
Hearing and Hearing Loss Progression in Patients with GJB2 Gene Mutations: A Long-Term Follow-Up
 and
and
Abstract
:1. Introduction
2. Results
2.1. Patients
2.2. Genetic Diagnosis
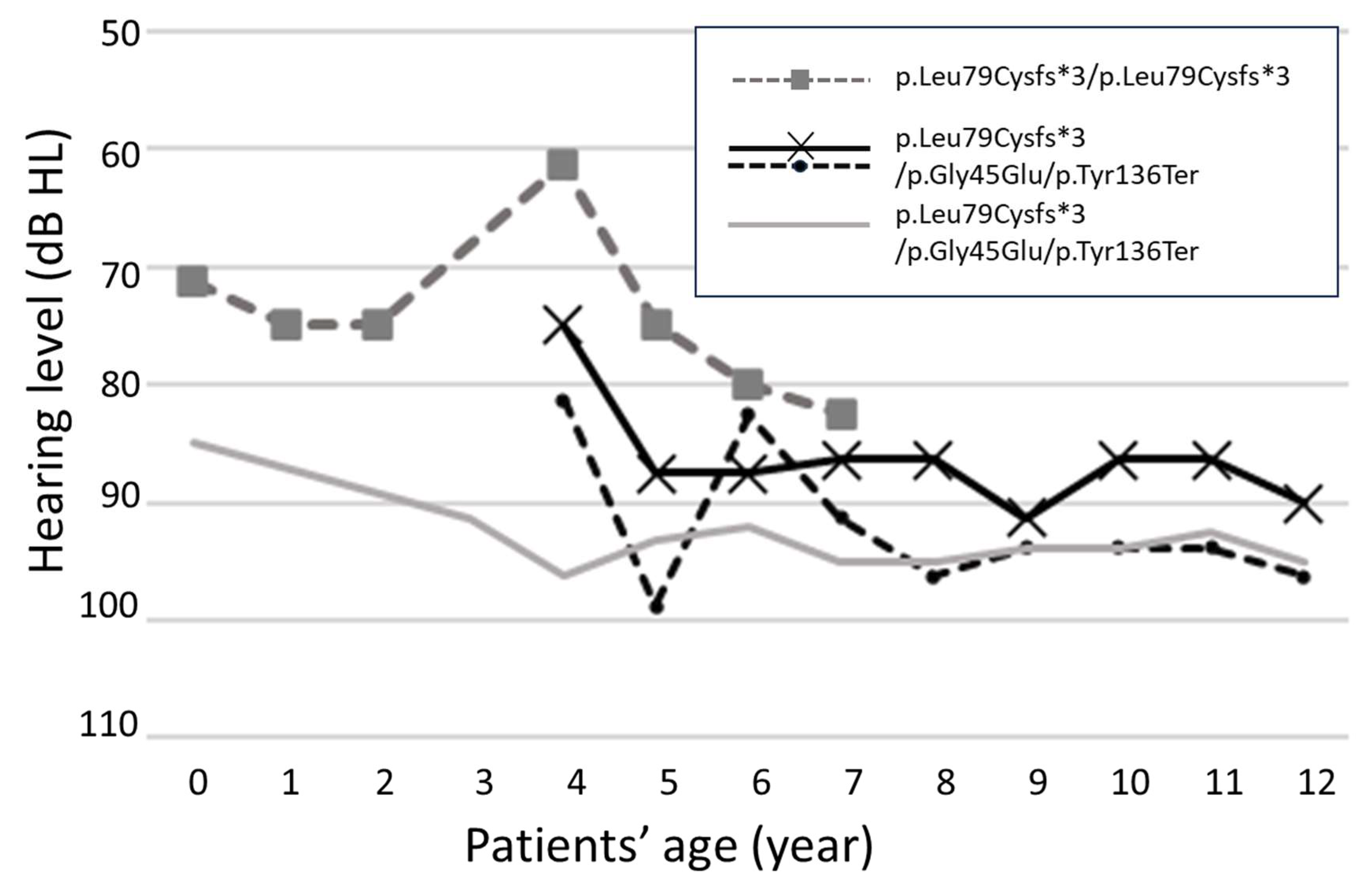
2.3. Longitudinal Analysis of Hearing Outcomes
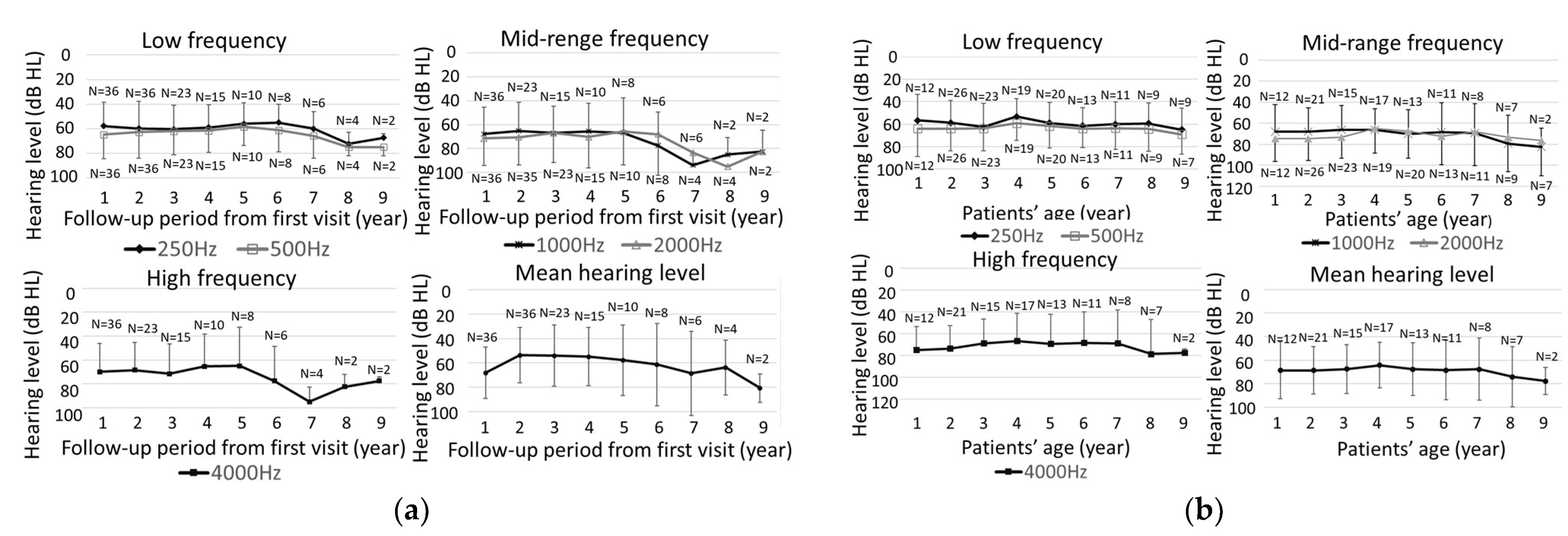
2.4. Evaluation of Hearing Threshold Changes at Each Frequency
3. Discussion
3.1. Genetic Phenotype and Initial Hearing Levels
3.2. Progression of Hearing Loss
3.3. Hearing Level at Initial Examination in Cases of Progressive Hearing Loss
4. Materials and Methods
4.1. Patients
4.2. Audiological Evaluations
4.3. Longitudinal Analysis of Hearing Outcomes
4.4. Genetic Testing
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morton, C.C.; Nance, W.E. Newborn hearing screening—A silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef]
- Leigh, J.; Dettman, S.; Dowell, R.; Briggs, R. Communication development in children who receive a cochlear implant by 12 months of age. Otol. Neurotol. 2013, 34, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Joint Committee on Infant Hearing. Year 2019 Position Statement: Principles and guidelines for early hearing detection and intervention programs. J. Early Hear. Detect. Intervention 2019, 4, 1–44. [Google Scholar]
- Kashio, A.; Takahashi, H.; Nishizaki, K.; Hara, A.; Yamasoba, T.; Moriyama, H. Cochlear implants in Japan: Results of cochlear implant reporting system over more than 30 years. Auris Nasus Larynx 2021, 48, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Hang, A.X.; Roush, P.A.; Teagle, H.F.; Zdanski, C.; Pillsbury, H.C.; Adunka, O.F.; Buchman, C.A. Is “no response” on diagnostic auditory brainstem response testing an indication for cochlear implantation in children? Ear Hear. 2015, 36, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Sininger, Y.S. Auditory brain stem response for objective measures of hearing. Ear Hear. 1993, 14, 23–30. [Google Scholar] [CrossRef]
- Suzuki, T.; Hirai, Y.; Horiuchi, K. Auditory brain stem responses to pure tone stimuli. Scand. Audiol. 1977, 6, 51–56. [Google Scholar] [CrossRef]
- Gorga, M.P.; Johnson, T.A.; Kaminski, J.R.; Beauchaine, K.L.; Garner, C.A.; Neely, S.T. Using a combination of click- and tone burst-evoked auditory brain stem response measurements to estimate pure-tone thresholds. Ear Hear. 2006, 27, 60–74. [Google Scholar] [CrossRef]
- Marttila, T.I.; Karikoski, J.O. Comparison between audiometric and ABR thresholds in children. Contradictory findings. Eur. Arch. Otorhinolaryngol. 2006, 263, 399–403. [Google Scholar] [CrossRef]
