
Multi-Omics of Familial Thoracic Aortic Aneurysm and Dissection: Calcium Transport Impairment Predisposes Aortas to Dissection
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Expression of Genes, Mutations of Which Increase the Risk of Aortic Dissection, Is Unaffected by Myh11 K1256 Deletion
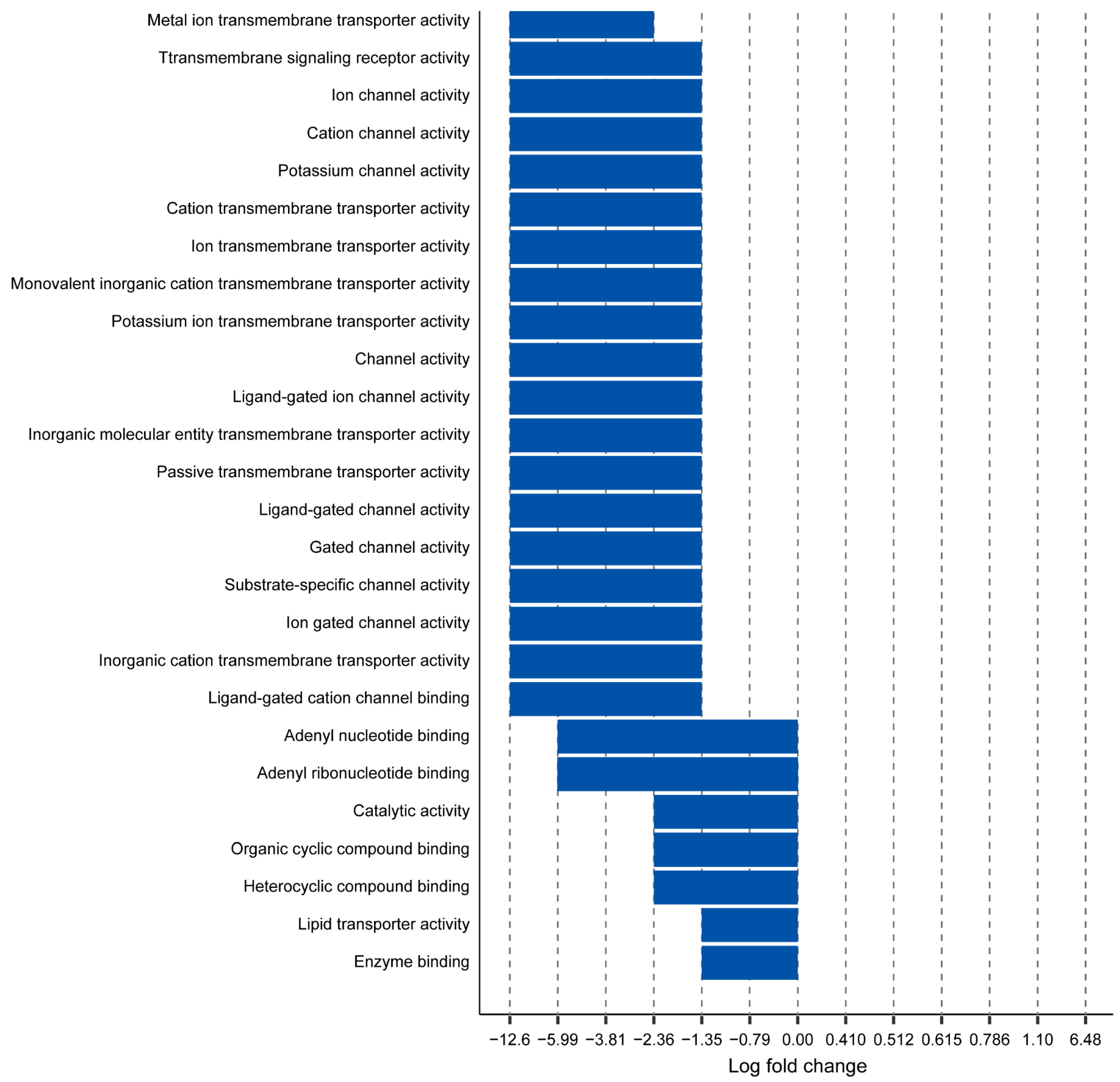
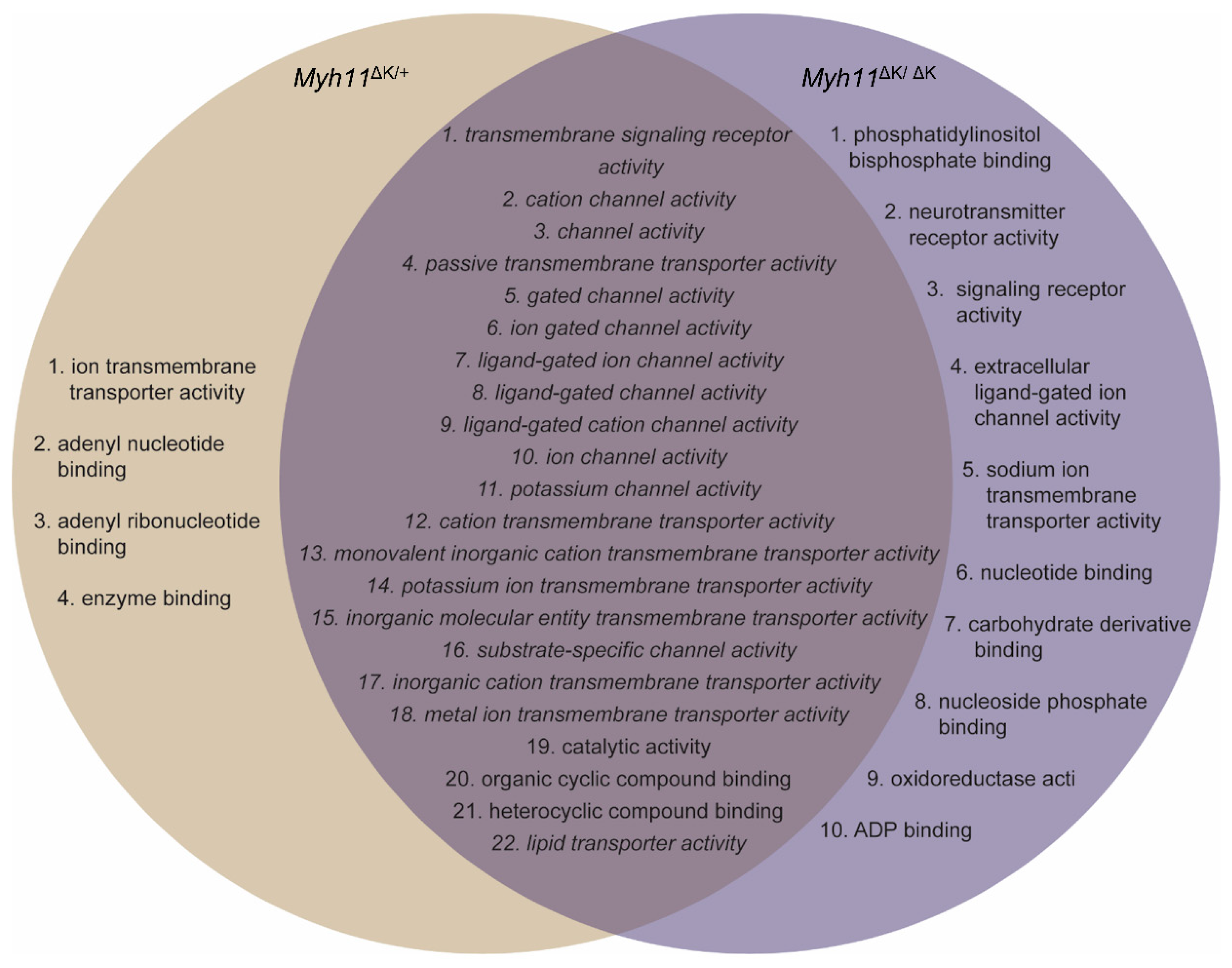
2.2. Myh11 K1256 Deletion Downregulates Transmembrane Transporters
2.3. Myh11 K1256 Deletion Downregulates Inward Calcium Channels
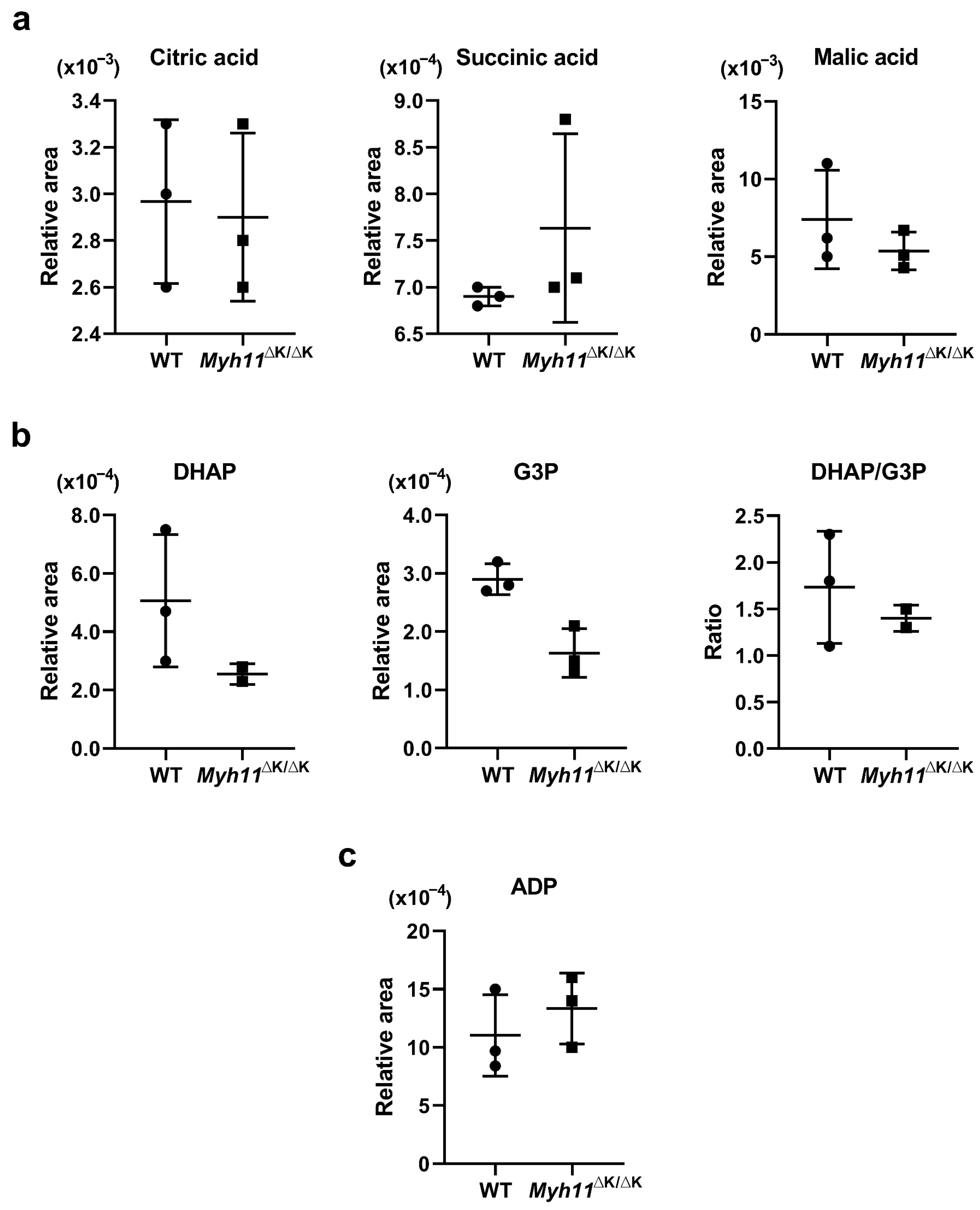
2.4. Attenuated ADP-Ribose (ADPR) Synthesis in Myh11ΔK/ΔK Aortas Leads to Reduced Cytosolic Ca2+
2.5. ATP Synthesis Is Not Attenuated in Myh11∆K/∆K Aortas
2.6. Potassium Channels Are Downregulated in Myh11ΔK/ΔK Aortas
2.7. Genes Involved in Nitric Oxide-Induced Smooth Muscle Relaxation Are Upregulated in Myh11ΔK/ΔK Aortas
3. Discussion
4. Materials and Methods
4.1. Fold-Change-Specific Enrichment Analysis
4.2. Animals
4.3. Metabolomic Analysis
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bradley, T.J.; Bowdin, S.C.; Morel, C.F.; Pyeritz, R.E. The Expanding Clinical Spectrum of Extracardiovascular and Cardiovascular Manifestations of Heritable Thoracic Aortic Aneurysm and Dissection. Can. J. Cardiol. 2016, 32, 86–99. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Regalado, E.S. Use of genetics for personalized management of heritable thoracic aortic disease: How do we get there? J. Thorac. Cardiovasc. Surg. 2015, 149, S3–S5. [Google Scholar] [CrossRef]
