
Cracking the Endothelial Calcium (Ca2+) Code: A Matter of Timing and Spacing
 , , and
, , and 
Abstract
:1. Introduction
2. An Introduction to Endothelial Ca2+ Signaling: From Global to Local Ca2+ Signals
2.1. General Mechanisms of Agonist-Induced Endothelial Ca2+ Signals
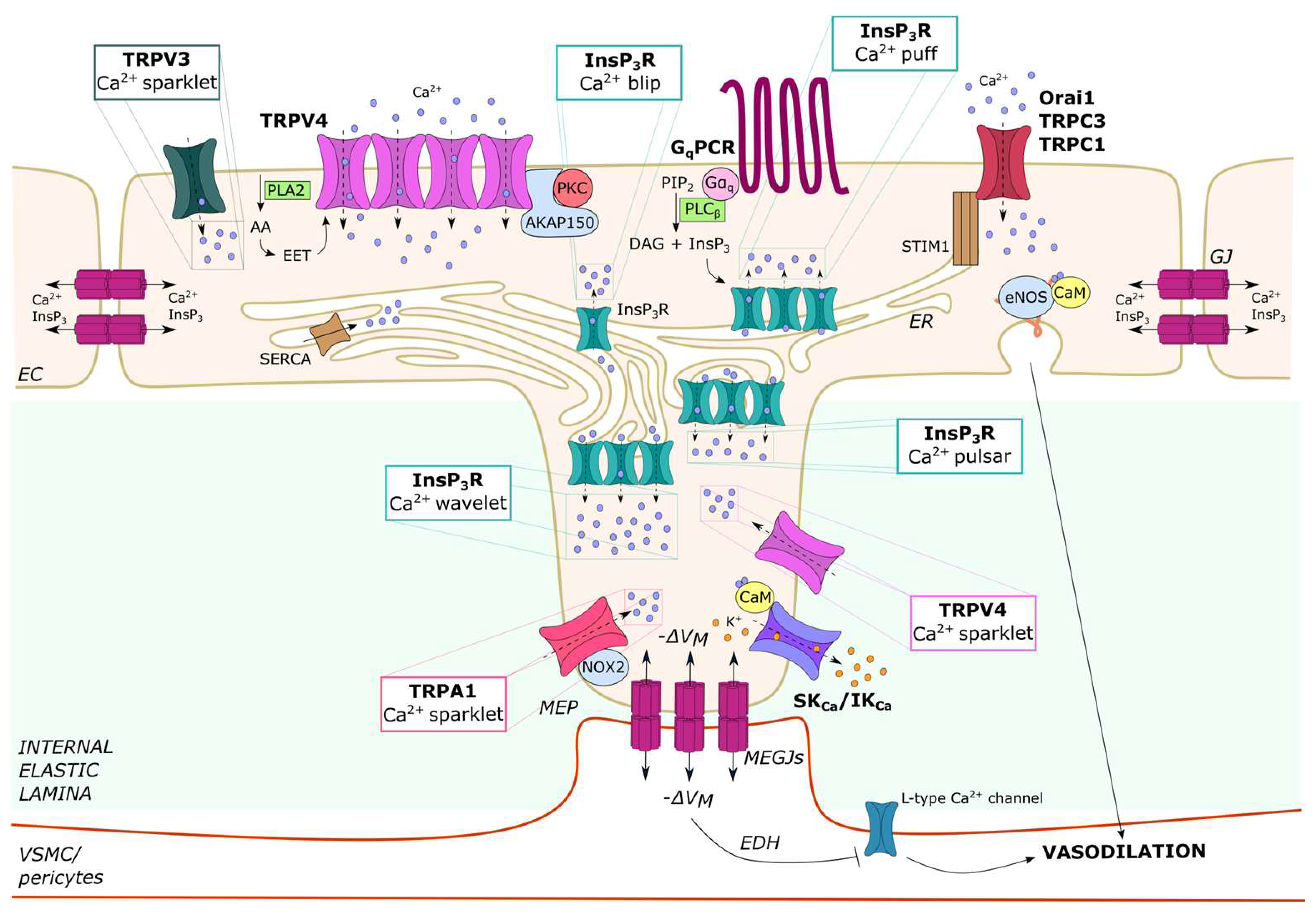
2.2. Endothelial Ca2+ Microdomains Generated by InsP3-Dependent ER Ca2+ Release
2.2.1. Ca2+ Blips and Puffs in Cultured Endothelial Cells
2.2.2. Ca2+ Puffs, Pulsars and Wavelets in Native Endothelial Cells

{kind=link}
{kind=link}
{kind=link}
| Ca2+ Signal | Source | Spatio-Temporal Features and Amplitude | Function | Reference |
|---|---|---|---|---|
| Ca2+ blips | InsP3Rs in the ER | Spatial spread: 1–3 µm; duration < 100 ms; amplitude: 23 nM | Building block of Ca2+ puffs | [84] |
| Ca2+ puffs | InsP3Rs in the ER | Spatial spread: 30 µm; duration: >>100 ms; amplitude: 50–100 nM | Building blocks of the intracellular Ca2+ waves | [84] |
| Ca2+ pulsars | InsP3Rs in the ER | Area: ~14 µm2; duration: ~250 ms; frequency: ~0.08 Hz | Recruitment of SKCa/IKCa channels at MEPs and, to be demonstrated, of eNOS to induce endothelium-dependent vasorelaxation | [48,49,80,94] |
| Ca2+ wavelets | InsP3Rs in the ER | Area: ~41 µm2; duration: ~470 ms; frequency: ~0.22 Hz | Recruitment of SKCa/IKCa channels at MEPs and, to be demonstrated, of eNOS to induce endothelium-dependent vasorelaxation | [50] |
| TRPV4-mediated Ca2+ sparklets | TRPV4 in the PM | Area: ~11 µm2; τ: ~37 ms; frequency: ~0.25 Hz | Recruitment of SKCa/IKCa channels at MEPs to induce endothelium-dependent vasodilation in systemic resistance arteries; recruitment of eNOS to induce vasodilation in pulmonary resistance vessels | [47,53,55,101,102,103] |
| TRPV3-mediated Ca2+ sparklets | TRPV3 in the PM | Area: ~1–2 µm2; duration: ~70 ms; frequency: ~0.6 Hz | SKCa/IKCa channels to induce endothelium-dependent vasodilation in cerebral parenchymal arterioles | [104,105] |
| TRPA1-mediated Ca2+ sparklets | TRPA1 in the PM | Area < 1 µm2; duration > 200 ms; frequency: ~0.3 Hz | SKCa/IKCa channels and TRPA1/Panx1/purinergic signaling to induce endothelium-dependent vasodilation in cerebral parenchymal arterioles | [106,107,108,109] |
| NMDAR-mediated Ca2+ sparklets | NMDARs in the PM | Area: 8 µm2; duration: 300 ms and 500 ms; frequency: ~0.15 Hz | SKCa/IKCa channels and eNOS to induce endothelium-dependent vasodilation in cerebral parenchymal arteries | [14,110] |
2.3. Endothelial Ca2+ Microdomains Generated by Extracellular Ca2+ Entry
2.3.1. Ca2+ Sparklets in Native Endothelial Cells: TRPV3, TRPV4 and TRPA1
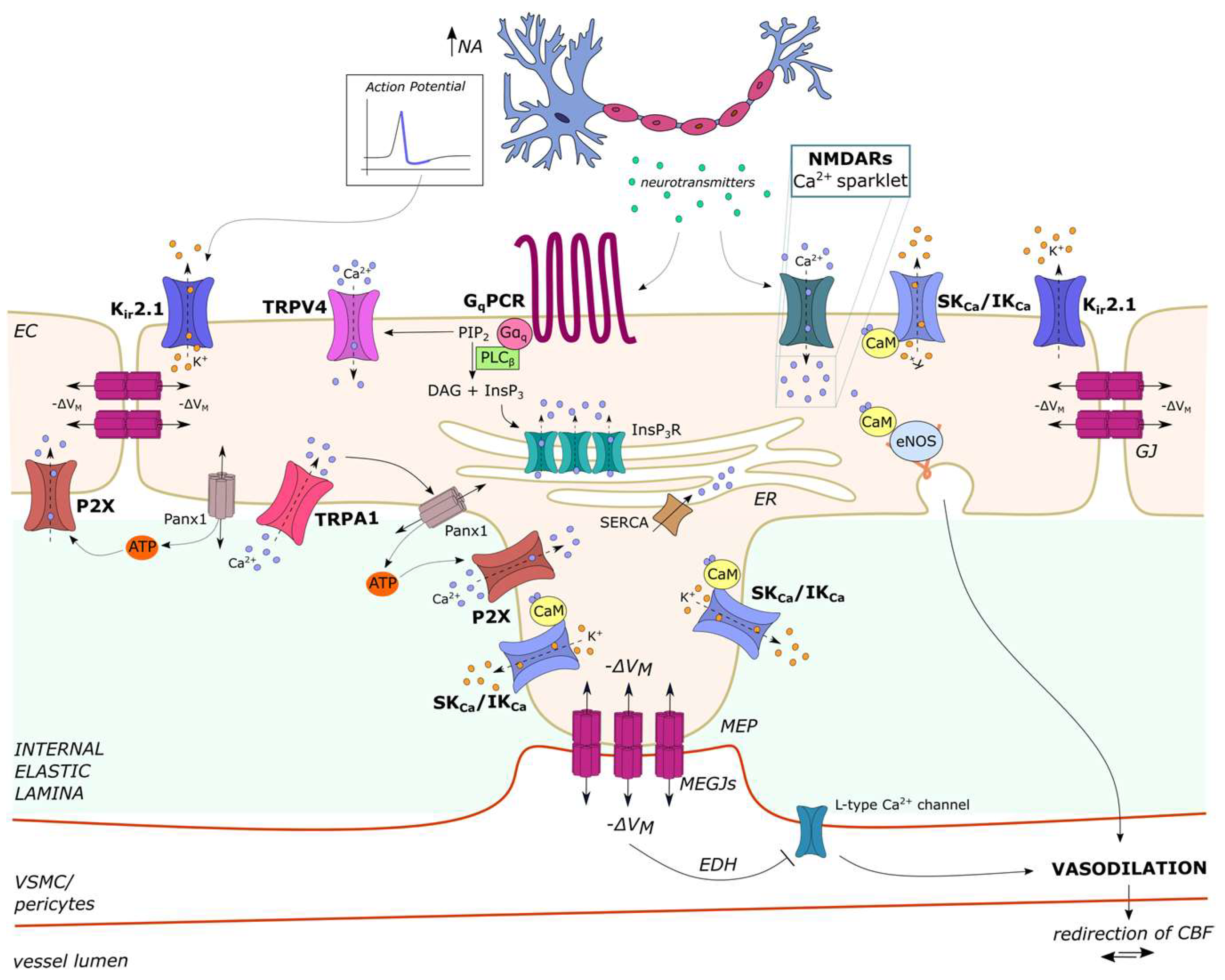
2.3.2. Ca2+ Sparklets in Native Cerebrovascular Endothelial Cells: NMDARs
2.3.3. Local Ca2+ Signals Activated by Store-Operated Ca2+ Entry (SOCE) in Vascular Endothelial Cells
2.4. Intra- and Intercellular Endothelial Ca2+ Waves
2.4.1. Intracellular Ca2+ Waves in Vascular Endothelial Cells
2.4.2. Intracellular Ca2+ Waves Induced by Neuronal Activity in Cerebrovascular Endothelial Cells
2.4.3. Intercellular Ca2+ Waves in Vascular Endothelial Cells
3. Vascular Endothelial Cells Use Ca2+ Signals to Interpret the Local Microenvironment and Transfer Information to Distant Sites
4. Local Endothelial Ca2+ Signals Regulate Mean Arterial Pressure and Local Blood Perfusion
4.1. The Selective Coupling of SOCE with eNOS: Indirect Evidence for Orai1-Mediated Ca2+ Sparklets in Vascular Endothelial Cells in Large Conduit Vessels
4.2. Ca2+ Sparklets and Ca2+ Pulsars Recruit SKCa/IKCa Channels to Induce EDH-Dependent Vasorelaxation
4.2.1. Ca2+ Sparklets Induce EDH-Dependent Vasorelaxation in Systemic Resistance Vessels and in Brain Parenchymal Arterioles