- Stapells, D.R. Threshold Estimation by the Tone-Evoked Auditory Brainstem Response: A Literature Meta-Analysis. J. Speech Lang. Pathol. Audiol 2000, 24, 74–83. [Google Scholar]
- Xiang, J.; Jin, Y.; Song, N.; Chen, S.; Shen, J.; Xie, W.; Sun, X.; Peng, Z.; Sun, Y. Comprehensive genetic testing improves the clinical diagnosis and medical management of pediatric patients with isolated hearing loss. BMC Med. Genom. 2022, 15, 142. [Google Scholar] [CrossRef] [PubMed]
- Oguchi, T.; Ohtsuka, A.; Hashimoto, S.; Oshima, A.; Abe, S.; Kobayashi, T.; Nagai, K.; Matsunaga, T.; Iwasaki, S.; Nakagawa, T.; et al. Clinical features of patients with GJB2 (connexin 26) mutations: Severity of hearing loss is correlated with genotypes and protein expression patterns. J. Hum. Genet./Jpn. Soc. Hum. Genet. 2005, 50, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Snoeckx, R.L.; Huygen, P.L.; Feldmann, D.; Marlin, S.; Denoyelle, F.; Waligora, J.; Mueller-Malesinska, M.; Pollak, A.; Ploski, R.; Murgia, A.; et al. GJB2 mutations and degree of hearing loss: Multicenter study. Am. J. Hum. Genet. 2005, 77, 945–957. [Google Scholar] [CrossRef]
- Kenna, M.A.; Feldman, H.A.; Neault, M.W.; Frangulov, A.; Wu, B.L.; Fligor, B.; Rehm, H.L. Audiologic phenotype and progression in GJB2 (Connexin 26) hearing loss. Arch. Otolaryngol. Head. Neck Surg. 2010, 136, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Matsuo, H.; Shimizu, S.; Nakayama, A.; Suzuki, K.; Hamajima, N.; Shinomiya, N.; Nishio, S.; Kosugi, S.; Usami, S.; et al. Carrier frequency of the GJB2 mutations that cause hereditary hearing loss in the Japanese population. J. Hum. Genet. 2015, 60, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, H.; Yan, C.; Guan, J.; Yin, L.; Lan, L.; Li, J.; Zhao, L.; Wang, Q. The Frequency of Common Deafness-Associated Variants Among 3,555,336 Newborns in China and 141,456 Individuals Across Seven Populations Worldwide. Ear Hear. 2023, 44, 232–241. [Google Scholar] [CrossRef]
- Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2014, 124, E34–E53. [Google Scholar] [CrossRef]
- Bruzzone, R.; White, T.W.; Paul, D.L. Connections with connexins: The molecular basis of direct intercellular signaling. Eur. J. Biochem. 1996, 238, 1–27. [Google Scholar] [CrossRef]
- Kikuchi, T.; Kimura, R.S.; Paul, D.L.; Takasaka, T.; Adams, J.C. Gap junction systems in the mammalian cochlea. Brain Res. Brain Res. Rev. 2000, 32, 163–166. [Google Scholar] [CrossRef]
