gating by bulk anionic phospholipids. J. Gen. Physiol. 2016, 148, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liman, E.R. Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc. Natl. Acad. Sci. USA 2003, 100, 15160–15165. [Google Scholar] [CrossRef] [PubMed]
- Vivas, O.; Castro, H.; Arenas, I.; Elías-Viñas, D.; García, D.E. PIP2 hydrolysis is responsible for voltage independent inhibition of CaV2.2 channels in sympathetic neurons. Biochem. Biophys. Res. Commun. 2013, 432, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cha, S.-K.; Sun, T.-J.; Huang, C.-L. PIP2 Activates TRPV5 and Releases Its Inhibition by Intracellular Mg2+. J. Gen. Physiol. 2005, 126, 439–451. [Google Scholar] [CrossRef] [PubMed]
- de Groot, T.; Bindels, R.J.M.; Hoenderop, J.G.J. TRPV5: An ingeniously controlled calcium channel. Kidney Int. 2008, 74, 1241–1246. [Google Scholar] [CrossRef]
- Thyagarajan, B.; Lukacs, V.; Rohacs, T. Hydrolysis of Phosphatidylinositol 4,5-Bisphosphate Mediates Calcium-induced Inactivation of TRPV6 Channels. J. Biol. Chem. 2008, 283, 14980–14987. [Google Scholar] [CrossRef]
- Thyagarajan, B.; Benn, B.S.; Christakos, S.; Rohacs, T. Phospholipase C-Mediated Regulation of Transient Receptor Potential Vanilloid 6 Channels: Implications in Active Intestinal Ca2+ Transport. Mol. Pharmacol. 2009, 75, 608. [Google Scholar] [CrossRef]
- Yelshanskaya, M.V.; Nadezhdin, K.D.; Kurnikova, M.G.; Sobolevsky, A.I. Structure and function of the calcium-selective TRP channel TRPV6. J. Physiol. 2021, 599, 2673–2697. [Google Scholar] [CrossRef] [PubMed]
- Vennekens, R.; Hoenderop, J.G.J.; Prenen, J.; Stuiver, M.; Willems, P.H.G.M.; Droogmans, G.; Nilius, B.; Bindels, R.J.M. Permeation and Gating Properties of the Novel Epithelial Ca2+ Channel. J. Biol. Chem. 2000, 275, 3963–3969. [Google Scholar] [CrossRef] [PubMed]
- Owsianik, G.; Talavera, K.; Voets, T.; Nilius, B. Permeation and selectivity of TRP channels. Annu. Rev. Physiol. 2006, 68, 685–717. [Google Scholar] [CrossRef] [PubMed]
- Khattar, V.; Wang, L.; Peng, J.-B. Calcium selective channel TRPV6: Structure, function, and implications in health and disease. Gene 2022, 817, 146192. [Google Scholar] [CrossRef] [PubMed]
- Voets, T.; Janssens, A.; Prenen, J.; Droogmans, G.; Nilius, B. Mg2+-dependent Gating and Strong Inward Rectification of the Cation Channel TRPV6. J. Gen. Physiol. 2003, 121, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Hoenderop, J.G.J.; van der Kemp, A.W.C.M.; Hartog, A.; van de Graaf, S.F.J.; van Os, C.H.; Willems, P.H.G.M.; Bindels, R.J.M. Molecular Identification of the Apical Ca2+ Channel in 1,25-Dihydroxyvitamin D3-responsive Epithelia. J. Biol. Chem. 1999, 274, 8375–8378. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.-B.; Chen, X.-Z.; Berger, U.V.; Vassilev, P.M.; Tsukaguchi, H.; Brown, E.M.; Hediger, M.A. Molecular Cloning and Characterization of a Channel-like Transporter Mediating Intestinal Calcium Absorption. J. Biol. Chem. 1999, 274, 22739–22746. [Google Scholar] [CrossRef] [PubMed]
- Hoenderop, J.G.J.; Nilius, B.; Bindels, R.J.M. Calcium Absorption Across Epithelia. Physiol. Rev. 2005, 85, 373–422. [Google Scholar] [CrossRef]
