
Profiling Novel Quinuclidine-Based Derivatives as Potential Anticholinesterase Drugs: Enzyme Inhibition and Effects on Cell Viability
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
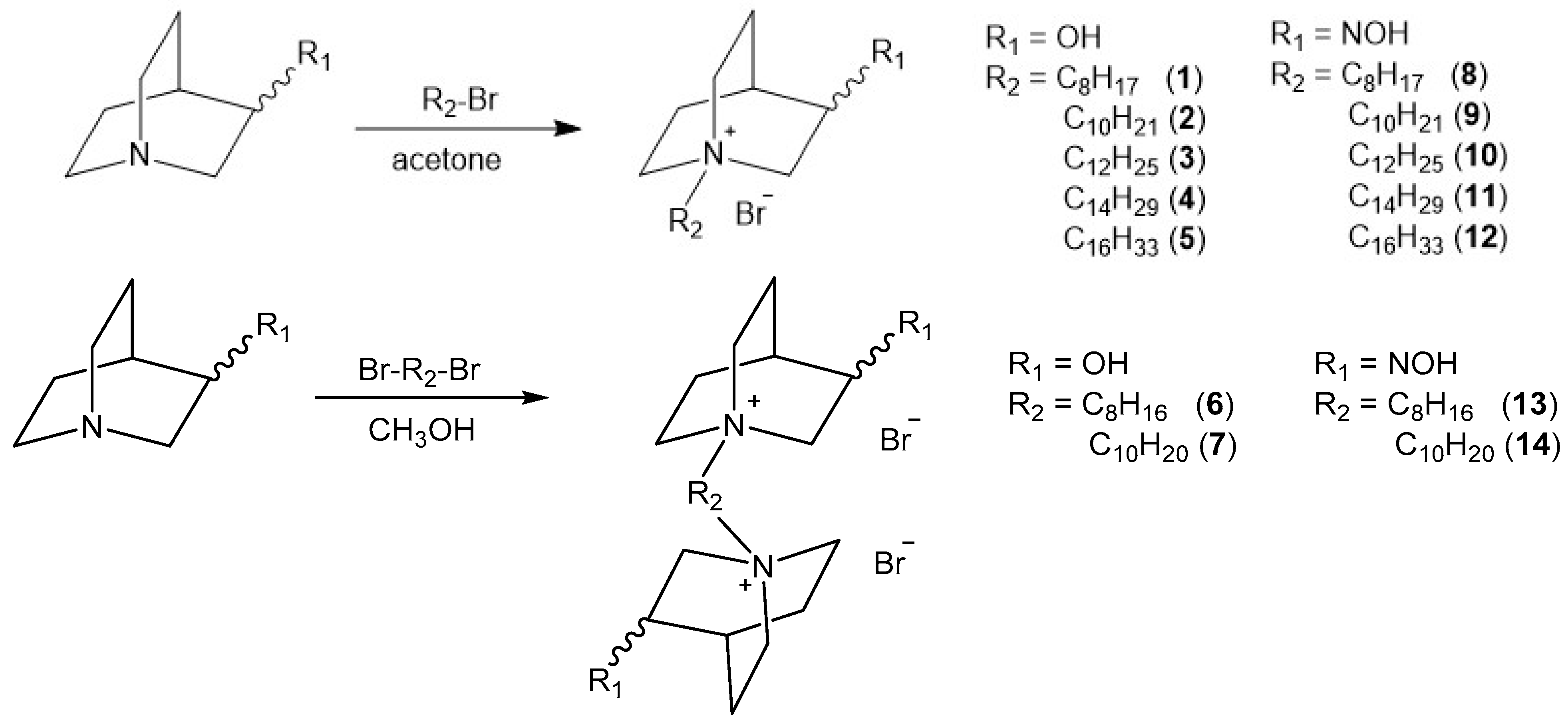
2.1. Synthesis of Compounds
2.2. Reversible Inhibition of AChE and BChE with N-Alkyl Quaternary Quinuclidines
2.3. Molecular Modeling of AChE and BChE in Complex with N-Alkyl Quaternary Quinuclidines

2.4. Effects of N-Alkyl Quaternary Quinuclidines on Cells’ Viability
2.4.1. Cytotoxic Effect of Quinuclidine Derivatives
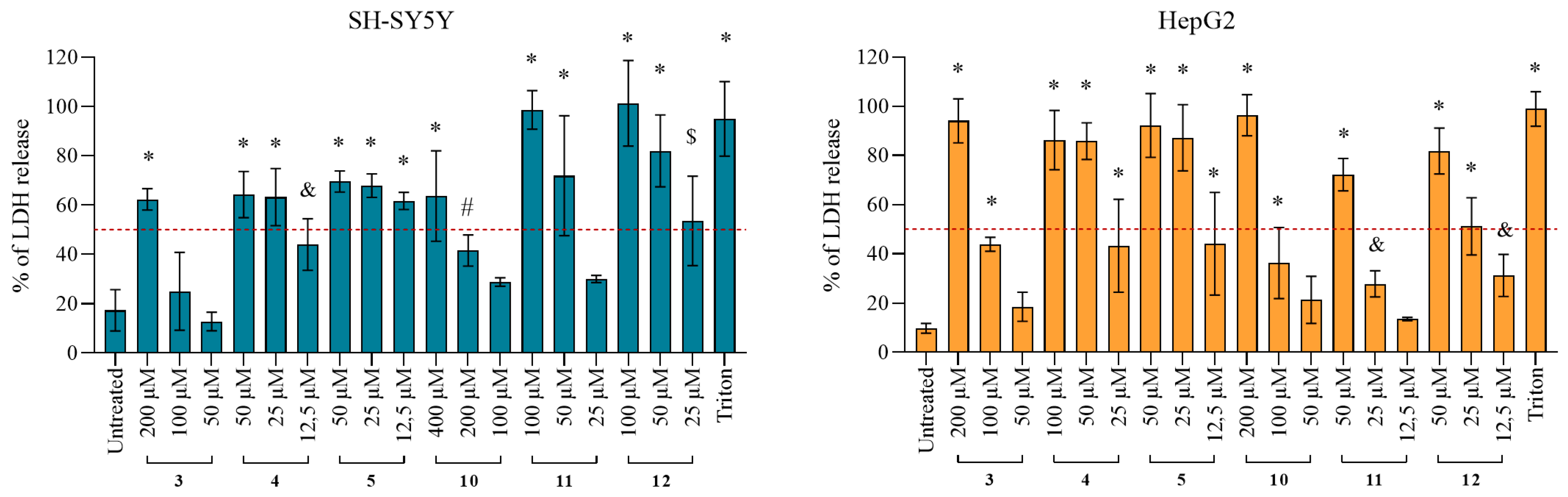
2.4.2. Damage of the Cell Membrane Integrity
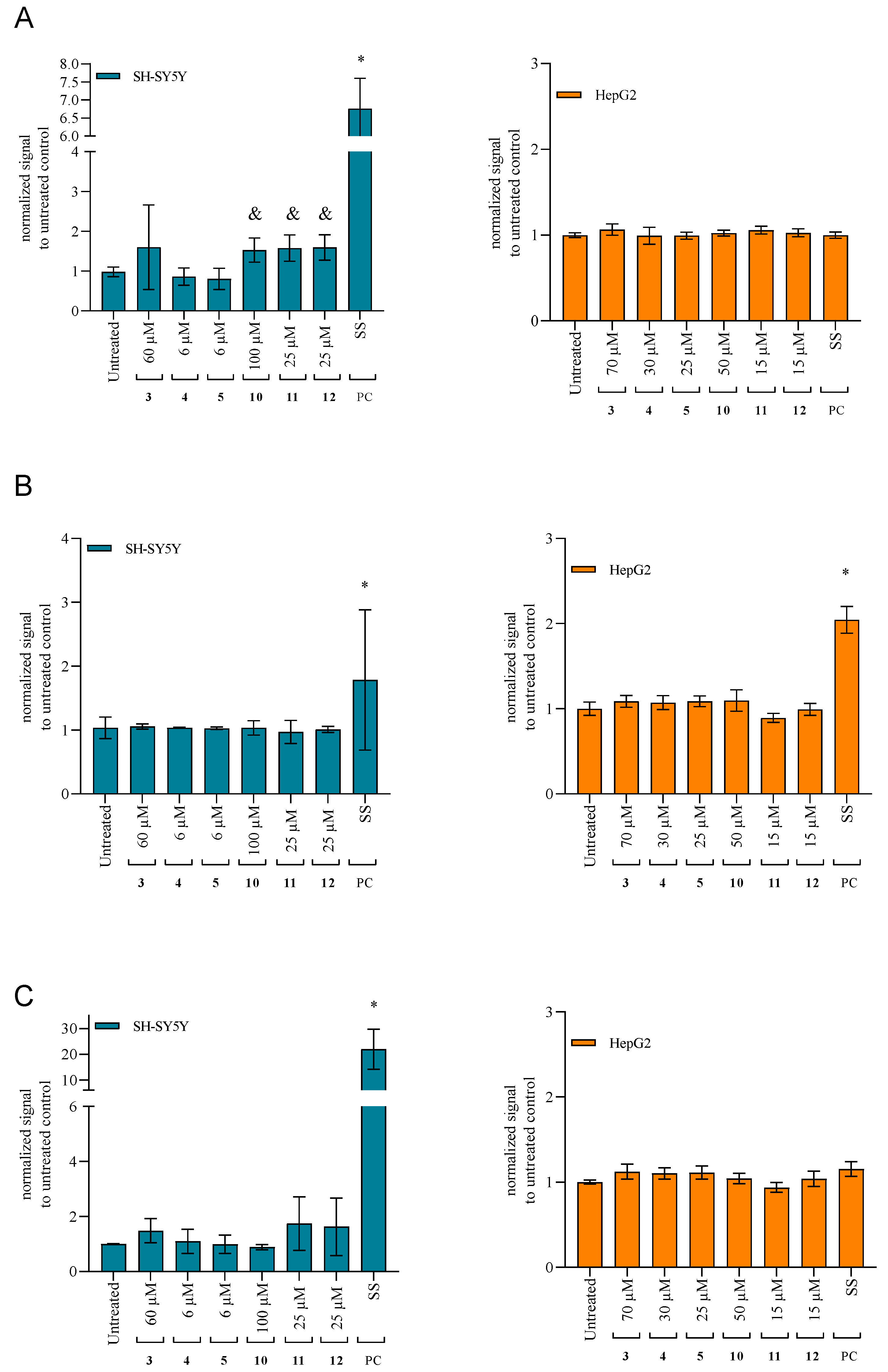
2.4.3. Changes in the Mitochondrial Membrane Potential
2.4.4. Activation of the Caspases as the Specific Apoptotic Markers
3. Discussion
4. Materials and Methods
4.1. Synthesis of N-Alkyl Quaternary Quinuclidines
4.1.1. Chemicals
4.1.2. General Procedure for Preparation of QOH and QNOH Monoquaternary Derivatives
4.1.3. General Procedure for Preparation of QOH and QNOH Bisquaternary Derivatives

4.2. AChE and BChE Reversible Inhibition
4.2.1. Chemicals and Enzymes
4.2.2. The Enzyme Activity Measurement and Determination of the Reversible Inhibition by Quinuclidine Compounds
4.3. In Silico Molecular Docking Studies
4.4. Cell Tests
4.4.1. Human Cells
4.4.2. Cytotoxicity Assay
4.4.3. Cell Membrane Integrity
4.4.4. Mitochondrial Membrane Potential
4.4.5. Caspases Activity
4.5. Statistics and Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwartz, J. Neurotransmitters. In Principles of Neuronal Science; Kandel, E.R., Jessell, T.M., Schwartz, J.H., Eds.; McGrae-Hill: New York, NY, USA, 2000; pp. 281–297. [Google Scholar]
- Giacobini, E. Cholinesterases and Cholinesterases Inhibitors, 3rd ed.; Informa Healthcare: London, UK, 2000. [Google Scholar]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- McHardy, S.F.; Wang, H.-Y.L.; McCowen, S.V.; Valdez, M.C. Recent advances in acetylcholinesterase inhibitors and Reactivators: An update on the patent literature (2012–2015). Expert Opin. Ther. Pat. 2017, 27, 455–476. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.; Moore, S.W. Why has butyrylcholinesterase been retained? Structural and functional diversification in a duplicated gene. Neurochem. Int. 2012, 61, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Lockridge, O. Review of human butyrylcholinesterase structure, function, genetic variants, history of use in the clinic, and potential therapeutic uses. Pharmacol. Ther. 2015, 148, 34–46. [Google Scholar] [CrossRef]