- Kamiya, K.; Yum, S.W.; Kurebayashi, N.; Muraki, M.; Ogawa, K.; Karasawa, K.; Miwa, A.; Guo, X.; Gotoh, S.; Sugitani, Y.; et al. Assembly of the cochlear gap junction macromolecular complex requires connexin 26. J. Clin. Investig. 2014, 124, 1598–1607. [Google Scholar] [CrossRef]
- Kikuchi, T.; Kimura, R.S.; Paul, D.L.; Adams, J.C. Gap junctions in the rat cochlea: Immunohistochemical and ultrastructural analysis. Anat. Embryol. 1995, 191, 101–118. [Google Scholar] [CrossRef]
- Denoyelle, F.; Weil, D.; Maw, M.A.; Wilcox, S.A.; Lench, N.J.; Allen-Powell, D.R.; Osborn, A.H.; Dahl, H.-H.M.; Middleton, A.; Houseman, M.J.; et al. Prelingual deafness: High prevalence of a 30delG mutation in the connexin 26 gene. Hum. Mol. Genet. 1997, 6, 2173–2177. [Google Scholar] [CrossRef]
- Morell, R.J.; Kim, H.J.; Hood, L.J.; Goforth, L.; Friderici, K.; Fisher, R.; Van Camp, G.; Berlin, C.I.; Oddoux, C.; Ostrer, H.; et al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with non-syndromic recessive deafness. N. Engl. J. Med. 1998, 339, 1500–1505. [Google Scholar] [CrossRef]
- Abe, S.; Usami, S.; Shinkawa, H.; Kelley, P.M.; Kimberling, W.J. Prevalent connexin 26 gene (GJB2) mutations in Japanese. J. Med. Genet. 2000, 37, 41–43. [Google Scholar] [CrossRef]
- Tsukada, K.; Nishio, S.Y.; Hattori, M.; Usami, S. Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: Their origin and a literature review. Ann. Otol. Rhinol. Laryngol. 2015, 124 (Suppl. 1), 61S–76S. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, K.; Nishio, S.; Usami, S. Deafness Gene Study Consortium. A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clin. Genet. 2010, 78, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.H.; Jones, M.N.; Adam, M.P.; Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Gripp, K.W.; Amemiya, A. Non-syndromic Hearing Loss and Deafness, DFNB1; GeneReviews [Internet]; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Norris, V.W.; Arnos, K.S.; Hanks, W.D.; Xia, X.; Nance, W.E.; Pandya, A. Does universal newborn hearing screening identify all children with GJB2 (Connexin 26) deafness? Penetrance of GJB2 deafness. Ear Hear. 2006, 27, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Pagarkar, W.; Bitner-Glindzicz, M.; Knight, J.; Sirimanna, T. Late postnatal onset of hearing loss due to GJB2 mutations. Int. J. Pediatr. Otorhinolaryngol. 2006, 70, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Imai, N.; Kumakawa, K.; Adachi, N.; Asamuna, S.; Ohashi, H.; Sakata, H.; Yamasoba, T.; Usami, S. Characteristics and genotype of GJB2 mutation with progressive hearing loss. Pediatr. Otorhinolaryngol. Japan 2013, 34, 352–359. [Google Scholar]