- Biddinger, A.; Rocklin, M.; Coselli, J.; Milewicz, D.M. Familial thoracic aortic dilatations and dissections: A case control study. J. Vasc. Surg. 1997, 25, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Coady, M.A.; Davies, R.R.; Roberts, M.; Goldstein, L.J.; Rogalski, M.J.; Rizzo, J.A.; Hammond, G.L.; Kopf, G.S.; Elefteriades, J.A. Familial patterns of thoracic aortic aneurysms. Arch. Surg. 1999, 134, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Albornoz, G.; Coady, M.A.; Roberts, M.; Davies, R.R.; Tranquilli, M.; Rizzo, J.A.; Elefteriades, J.A. Familial thoracic aortic aneurysms and dissections-incidence, modes of inheritance, and phenotypic patterns. Ann. Thorac. Surg. 2006, 82, 1400–1405. [Google Scholar] [CrossRef]
- Imai, Y.; Morita, H.; Takeda, N.; Miya, F.; Hyodo, H.; Fujita, D.; Tajima, T.; Tsunoda, T.; Nagai, R.; Kubo, M.; et al. A deletion mutation in myosin heavy chain 11 causing familial thoracic aortic dissection in two Japanese pedigrees. Int. J. Cardiol. 2015, 195, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Negishi, K.; Aizawa, K.; Shindo, T.; Suzuki, T.; Sakurai, T.; Saito, Y.; Miyakawa, T.; Tanokura, M.; Kataoka, Y.; Maeda, M.; et al. An Myh11 single lysine deletion causes aortic dissection by reducing aortic structural integrity and contractility. Sci. Rep. 2022, 12, 8844. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299–310. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Leon-Mimila, P.; Wang, J.; Huertas-Vazquez, A. Relevance of Multi-Omics Studies in Cardiovascular Diseases. Front Cardiovasc. Med. 2019, 6, 91. [Google Scholar] [CrossRef]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res. 2019, 124, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H.; Lemaire, S.A. Molecular pathogenesis of genetic and sporadic aortic aneurysms and dissections. Curr. Probl. Surg. 2017, 54, 95–155. [Google Scholar] [CrossRef] [PubMed]