4.2.2. InsP3-Driven Ca2+ Pulsars and Wavelets Induce EDH-Dependent Vasorelaxation in Systemic Resistance Vessels
4.2.3. Why Do Local Ca2+ Signals at MEPs in Systemic Resistance Vessels Fail to Stimulate Robust NO Release?
4.3. TRPV4-Mediated Ca2+ Sparklets Induce NO Release in Pulmonary Resistance Arteries
4.4. The Complex Regulation of Cerebral Blood Flow (CBF) by Endothelial Ca2+ Signaling
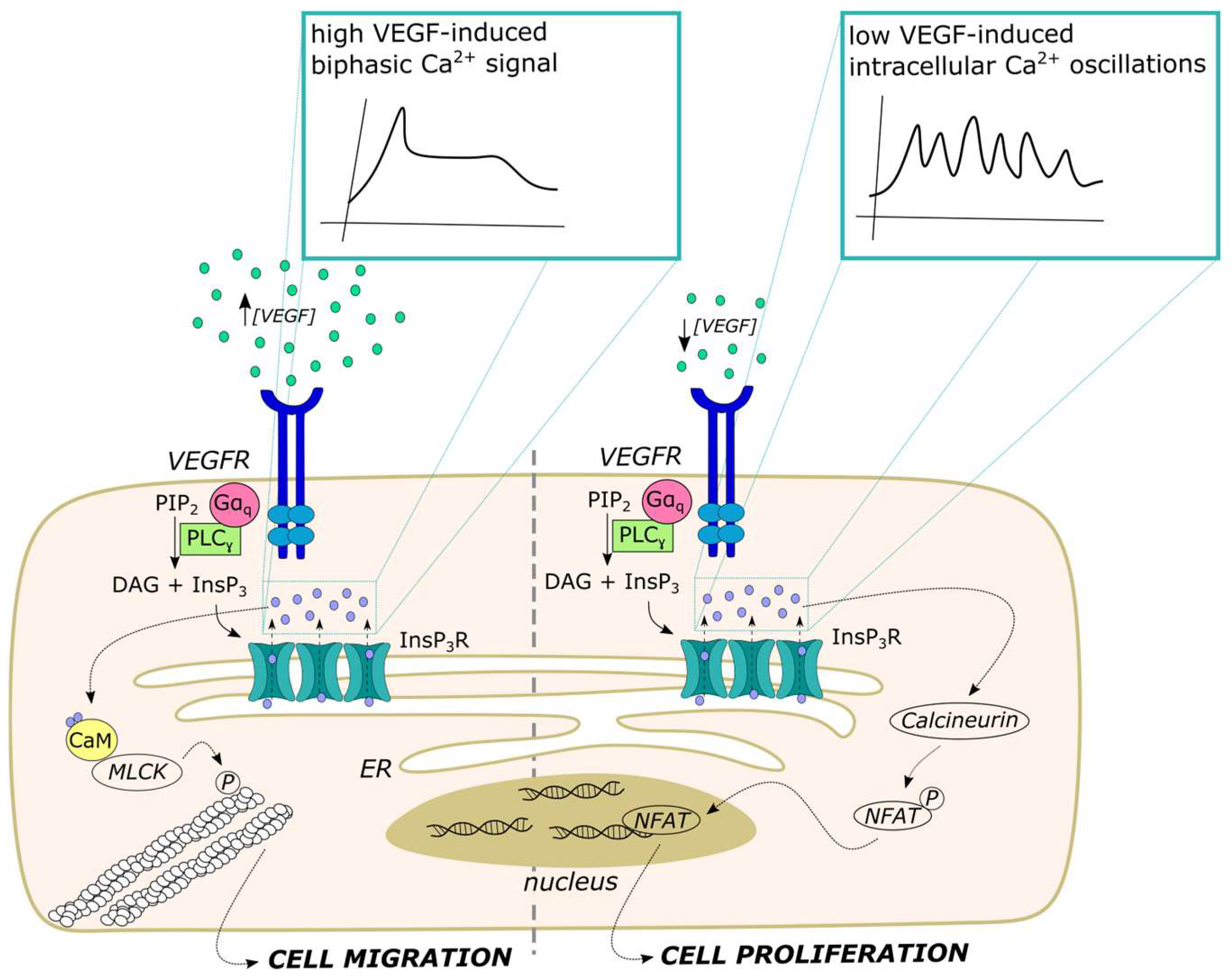
5. Distinct Spatio-Temporal Patterns of Endothelial Ca2+ Signals Regulate Angiogenesis
5.1. VEGF-Induced Intracellular Ca2+ Oscillations Select Endothelial Tip and Stalk Cells
5.2. VEGF Stimulates Endothelial Cell Proliferation and Migration through Distinct Ca2+ Signatures
5.3. Distinct Ca2+ Signatures Control ECFC Proliferation and Migration
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kruger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef]
- Bkaily, G.; Jacques, D. Morphological and Functional Remodeling of Vascular Endothelium in Cardiovascular Diseases. Int. J. Mol. Sci. 2023, 24, 1998. [Google Scholar] [CrossRef] [PubMed]
- Marzoog, B.A. Tree of life: Endothelial cell in norm and disease, the good guy is a partner in crime! Anat. Cell Biol. 2023, 56, 166–178. [Google Scholar] [CrossRef] [PubMed]
- McCarron, J.G.; Lee, M.D.; Wilson, C. The Endothelium Solves Problems That Endothelial Cells Do Not Know Exist. Trends Pharmacol. Sci. 2017, 38, 322–338. [Google Scholar] [CrossRef]
- Murphy, T.V.; Sandow, S.L. Agonist-evoked endothelial Ca2+ signalling microdomains. Curr. Opin. Pharmacol. 2019, 45, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, M.; Hong, K.; Sonkusare, S.K. Calcium signals that determine vascular resistance. Wiley Interdiscip. Rev. Syst. Biol. Med. 2019, 11, e1448. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Soda, T.; Moccia, F. Endothelial signaling at the core of neurovascular coupling: The emerging role of endothelial inward-rectifier K(+) (Kir2.1) channels and N-methyl-d-aspartate receptors in the regulation of cerebral blood flow. Int. J. Biochem. Cell Biol. 2021, 135, 105983. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca2+ Signaling, Angiogenesis and Vasculogenesis: Just What It Takes to Make a Blood Vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef]
- Jackson, W.F. Calcium-Dependent Ion Channels and the Regulation of Arteriolar Myogenic Tone. Front. Physiol. 2021, 12, 770450. [Google Scholar] [CrossRef]
- Lemmey, H.A.L.; Garland, C.J.; Dora, K.A. Intrinsic regulation of microvascular tone by myoendothelial feedback circuits. Curr. Top. Membr. 2020, 85, 327–355. [Google Scholar] [CrossRef]
- Moccia, F. Calcium Signaling in Endothelial Colony Forming Cells in Health and Disease. Adv. Exp. Med. Biol. 2020, 1131, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Yang, J.; Santulli, G.; Reiken, S.R.; Wronska, A.; Kim, M.M.; Osborne, B.W.; Lacampagne, A.; Yin, Y.; Marks, A.R. Maintenance of normal blood pressure is dependent on IP3R1-mediated regulation of eNOS. Proc. Natl. Acad. Sci. USA 2016, 113, 8532–8537. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Zhao, L.; Jing, R.; Trexler, C.; Wang, H.; Li, Y.; Tang, H.; Huang, F.; Zhang, F.; Fang, X.; et al. Inositol 1,4,5-Trisphosphate Receptors in Endothelial Cells Play an Essential Role in Vasodilation and Blood Pressure Regulation. J. Am. Heart Assoc. 2019, 8, e011704. [Google Scholar] [CrossRef] [PubMed]
- Peters, E.C.; Gee, M.T.; Pawlowski, L.N.; Kath, A.M.; Polk, F.D.; Vance, C.J.; Sacoman, J.L.; Pires, P.W. Amyloid-beta disrupts unitary calcium entry through endothelial NMDA receptors in mouse cerebral arteries. J. Cereb. Blood Flow. Metab. 2021, 42, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Scarpellino, G.; Genova, T.; Avanzato, D.; Bernardini, M.; Bianco, S.; Petrillo, S.; Tolosano, E.; de Almeida Vieira, J.R.; Bussolati, B.; Fiorio Pla, A.; et al. Purinergic Calcium Signals in Tumor-Derived Endothelium. Cancers 2019, 11, 766. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef]
- Wilson, C.; Zhang, X.; Lee, M.D.; MacDonald, M.; Heathcote, H.R.; Alorfi, N.M.N.; Buckley, C.; Dolan, S.; McCarron, J.G. Disrupted Endothelial Cell Heterogeneity and Network Organization Impair Vascular Function in Prediabetic Obesity. Metabolism 2020, 111, 154340. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Guzman-Silva, A.; Vargaz-Guadarrama, A.; Flores-Alonso, J.C.; Alonso-Romero, J.; Trevino, S.; Sanchez-Gomez, J.; Coyotl-Santiago, N.; Garcia-Carrasco, M.; Moccia, F. Type 2 Diabetes Alters Intracellular Ca2+ Handling in Native Endothelium of Excised Rat Aorta. Int. J. Mol. Sci. 2019, 21, 250. [Google Scholar] [CrossRef]
- Noy, P.J.; Gavin, R.L.; Colombo, D.; Haining, E.J.; Reyat, J.S.; Payne, H.; Thielmann, I.; Lokman, A.B.; Neag, G.; Yang, J.; et al. Tspan18 is a novel regulator of the Ca2+ channel Orai1 and von Willebrand factor release in endothelial cells. Haematologica 2019, 104, 1892–1905. [Google Scholar] [CrossRef]
- Gandhirajan, R.K.; Meng, S.; Chandramoorthy, H.C.; Mallilankaraman, K.; Mancarella, S.; Gao, H.; Razmpour, R.; Yang, X.F.; Houser, S.R.; Chen, J.; et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Investig. 2013, 123, 887–902. [Google Scholar] [CrossRef]
- Hakim, M.A.; Chum, P.P.; Buchholz, J.N.; Behringer, E.J. Aging Alters Cerebrovascular Endothelial GPCR and K+ Channel Function: Divergent Role of Biological Sex. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2064–2073. [Google Scholar] [CrossRef]
- Hakim, M.A.; Behringer, E.J. Development of Alzheimer’s Disease Progressively Alters Sex-Dependent KCa and Sex-Independent KIR Channel Function in Cerebrovascular Endothelium. J. Alzheimers Dis. 2020, 76, 1423–1442. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.; Sellitto, C.; Komici, K.; Hay, E.; Rocca, A.; De Blasiis, P.; Lucariello, A.; Moccia, F.; Guerra, G. Transient Receptor Potential (TRP) Channels in Tumor Vascularization. Int. J. Mol. Sci. 2022, 23, 14253. [Google Scholar] [CrossRef] [PubMed]
- Behringer, E.J.; Segal, S.S. Impact of Aging on Calcium Signaling and Membrane Potential in Endothelium of Resistance Arteries: A Role for Mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Francis, M. Decoding dynamic Ca2+ signaling in the vascular endothelium. Front. Physiol. 2014, 5, 447. [Google Scholar] [CrossRef] [PubMed]
- McCarron, J.G.; Wilson, C.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Lee, M.D. Heterogeneity and emergent behaviour in the vascular endothelium. Curr. Opin. Pharmacol. 2019, 45, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, M.; Sonkusare, S.K. The Calcium Signaling Mechanisms in Arterial Smooth Muscle and Endothelial Cells. Compr. Physiol. 2021, 11, 1831–1869. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Saunter, C.D.; Girkin, J.M.; McCarron, J.G. Clusters of specialized detector cells provide sensitive and high fidelity receptor signaling in the intact endothelium. FASEB J. 2016, 30, 2000–2013. [Google Scholar] [CrossRef]
- Wilson, C.; Lee, M.D.; McCarron, J.G. Acetylcholine released by endothelial cells facilitates flow-mediated dilatation. J. Physiol. 2016, 594, 7267–7307. [Google Scholar] [CrossRef]
- Boittin, F.X.; Alonso, F.; Le Gal, L.; Allagnat, F.; Beny, J.L.; Haefliger, J.A. Connexins and M3 muscarinic receptors contribute to heterogeneous Ca2+ signaling in mouse aortic endothelium. Cell Physiol. Biochem. 2013, 31, 166–178. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef] [PubMed]
- Scarpellino, G.; Genova, T.; Quarta, E.; Distasi, C.; Dionisi, M.; Fiorio Pla, A.; Munaron, L. P2X Purinergic Receptors Are Multisensory Detectors for Micro-Environmental Stimuli That Control Migration of Tumoral Endothelium. Cancers 2022, 14, 2743. [Google Scholar] [CrossRef] [PubMed]
- Bintig, W.; Begandt, D.; Schlingmann, B.; Gerhard, L.; Pangalos, M.; Dreyer, L.; Hohnjec, N.; Couraud, P.O.; Romero, I.A.; Weksler, B.B.; et al. Purine receptors and Ca2+ signalling in the human blood-brain barrier endothelial cell line hCMEC/D3. Purinergic Signal 2012, 8, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Bachkoenig, O.A.; Gottschalk, B.; Malli, R.; Graier, W.F. An unexpected effect of risperidone reveals a nonlinear relationship between cytosolic Ca2+ and mitochondrial Ca2+ uptake. Curr. Top. Membr. 2022, 90, 13–35. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Faris, P.; Pellavio, G.; Orgiu, M.; Negri, S.; Forcaia, G.; Var-Gaz-Guadarrama, V.; Garcia-Carrasco, M.; Botta, L.; Sancini, G.; et al. Histamine induces intracellular Ca2+ oscillations and nitric oxide release in endothelial cells from brain microvascular circulation. J. Cell Physiol. 2020, 235, 1515–1530. [Google Scholar] [CrossRef] [PubMed]