- Palmada, M.; Schmalisch, K.; Böhmer, C.; Schug, N.; Pfister, M.; Lang, F.; Blin, N. Loss of function mutations of the GJB2 gene detected in patients with DFNB1-associated hearing impairment. Neurobiol. Dis. 2006, 22, 112–118. [Google Scholar] [CrossRef]
- Lee, S.; Han, S.; Han, J.; Kim, M.; Oh, D.; Kim, N.; Song, J.; Koo, J.; Lee, J.; Oh, S.; et al. Natural Course of Residual Hearing with Reference to GJB2 and SLC26A4 Genotypes: Clinical Implications for Hearing Rehabilitation. Ear Hear. 2021, 42, 644–653. [Google Scholar] [CrossRef]
- Chan, D.K.; Schrijver, I.; Chang, K.W. Connexin-26-associated deafness: Phenotypic variability and progression of hearing loss. Genet. Med. 2010, 12, 174–181. [Google Scholar] [CrossRef]
- Hosoya, M.; Fujioka, M.; Nara, K.; Morimoto, N.; Masuda, S.; Sugiuchi, T.; Katsunuma, S.; Takagi, A.; Morita, N.; Ogawa, K.; et al. Investigation of the hearing levels of siblings affected by a single GJB2 variant: Possibility of genetic modifiers. Int. J. Pediatr. Otorhinolaryngol. 2021, 149, 110840. [Google Scholar] [CrossRef]
- Wang, H.; Gao, Y.; Guan, J.; Lan, K.; Yang, J.; Xiong, W.; Zhao, C.; Xie, L.; Yu, L.; Wang, D.; et al. Phenotypic heterogeneity of post-lingual and/or milder hearing loss for the patients with the GJB2 c.235delC homozygous mutation. Front. Cell Dev. Biol. 2021, 9, 647240. [Google Scholar] [CrossRef]
- Hilgert, N.; Huentelman, M.J.; Thorburn, A.Q.; Fransen, E.; Dieltjens, N.; Mueller-Malesinska, M.; Pollak, A.; Skorka, A.; Waligora, J.; Ploski, R.; et al. Phenotypic variability of patients homozygous for the GJB2 mutation 35delG cannot be explained by the influence of one major modifier gene. Eur. J. Hum. Genet. 2009, 17, 517–524. [Google Scholar] [CrossRef]
- Fujioka, M.; Hosoya, M.; Nara, K.; Morimoto, N.; Sakamoto, H.; Otsu, M.; Nakano, A.; Arimoto, Y.; Masuda, S.; Sugiuchi, T.; et al. Differences in hearing levels between siblings with hearing loss caused by GJB2 mutations. Auris Nasus Larynx 2020, 47, 938–942. [Google Scholar] [CrossRef]