- Karimi, A.; Milewicz, D.M. Structure of the Elastin-Contractile Units in the Thoracic Aorta and How Genes That Cause Thoracic Aortic Aneurysms and Dissections Disrupt This Structure. Can. J. Cardiol. 2016, 32, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Scherer, S.; Liu, Y.; Presley, C.; Guo, D.; Estrera, A.L.; Safi, H.J.; Brasier, A.R.; et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum. Mol. Genet. 2007, 16, 2453–2462. [Google Scholar] [CrossRef]
- Chung, A.W.Y.; Au Yeung, K.; Sandor, G.G.S.; Judge, D.P.; Dietz, H.C.; Van Breemen, C. Loss of Elastic Fiber Integrity and Reduction of Vascular Smooth Muscle Contraction Resulting From the Upregulated Activities of Matrix Metalloproteinase-2 and -9 in the Thoracic Aortic Aneurysm in Marfan Syndrome. Circ. Res. 2007, 101, 512–522. [Google Scholar] [CrossRef]
- Warren, M.W.; Zheng, W.; Kobeissy, F.H.; Cheng Liu, M.; Hayes, R.L.; Gold, M.S.; Larner, S.F.; Wang, K.K. Calpain- and caspase-mediated alphaII-spectrin and tau proteolysis in rat cerebrocortical neuronal cultures after ecstasy or methamphetamine exposure. Int. J. Neuropsychopharmacol. 2007, 10, 479–489. [Google Scholar] [CrossRef]
- Yu, P.; Cai, X.; Liang, Y.; Wang, M.; Yang, W. Roles of NAD+ and Its Metabolites Regulated Calcium Channels in Cancer. Molecules 2020, 25, 4826. [Google Scholar] [CrossRef]
- Lee, H.C. Mechanisms of calcium signaling by cyclic ADP-ribose and NAADP. Physiol. Rev. 1997, 77, 1133–1164. [Google Scholar] [CrossRef]
- Koch-Nolte, F.; Fischer, S.; Haag, F.; Ziegler, M. Compartmentation of NAD+ -dependent signalling. FEBS Lett. 2011, 585, 1651–1656. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Deng, J.T.; Van Lierop, J.E.; Sutherland, C.; Walsh, M.P. Ca2+-independent Smooth Muscle Contraction. J. Biol. Chem. 2001, 276, 16365–16373. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Denton, R.M. Mitochondrial Ca2+ transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Dev. Neurosci. 