- Dora, K.A.; Lin, J.; Borysova, L.; Beleznai, T.; Taggart, M.; Ascione, R.; Garland, C. Signaling and structures underpinning conducted vasodilation in human and porcine intramyocardial coronary arteries. Front. Cardiovasc. Med. 2022, 9, 980628. [Google Scholar] [CrossRef] [PubMed]
- Noren, D.P.; Chou, W.H.; Lee, S.H.; Qutub, A.A.; Warmflash, A.; Wagner, D.S.; Popel, A.S.; Levchenko, A. Endothelial cells decode VEGF-mediated Ca2+ signaling patterns to produce distinct functional responses. Sci. Signal 2016, 9, ra20. [Google Scholar] [CrossRef]
- Yokota, Y.; Nakajima, H.; Wakayama, Y.; Muto, A.; Kawakami, K.; Fukuhara, S.; Mochizuki, N. Endothelial Ca2+ oscillations reflect VEGFR signaling-regulated angiogenic capacity in vivo. Elife 2015, 4, e08817. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef]
- Pafumi, I.; Favia, A.; Gambara, G.; Papacci, F.; Ziparo, E.; Palombi, F.; Filippini, A. Regulation of Angiogenic Functions by Angiopoietins through Calcium-Dependent Signaling Pathways. Biomed. Res. Int. 2015, 2015, 965271. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Tullii, G.; Vismara, M.; Pellegata, A.F.; Lodola, F.; Guidetti, G.; Rosti, V.; Antognazza, M.R.; Moccia, F. Conjugated polymers mediate intracellular Ca2+ signals in circulating endothelial colony forming cells through the reactive oxygen species-dependent activation of Transient Receptor Potential Vanilloid 1 (TRPV1). Cell Calcium 2022, 101, 102502. [Google Scholar] [CrossRef]
- Hu, Q.; Corda, S.; Zweier, J.L.; Capogrossi, M.C.; Ziegelstein, R.C. Hydrogen peroxide induces intracellular calcium oscillations in human aortic endothelial cells. Circulation 1998, 97, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca2+ oscillations and nitric oxide release by inducing NAADP- and InsP3-dependent Ca2+ release in mouse brain endothelial cells. J. Cell Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef] [PubMed]
- Soda, T.; Brunetti, V.; Berra-Romani, R.; Moccia, F. The Emerging Role of N-Methyl-D-Aspartate (NMDA) Receptors in the Cardiovascular System: Physiological Implications, Pathological Consequences, and Therapeutic Perspectives. Int. J. Mol. Sci. 2023, 24, 3914. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kumar, T.P.; Joshee, S.; Kirschstein, T.; Subburaju, S.; Khalili, J.S.; Kloepper, J.; Du, C.; Elkhal, A.; Szabo, G.; et al. Endothelial cell-derived GABA signaling modulates neuronal migration and postnatal behavior. Cell Res. 2018, 28, 221–248. [Google Scholar] [CrossRef]
- Negri, S.; Scolari, F.; Vismara, M.; Brunetti, V.; Faris, P.; Terribile, G.; Sancini, G.; Berra-Romani, R.; Moccia, F. GABA(A) and GABA(B) Receptors Mediate GABA-Induced Intracellular Ca2+ Signals in Human Brain Microvascular Endothelial Cells. Cells 2022, 11, 3860. [Google Scholar] [CrossRef]
- Hong, K.; Cope, E.L.; DeLalio, L.J.; Marziano, C.; Isakson, B.E.; Sonkusare, S.K. TRPV4 (Transient Receptor Potential Vanilloid 4) Channel-Dependent Negative Feedback Mechanism Regulates G(q) Protein-Coupled Receptor-Induced Vasoconstriction. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 542–554. [Google Scholar] [CrossRef]
- Garland, C.J.; Bagher, P.; Powell, C.; Ye, X.; Lemmey, H.A.L.; Borysova, L.; Dora, K.A. Voltage-dependent Ca2+ entry into smooth muscle during contraction promotes endothelium-mediated feedback vasodilation in arterioles. Sci. Signal 2017, 10, eaal3806. [Google Scholar] [CrossRef]
- Nausch, L.W.; Bonev, A.D.; Heppner, T.J.; Tallini, Y.; Kotlikoff, M.I.; Nelson, M.T. Sympathetic nerve stimulation induces local endothelial Ca2+ signals to oppose vasoconstriction of mouse mesenteric arteries. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H594–H602. [Google Scholar] [CrossRef]
- Tran, C.H.; Taylor, M.S.; Plane, F.; Nagaraja, S.; Tsoukias, N.M.; Solodushko, V.; Vigmond, E.J.; Furstenhaupt, T.; Brigdan, M.; Welsh, D.G. Endothelial Ca2+ wavelets and the induction of myoendothelial feedback. Am. J. Physiol. Cell Physiol. 2012, 302, C1226–C1242. [Google Scholar] [CrossRef]
- Wier, W.G.; Mauban, J.R.H. Imaging sympathetic neurogenic Ca2+ signaling in blood vessels. Auton. Neurosci. 2017, 207, 59–66. [Google Scholar] [CrossRef]
- Swain, S.M.; Liddle, R.A. Piezo1 acts upstream of TRPV4 to induce pathological changes in endothelial cells due to shear stress. J. Biol. Chem. 2021, 296, 100171. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Zhang, C.; He, C.; Zhang, K.; Kan, H.; Mao, A.; Ma, X. Physiological levels of fluid shear stress modulate vascular function through TRPV4 sparklets. Acta Biochim Biophys Sin 2022, 54, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, B.; Tumova, S.; Muraki, K.; Bruns, A.; Ludlow, M.J.; Sedo, A.; Hyman, A.J.; McKeown, L.; Young, R.S.; et al. Piezo1 integration of vascular architecture with physiological force. Nature 2014, 515, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Bagher, P.; Beleznai, T.; Kansui, Y.; Mitchell, R.; Garland, C.J.; Dora, K.A. Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proc. Natl. Acad. Sci. USA 2012, 109, 18174–18179. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Raqeeb, A.; Avelino-Cruz, J.E.; Moccia, F.; Oldani, A.; Speroni, F.; Taglietti, V.; Tanzi, F. Ca2+ signaling in injured in situ endothelium of rat aorta. Cell Calcium 2008, 44, 298–309. [Google Scholar] [CrossRef]
- Zhao, Z.; Walczysko, P.; Zhao, M. Intracellular Ca2+ stores are essential for injury induced Ca2+ signaling and re-endothelialization. J. Cell Physiol. 2008, 214, 595–603. [Google Scholar] [CrossRef]
- Luo, H.; Rossi, E.; Saubamea, B.; Chasseigneaux, S.; Cochois, V.; Choublier, N.; Smirnova, M.; Glacial, F.; Perriere, N.; Bourdoulous, S.; et al. Cannabidiol Increases Proliferation, Migration, Tubulogenesis, and Integrity of Human Brain Endothelial Cells through TRPV2 Activation. Mol. Pharm. 2019, 16, 1312–1326. [Google Scholar] [CrossRef]
- Taylor, M.S.; Francis, M.; Qian, X.; Solodushko, V. Dynamic Ca2+ signal modalities in the vascular endothelium. Microcirculation 2012, 19, 423–429. [Google Scholar] [CrossRef]
- Thakore, P.; Earley, S. Transient Receptor Potential Channels and Endothelial Cell Calcium Signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar] [CrossRef]
- Moccia, F.; Brunetti, V.; Perna, A.; Guerra, G.; Soda, T.; Berra-Romani, R. The Molecular Heterogeneity of Store-Operated Ca2+ Entry in Vascular Endothelial Cells: The Different roles of Orai1 and TRPC1/TRPC4 Channels in the Transition from Ca2+-Selective to Non-Selective Cation Currents. Int. J. Mol. Sci. 2023, 24, 3259. [Google Scholar] [CrossRef]
- Sun, M.Y.; Geyer, M.; Komarova, Y.A. IP3 receptor signaling and endothelial barrier function. Cell Mol. Life Sci. 2017, 74, 4189–4207. [Google Scholar] [CrossRef] [PubMed]
- Prole, D.L.; Taylor, C.W. Structure and Function of IP3 Receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035063. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Perna, A.; De Luca, A.; Soda, T.; Romani, R.B.; Guerra, G. Targeting Endolysosomal Two-Pore Channels to Treat Cardiovascular Disorders in the Novel COronaVIrus Disease 2019. Front Physiol 2021, 12, 629119. [Google Scholar] [CrossRef]
- Galione, A.; Davis, L.C.; Martucci, L.L.; Morgan, A.J. NAADP-Mediated Ca2+ Signalling. Handb. Exp. Pharmacol. 2023, 278, 3–34. [Google Scholar] [CrossRef] [PubMed]
- Blatter, L.A. Tissue Specificity: SOCE: Implications for Ca2+ Handling in Endothelial Cells. Adv. Exp. Med. Biol. 2017, 993, 343–361. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial Transient Receptor Potential Channels and Vascular Remodeling: Extracellular Ca2+ Entry for Angiogenesis, Arteriogenesis and Vasculogenesis. Front. Physiol. 2019, 10, 1618. [Google Scholar] [CrossRef]
- Smani, T.; Gomez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.M.; Rosado, J.A. TRP Channels in Angiogenesis and Other Endothelial Functions. Front. Physiol. 2018, 9, 1731. [Google Scholar] [CrossRef]
- Muzorewa, T.T.; Buerk, D.G.; Jaron, D.; Barbee, K.A. TRPC channel-derived calcium fluxes differentially regulate ATP and flow-induced activation of eNOS. Nitric Oxide 2021, 111–112, 1–13. [Google Scholar] [CrossRef]
- Andrikopoulos, P.; Eccles, S.A.; Yaqoob, M.M. Coupling between the TRPC3 ion channel and the NCX1 transporter contributed to VEGF-induced ERK1/2 activation and angiogenesis in human primary endothelial cells. Cell Signal 2017, 37, 12–30. [Google Scholar] [CrossRef]
- Moccia, F.; Lucariello, A.; Guerra, G. TRPC3-mediated Ca2+ signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: The role of autologous endothelial colony forming cells. J. Cell Physiol. 2018, 233, 3901–3917. [Google Scholar] [CrossRef]