- Blondet, B.; Carpentier, G.; Ferry, A.; Chatonnet, A.; José, C. Localization of butyrylcholinesterase at the neuromuscular junction of normal and acetylcholinesterase knockout mice. J. Histochem. Cytochem. 2010, 58, 1075–1082. [Google Scholar] [CrossRef]
- Greig, N.; Reale, M.; Tata, A. New pharmacological approaches to the cholinergic system: An overview on muscarinic receptor ligands and cholinesterase inhibitors. Recent Pat. CNS Drug Discov. 2013, 8, 123–141. [Google Scholar] [CrossRef]
- Tabet, N. Acetylcholinesterase inhibitors for Alzheimer’s disease: Anti-inflammatories in acetylcholine clothing. Age Ageing 2006, 35, 336–338. [Google Scholar] [CrossRef]
- Ofek, K.; Soreq, H. Cholinergic involvement and manipulation approaches in multiple system disorders. Chem. Biol. Interact. 2013, 203, 113–119. [Google Scholar] [CrossRef]
- VC Guido, R.; Oliva, G.; D Andricopulo, A. Modern drug discovery technologies: Opportunities and challenges in lead discovery. Comb. Chem. High Throughput Screen. 2011, 14, 830–839. [Google Scholar] [CrossRef]
- Mashkovsky, M.D.; Yakhontov, L.N. Relationships between the chemical structure and pharmacological activity in a series of synthetic quinuclidine derivatives. In Progress in Drug Research/Fortschritte der Arzneimittelforschung/Progrès des Recherches Pharmaceutiques; Birkhäuser: Basel, Switzerland, 1969; pp. 293–339. [Google Scholar]
- Mashkovsky, M.D.; Yakhontov, L.N.; Kaminka, M.E.; Mikhlina, E.E. Further developments in research on the chemistry and pharmacology of synthetic quinuclidine derivatives. In Progress in Drug Research/Fortschritte der Arzneimittelforschung/Progrès des Recherches Pharmaceutiques; Birkhäuser: Basel, Switzerland, 1983; pp. 9–61. [Google Scholar]
- Kacprzak, K.M. Chemistry and biology of Cinchona alkaloids. In Natural Products; Springer: Berlin/Heidelberg, Germany, 2013; pp. 605–641. [Google Scholar]
- Venkateswaran, A.; Reddy, Y.T.; Sonar, V.N.; Muthusamy, V.; Crooks, P.A.; Freeman, M.L.; Sekhar, K.R. Antiangiogenic properties of substituted (Z)-(±)-2-(N-benzylindol-3-ylmethylene)quinuclidin-3-ol/one analogs and their derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 7323–7326. [Google Scholar] [CrossRef]
- Cammerer, S.B.; Jimenez, C.; Jones, S.; Gros, L.; Lorente, S.O.; Rodrigues, C.; Rodrigues, J.C.F.; Caldera, A.; Ruiz Perez, L.M.; da Souza, W.; et al. Quinuclidine derivatives as potential antiparasitics. Antimicrob. Agents Chemother. 2007, 51, 4049–4061. [Google Scholar] [CrossRef] [PubMed]