1993, 15, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Mracek, T.; Drahota, Z.; Houstek, J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 2013, 1827, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Quayle, J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 1995, 268, C799–C822. [Google Scholar] [CrossRef]
- Denninger, J.W.; Marletta, M.A. Guanylate cyclase and the ⋅NO/cGMP signaling pathway. Biochim. Biophys. Acta (BBA) Bioenerg. 1999, 1411, 334–350. [Google Scholar] [CrossRef]
- Wilson, D.P. Vascular Smooth Muscle Structure and Function. In Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists; Fitridge, R., Thompson, M., Eds.; University of Adelaide Press: Adelaide, SA, Australia, 2011. [Google Scholar]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Lacinová, L.; Hofmann, F. Ca2+- and voltage-dependent inactivation of the expressed L-type Cav1.2 calcium channel. Arch. Biochem. Biophys. 2005, 437, 42–50. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Guo, D.-C.; Tran-Fadulu, V.; Lafont, A.L.; Papke, C.L.; Inamoto, S.; Kwartler, C.S.; Pannu, H. Genetic Basis of Thoracic Aortic Aneurysms and Dissections: Focus on Smooth Muscle Cell Contractile Dysfunction. Annu. Rev. Genom. Hum. Genet. 2008, 9, 283–302. [Google Scholar] [CrossRef]
- Ferruzzi, J.; Murtada, S.-I.; Li, G.; Jiao, Y.; Uman, S.; Ting, M.Y.L.; Tellides, G.; Humphrey, J.D. Pharmacologically Improved Contractility Protects Against Aortic Dissection in Mice With Disrupted Transforming Growth Factor-β Signaling Despite Compromised Extracellular Matrix Properties. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 919–927. [Google Scholar] [CrossRef]