- Adapala, R.K.; Talasila, P.K.; Bratz, I.N.; Zhang, D.X.; Suzuki, M.; Meszaros, J.G.; Thodeti, C.K. PKCalpha mediates acetylcholine-induced activation of TRPV4-dependent calcium influx in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H757–H765. [Google Scholar] [CrossRef]
- Zuccolo, E.; Dragoni, S.; Poletto, V.; Catarsi, P.; Guido, D.; Rappa, A.; Reforgiato, M.; Lodola, F.; Lim, D.; Rosti, V.; et al. Arachidonic acid-evoked Ca2+ signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vascul Pharmacol. 2016, 87, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Sonkusare, S.K. Endothelial TRPV4 channels and vasodilator reactivity. Curr. Top. Membr. 2020, 85, 89–117. [Google Scholar] [CrossRef]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef]
- Miyakawa, T.; Maeda, A.; Yamazawa, T.; Hirose, K.; Kurosaki, T.; Iino, M. Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 1999, 18, 1303–1308. [Google Scholar] [CrossRef]
- Zhang, S.; Fritz, N.; Ibarra, C.; Uhlen, P. Inositol 1,4,5-trisphosphate receptor subtype-specific regulation of calcium oscillations. Neurochem. Res. 2011, 36, 1175–1185. [Google Scholar] [CrossRef]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca2+ oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Pellavio, G.; Botta, L.; Orgiu, M.; Forcaia, G.; Sancini, G.; Laforenza, U.; Moccia, F. Group 1 metabotropic glutamate receptors trigger glutamate-induced intracellular Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. Cell Mol. Life Sci. 2020, 77, 2235–2253. [Google Scholar] [CrossRef]
- Ledoux, J.; Taylor, M.S.; Bonev, A.D.; Hannah, R.M.; Solodushko, V.; Shui, B.; Tallini, Y.; Kotlikoff, M.I.; Nelson, M.T. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc. Natl. Acad. Sci. USA 2008, 105, 9627–9632. [Google Scholar] [CrossRef]
- Kansui, Y.; Garland, C.J.; Dora, K.A. Enhanced spontaneous Ca2+ events in endothelial cells reflect signalling through myoendothelial gap junctions in pressurized mesenteric arteries. Cell Calcium 2008, 44, 135–146. [Google Scholar] [CrossRef]
- Burdyga, T.; Shmygol, A.; Eisner, D.A.; Wray, S. A new technique for simultaneous and in situ measurements of Ca2+ signals in arteriolar smooth muscle and endothelial cells. Cell Calcium 2003, 34, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Borisova, L.; Wray, S.; Eisner, D.A.; Burdyga, T. How structure, Ca signals, and cellular communications underlie function in precapillary arterioles. Circ. Res. 2009, 105, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Huser, J.; Blatter, L.A. Elementary events of agonist-induced Ca2+ release in vascular endothelial cells. Am. J. Physiol. 1997, 273, C1775–C1782. [Google Scholar] [CrossRef]
- Moccia, F.; Berra-Romani, R.; Tritto, S.; Signorelli, S.; Taglietti, V.; Tanzi, F. Epidermal growth factor induces intracellular Ca2+ oscillations in microvascular endothelial cells. J. Cell Physiol. 2003, 194, 139–150. [Google Scholar] [CrossRef]
- Ikeda, M.; Ariyoshi, H.; Kambayashi, J.; Fujitani, K.; Shinoki, N.; Sakon, M.; Kawasaki, T.; Monden, M. Separate analysis of nuclear and cytosolic Ca2+ concentrations in human umbilical vein endothelial cells. J. Cell Biochem. 1996, 63, 23–36. [Google Scholar] [CrossRef]
- Garnier-Raveaud, S.; Usson, Y.; Cand, F.; Robert-Nicoud, M.; Verdetti, J.; Faury, G. Identification of membrane calcium channels essential for cytoplasmic and nuclear calcium elevations induced by vascular endothelial growth factor in human endothelial cells. Growth Factors 2001, 19, 35–48. [Google Scholar] [CrossRef]
- McSherry, I.N.; Spitaler, M.M.; Takano, H.; Dora, K.A. Endothelial cell Ca2+ increases are independent of membrane potential in pressurized rat mesenteric arteries. Cell Calcium 2005, 38, 23–33. [Google Scholar] [CrossRef]
- Mumtaz, S.; Burdyga, G.; Borisova, L.; Wray, S.; Burdyga, T. The mechanism of agonist induced Ca2+ signalling in intact endothelial cells studied confocally in in situ arteries. Cell Calcium 2011, 49, 66–77. [Google Scholar] [CrossRef]
- Socha, M.J.; Domeier, T.L.; Behringer, E.J.; Segal, S.S. Coordination of intercellular Ca2+ signaling in endothelial cell tubes of mouse resistance arteries. Microcirculation 2012, 19, 757–770. [Google Scholar] [CrossRef]
- Duza, T.; Sarelius, I.H. Localized transient increases in endothelial cell Ca2+ in arterioles in situ: Implications for coordination of vascular function. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2322–H2331. [Google Scholar] [CrossRef] [PubMed]
- Duza, T.; Sarelius, I.H. Increase in endothelial cell Ca2+ in response to mouse cremaster muscle contraction. J. Physiol. 2004, 555, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.J.; Dora, K.A. Endothelium-Dependent Hyperpolarization: The Evolution of Myoendothelial Microdomains. J. Cardiovasc. Pharmacol. 2021, 78, S3–S12. [Google Scholar] [CrossRef]
- Toussaint, F.; Charbel, C.; Blanchette, A.; Ledoux, J. CaMKII regulates intracellular Ca2+ dynamics in native endothelial cells. Cell Calcium 2015, 58, 275–285. [Google Scholar] [CrossRef]
- Francis, M.; Waldrup, J.R.; Qian, X.; Solodushko, V.; Meriwether, J.; Taylor, M.S. Functional Tuning of Intrinsic Endothelial Ca2+ Dynamics in Swine Coronary Arteries. Circ. Res. 2016, 118, 1078–1090. [Google Scholar] [CrossRef]
- Hennessey, J.C.; Stuyvers, B.D.; McGuire, J.J. Small caliber arterial endothelial cells calcium signals elicited by PAR2 are preserved from endothelial dysfunction. Pharmacol. Res. Perspect. 2015, 3, e00112. [Google Scholar] [CrossRef]
- Wilson, C.; Saunter, C.D.; Girkin, J.M.; McCarron, J.G. Pressure-dependent regulation of Ca2+ signalling in the vascular endothelium. J. Physiol. 2015, 593, 5231–5253. [Google Scholar] [CrossRef]
- Wilson, C.; Saunter, C.D.; Girkin, J.M.; McCarron, J.G. Advancing Age Decreases Pressure-Sensitive Modulation of Calcium Signaling in the Endothelium of Intact and Pressurized Arteries. J. Vasc. Res. 2016, 53, 358–369. [Google Scholar] [CrossRef]
- Jackson, W.F.; Boerman, E.M.; Lange, E.J.; Lundback, S.S.; Cohen, K.D. Smooth muscle alpha1D-adrenoceptors mediate phenylephrine-induced vasoconstriction and increases in endothelial cell Ca2+ in hamster cremaster arterioles. Br. J. Pharmacol. 2008, 155, 514–524. [Google Scholar] [CrossRef]
- Biwer, L.A.; Good, M.E.; Hong, K.; Patel, R.K.; Agrawal, N.; Looft-Wilson, R.; Sonkusare, S.K.; Isakson, B.E. Non-Endoplasmic Reticulum-Based Calr (Calreticulin) Can Coordinate Heterocellular Calcium Signaling and Vascular Function. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 120–130. [Google Scholar] [CrossRef]
- Sonkusare, S.K.; Bonev, A.D.; Ledoux, J.; Liedtke, W.; Kotlikoff, M.I.; Heppner, T.J.; Hill-Eubanks, D.C.; Nelson, M.T. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012, 336, 597–601. [Google Scholar] [CrossRef]
- Sonkusare, S.K.; Dalsgaard, T.; Bonev, A.D.; Hill-Eubanks, D.C.; Kotlikoff, M.I.; Scott, J.D.; Santana, L.F.; Nelson, M.T. AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension. Sci. Signal 2014, 7, ra66. [Google Scholar] [CrossRef]
- Ottolini, M.; Daneva, Z.; Chen, Y.L.; Cope, E.L.; Kasetti, R.B.; Zode, G.S.; Sonkusare, S.K. Mechanisms underlying selective coupling of endothelial Ca2+ signals with eNOS vs. IK/SK channels in systemic and pulmonary arteries. J. Physiol. 2020, 598, 3577–3596. [Google Scholar] [CrossRef]
- Earley, S. Endothelium-dependent cerebral artery dilation mediated by transient receptor potential and Ca2+-activated K+ channels. J. Cardiovasc. Pharmacol. 2011, 57, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.W.; Sullivan, M.N.; Pritchard, H.A.; Robinson, J.J.; Earley, S. Unitary TRPV3 channel Ca2+ influx events elicit endothelium-dependent dilation of cerebral parenchymal arterioles. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H2031–H2041. [Google Scholar] [CrossRef]
- Qian, X.; Francis, M.; Solodushko, V.; Earley, S.; Taylor, M.S. Recruitment of dynamic endothelial Ca2+ signals by the TRPA1 channel activator AITC in rat cerebral arteries. Microcirculation 2013, 20, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.N.; Gonzales, A.L.; Pires, P.W.; Bruhl, A.; Leo, M.D.; Li, W.; Oulidi, A.; Boop, F.A.; Feng, Y.; Jaggar, J.H.; et al. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci. Signal 2015, 8, ra2. [Google Scholar] [CrossRef]
- Pires, P.W.; Earley, S. Neuroprotective effects of TRPA1 channels in the cerebral endothelium following ischemic stroke. Elife 2018, 7, e35316. [Google Scholar] [CrossRef]
- Thakore, P.; Alvarado, M.G.; Ali, S.; Mughal, A.; Pires, P.W.; Yamasaki, E.; Pritchard, H.A.; Isakson, B.E.; Tran, C.H.T.; Earley, S. Brain endothelial cell TRPA1 channels initiate neurovascular coupling. Elife 2021, 10, e63040. [Google Scholar] [CrossRef] [PubMed]