- de Macedo-Silva, S.T.; Visbal, G.; Urbina, J.A.; de Souza, W.; Rodrigues, J.C.F. Potent in vitro antiproliferative synergism of combinations of ergosterol biosynthesis inhibitors against Leishmania amazonensis. Antimicrob. Agents Chemother. 2015, 59, 6402–6418. [Google Scholar] [CrossRef] [PubMed]
- de Souza, B.F.; dos Santos, M.R.; de Souza, R.P.; Cämmerer, S.B.; D´Angelo, N.A.; Franco, C.H.; Moraes, C.B.; Freitas-Junior, L.H. Synthesis of chiral 3-[[(aryl) methyl] amino]- and 3-[[(heteroaryl)-methyl]-amino]-quinuclidines with high biological activity against intracellular Trypansoma cruzi amastigotes. Int. J. Chem. Pharm. Sci. 2016, 4, 272–278. [Google Scholar]
- Simeon-Rudolf, V.; Reiner, E.; Škrinjarić-Špoljar, M.; Radić, B.; Lucić, A.; Primožič, I.; Tomić, S. Quinuclidinium-imidazolium compounds: Synthesis, mode of interaction with acetylcholinesterase and effect upon Soman intoxicated mice. Arch. Toxicol. 1998, 72, 289–295. [Google Scholar] [CrossRef]
- Reiner, E.; Škrinjarić-Špoljar, M.; Dunaj, S.; Simeon-Rudolf, V.; Primožič, I.; Tomić, S. 3-Hydroxyquinuclidinium derivatives: Synthesis of compounds and inhibition of acetylcholinesterase. Chem. Biol. Interact. 1999, 119–120, 173–181. [Google Scholar] [CrossRef]
- Skočibušić, M.; Odžak, R.; Štefanić, Z.; Križić, I.; Krišto, L.; Jović, O.; Hrenar, T.; Primožič, I.; Jurašin, D. Structure–property relationship of quinuclidinium surfactants—Towards multifunctional biologically active molecules. Colloids Surfaces B Biointerfaces 2016, 140, 548–559. [Google Scholar] [CrossRef]
- Bazina, L.; Maravić, A.; Krce, L.; Soldo, B.; Odžak, R.; Popović, V.B.; Aviani, I.; Primožič, I.; Šprung, M. Discovery of novel quaternary ammonium compounds based on quinuclidine-3-ol as new potential antimicrobial candidates. Eur. J. Med. Chem. 2019, 163, 626–635. [Google Scholar] [CrossRef]
- Primožič, I.; Hrenar, T.; Baumann, K.; Krišto, L. Mechanochemical and Conformational Study of iV-heterocyclic Carbonyl-Oxime Transformations. Croat. Chem. Acta 2014, 87, 153–160. [Google Scholar] [CrossRef]
- Saxena, A.; Redman, A.M.G.; Jiang, X.; Lockridge, O.; Doctor, B.P. Differences in active-site gorge dimensions of cholinesterases revealed by binding of inhibitors to human butyrylcholinesterase. Chem. Biol. Interact. 1999, 119–120, 61–69. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Calas, A.-G.; Dias, J.; Rousseau, C.; Arboléas, M.; Touvrey-Loiodice, M.; Mercey, G.; Jean, L.; Renard, P.-Y.; Nachon, F. An easy method for the determination of active concentrations of cholinesterase reactivators in blood samples: Application to the efficacy assessment of non quaternary reactivators compared to HI-6 and pralidoxime in VX-poisoned mice. Chem. Biol. Interact. 2017, 267, 11–16. [Google Scholar] [CrossRef]