- Moriguchi, S.; Shioda, N.; Yamamoto, Y.; Tagashira, H.; Fukunaga, K. The T-type voltage-gated calcium channel as a molecular target of the novel cognitive enhancer ST101: Enhancement of long-term potentiation and CaMKII autophosphorylation in rat cortical slices. J. Neurochem. 2012, 121, 44–53. [Google Scholar] [CrossRef]
- Ito, Y.; Takuma, K.; Mizoguchi, H.; Nagai, T.; Yamada, K. A Novel Azaindolizinone Derivative ZSET1446 (Spiro[imidazo[1,2-a]pyridine-3,2-indan]-2(3H)-one) Improves Methamphetamine-Induced Impairment of Recognition Memory in Mice by Activating Extracellular Signal-Regulated Kinase 1/2. J. Pharmacol. Exp. Ther. 2007, 320, 819–827. [Google Scholar] [CrossRef]
- Takeda, K.; Yamaguchi, Y.; Hino, M.; Kato, F. Potentiation of Acetylcholine-Mediated Facilitation of Inhibitory Synaptic Transmission by an Azaindolizione Derivative, ZSET1446 (ST101), in the Rat Hippocampus. J. Pharmacol. Exp. Ther. 2016, 356, 446–456. [Google Scholar] [CrossRef]
- De Matteis, M.A.; Morrow, J.S. Spectrin tethers and mesh in the biosynthetic pathway. J. Cell Sci. 2000, 113, 2331–2343. [Google Scholar] [CrossRef] [PubMed]
- Kunnen, S.J.; Malas, T.B.; Semeins, C.M.; Bakker, A.D.; Peters, D.J.M. Comprehensive transcriptome analysis of fluid shear stress altered gene expression in renal epithelial cells. J. Cell Physiol. 2018, 233, 3615–3628. [Google Scholar] [CrossRef] [PubMed]
- Häger, S.C.; Dias, C.; Sønder, S.L.; Olsen, A.V.; da Piedade, I.; Heitmann, A.S.B.; Papaleo, E.; Nylandsted, J. Short-term transcriptomic response to plasma membrane injury. Sci. Rep. 2021, 11, 19141. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.S.; Nowak, R.B.; Zhou, S.; Giannetto, M.; Gokhin, D.S.; Papoin, J.; Ghiran, I.C.; Blanc, L.; Wan, J.; Fowler, V.M. Myosin IIA interacts with the spectrin-actin membrane skeleton to control red blood cell membrane curvature and deformability. Proc Natl. Acad. Sci. USA 2018, 115, E4377–E4385. [Google Scholar] [CrossRef]