- Suzuki, Y.; Landowski, C.P.; Hediger, M.A. Mechanisms and Regulation of Epithelial Ca2+ Absorption in Health and Disease. Annu. Rev. Physiol. 2008, 70, 257–271. [Google Scholar] [CrossRef]
- Suzuki, Y.; Sawada, H.; Tokumasu, T.; Suzuki, S.; Ninomiya, S.; Shirai, M.; Mukai, T.; Saito, C.T.; Nishimura, G.; Tominaga, M. Novel TRPV6 mutations in the spectrum of transient neonatal hyperparathyroidism. J. Physiol. Sci. 2020, 70, 33. [Google Scholar] [CrossRef]
- Yamashita, S.; Mizumoto, H.; Sawada, H.; Suzuki, Y.; Hata, D. TRPV6 Gene Mutation in a Dizygous Twin with Transient Neonatal Hyperparathyroidism. J. Endocr. Soc. 2019, 3, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Chitayat, D.; Sawada, H.; Deardorff, M.A.; McLaughlin, H.M.; Begtrup, A.; Millar, K.; Harrington, J.; Chong, K.; Roifman, M.; et al. TRPV6 Variants Interfere with Maternal-Fetal Calcium Transport through the Placenta and Cause Transient Neonatal Hyperparathyroidism. Am. J. Hum. Genet. 2018, 102, 1104–1114. [Google Scholar] [CrossRef]
- Zhuang, L.; Peng, J.; Tou, L.; Takanaga, H.; Adam, R.M.; Hediger, M.A.; Freeman, M.R. Calcium-selective ion channel, CaT1, is apically localized in gastrointestinal tract epithelia and is aberrantly expressed in human malignancies. Lab. Investig. 2002, 82, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Wissenbach, U.; Niemeyer, B.A.; Fixemer, T.; Schneidewind, A.; Trost, C.; Cavalié, A.; Reus, K.; Meese, E.; Bonkhoff, H.; Flockerzi, V. Expression of CaT-like, a Novel Calcium-selective Channel, Correlates with the Malignancy of Prostate Cancer. J. Biol. Chem. 2001, 276, 19461–19468. [Google Scholar] [CrossRef]
- Landowski, C.P.; Bolanz, K.A.; Suzuki, Y.; Hediger, M.A. Chemical Inhibitors of the Calcium Entry Channel TRPV6. Pharm. Res. 2011, 28, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Wissenbach, U.; Niemeyer, B.; Himmerkus, N.; Fixemer, T.; Bonkhoff, H.; Flockerzi, V. TRPV6 and prostate cancer: Cancer growth beyond the prostate correlates with increased TRPV6 Ca2+ channel expression. Biochem. Biophys. Res. Commun. 2004, 322, 1359–1363. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.M. TRPV6 as A Target for Cancer Therapy. J. Cancer 2020, 11, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Gou, H.; Zhu, J.; Tian, S.; Yu, L. Lidocaine inhibits the invasion and migration of TRPV6-expressing cancer cells by TRPV6 downregulation Corrigendum in /10.3892/ol.2023.13957. Oncol. Lett. 2016, 12, 1164–1170. [Google Scholar] [CrossRef]
- Xue, H.; Wang, Y.; MacCormack, T.J.; Lutes, T.; Rice, C.; Davey, M.; Dugourd, D.; Ilenchuk, T.T.; Stewart, J.M. Inhibition of Transient Receptor Potential Vanilloid 6 channel, elevated in human ovarian cancers, reduces tumour growth in a xenograft model. J. Cancer 2018, 9, 3196–3207. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kim, S.H.; Yang, E.K. RNA interference mediated suppression of TRPV6 inhibits the progression of prostate cancer in vitro by modulating cathepsin B and MMP9 expression. Investig. Clin. Urol. 2021, 62, 447–454. [Google Scholar] [CrossRef]