- Sit, R.K.; Kovarik, Z.; Maček Hrvat, N.; Žunec, S.; Green, C.; Fokin, V.V.; Sharpless, K.B.; Radić, Z.; Taylor, P. Pharmacology, pharmacokinetics, and tissue disposition of zwitterionic hydroxyiminoacetamido alkylamines as reactivating antidotes for organophosphate exposure. J. Pharmacol. Exp. Ther. 2018, 367, 363–372. [Google Scholar] [CrossRef]
- Zorbaz, T.; Mišetić, P.; Probst, N.; Žunec, S.; Zandona, A.; Mendaš, G.; Micek, V.; Maček Hrvat, N.; Katalinić, M.; Braïki, A.; et al. Pharmacokinetic evaluation of brain penetrating morpholine-3-hydroxy-2-pyridine oxime as an antidote for nerve agent poisoning. ACS Chem. Neurosci. 2020, 11, 1072–1084. [Google Scholar] [CrossRef] [PubMed]
- Zandona, A.; Katalinić, M.; Šinko, G.; Radman Kastelic, A.; Primožič, I.; Kovarik, Z. Targeting organophosphorus compounds poisoning by novel quinuclidine-3 oximes: Development of butyrylcholinesterase-based bioscavengers. Arch. Toxicol. 2020, 94, 3157–3171. [Google Scholar] [CrossRef] [PubMed]
- Zandona, A.; Maraković, N.; Mišetić, P.; Madunić, J.; Miš, K.; Padovan, J.; Pirkmajer, S.; Katalinić, M. Activation of (un)regulated cell death as a new perspective for bispyridinium and imidazolium oximes. Arch. Toxicol. 2021, 95, 2737–2754. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.N.; Brimijoin, S. Cholinesterases and the fine line between poison and remedy. Biochem. Pharmacol. 2018, 153, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, Z.; Radić, Z.; Berman, H.A.; Simeon-Rudolf, V.; Reiner, E.; Taylor, P. Acetylcholinesterase active centre and gorge conformations analysed by combinatorial mutations and enantiomeric phosphonates. Biochem. J. 2003, 373, 33–40. [Google Scholar] [CrossRef]
- Bosak, A.; Knežević, A.; Gazić Smilović, I.; Šinko, G.; Kovarik, Z. Resorcinol-, catechol- and saligenin-based bronchodilating β2-agonists as inhibitors of human cholinesterase activity. J. Enzyme Inhib. Med. Chem. 2017, 32, 789–797. [Google Scholar] [CrossRef]
- Matošević, A.; Opsenica, D.M.; Spasić, M.; Maraković, N.; Zandona, A.; Žunec, S.; Bartolić, M.; Kovarik, Z.; Bosak, A. Evaluation of 4-aminoquinoline derivatives with an n-octylamino spacer as potential multi-targeting ligands for the treatment of Alzheimer’s disease. Chem. Biol. Interact. 2023, 382, 110620. [Google Scholar] [CrossRef]