- LeMaistre, J.L.; Sanders, S.A.; Stobart, M.J.; Lu, L.; Knox, J.D.; Anderson, H.D.; Anderson, C.M. Coactivation of NMDA receptors by glutamate and D-serine induces dilation of isolated middle cerebral arteries. J. Cereb. Blood Flow. Metab. 2012, 32, 537–547. [Google Scholar] [CrossRef]
- Moccia, F.; Zuccolo, E.; Di Nezza, F.; Pellavio, G.; Faris, P.S.; Negri, S.; De Luca, A.; Laforenza, U.; Ambrosone, L.; Rosti, V.; et al. Nicotinic acid adenine dinucleotide phosphate activates two-pore channel TPC1 to mediate lysosomal Ca2+ release in endothelial colony-forming cells. J. Cell Physiol. 2021, 236, 688–705. [Google Scholar] [CrossRef]
- Straub, A.C.; Billaud, M.; Johnstone, S.R.; Best, A.K.; Yemen, S.; Dwyer, S.T.; Looft-Wilson, R.; Lysiak, J.J.; Gaston, B.; Palmer, L.; et al. Compartmentalized connexin 43 s-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 399–407. [Google Scholar] [CrossRef]
- Straub, A.C.; Zeigler, A.C.; Isakson, B.E. The myoendothelial junction: Connections that deliver the message. Physiology (Bethesda) 2014, 29, 242–249. [Google Scholar] [CrossRef]
- Wang, S.Q.; Song, L.S.; Lakatta, E.G.; Cheng, H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature 2001, 410, 592–596. [Google Scholar] [CrossRef]
- Harraz, O.F.; Longden, T.A.; Hill-Eubanks, D.; Nelson, M.T. PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. Elife 2018, 7, e38689. [Google Scholar] [CrossRef]
- Dragoni, S.; Guerra, G.; Fiorio Pla, A.; Bertoni, G.; Rappa, A.; Poletto, V.; Bottino, C.; Aronica, A.; Lodola, F.; Cinelli, M.P.; et al. A functional Transient Receptor Potential Vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J. Cell Physiol. 2015, 230, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.; Ali, S.; Earley, S. Regulation of vascular tone by transient receptor potential ankyrin 1 channels. Curr. Top. Membr. 2020, 85, 119–150. [Google Scholar] [CrossRef]
- Alvarado, M.G.; Thakore, P.; Earley, S. Transient Receptor Potential Channel Ankyrin 1: A Unique Regulator of Vascular Function. Cells 2021, 10, 1167. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Brunetti, V.; Pellavio, G.; Soda, T.; Laforenza, U.; Scarpellino, G.; Moccia, F. Allyl Isothiocianate Induces Ca2+ Signals and Nitric Oxide Release by Inducing Reactive Oxygen Species Production in the Human Cerebrovascular Endothelial Cell Line hCMEC/D3. Cells 2023, 12, 1732. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Maniezzi, C.; Pellavio, G.; Spaiardi, P.; Botta, L.; Laforenza, U.; Biella, G.; Moccia, D.F. NMDA receptors elicit flux-independent intracellular Ca2+ signals via metabotropic glutamate receptors and flux-dependent nitric oxide release in human brain microvascular endothelial cells. Cell Calcium 2021, 99, 102454. [Google Scholar] [CrossRef]
- Kassan, M.; Zhang, W.; Aissa, K.A.; Stolwijk, J.; Trebak, M.; Matrougui, K. Differential role for stromal interacting molecule 1 in the regulation of vascular function. Pflugers Arch. 2015, 467, 1195–1202. [Google Scholar] [CrossRef]
- Nishimoto, M.; Mizuno, R.; Fujita, T.; Isshiki, M. Stromal interaction molecule 1 modulates blood pressure via NO production in vascular endothelial cells. Hypertens. Res. 2018, 41, 506. [Google Scholar] [CrossRef]
- Freichel, M.; Suh, S.H.; Pfeifer, A.; Schweig, U.; Trost, C.; Weissgerber, P.; Biel, M.; Philipp, S.; Freise, D.; Droogmans, G.; et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat. Cell Biol. 2001, 3, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.C.; Seki, A.; Yang, H.W.; Hayer, A.; Carrasco, S.; Malmersjo, S.; Meyer, T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 2014, 16, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Huser, J.; Holda, J.R.; Kockskamper, J.; Blatter, L.A. Focal agonist stimulation results in spatially restricted Ca2+ release and capacitative Ca2+ entry in bovine vascular endothelial cells. J. Physiol. 1999, 514 Pt 1, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Choi, C.S.; Bayazid, L.; Glosemeyer, K.E.; Baker, C.C.P.; Weber, D.S. Changes in vascular reactivity and endothelial Ca2+ dynamics with chronic low flow. Microcirculation 2017, 24, e12354. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Minamiya, Y.; Fu, C.; Bhattacharya, J. Ca2+ waves in lung capillary endothelium. Circ. Res. 1996, 79, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Escue, R.; Kandasamy, K.; Parthasarathi, K. Thrombin Induces Inositol Trisphosphate-Mediated Spatially Extensive Responses in Lung Microvessels. Am. J. Pathol. 2017, 187, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Savage, A.M.; Kurusamy, S.; Chen, Y.; Jiang, Z.; Chhabria, K.; MacDonald, R.B.; Kim, H.R.; Wilson, H.L.; van Eeden, F.J.M.; Armesilla, A.L.; et al. tmem33 is essential for VEGF-mediated endothelial calcium oscillations and angiogenesis. Nat. Commun. 2019, 10, 732. [Google Scholar] [CrossRef]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef]
- Longden, T.A.; Mughal, A.; Hennig, G.W.; Harraz, O.F.; Shui, B.; Lee, F.K.; Lee, J.C.; Reining, S.; Kotlikoff, M.I.; Konig, G.M.; et al. Local IP3 receptor-mediated Ca2+ signals compound to direct blood flow in brain capillaries. Sci. Adv. 2021, 7, eabh0101. [Google Scholar] [CrossRef] [PubMed]
- Harraz, O.F.; Longden, T.A.; Dabertrand, F.; Hill-Eubanks, D.; Nelson, M.T. Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion. Proc. Natl. Acad. Sci. USA 2018, 115, E3569–E3577. [Google Scholar] [CrossRef] [PubMed]
- Rosehart, A.C.; Longden, T.A.; Weir, N.; Fontaine, J.T.; Joutel, A.; Dabertrand, F. Prostaglandin E2 Dilates Intracerebral Arterioles When Applied to Capillaries: Implications for Small Vessel Diseases. Front. Aging Neurosci. 2021, 13, 695965. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Angelone, T. Targeting endothelial ion signalling to rescue cerebral blood flow in cerebral disorders. Vascul Pharmacol. 2022, 145, 106997. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.F. Endothelial Ion Channels and Cell-Cell Communication in the Microcirculation. Front. Physiol. 2022, 13, 805149. [Google Scholar] [CrossRef] [PubMed]
- Bagher, P.; Davis, M.J.; Segal, S.S. Visualizing calcium responses to acetylcholine convection along endothelium of arteriolar networks in Cx40BAC-GCaMP2 transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H794–H802. [Google Scholar] [CrossRef] [PubMed]
- Tallini, Y.N.; Brekke, J.F.; Shui, B.; Doran, R.; Hwang, S.M.; Nakai, J.; Salama, G.; Segal, S.S.; Kotlikoff, M.I. Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: Measurements in Cx40BAC GCaMP2 transgenic mice. Circ. Res. 2007, 101, 1300–1309. [Google Scholar] [CrossRef]
- Zhang, X.; Lee, M.D.; Buckley, C.; Hollenberg, M.D.; Wilson, C.; McCarron, J.G. Endothelial PAR2 activation evokes resistance artery relaxation. J. Cell Physiol. 2023, 238, 776–789. [Google Scholar] [CrossRef]
- Domenighetti, A.A.; Beny, J.L.; Chabaud, F.; Frieden, M. An intercellular regenerative calcium wave in porcine coronary artery endothelial cells in primary culture. J. Physiol. 1998, 513 Pt 1, 103–116. [Google Scholar] [CrossRef]
- De Bock, M.; Culot, M.; Wang, N.; Bol, M.; Decrock, E.; De Vuyst, E.; da Costa, A.; Dauwe, I.; Vinken, M.; Simon, A.M.; et al. Connexin channels provide a target to manipulate brain endothelial calcium dynamics and blood-brain barrier permeability. J. Cereb. Blood Flow. Metab. 2011, 31, 1942–1957. [Google Scholar] [CrossRef]
- De Bock, M.; Culot, M.; Wang, N.; da Costa, A.; Decrock, E.; Bol, M.; Bultynck, G.; Cecchelli, R.; Leybaert, L. Low extracellular Ca2+ conditions induce an increase in brain endothelial permeability that involves intercellular Ca2+ waves. Brain Res. 2012, 1487, 78–87. [Google Scholar] [CrossRef]
- Lamb, I.R.; Novielli-Kuntz, N.M.; Murrant, C.L. Capillaries communicate with the arteriolar microvascular network by a pannexin/purinergic-dependent pathway in hamster skeletal muscle. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1699–H1711. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.D.; Wilson, C.; Saunter, C.D.; Kennedy, C.; Girkin, J.M.; McCarron, J.G. Spatially structured cell populations process multiple sensory signals in parallel in intact vascular endothelium. Sci. Signal 2018, 11, eaar4411. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Chu, T.F.; Chen, H.I.; Jen, C.J. Heterogeneity of [Ca2+](i) signaling in intact rat aortic endothelium. FASEB J. 2000, 14, 797–804. [Google Scholar] [CrossRef]
- Marie, I.; Beny, J.L. Calcium imaging of murine thoracic aorta endothelium by confocal microscopy reveals inhomogeneous distribution of endothelial cells responding to vasodilator agents. J. Vasc. Res. 2002, 39, 260–267. [Google Scholar] [CrossRef]