- Minami, S.B.; Mutai, H.; Nakano, A.; Arimoto, Y.; Taiji, H.; Morimoto, N.; Sakata, H.; Adachi, N.; Masuda, S.; Sakamoto, H.; et al. GJB2-associated hearing loss undetected by hearing screening of newborns. Gene 2013, 532, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Carlson, R.J.; Walsh, T.; Mandell, J.B.; Aburayyan, A.; Lee, M.K.; Gulsuner, S.; Horn, D.L.; Ou, H.C.; Sie, K.C.Y.; Mancl, L.; et al. Association of genetic diagnoses of childhood-onset hearing loss with cochlear implant outcomes. JAMA Otolaryngol. Head. Neck Surg. 2023, 149, 212–222. [Google Scholar] [CrossRef]
- Chen, P.; Lin, Y.; Liu, T.; Lin, Y.; Tseng, L.; Yang, T.; Chen, P.; Wu, C.; Hsu, C. Prediction model for audiological outcomes in patients with GJB2 mutations. Ear Hear. 2020, 41, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Arimoto, Y.; Mutai, H.; Matsunaga, T. Clinical features of GJB2 associated hearing loss children. Nihon. Jibiinkoka Gakkai Kaiho 2020, 123, 1225–1230. [Google Scholar] [CrossRef]
- Maxon, A.B.; Hochberg, I. Development of psychoacoustic behavior: Sensitivity and discrimination. Ear Hear. 1982, 3, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Sakata, A.; Kashio, A.; Koyama, H.; Uranaka, T.; Iwasaki, S.; Fujimoto, C.; Kinoshita, M.; Yamasoba, T. Long-Term Progression and Rapid Decline in Hearing Loss in Patients with a Point Mutation at Nucleotide 3243 of the Mitochondrial DNA. Life 2022, 12, 543. [Google Scholar] [CrossRef] [PubMed]
- Usami, S.; Nishio, S.Y.; Nagano, M.; Abe, S.; Yamaguchi, T. Simultaneous screening of multiple mutations by invader assay improves molecular diagnosis of hereditary hearing loss: A multicenter study. PLoS ONE 2012, 7, e31276. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Variation | NT/T | Number of Alleles | Hearing Level of Better Ear at First Visit | |||
|---|---|---|---|---|---|---|
| Profound | Severe | Moderate | Mild | |||
| ≧90 dB | 70–90 dB | 40–70 dB | 25–40 dB | |||
| p.Leu79Cysfs*3 | T | 77 | 48 | 10 | 19 | |
| p.Gly45Glu p.Tyr136Ter | T | 25 | 18 | 2 | 3 | 2 |
| c.176_191del16bp | T | 12 | 9 | 1 | 1 | 1 |
| c.299-300delAT | T | 6 | 5 | 1 | ||
| p.Typ134Ter | T | 1 | 1 | |||
| p.Ala171Ter | T | 1 | 1 | |||
| p.Arg143Trp | NT | 16 | 16 | |||
| p.Val37Ile | NT | 7 | 2 | 5 | ||
| p.Thr86Arg | NT | 3 | 2 | 1 | ||
| p.Arg127Cys | NT | 2 | 2 | |||
| Total | 150 | 102 (102/150 = 68.0%) | 14 (14/150 = 9.3%) | 26 (26/150 = 17.3%) | 8 (8/150 = 5.3%) | |
| Number of Alleles | Hearing Level of Better Ear at First Visit | ||||
|---|---|---|---|---|---|
| Profound | Severe | Moderate | Mild | ||
| Truncating mutation | 122 | 82 | 12 | 23 | 5 |
| 82/102 = 80% | 12/12 = 100% | 23/26 = 88% | 5/10 = 50% | ||
| Non-truncating mutation | 28 | 20 | 0 | 3 | 5 |
| 28/102 = 27% | 3/26 = 12% | 5/10 = 50% | |||