- Matošević, A.; Radman Kastelic, A.; Mikelić, A.; Zandona, A.; Katalinić, M.; Primožič, I.; Bosak, A.; Hrenar, T. Quinuclidine-based carbamates as potential CNS active compounds. Pharmaceutics 2021, 13, 420. [Google Scholar] [CrossRef] [PubMed]
- Reiner, E.; Radić, Z. Mechanism of action of cholinesterase inhibitors. In Cholinesterase and Cholinesterase Inhibitors; Giacobini, E., Ed.; Martin Dunitz Ltd.: London, UK, 2000; pp. 103–120. [Google Scholar]
- Bušić, V.; Katalinić, M.; Šinko, G.; Kovarik, Z.; Gašo-Sokač, D. Pyridoxal oxime derivative potency to reactivate cholinesterases inhibited by organophosphorus compounds. Toxicol. Lett. 2016, 262, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Fedorko, J.M.; Vinayaka, C.R.; Medhekar, R.; Radić, Z.; Taylor, P.; Lockridge, O.; Doctor, B.P. Aromatic amino-acid residues at the active and peripheral anionic sites control the binding of E2020 (Aricept®) to cholinesterases. Eur. J. Biochem. 2003, 270, 4447–4458. [Google Scholar] [CrossRef] [PubMed]
- Zandona, A.; Lihtar, G.; Maraković, N.; Miš, K.; Bušić, V.; Gašo-Sokač, D.; Pirkmajer, S.; Katalinić, M. Vitamin B3-based biologically active compounds as inhibitors of human cholinesterases. Int. J. Mol. Sci. 2020, 21, 8088. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Radić, Z.; Sulzenbacher, G.; Kim, E.; Taylor, P.; Marchot, P. Substrate and product trafficking through the active center gorge of acetylcholinesterase analyzed by crystallography and equilibrium binding. J. Biol. Chem. 2006, 281, 29256–29267. [Google Scholar] [CrossRef] [PubMed]
- Colletier, J.-P.; Fournier, D.; Greenblatt, H.M.; Stojan, J.; Sussman, J.L.; Zaccai, G.; Silman, I.; Weik, M. Structural insights into substrate traffic and inhibition in acetylcholinesterase. EMBO J. 2006, 25, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Burilova, E.A.; Pashirova, T.N.; Lukashenko, S.S.; Sapunova, A.S.; Voloshina, A.D.; Zhiltsova, E.P.; Campos, J.R.; Souto, E.B.; Zakharova, L.Y. Synthesis, biological evaluation and structure-activity relationships of self-assembled and solubilization properties of amphiphilic quaternary ammonium derivatives of quinuclidine. J. Mol. Liq. 2018, 272, 722–730. [Google Scholar] [CrossRef]
- Liu, W.; Wang, C.; Sun, J.; Hou, G.; Wang, Y.; Qu, R. Synthesis, Characterization and Antibacterial Properties of Dihydroxy Quaternary Ammonium Salts with Long Chain Alkyl Bromides. Chem. Biol. Drug Des. 2015, 85, 91–97. [Google Scholar] [CrossRef]