- Lee, M.D.; Buckley, C.; Zhang, X.; Louhivuori, L.; Uhlen, P.; Wilson, C.; McCarron, J.G. Small-world connectivity dictates collective endothelial cell signaling. Proc. Natl. Acad. Sci. USA 2022, 119, e2118927119. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef]
- Khaddaj Mallat, R.; Mathew John, C.; Kendrick, D.J.; Braun, A.P. The vascular endothelium: A regulator of arterial tone and interface for the immune system. Crit. Rev. Clin. Lab. Sci. 2017, 54, 458–470. [Google Scholar] [CrossRef]
- Sonkusare, S.K.; Laubach, V.E. Endothelial TRPV4 channels in lung edema and injury. Curr. Top. Membr. 2022, 89, 43–62. [Google Scholar] [CrossRef]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Joannides, R.; Haefeli, W.E.; Linder, L.; Richard, V.; Bakkali, E.H.; Thuillez, C.; Luscher, T.F. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation 1995, 91, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Hyre, C.E.; Unthank, J.L.; Dalsing, M.C. Direct in vivo measurement of flow-dependent nitric oxide production in mesenteric resistance arteries. J. Vasc. Surg. 1998, 27, 726–732. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Zhao, Y.; Xu, A.; Leung, S.W. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ. Res. 2016, 119, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.W.; Nunez, V.; Kaplan, L.; Granger, A.J.; Bistrong, K.; Zucker, H.L.; Kumar, P.; Sabatini, B.L.; Gu, C. Caveolae in CNS arterioles mediate neurovascular coupling. Nature 2020, 579, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Nitric oxide inhibits capacitative Ca2+ entry and enhances endoplasmic reticulum Ca2+ uptake in bovine vascular endothelial cells. J. Physiol. 2002, 539, 77–91. [Google Scholar] [CrossRef]
- Murata, T.; Lin, M.I.; Stan, R.V.; Bauer, P.M.; Yu, J.; Sessa, W.C. Genetic evidence supporting caveolae microdomain regulation of calcium entry in endothelial cells. J. Biol. Chem. 2007, 282, 16631–16643. [Google Scholar] [CrossRef]
- Barbee, K.A.; Parikh, J.B.; Liu, Y.; Buerk, D.G.; Jaron, D. Effect of spatial heterogeneity and colocalization of eNOS and capacitative calcium entry channels on shear stress-induced NO production by endothelial cells: A modeling approach. Cell Mol. Bioeng. 2018, 11, 143–155. [Google Scholar] [CrossRef]
- Zhang, B.; Naik, J.S.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Reduced membrane cholesterol after chronic hypoxia limits Orai1-mediated pulmonary endothelial Ca2+ entry. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H359–H369. [Google Scholar] [CrossRef]
- Hirano, K.; Hirano, M.; Hanada, A. Involvement of STIM1 in the proteinase-activated receptor 1-mediated Ca2+ influx in vascular endothelial cells. J. Cell Biochem. 2009, 108, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Erac, Y.; Selli, C.; Tosun, M. Alterations of store-operated calcium entry and cyclopiazonic acid-induced endothelium-derived relaxations in aging rat thoracic aorta. Physiol. Int. 2016, 103, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Ballejo, G. Pharmacological characterization of the calcium influx pathways involved in nitric oxide production by endothelial cells. Einstein 2019, 17, eAO4600. [Google Scholar] [CrossRef] [PubMed]
- Luik, R.M.; Wu, M.M.; Buchanan, J.; Lewis, R.S. The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006, 174, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Baker, T.M.; Lee, F.; Shui, B.; Lee, J.C.; Tvrdik, P.; Kotlikoff, M.I.; Sonkusare, S.K. Calcium Signal Profiles in Vascular Endothelium from Cdh5-GCaMP8 and Cx40-GCaMP2 Mice. J. Vasc. Res. 2021, 58, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Hannah, R.M.; Dunn, K.M.; Bonev, A.D.; Nelson, M.T. Endothelial SK(Ca) and IK(Ca) channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J. Cereb. Blood Flow. Metab. 2011, 31, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Longden, T.A.; Dabertrand, F.; Koide, M.; Gonzales, A.L.; Tykocki, N.R.; Brayden, J.E.; Hill-Eubanks, D.; Nelson, M.T. Capillary K(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 2017, 20, 717–726. [Google Scholar] [CrossRef]
- Willette, R.N.; Bao, W.; Nerurkar, S.; Yue, T.L.; Doe, C.P.; Stankus, G.; Turner, G.H.; Ju, H.; Thomas, H.; Fishman, C.E.; et al. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J. Pharmacol. Exp. Ther. 2008, 326, 443–452. [Google Scholar] [CrossRef]
- Zhang, D.X.; Mendoza, S.A.; Bubolz, A.H.; Mizuno, A.; Ge, Z.D.; Li, R.; Warltier, D.C.; Suzuki, M.; Gutterman, D.D. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 2009, 53, 532–538. [Google Scholar] [CrossRef]
- Ottolini, M.; Hong, K.; Cope, E.L.; Daneva, Z.; DeLalio, L.J.; Sokolowski, J.D.; Marziano, C.; Nguyen, N.Y.; Altschmied, J.; Haendeler, J.; et al. Local Peroxynitrite Impairs Endothelial Transient Receptor Potential Vanilloid 4 Channels and Elevates Blood Pressure in Obesity. Circulation 2020, 141, 1318–1333. [Google Scholar] [CrossRef]
- Earley, S.; Gonzales, A.L.; Garcia, Z.I. A dietary agonist of transient receptor potential cation channel V3 elicits endothelium-dependent vasodilation. Mol. Pharmacol. 2010, 77, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Wallis, L.; Donovan, L.; Johnston, A.; Phillips, L.C.; Lin, J.; Garland, C.J.; Dora, K.A. Tracking endothelium-dependent NO release in pressurized arteries. Front. Physiol. 2023, 14, 1108943. [Google Scholar] [CrossRef] [PubMed]
- MacKay, C.E.; Leo, M.D.; Fernandez-Pena, C.; Hasan, R.; Yin, W.; Mata-Daboin, A.; Bulley, S.; Gammons, J.; Mancarella, S.; Jaggar, J.H. Intravascular flow stimulates PKD2 (polycystin-2) channels in endothelial cells to reduce blood pressure. eLife 2020, 9, e56655. [Google Scholar] [CrossRef] [PubMed]
- Billaud, M.; Lohman, A.W.; Johnstone, S.R.; Biwer, L.A.; Mutchler, S.; Isakson, B.E. Regulation of cellular communication by signaling microdomains in the blood vessel wall. Pharmacol. Rev. 2014, 66, 513–569. [Google Scholar] [CrossRef] [PubMed]
- Straub, A.C.; Lohman, A.W.; Billaud, M.; Johnstone, S.R.; Dwyer, S.T.; Lee, M.Y.; Bortz, P.S.; Best, A.K.; Columbus, L.; Gaston, B.; et al. Endothelial cell expression of haemoglobin alpha regulates nitric oxide signalling. Nature 2012, 491, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Ruddiman, C.A.; Keller, T.C.S.t.; Keller, A.S.; Yang, Y.; Good, M.E.; Best, A.K.; Columbus, L.; Isakson, B.E. Heterocellular Contact Can Dictate Arterial Function. Circ. Res. 2019, 124, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Senadheera, S.; Kim, Y.; Grayson, T.H.; Toemoe, S.; Kochukov, M.Y.; Abramowitz, J.; Housley, G.D.; Bertrand, R.L.; Chadha, P.S.; Bertrand, P.P.; et al. Transient receptor potential canonical type 3 channels facilitate endothelium-derived hyperpolarization-mediated resistance artery vasodilator activity. Cardiovasc. Res. 2012, 95, 439–447. [Google Scholar] [CrossRef]
- Kerr, P.M.; Wei, R.; Tam, R.; Sandow, S.L.; Murphy, T.V.; Ondrusova, K.; Lunn, S.E.; Tran, C.H.T.; Welsh, D.G.; Plane, F. Activation of endothelial IKCa channels underlies NO-dependent myoendothelial feedback. Vascul Pharmacol. 2015, 74, 130–138. [Google Scholar] [CrossRef]
- Yeon, S.I.; Kim, J.Y.; Yeon, D.S.; Abramowitz, J.; Birnbaumer, L.; Muallem, S.; Lee, Y.H. Transient receptor potential canonical type 3 channels control the vascular contractility of mouse mesenteric arteries. PLoS ONE 2014, 9, e110413. [Google Scholar] [CrossRef]
- Shesely, E.G.; Maeda, N.; Kim, H.S.; Desai, K.M.; Krege, J.H.; Laubach, V.E.; Sherman, P.A.; Sessa, W.C.; Smithies, O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 13176–13181. [Google Scholar] [CrossRef]
- Van Vliet, B.N.; Chafe, L.L.; Montani, J.P. Characteristics of 24 h telemetered blood pressure in eNOS-knockout and C57Bl/6J control mice. J. Physiol. 2003, 549, 313–325. [Google Scholar] [CrossRef]
- Monica, F.Z.; Bian, K.; Murad, F. The Endothelium-Dependent Nitric Oxide-cGMP Pathway. Adv. Pharmacol. 2016, 77, 1–27. [Google Scholar] [CrossRef]
- Marziano, C.; Hong, K.; Cope, E.L.; Kotlikoff, M.I.; Isakson, B.E.; Sonkusare, S.K. Nitric Oxide-Dependent Feedback Loop Regulates Transient Receptor Potential Vanilloid 4 (TRPV4) Channel Cooperativity and Endothelial Function in Small Pulmonary Arteries. J. Am. Heart Assoc. 2017, 6, e007157. [Google Scholar] [CrossRef] [PubMed]
- Daneva, Z.; Marziano, C.; Ottolini, M.; Chen, Y.L.; Baker, T.M.; Kuppusamy, M.; Zhang, A.; Ta, H.Q.; Reagan, C.E.; Mihalek, A.D.; et al. Caveolar peroxynitrite formation impairs endothelial TRPV4 channels and elevates pulmonary arterial pressure in pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2021, 118, e2023130118. [Google Scholar] [CrossRef] [PubMed]
- Daneva, Z.; Ottolini, M.; Chen, Y.L.; Klimentova, E.; Kuppusamy, M.; Shah, S.A.; Minshall, R.D.; Seye, C.I.; Laubach, V.E.; Isakson, B.E.; et al. Endothelial pannexin 1-TRPV4 channel signaling lowers pulmonary arterial pressure in mice. Elife 2021, 10, e67777. [Google Scholar] [CrossRef] [PubMed]