- Liu, J.-H.; Cao, Y.-M.; Rong, Z.-P.; Ding, J.; Pan, X. Trichostatin A Induces Autophagy in Cervical Cancer Cells by Regulating the PRMT5-STC1-TRPV6-JNK Pathway. Pharmacology 2020, 106, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Bolanz, K.A.; Kovacs, G.G.; Landowski, C.P.; Hediger, M.A. Tamoxifen Inhibits TRPV6 Activity via Estrogen Receptor–Independent Pathways in TRPV6-Expressing MCF-7 Breast Cancer Cells. Mol. Cancer Res. 2009, 7, 2000–2010. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Hirte, H.; Welch, S.; Ilenchuk, T.T.; Lutes, T.; Rice, C.; Fields, N.; Nemet, A.; Dugourd, D.; Piha-Paul, S.; et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Saotome, K.; Singh, A.K.; Yelshanskaya, M.V.; Sobolevsky, A.I. Crystal structure of the epithelial calcium channel TRPV6. Nature 2016, 534, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Saotome, K.; Sobolevsky, A.I. Swapping of transmembrane domains in the epithelial calcium channel TRPV6. Sci. Rep. 2017, 7, 10669. [Google Scholar] [CrossRef] [PubMed]
- McGoldrick, L.L.; Singh, A.K.; Saotome, K.; Yelshanskaya, M.V.; Twomey, E.C.; Grassucci, R.A.; Sobolevsky, A.I. Opening of the human epithelial calcium channel TRPV6. Nature 2018, 553, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Lindinger, S.; Neuberger, A.; Nadezhdin, K.D.; Singh, A.K.; Cunha, M.R.; Derler, I.; Gyimesi, G.; Reymond, J.-L.; Hediger, M.A.; et al. Inactivation-mimicking block of the epithelial calcium channel TRPV6. Sci. Adv. 2020, 6, eabe1508. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, A.; Nadezhdin, K.D.; Sobolevsky, A.I. Structural mechanisms of TRPV6 inhibition by ruthenium red and econazole. Nat. Commun. 2021, 12, 6284. [Google Scholar] [CrossRef]
- Neuberger, A.; Trofimov, Y.A.; Yelshanskaya, M.V.; Nadezhdin, K.D.; Krylov, N.A.; Efremov, R.G.; Sobolevsky, A.I. Structural mechanism of human oncochannel TRPV6 inhibition by the natural phytoestrogen genistein. Nat. Commun. 2023, 14, 2659. [Google Scholar] [CrossRef]
- Singh, A.K.; Saotome, K.; McGoldrick, L.L.; Sobolevsky, A.I. Structural bases of TRP channel TRPV6 allosteric modulation by 2-APB. Nat. Commun. 2018, 9, 2465. [Google Scholar] [CrossRef]
- Zhao, C.; MacKinnon, R. Structural and functional analyses of a GPCR-inhibited ion channel TRPM3. Neuron 2023, 111, 81–91.e87. [Google Scholar] [CrossRef]
- Zhou, X.; Li, M.; Su, D.; Jia, Q.; Li, H.; Li, X.; Yang, J. Cryo-EM structures of the human endolysosomal TRPML3 channel in three distinct states. Nat. Struct. Mol. Biol. 2017, 24, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Tao, X.; Touhara, K.K.; MacKinnon, R. Cryo-EM analysis of PIP2 regulation in mammalian GIRK channels. eLife 2020, 9, e60552. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.E.T.; Pumroy, R.A.; Yazici, A.T.; Kasimova, M.A.; Fluck, E.C.; Huynh, K.W.; Samanta, A.; Molugu, S.K.; Zhou, Z.H.; Carnevale, V.; et al. Structural insights on TRPV5 gating by endogenous modulators. Nat. Commun. 2018, 9, 4198. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, A.; Trofimov, Y.A.; Yelshanskaya, M.V.; Khau, J.; Nadezhdin, K.D.; Khosrof, L.S.; Krylov, N.A.; Efremov, R.G.; Sobolevsky, A.I. Molecular pathway and structural mechanism of human oncochannel TRPV6 inhibition by the phytocannabinoid tetrahydrocannabivarin. Nat. Commun. 2023, 14, 4630. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cai, R.; Chen, X.Z.; Peng, J.B. Molecular insights into the structural and dynamical changes of calcium channel TRPV6 induced by its interaction with phosphatidylinositol 4,5-bisphosphate. J. Biol. Struct. Dyn. 2023, 41, 6559–6568. [Google Scholar] [CrossRef] [PubMed]