- Zandona, A.; Madunić, J.; Miš, K.; Maraković, N.; Dubois-Geoffroy, P.; Cavaco, M.; Mišetić, P.; Padovan, J.; Castanho, M.; Jean, L.; et al. Biological response and cell death signaling pathways modulated by tetrahydroisoquinoline-based aldoximes in human cells. Toxicology 2023, 494, 153588. [Google Scholar] [CrossRef]
- Dymond, M.K.; Attard, G.S. Cationic type I amphiphiles as modulators of membrane curvature elastic stress in vivo. Langmuir 2008, 24, 11743–11751. [Google Scholar] [CrossRef]
- Worek, F.; Thiermann, H.; Wille, T. Oximes in organophosphate poisoning: 60 years of hope and despair. Chem. Biol. Interact. 2016, 259, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Plotnikov, M.B.; Khlebnikov, A.I.; Plotnikova, T.M.; Quinn, M.T. Oximes: Novel therapeutics with anticancer and anti-inflammatorypotential. Biomolecules 2021, 11, 777. [Google Scholar] [CrossRef] [PubMed]
- Čalić, M.; Vrdoljak, A.L.; Radić, B.; Jelić, D.; Jun, D.; Kuča, K.; Kovarik, Z. In vitro and in vivo evaluation of pyridinium oximes: Mode of interaction with acetylcholinesterase, effect on tabun- and soman-poisoned mice and their cytotoxicity. Toxicology 2006, 219, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Reiner, E.; Bosak, A.; Simeon-Rudolf, V. Activity of cholinesterases in human whole blood measured with acetylthiocholine as substrate and ethopropazine as selective inhibitor of plasma butyrylcholinesterase. Arh. Hig. Rada Toksikol. 2004, 55, 1–4. [Google Scholar] [PubMed]
- Reiner, E.; Šinko, G.; Škrinjarić-Špoljar, M.; Simeon-Rudolf, V. Comparison of protocols for measuring activities of human blood cholinesterases by the Ellman method. Arh. Hig. Rada Toksikol. 2000, 51, 13–18. [Google Scholar] [PubMed]
- Zorbaz, T.; Malinak, D.; Maraković, N.; Maček Hrvat, N.; Zandona, A.; Novotny, M.; Skarka, A.; Andrys, R.; Benkova, M.; Soukup, O.; et al. Pyridinium Oximes with Ortho -Positioned Chlorine Moiety Exhibit Improved Physicochemical Properties and Efficient Reactivation of Human Acetylcholinesterase Inhibited by Several Nerve Agents. J. Med. Chem. 2018, 61, 10753–10766. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Worek, F.; Mast, U.; Kiderlen, D.; Diepold, C.; Eyer, P. Improved determination of acetylcholinesterase activity in human whole blood. Clin. Chim. Acta 1999, 288, 73–90. [Google Scholar] [CrossRef]
- Šinko, G.; Čalić, M.; Kovarik, Z. para- and ortho -Pyridinium aldoximes in reaction with acetylthiocholine. FEBS Lett. 2006, 580, 3167–3172. [Google Scholar] [CrossRef]
- Hunter, A.; Downs, C.E. The inhibition of arginase by amino acids. J. Biol. Chem. 1945, 157, 427–446. [Google Scholar] [CrossRef]