- Hogan-Cann, A.D.; Lu, P.; Anderson, C.M. Endothelial NMDA receptors mediate activity-dependent brain hemodynamic responses in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 10229–10231. [Google Scholar] [CrossRef] [PubMed]
- Freeman, K.; Sackheim, A.; Mughal, A.; Koide, M.; Bonson, G.; Ebner, G.; Hennig, G.; Lockette, W.; Nelson, M.T. Pathogenic soluble tau peptide disrupts endothelial calcium signaling and vasodilation in the brain microvasculature. bioRxiv 2023. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Berra-Romani, R. Targeting the endothelial Ca2+ toolkit to rescue endothelial dysfunction in obesity associated-hypertension. Curr. Med. Chem. 2019, 27, 240–257. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef]
- Moccia, F.; Bonetti, E.; Dragoni, S.; Fontana, J.; Lodola, F.; Romani, R.B.; Laforenza, U.; Rosti, V.; Tanzi, F. Hematopoietic progenitor and stem cells circulate by surfing on intracellular Ca2+ waves: A novel target for cell-based therapy and anti-cancer treatment? Curr. Signal Transd T 2012, 7, 161–176. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Rosti, V.; Antognazza, M.R.; Lodola, F.; Moccia, F. Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis. Cells 2020, 9, 1341. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Negri, S.; Perna, A.; Rosti, V.; Guerra, G.; Moccia, F. Therapeutic Potential of Endothelial Colony-Forming Cells in Ischemic Disease: Strategies to Improve their Regenerative Efficacy. Int. J. Mol. Sci. 2020, 21, 7406. [Google Scholar] [CrossRef] [PubMed]
- Eilken, H.M.; Adams, R.H. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr. Opin. Cell Biol. 2010, 22, 617–625. [Google Scholar] [CrossRef]
- Norton, K.A.; Popel, A.S. Effects of endothelial cell proliferation and migration rates in a computational model of sprouting angiogenesis. Sci. Rep. 2016, 6, 36992. [Google Scholar] [CrossRef] [PubMed]
- Debir, B.; Meaney, C.; Kohandel, M.; Unlu, M.B. The role of calcium oscillations in the phenotype selection in endothelial cells. Sci. Rep. 2021, 11, 23781. [Google Scholar] [CrossRef] [PubMed]
- Dolmetsch, R.E.; Xu, K.; Lewis, R.S. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 1998, 392, 933–936. [Google Scholar] [CrossRef]
- Tomida, T.; Hirose, K.; Takizawa, A.; Shibasaki, F.; Iino, M. NFAT functions as a working memory of Ca2+ signals in decoding Ca2+ oscillation. EMBO J. 2003, 22, 3825–3832. [Google Scholar] [CrossRef]
- Lewis, R.S. Calcium oscillations in T-cells: Mechanisms and consequences for gene expression. Biochem. Soc. Trans. 2003, 31, 925–929. [Google Scholar] [CrossRef]
- Barak, P.; Parekh, A.B. Signaling through Ca2+ Microdomains from Store-Operated CRAC Channels. Cold Spring Harb. Perspect. Biol. 2020, 12, a035097. [Google Scholar] [CrossRef]
- Zhou, M.H.; Zheng, H.; Si, H.; Jin, Y.; Peng, J.M.; He, L.; Zhou, Y.; Munoz-Garay, C.; Zawieja, D.C.; Kuo, L.; et al. Stromal interaction molecule 1 (STIM1) and Orai1 mediate histamine-evoked calcium entry and nuclear factor of activated T-cells (NFAT) signaling in human umbilical vein endothelial cells. J. Biol. Chem. 2014, 289, 29446–29456. [Google Scholar] [CrossRef]
- Suehiro, J.; Kanki, Y.; Makihara, C.; Schadler, K.; Miura, M.; Manabe, Y.; Aburatani, H.; Kodama, T.; Minami, T. Genome-wide approaches reveal functional vascular endothelial growth factor (VEGF)-inducible nuclear factor of activated T cells (NFAT) c1 binding to angiogenesis-related genes in the endothelium. J. Biol. Chem. 2014, 289, 29044–29059. [Google Scholar] [CrossRef]
- Moccia, F.; Fotia, V.; Tancredi, R.; Della Porta, M.G.; Rosti, V.; Bonetti, E.; Poletto, V.; Marchini, S.; Beltrame, L.; Gallizzi, G.; et al. Breast and renal cancer-Derived endothelial colony forming cells share a common gene signature. Eur. J. Cancer 2017, 77, 155–164. [Google Scholar] [CrossRef]
- Zhu, L.P.; Luo, Y.G.; Chen, T.X.; Chen, F.R.; Wang, T.; Hu, Q. Ca2+ oscillation frequency regulates agonist-stimulated gene expression in vascular endothelial cells. J. Cell Sci. 2008, 121, 2511–2518. [Google Scholar] [CrossRef]
- Hu, Q.; Deshpande, S.; Irani, K.; Ziegelstein, R.C. [Ca2+](i) oscillation frequency regulates agonist-stimulated NF-kappaB transcriptional activity. J. Biol. Chem. 1999, 274, 33995–33998. [Google Scholar] [CrossRef]
- Lodola, F.; Laforenza, U.; Bonetti, E.; Lim, D.; Dragoni, S.; Bottino, C.; Ong, H.L.; Guerra, G.; Ganini, C.; Massa, M.; et al. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS ONE 2012, 7, e42541. [Google Scholar] [CrossRef]
- Lodola, F.; Laforenza, U.; Cattaneo, F.; Ruffinatti, F.A.; Poletto, V.; Massa, M.; Tancredi, R.; Zuccolo, E.; Khdar, A.D.; Riccardi, A.; et al. VEGF-induced intracellular Ca2+ oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 2017, 8, 95223–95246. [Google Scholar] [CrossRef] [PubMed]
- Bair, A.M.; Thippegowda, P.B.; Freichel, M.; Cheng, N.; Ye, R.D.; Vogel, S.M.; Yu, Y.; Flockerzi, V.; Malik, A.B.; Tiruppathi, C. Ca2+ entry via TRPC channels is necessary for thrombin-induced NF-kappaB activation in endothelial cells through AMP-activated protein kinase and protein kinase Cdelta. J. Biol. Chem. 2009, 284, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Thippegowda, P.B.; Singh, V.; Sundivakkam, P.C.; Xue, J.; Malik, A.B.; Tiruppathi, C. Ca2+ influx via TRPC channels induces NF-kappaB-dependent A20 expression to prevent thrombin-induced apoptosis in endothelial cells. Am. J. Physiol. Cell Physiol. 2010, 298, C656–C664. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Li, J.; Zhu, L.; Cai, L.; Xu, Q.; Ling, C.; Su, Y.; Hu, Q. Irregular Ca2+ oscillations regulate transcription via cumulative spike duration and spike amplitude. J. Biol. Chem. 2012, 287, 40246–40255. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Song, S.; Pi, Y.; Yu, Y.; She, W.; Ye, H.; Su, Y.; Hu, Q. Cumulated Ca2+ spike duration underlies Ca2+ oscillation frequency-regulated NFkappaB transcriptional activity. J. Cell Sci. 2011, 124, 2591–2601. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Rosti, V. Manipulating Intracellular Ca2+ Signals to Stimulate Therapeutic Angiogenesis in Cardiovascular Disorders. Curr. Pharm. Biotechnol. 2018, 19, 686–699. [Google Scholar] [CrossRef]
- Balbi, C.; Lodder, K.; Costa, A.; Moimas, S.; Moccia, F.; van Herwaarden, T.; Rosti, V.; Campagnoli, F.; Palmeri, A.; De Biasio, P.; et al. Reactivating endogenous mechanisms of cardiac regeneration via paracrine boosting using the human amniotic fluid stem cell secretome. Int. J. Cardiol. 2019, 287, 87–95. [Google Scholar] [CrossRef]
- Balbi, C.; Lodder, K.; Costa, A.; Moimas, S.; Moccia, F.; van Herwaarden, T.; Rosti, V.; Campagnoli, F.; Palmeri, A.; De Biasio, P.; et al. Supporting data on in vitro cardioprotective and proliferative paracrine effects by the human amniotic fluid stem cell secretome. Data Brief. 2019, 25, 104324. [Google Scholar] [CrossRef]
- Maghin, E.; Garbati, P.; Quarto, R.; Piccoli, M.; Bollini, S. Young at Heart: Combining Strategies to Rejuvenate Endogenous Mechanisms of Cardiac Repair. Front. Bioeng. Biotechnol. 2020, 8, 447. [Google Scholar] [CrossRef]
- Balducci, V.; Faris, P.; Balbi, C.; Costa, A.; Negri, S.; Rosti, V.; Bollini, S.; Moccia, F. The human amniotic fluid stem cell secretome triggers intracellular Ca2+ oscillations, NF-kappaB nuclear translocation and tube formation in human endothelial colony-forming cells. J. Cell Mol. Med. 2021, 25, 8074–8086. [Google Scholar] [CrossRef]
- Lodola, F.; Rosti, V.; Tullii, G.; Desii, A.; Tapella, L.; Catarsi, P.; Lim, D.; Moccia, F.; Antognazza, M.R. Conjugated polymers optically regulate the fate of endothelial colony-forming cells. Sci. Adv. 2019, 5, eaav4620. [Google Scholar] [CrossRef]
- Moccia, F.; Antognazza, M.R.; Lodola, F. Towards Novel Geneless Approaches for Therapeutic Angiogenesis. Front. Physiol. 2020, 11, 616189. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Ronchi, C.; Lodola, F. Optical excitation of organic semiconductors as a highly selective strategy to induce vascular regeneration and tissue repair. Vascul. Pharmacol. 2022, 144, 106998. [Google Scholar] [CrossRef] [PubMed]
- Di Maria, F.; Lodola, F.; Zucchetti, E.; Benfenati, F.; Lanzani, G. The evolution of artificial light actuators in living systems: From planar to nanostructured interfaces. Chem. Soc. Rev. 2018, 47, 4757–4780. [Google Scholar] [CrossRef] [PubMed]