- Vachel, L.; Norez, C.; Jayle, C.; Becq, F.; Vandebrouck, C. The low PLC-δ1 expression in cystic fibrosis bronchial epithelial cells induces upregulation of TRPV6 channel activity. Cell Calcium 2015, 57, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Liu, X.; Zhang, R.; Hofmann, L.; Zheng, W.; Amin, M.R.; Wang, L.; Hu, Q.; Peng, J.-B.; Michalak, M.; et al. Autoinhibition of TRPV6 Channel and Regulation by PIP2. iScience 2020, 23, 101444. [Google Scholar] [CrossRef]
- Velisetty, P.; Borbiro, I.; Kasimova, M.A.; Liu, L.; Badheka, D.; Carnevale, V.; Rohacs, T. A molecular determinant of phosphoinositide affinity in mammalian TRPV channels. Sci. Rep. 2016, 6, 27652. [Google Scholar] [CrossRef]
- Jon, R.; Stephen, J.M.; David, A.B. Probing the Regulation of M (Kv7) Potassium Channels in Intact Neurons with Membrane-Targeted Peptides. J. Neurosci. 2006, 26, 7950. [Google Scholar] [CrossRef]
- Buchmayer, F.; Schicker, K.; Steinkellner, T.; Geier, P.; Stübiger, G.; Hamilton, P.J.; Jurik, A.; Stockner, T.; Yang, J.-W.; Montgomery, T.; et al. Amphetamine actions at the serotonin transporter rely on the availability of phosphatidylinositol-4,5-bisphosphate. Proc. Natl. Acad. Sci. USA 2013, 110, 11642–11647. [Google Scholar] [CrossRef]
- Humer, C.; Lindinger, S.; Carrel, A.L.; Romanin, C.; Höglinger, C. TRPV6 Regulation by Cis-22a and Cholesterol. Biomolecules 2022, 12, 804. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, L.; Wang, H.; Zheng, W.; Philipp, S.E.; Hidalgo, P.; Cavalié, A.; Chen, X.-Z.; Beck, A.; Flockerzi, V. The S4–S5 linkergearbox of TRP channel gating. Cell Calcium 2017, 67, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Sallinger, M.; Tiffner, A.; Schmidt, T.; Bonhenry, D.; Waldherr, L.; Frischauf, I.; Lunz, V.; Derler, I.; Schober, R.; Schindl, R. Luminal STIM1 Mutants that Cause Tubular Aggregate Myopathy Promote Autophagic Processes. Int. J. Mol. Sci. 2020, 21, 4410. [Google Scholar] [CrossRef] [PubMed]
- Schober, R.; Bonhenry, D.; Lunz, V.; Zhu, J.; Krizova, A.; Frischauf, I.; Fahrner, M.; Zhang, M.; Waldherr, L.; Schmidt, T.; et al. Sequential activation of STIM1 links Ca2+ with luminal domain unfolding. Sci. Signal. 2019, 12, eaax3194. [Google Scholar] [CrossRef] [PubMed]
- Sheth, H.; Gorey, C.; Roush, N.; Smallman, S.; Collantes, E.; Santoro, M.; Olson, B.; Fitzgerald, L.; Lee, P.H.; Shen, X.J. A Multiplexed Fluorescent Calcium and NFAT Reporter Gene Assay to Identify GPCR Agonists. Curr. Chem. Genom. Transl. Med. 2013, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rydzek, J.; Nerreter, T.; Peng, H.; Jutz, S.; Leitner, J.; Steinberger, P.; Einsele, H.; Rader, C.; Hudecek, M. Chimeric Antigen Receptor Library Screening Using a Novel NF-κB/NFAT Reporter Cell Platform. Mol. Ther. 2019, 27, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Baldari, C.T.; Di Somma, M.M.; Majolini, M.B.; Ulivieri, C.; Milia, E.; Telford, J.L. NF-AT-Luciferase Reporter T Cell Lines as Tools to Screen Immunosuppressive Drugs. Biologicals 1998, 26, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Berdigaliyev, N.; Aljofan, M. An overview of drug discovery and development. Future Med. Chem. 2020, 12, 939–947. [Google Scholar] [CrossRef]