- Koska, J.; Spassov, V.Z.; Maynard, A.J.; Yan, L.; Austin, N.; Flook, P.K.; Venkatachalam, C.M. Fully automated molecular mechanics based induced fit protein−ligand docking method. J. Chem. Inf. Model. 2008, 48, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Colletier, J.-P.; Weik, M.; Jiang, H.; Moult, J.; Silman, I.; Sussman, J.L. Flexibility of aromatic residues in the active-site gorge of acetylcholinesterase: X-ray versus molecular dynamics. Biophys. J. 2008, 95, 2500–2511. [Google Scholar] [CrossRef] [PubMed]
- Šinko, G. Assessment of scoring functions and in silico parameters for AChE-ligand interactions as a tool for predicting inhibition potency. Chem. Biol. Interact. 2019, 308, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Rosenberry, T.; Brazzolotto, X.; Macdonald, I.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the binding of reversible inhibitors to human butyrylcholinesterase and acetylcholinesterase: A crystallographic, kinetic and calorimetric study. Molecules 2017, 22, 2098. [Google Scholar] [CrossRef]
- Gupta, R.C. Handbook of Toxicology of Chemical Warfare Agents, 2nd ed.; Academic Press: San Diego, CA, USA, 2015; ISBN 9780128001592. [Google Scholar]
- Katalinić, M.; Maček Hrvat, N.; Baumann, K.; Morasi Piperčić, S.; Makarić, S.; Tomić, S.; Jović, O.; Hrenar, T.; Miličević, A.; Jelić, D.; et al. A comprehensive evaluation of novel oximes in creation of butyrylcholinesterase-based nerve agent bioscavengers. Toxicol. Appl. Pharmacol. 2016, 310, 195–204. [Google Scholar] [CrossRef]
- Katalinić, M.; Zandona, A.; Ramić, A.; Zorbaz, T.; Primožič, I.; Kovarik, Z. New Cinchona Oximes Evaluated as Reactivators of Acetylcholinesterase and Butyrylcholinesterase Inhibited by Organophosphorus Compounds. Molecules 2017, 22, 1234. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Concentration Range (μM) | Ki (μM) | Ki (AChE)/Ki (BChE) | |
|---|---|---|---|---|
| AChE | BChE | |||
| 1 (OH-C8) | 200–400 | 156.2 ± 26.1 | 85.1 ± 22.1 | 1.8 |
| 2 (OH-C10) | 40–60 | 63.8 ± 5.1 | 11.8 ± 1.1 | 5.4 |
| 3 (OH-C12) | 35–65 | 13.2 ± 0.6 | 9.0 ± 0.8 | 1.5 |
| 4 (OH-C14) | 2–10 | 4.2 ± 0.5 | 7.9 ± 0.7 | 0.5 |
| 5 (OH-C16) | 20–40 | 9.0 ± 1.3 | 26.1 ± 2.4 | 0.3 |
| 6 (bisOH-C8) | 20–40 | 24.0 ± 1.1 | 12.9 ± 1.0 | 1.9 |
| 7 (bisOH-C10) | 0.2–1 | 0.52 ± 0.05 | 1.6 ± 0.2 | 0.3 |
| 8 (NOH-C8) | 50–200 | 82.7 ± 3.5 | 37.9 ± 10.4 | 2.2 |
| 9 (NOH-C10) | 5–50 | 23.8 ± 0.8 | 5.2 ± 1.1 | 4.6 |
| 10 (NOH-C12) | 5–15 | 14.3 ± 1.8 | 5.4 ± 0.8 | 2.6 |