- Maeng, Y.S.; Choi, H.J.; Kwon, J.Y.; Park, Y.W.; Choi, K.S.; Min, J.K.; Kim, Y.H.; Suh, P.G.; Kang, K.S.; Won, M.H.; et al. Endothelial progenitor cell homing: Prominent role of the IGF2-IGF2R-PLCbeta2 axis. Blood 2009, 113, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.D.; Huang, Y.; Liu, D.; Hickey, R.; Krause, D.S.; Giordano, F.J. Stromal cell-derived factor-1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation 2004, 110, 3300–3305. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Di Buduo, C.; Lodola, F.; Orecchioni, S.; Scarpellino, G.; Kheder, D.A.; Poletto, V.; Guerra, G.; Bertolini, F.; Balduini, A.; et al. Stromal Cell-Derived Factor-1alpha Promotes Endothelial Colony-Forming Cell Migration Through the Ca2+-Dependent Activation of the Extracellular Signal-Regulated Kinase 1/2 and Phosphoinositide 3-Kinase/AKT Pathways. Stem Cells Dev. 2018, 27, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Bromage, D.I.; Taferner, S.; He, Z.; Ziff, O.J.; Yellon, D.M.; Davidson, S.M. Stromal cell-derived factor-1alpha signals via the endothelium to protect the heart against ischaemia-reperfusion injury. J. Mol. Cell Cardiol. 2019, 128, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Guerra, G.; Borghesi, A.; Stronati, M.; Rosti, V.; Tanzi, F.; et al. Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor-induced intracellular Ca2+ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 2013, 22, 2561–2580. [Google Scholar] [CrossRef]
- Moccia, F.; Ruffinatti, F.A.; Zuccolo, E. Intracellular Ca2+ Signals to Reconstruct A Broken Heart: Still A Theoretical Approach? Curr. Drug Targets 2015, 16, 793–815. [Google Scholar] [CrossRef]
- Ingram, D.A.; Mead, L.E.; Tanaka, H.; Meade, V.; Fenoglio, A.; Mortell, K.; Pollok, K.; Ferkowicz, M.J.; Gilley, D.; Yoder, M.C. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 2004, 104, 2752–2760. [Google Scholar] [CrossRef] [PubMed]
- Smedlund, K.; Bah, M.; Vazquez, G. On the role of endothelial TRPC3 channels in endothelial dysfunction and cardiovascular disease. Cardiovasc. Hematol. Agents Med. Chem. 2012, 10, 265–274. [Google Scholar] [CrossRef]
- Varberg, K.M.; Garretson, R.O.; Blue, E.K.; Chu, C.; Gohn, C.R.; Tu, W.; Haneline, L.S. Transgelin induces dysfunction of fetal endothelial colony-forming cells from gestational diabetic pregnancies. Am. J. Physiol. Cell Physiol. 2018, 315, C502–C515. [Google Scholar] [CrossRef]
- Wang, R. Roles of Hydrogen Sulfide in Hypertension Development and Its Complications: What, So What, Now What. Hypertension 2023, 80, 936–944. [Google Scholar] [CrossRef]
- Baylie, R.L.; Brayden, J.E. TRPV channels and vascular function. Acta Physiol 2011, 203, 99–116. [Google Scholar] [CrossRef]
- Filippini, A.; D’Amore, A.; D’Alessio, A. Calcium Mobilization in Endothelial Cell Functions. Int. J. Mol. Sci. 2019, 20, 4525. [Google Scholar] [CrossRef]
- Alves-Lopes, R.; Lacchini, S.; Neves, K.B.; Harvey, A.; Montezano, A.C.; Touyz, R.M. Vasoprotective effects of NOX4 are mediated via polymerase and transient receptor potential melastatin 2 cation channels in endothelial cells. J. Hypertens. 2023, 41, 1389–1400. [Google Scholar] [CrossRef]
- Zielinska, W.; Zabrzynski, J.; Gagat, M.; Grzanka, A. The Role of TRPM2 in Endothelial Function and Dysfunction. Int. J. Mol. Sci. 2021, 22, 7635. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Nepal, S.; Tsukasaki, Y.; Hecquet, C.M.; Soni, D.; Rehman, J.; Tiruppathi, C.; Malik, A.B. Neutrophil Activation of Endothelial Cell-Expressed TRPM2 Mediates Transendothelial Neutrophil Migration and Vascular Injury. Circ. Res. 2017, 121, 1081–1091. [Google Scholar] [CrossRef]
- Woudenberg-Vrenken, T.E.; Bindels, R.J.; Hoenderop, J.G. The role of transient receptor potential channels in kidney disease. Nat. Rev. Nephrol. 2009, 5, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Hamzaoui, M.; Groussard, D.; Nezam, D.; Djerada, Z.; Lamy, G.; Tardif, V.; Dumesnil, A.; Renet, S.; Brunel, V.; Peters, D.J.M.; et al. Endothelium-Specific Deficiency of Polycystin-1 Promotes Hypertension and Cardiovascular Disorders. Hypertension 2022, 79, 2542–2551. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Endolysosomal Ca2+ signaling in cardiovascular health and disease. Int. Rev. Cell Mol. Biol. 2021, 363, 203–269. [Google Scholar] [CrossRef]
- Moccia, F.; Fiorio Pla, A.; Lim, D.; Lodola, F.; Gerbino, A. Intracellular Ca2+ signalling: Unexpected new roles for the usual suspect. Front. Physiol. 2023, 14, 1210085. [Google Scholar] [CrossRef] [PubMed]
- King, O.; Cruz-Moreira, D.; Sayed, A.; Kermani, F.; Kit-Anan, W.; Sunyovszki, I.; Wang, B.X.; Downing, B.; Fourre, J.; Hachim, D.; et al. Functional microvascularization of human myocardium in vitro. Cell Rep. Methods 2022, 2, 100280. [Google Scholar] [CrossRef]
- Pitoulis, F.G.; Smith, J.J.; Pamias-Lopez, B.; de Tombe, P.P.; Hayman, D.; Terracciano, C.M. MyoLoop: Design, development and validation of a standalone bioreactor for pathophysiological electromechanical in vitro cardiac studies. Exp. Physiol. 2023. [Google Scholar] [CrossRef]
- Guan, N.N.; Sharma, N.; Hallen-Grufman, K.; Jager, E.W.H.; Svennersten, K. The role of ATP signalling in response to mechanical stimulation studied in T24 cells using new microphysiological tools. J. Cell Mol. Med. 2018, 22, 2319–2328. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Wang, Y.; Chen, W.; Li, Z.; Su, W.; Guo, Y.; Deng, P.; Qin, J. Engineering human islet organoids from iPSCs using an organ-on-chip platform. Lab. Chip 2019, 19, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Mussano, F.; Genova, T.; Laurenti, M.; Gaglioti, D.; Scarpellino, G.; Rivolo, P.; Faga, M.G.; Fiorio Pla, A.; Munaron, L.; Mandracci, P.; et al. Beta1-integrin and TRPV4 are involved in osteoblast adhesion to different titanium surface topographies. Appl. Surf. Sci. 2020, 507, 145112. [Google Scholar] [CrossRef]
- Taylor, M.S.; Lowery, J.; Choi, C.S.; Francis, M. Restricted Intimal Ca2+ Signaling Associated With Cardiovascular Disease. Front. Physiol. 2022, 13, 848681. [Google Scholar] [CrossRef] [PubMed]



| Agonist | Vascular Bed | Function | Mechanism | Reference |
|---|---|---|---|---|
| Acetylcholine | Rat carotid artery (pressurized) | Flow-dependent vasodilation | InsP3-induced ER Ca2+ release and an SKF-sensitive Ca2+ entry pathway | [29] |
| Acetylcholine | Mouse carotid artery (en face) | NO-mediated vasodilation | InsP3-induced ER Ca2+ release, Ca2+ entry not assessed | [126] |
| Acetylcholine | Mouse second-order mesenteric artery (pressurized) | EDH-mediated vasodilation | InsP3-induced ER Ca2+ release and Ca2+ entry | [88] |
| Acetylcholine | Rat third-order mesenteric artery (pressurized) | Flow-dependent vasodilation | InsP3-induced ER Ca2+ release and an SKF-sensitive Ca2+ entry pathway | [29] |
| Acetylcholine | Mouse superior epigastric arteries (en face) | Not defined | InsP3-induced ER Ca2+ release, no Ca2+ entry | [90] |
| Carbachol | Rat tail artery (en face) | Not defined | InsP3-induced ER Ca2+ release and STIM1-mediated SOCE | [89] |
| Carbachol | Rat ureteric precapillary arterioles (en face) | NO- and EDH-mediated vasodilation | InsP3-induced ER Ca2+ release, no Ca2+ entry | [83] |
| ATP | Mouse carotid artery (pressurized) | Not defined | InsP3-induced ER Ca2+ release and an SKF-sensitive Ca2+ entry pathway | [29] |
| Adenosine | Mouse cremaster muscle arterioles (pressurized) | Not defined | Not defined | [91] |
| Histamine | Rat lung capillaries (pressurized) | Not defined | InsP3-induced ER Ca2+ release and Ca2+ entry, likely mediated by SOCE | [35,127] |
| Thrombin | Rat lung capillaries and venules (pressurized) | Lung microvascular permeability | InsP3-induced ER Ca2+ release, Ca2+ entry not assessed | [128] |
| LPS | Mouse lung microvessels | Lung microvascular permeability | ER Ca2+ release through InsP3R2 and STIM1/Orai1-mediated SOCE | [20] |
| VEGF | Zebrafish dorsal aorta and posterior cardinal vein | Angiogenesis | ER Ca2+ release through InsP3R and STIM1/Orai1-mediated SOCE; plausible contribution of lysosomal Ca2+ release via TPC2 | [8,38,129,130] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moccia, F.; Brunetti, V.; Soda, T.; Berra-Romani, R.; Scarpellino, G. Cracking the Endothelial Calcium (Ca2+) Code: A Matter of Timing and Spacing. Int. J. Mol. Sci. 2023, 24, 16765. https://doi.org/10.3390/ijms242316765
Moccia F, Brunetti V, Soda T, Berra-Romani R, Scarpellino G. Cracking the Endothelial Calcium (Ca2+) Code: A Matter of Timing and Spacing. International Journal of Molecular Sciences. 2023; 24(23):16765. https://doi.org/10.3390/ijms242316765
Chicago/Turabian StyleMoccia, Francesco, Valentina Brunetti, Teresa Soda, Roberto Berra-Romani, and Giorgia Scarpellino. 2023. "Cracking the Endothelial Calcium (Ca2+) Code: A Matter of Timing and Spacing" International Journal of Molecular Sciences 24, no. 23: 16765. https://doi.org/10.3390/ijms242316765