- Wiebe, D.S.; Omelyanchuk, N.A.; Mukhin, A.M.; Grosse, I.; Lashin, S.A.; Zemlyanskaya, E.V.; Mironova, V.V. Fold-Change-Specific Enrichment Analysis (FSEA): Quantification of Transcriptional Response Magnitude for Functional Gene Groups. Genes 2020, 11, 434. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Animal Research: Reporting in vivo Experiments—The ARRIVE Guidelines. J. Cereb. Blood Flow Metab. 2011, 31, 991–993. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Love, M.I.; Anders, S.; Huber, W. Analyzing RNA-seq Data with DESeq2. Available online: https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html#pvaluesNA (accessed on 6 October 2023).





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomida, S.; Ishima, T.; Sawaki, D.; Imai, Y.; Nagai, R.; Aizawa, K. Multi-Omics of Familial Thoracic Aortic Aneurysm and Dissection: Calcium Transport Impairment Predisposes Aortas to Dissection. Int. J. Mol. Sci. 2023, 24, 15213. https://doi.org/10.3390/ijms242015213
Tomida S, Ishima T, Sawaki D, Imai Y, Nagai R, Aizawa K. Multi-Omics of Familial Thoracic Aortic Aneurysm and Dissection: Calcium Transport Impairment Predisposes Aortas to Dissection. International Journal of Molecular Sciences. 2023; 24(20):15213. https://doi.org/10.3390/ijms242015213
Chicago/Turabian StyleTomida, Shota, Tamaki Ishima, Daigo Sawaki, Yasushi Imai, Ryozo Nagai, and Kenichi Aizawa. 2023. "Multi-Omics of Familial Thoracic Aortic Aneurysm and Dissection: Calcium Transport Impairment Predisposes Aortas to Dissection" International Journal of Molecular Sciences 24, no. 20: 15213. https://doi.org/10.3390/ijms242015213