| 11 (NOH-C14) | 5–10 | 7.0 ± 0.9 | 8.8 ± 1.0 | 0.8 |
| 12 (NOH-C16) | 40–60 | 19.0 ± 1.6 | 48.6 ± 3.8 | 0.4 |
| 13 (bisNOH-C8) | 5–20 | 1.8 ± 0.07 | 3.4 ± 0.2 | 0.5 |
| 14 (bisNOH-C10) | 0.2–2 | 0.26 ± 0.02 | 1.2 ± 0.06 | 0.2 |
| IC50 ± SE (μM) | ||||||
|---|---|---|---|---|---|---|
| Compound | SH-SY5Y | HepG2 | ||||
| 1 h | 4 h | 24 h | 1 h | 4 h | 24 h | |
| 1 (OH-C8) | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 |
| 2 (OH-C10) | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 | ≥537 |
| 3 (OH-C12) | 145 ± 1 | 74 ± 1 | 56 ± 1 | 135 ± 1 | 81 ± 1 | 66 ± 1 |
| 4 (OH-C14) | 16 ± 1 | 9 ± 1 | 7 ± 1 | 35 ± 1 | 32 ± 1 | 43 ± 1 |
| 5 (OH-C16) | 11 ± 1 | 8 ± 1 | 7 ± 1 | 46 ± 1 | 26 ± 1 | 28 ± 1 |
| 6 (bisOH-C8) | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 |
| 7 (bisOH-C10) | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 |
| 8 (NOH-C8) | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 |
| 9 (NOH-C10) | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 |
| 10 (NOH-C12) | 208 ± 3 | 174 ± 1 | 147 ± 1 a | 218 ± 1 | 81 ± 1 | 15 ± 1 |
| 11 (NOH-C14) | 125 ± 2 | 83 ± 3 | 60 ± 1 | 37 ± 1 | 18 ± 1 | 11 ± 1 |
| 12 (NOH-C16) | 89 ± 1 | 78 ± 1 | 54 ± 1 | 40 ± 1 | 20 ± 1 | 7 ±1 |
| 13 (bisNOH-C8) | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 |
| 14 (bisNOH-C10) | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 |
| HI-6 b | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 | ≥800 |
| SS b | ≥4 | 3 ± 0.01 | 0.12 ± 0.01 | ≥2 | ≥2 | ≥2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Žunec, S.; Vadlja, D.; Ramić, A.; Zandona, A.; Maraković, N.; Brekalo, I.; Primožič, I.; Katalinić, M. Profiling Novel Quinuclidine-Based Derivatives as Potential Anticholinesterase Drugs: Enzyme Inhibition and Effects on Cell Viability. Int. J. Mol. Sci. 2024, 25, 155. https://doi.org/10.3390/ijms25010155
Žunec S, Vadlja D, Ramić A, Zandona A, Maraković N, Brekalo I, Primožič I, Katalinić M. Profiling Novel Quinuclidine-Based Derivatives as Potential Anticholinesterase Drugs: Enzyme Inhibition and Effects on Cell Viability. International Journal of Molecular Sciences. 2024; 25(1):155. https://doi.org/10.3390/ijms25010155
Chicago/Turabian StyleŽunec, Suzana, Donna Vadlja, Alma Ramić, Antonio Zandona, Nikola Maraković, Iva Brekalo, Ines Primožič, and Maja Katalinić. 2024. "Profiling Novel Quinuclidine-Based Derivatives as Potential Anticholinesterase Drugs: Enzyme Inhibition and Effects on Cell Viability" International Journal of Molecular Sciences 25, no. 1: 155. https://doi.org/10.3390/ijms25010155