| Total | 150 | 102 | 12 | 26 | 10 |
| Number of Cases | Hearing Level of Better Ear at First Visit | ||||
|---|---|---|---|---|---|
| Profound | Severe | Moderate | Mild | ||
| T/T | 52 | 31 | 11 | 9 | 1 |
| 31/49 = 63% | 11/11 = 100% | 9/12 = 75% | 1/3 = 33% | ||
| T/NT | 18 | 16 | 0 | 1 | 1 |
| 16/49 = 33% | 1/12 = 8% | 1/3 = 33% | |||
| NT/NT | 5 | 2 | 0 | 2 | 1 |
| 2/49 = 4% | 2/12 = 17% | 1/3 = 33% | |||
| Total | 75 | 49 | 11 | 12 | 3 |
| Frequency (Hz) | 250 | 500 | 1000 | 2000 | 4000 |
|---|---|---|---|---|---|
| 10 dB or more deterioration | 9 | 4 | 3 | 2 | 5 |
| No change | 23 | 24 | 26 | 28 | 28 |
| 10 dB or more improvement | 5 | 9 | 8 | 7 | 4 |
| Total (ear) | 37 | 37 | 37 | 37 | 37 |
| Author | Publication Year | Criteria of Progression | Number of Cases | Progression Cases | Ratio | Follow-Up Period |
|---|---|---|---|---|---|---|
| Chan et al. [33] | 2010 | 5 dB deterioration or 1 dB/year | 33 | 8 | 8/33 = 24% | 5.3-year average |
| Tsukada et al. [26] | 2010 | 26 | 4 | 4/26 = 15% | 2 years or more | |
| Kenna et al. [14] | 2010 | 10 dB or more deterioration at 2 frequencies 15 dB or more deterioration at a frequency of | 84 | 47 | 47/84 = 56% | 13-month average |
| Imai et al. [30] | 2013 | 1 dB/year or more | 32 | 1 | 1/32 = 3% | 1–24 years |
| Chan et al. [17] | 2014 | (Systematic review) | 18.70% | |||
| Nakano et al. [41] | 2020 | 10 dB or more deterioration | 34 | 4 | 4/34 = 12% | 1–17 years |
| Chen et al. [40] | 2020 | 0.5 dB/year or more | 227 | 38 | 38/227 = 17% | 0.3~21 years |
| Sakata | This paper | 10 dB or more deterioration | 48 | 3 | 3/48 = 6% | 2–24 years |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakata, A.; Kashio, A.; Koyama, M.; Urata, S.; Koyama, H.; Yamasoba, T. Hearing and Hearing Loss Progression in Patients with GJB2 Gene Mutations: A Long-Term Follow-Up. Int. J. Mol. Sci. 2023, 24, 16763. https://doi.org/10.3390/ijms242316763
Sakata A, Kashio A, Koyama M, Urata S, Koyama H, Yamasoba T. Hearing and Hearing Loss Progression in Patients with GJB2 Gene Mutations: A Long-Term Follow-Up. International Journal of Molecular Sciences. 2023; 24(23):16763. https://doi.org/10.3390/ijms242316763
Chicago/Turabian StyleSakata, Aki, Akinori Kashio, Misaki Koyama, Shinji Urata, Hajime Koyama, and Tatsuya Yamasoba. 2023. "Hearing and Hearing Loss Progression in Patients with GJB2 Gene Mutations: A Long-Term Follow-Up" International Journal of Molecular Sciences 24, no. 23: 16763. https://doi.org/10.3390/ijms242316763