- Lehen’Kyi, V.; Flourakis, M.; Skryma, R.; Prevarskaya, N. TRPV6 channel controls prostate cancer cell proliferation via Ca2+/NFAT-dependent pathways. Oncogene 2007, 26, 7380–7385. [Google Scholar] [CrossRef]
- Skrzypski, M.; Kołodziejski, P.A.; Mergler, S.; Khajavi, N.; Nowak, K.W.; Strowski, M.Z. TRPV6 modulates proliferation of human pancreatic neuroendocrine BON-1 tumour cells. Biosci. Rep. 2016, 36, e00372. [Google Scholar] [CrossRef] [PubMed]
- Skrzypski, M.; Khajavi, N.; Mergler, S.; Szczepankiewicz, D.; Kołodziejski, P.A.; Metzke, D.; Wojciechowicz, T.; Billert, M.; Nowak, K.W.; Strowski, M.Z. TRPV6 channel modulates proliferation of insulin secreting INS-1E beta cell line. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 3202–3210. [Google Scholar] [CrossRef] [PubMed]
- Frischauf, I.; Litviňuková, M.; Schober, R.; Zayats, V.; Svobodová, B.; Bonhenry, D.; Lunz, V.; Cappello, S.; Tociu, L.; Reha, D.; et al. Transmembrane helix connectivity in Orai1 controls two gates for calcium-dependent transcription. Sci. Signal. 2017, 10, eaao0358. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.-F.; Yang, H.; Zhao, Y.-Z.; Xue, M. Metabolism of Peptide Drugs and Strategies to Improve their Metabolic Stability. Curr. Drug Metab. 2018, 19, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Myeong, J.; Shin, Y.-C.; So, I. Differential PI(4,5)P2 sensitivities of TRPC4, C5 homomeric and TRPC1/4, C1/5 heteromeric channels. Sci. Rep. 2019, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Ningoo, M.; Plant, L.D.; Greka, A.; Logothetis, D.E. PIP2 regulation of TRPC5 channel activation and desensitization. J. Biol. Chem. 2021, 296, 100726. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aziz, T.M.; Kaur, A.; Shapiro, M.S.; Stockand, J.D.; Archer, C.R. Optogenetic Control of PIP2 Interactions Shaping ENaC Activity. Int. J. Mol. Sci. 2022, 23, 3884. [Google Scholar] [CrossRef] [PubMed]
- Archer, C.R.; Enslow, B.T.; Carver, C.M.; Stockand, J.D. Phosphatidylinositol 4,5-bisphosphate directly interacts with the β and γ subunits of the sodium channel ENaC. J. Biol. Chem. 2020, 295, 7958–7969. [Google Scholar] [CrossRef]
- Gada, K.D.; Kamuene, J.M.; Chandrashekar, A.; Kissell, R.C.; Yauch, A.K.; Plant, L.D. PI(4,5)P2 regulates the gating of NaV1.4 channels. J. Gen. Physiol. 2023, 155, e202213255. [Google Scholar] [CrossRef]
- Wu, G.; Zhang, W.; Na, T.; Jing, H.; Wu, H.; Peng, J.-B. Suppression of intestinal calcium entry channel TRPV6 by OCRL, a lipid phosphatase associated with Lowe syndrome and Dent disease. Am. J. Physiol. Cell Physiol. 2012, 302, C1479–C1491. [Google Scholar] [CrossRef]
- Liu, H.; Yu, X.; Li, K.; Klejnot, J.; Yang, H.; Lisiero, D.; Lin, C. Photoexcited CRY2 Interacts with CIB1 to Regulate Transcription and Floral Initiation in Arabidopsis. Science 2008, 322, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Grancher, N.; Heil, M.; Black, R.C.; Giovani, B.; Galland, P.; Lardemer, D. Action Spectrum for Cryptochrome-Dependent Hypocotyl Growth Inhibition in Arabidopsis. Plant Physiol. 2002, 129, 774–785. [Google Scholar] [CrossRef] [PubMed]
- van der Wijst, J.; Leunissen, E.H.; Blanchard, M.G.; Venselaar, H.; Verkaart, S.; Paulsen, C.E.; Bindels, R.J.; Hoenderop, J.G. A Gate Hinge Controls the Epithelial Calcium Channel TRPV5. Sci. Rep. 2017, 7, 45489. [Google Scholar] [CrossRef] [PubMed]
- Dang, S.; van Goor, M.K.; Asarnow, D.; Wang, Y.; Julius, D.; Cheng, Y.; van der Wijst, J. Structural insight into TRPV5 channel function and modulation. Proc. Natl. Acad. Sci. USA 2019, 116, 8869–8878. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; McGoldrick, L.L.; Twomey, E.C.; Sobolevsky, A.I. Mechanism of calmodulin inactivation of the calcium-selective TRP channel TRPV6. Sci. Adv. 2018, 4, eaau6088. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-H.; De Jesús Pérez, J.J.; Moiseenkova-Bell, V.; Rohacs, T. Structural basis of the activation of TRPV5 channels by long-chain acyl-Coenzyme-A. Nat. Commun. 2023, 14, 5883. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Wang, L.; Liu, X.; Michalak, M.; Tang, J.; Peng, J.-B.; Chen, X.-Z. Auto-inhibitory intramolecular S5/S6 interaction in the TRPV6 channel regulates breast cancer cell migration and invasion. Commun. Biol. 2021, 4, 990. [Google Scholar] [CrossRef]
- Yin, X.; Xu, R.; Song, J.; Ruze, R.; Chen, Y.; Wang, C.; Xu, Q. Lipid metabolism in pancreatic cancer: Emerging roles and potential targets. Cancer Commun. 2022, 42, 1234–1256. [Google Scholar] [CrossRef]
- Kopecka, J.; Trouillas, P.; Gašparović, A.Č.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and cholesterol: Inducers of cancer multidrug resistance and therapeutic targets. Drug Resist. Updates 2020, 49, 100670. [Google Scholar] [CrossRef]
- Skotland, T.; Kavaliauskiene, S.; Sandvig, K. The role of lipid species in membranes and cancer-related changes. Cancer Metastasis Rev. 2020, 39, 343–360. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Peptide Sequence | Mmo (Calculated) * | Mmo (Measured) ß | Rt (min) × |
|---|---|---|---|---|
| Pal-PIP2 peptide | Palmitoyl-HRQKHFEKRR | 1659.0209 | 1659.0198 | 16.6 |
| Control peptide | Palmitoyl-HAQKHFEAAA | 1346.7711 | 1346.7700 | 18.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Humer, C.; Radiskovic, T.; Horváti, K.; Lindinger, S.; Groschner, K.; Romanin, C.; Höglinger, C. Bidirectional Allosteric Coupling between PIP2 Binding and the Pore of the Oncochannel TRPV6. Int. J. Mol. Sci. 2024, 25, 618. https://doi.org/10.3390/ijms25010618
Humer C, Radiskovic T, Horváti K, Lindinger S, Groschner K, Romanin C, Höglinger C. Bidirectional Allosteric Coupling between PIP2 Binding and the Pore of the Oncochannel TRPV6. International Journal of Molecular Sciences. 2024; 25(1):618. https://doi.org/10.3390/ijms25010618
Chicago/Turabian StyleHumer, Christina, Tamara Radiskovic, Kata Horváti, Sonja Lindinger, Klaus Groschner, Christoph Romanin, and Carmen Höglinger. 2024. "Bidirectional Allosteric Coupling between PIP2 Binding and the Pore of the Oncochannel TRPV6" International Journal of Molecular Sciences 25, no. 1: 618. https://doi.org/10.3390/ijms25010618