

New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death
by

Lan-Lan Bu
1,
Huan-Huan Yuan
2,
Ling-Li Xie
2,3,
Min-Hua Guo
2,
Duan-Fang Liao
1 and
Xi-Long Zheng
3,* 1
School of Pharmacy, Hunan University of Chinese Medicine, Changsha 410208, China
2
College of Integrated Chinese and Western Medicine, Hunan University of Chinese Medicine, Changsha 410208, China
3
Departments of Biochemistry and Molecular Biology and Physiology and Pharmacology, Cumming School of Medicine, University of Calgary, Calgary, AB T2N 1N4, Canada
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(20), 15160; https://doi.org/10.3390/ijms242015160
Submission received: 1 September 2023
/
Revised: 1 October 2023
/
Accepted: 5 October 2023
/
Published: 13 October 2023
(This article belongs to the Special Issue Cell Death in Cardiovascular Disease)
Abstract
:Endothelial cells (ECs) form the inner linings of blood vessels, and are directly exposed to endogenous hazard signals and metabolites in the circulatory system. The senescence and death of ECs are not only adverse outcomes, but also causal contributors to endothelial dysfunction, an early risk marker of atherosclerosis. The pathophysiological process of EC senescence involves both structural and functional changes and has been linked to various factors, including oxidative stress, dysregulated cell cycle, hyperuricemia, vascular inflammation, and aberrant metabolite sensing and signaling. Multiple forms of EC death have been documented in atherosclerosis, including autophagic cell death, apoptosis, pyroptosis, NETosis, necroptosis, and ferroptosis. Despite this, the molecular mechanisms underlying EC senescence or death in atherogenesis are not fully understood. To provide a comprehensive update on the subject, this review examines the historic and latest findings on the molecular mechanisms and functional alterations associated with EC senescence and death in different stages of atherosclerosis.
1. Introduction
Atherosclerosis is a chronic cardiovascular disease (CVD) that poses significant risks to human health, and is the underlying cause of peripheral vascular disease, coronary heart disease, and stroke. The pathogenesis of atherosclerosis is complex and involves various cell types, including endothelial cells (ECs), vascular smooth muscle cells (SMCs), adventitial fibroblasts, macrophages, and other immune cells. Key factors in the development of atherosclerosis include endothelial dysfunction, leukocyte adhesion, foam macrophage formation, and SMC phenotypic transition. Endothelial dysfunction is considered the initial step in atherosclerosis, and in its broadest sense, it encompasses a constellation of nonadaptive alterations in functional phenotype, which have important implications for the regulation of hemostasis and thrombosis, local vascular tone, redox balance, and the orchestration of acute and chronic inflammatory reactions within the arterial wall [1]. ECs that line elastic arteries, such as the aorta, carotid artery, and femoral artery, have critical functions in maintaining vascular homeostasis. The primary function of the endothelium is to produce nitric oxide (NO) and other vasoactive substances, including hydrogen sulfide (H2S), carbon monoxide (CO), endothelial-derived hyperpolarizing factor (EDHF), prostacyclin, endothelin, and hydrogen peroxide (H2O2), to regulate vascular tone. ECs form a continuous monolayer barrier that controls substance exchange among the lumen, vascular wall, and parenchyma. A specialized barrier function of the endothelium involves its immunoregulatory effects on leukocyte recruitment. Quiescent endothelium is immunosuppressive, with a surface glycoprotein profile that prevents leukocyte adhesion, crawling, and extravasation. Upon tissue injury and inflammatory stress, activated ECs present adhesive molecules, such as vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and E-selection, to the cell surface to facilitate the transendothelial migration of leukocytes. The reactive, pro-inflammatory phenotype of ECs is indispensable for tissue repair after acute injury. However, in the context of chronic tissue damage, such as atherosclerosis, persistent endothelial inflammation becomes pathogenic. Moreover, the regenerative capacity of ECs is intrinsically critical to the re-endothelialization of the surface-eroded arterial lumen and the stabilization of atherosclerotic lesions. Notably, most of these endothelial dysfunctions are associated with endothelial cell senescence and death.
Cellular senescence is a process in which cells undergo permanent cell cycle arrest, with an altered secretome to remodel neighboring cells and the extracellular matrix (ECM) microenvironment [2]. Notably, vascular aging in animal models and humans is characterized by impaired endothelium-dependent dilation (EDD) [3], perturbed fibrinolysis [4], enhanced permeability [5], and aberrant angiogenesis [6]. In humans, endothelium-dependent vasodilation, usually measured as flow-mediated dilation of the radial artery, serves as a non-invasive marker of vascular aging and cardiovascular damage, even in the absence of clinical symptoms [7]. Importantly, cellular senescence of the endothelium is an integral component of vascular aging, as well as atherosclerosis [8]. EC senescence triggers structural and functional deterioration of the vascular wall by not only deterring re-endothelialization and barrier reconstitution at the injury zone, but also promoting an inflammatory and thrombotic niche via the senescence secretome, thereby contributing to the development and progression of CVD [9]. Similar to senescence, multiple forms of endothelial cell death have been associated with the progression of atherosclerosis and the stability of plaques [10]. The death of ECs not only directly causes vessel denudation, but also impacts the neighboring environment via damage-associated molecular patterns (DAMPs) from dying or dead cells. Therefore, investigating the functional impacts of endothelial cell senescence and death will enhance our understanding of the pathogenesis of atherosclerosis and provide theoretical frameworks to develop more effective therapeutic strategies.
2. Vascular Aging and EC Senescence in Atherosclerosis
2.1. What Is Vascular Aging?
2.1.1. Vascular Aging
Vascular aging is a specialized type of aging process involving the structural and functional deterioration of the vascular system. The clinically assessable features of vascular aging include reduced endothelium-dependent dilation and increased arterial stiffness, pulse wave velocity, systolic blood pressure, and central venous pressure, involving a spectrum of perturbed behaviors of ECs, SMCs, infiltrated immune cells, and the ECM. Importantly, vascular aging is a predictor of degenerative vascular diseases, including hypertension, aortic aneurysm, and atherosclerosis [11].
Aged ECs lose their balance in the production of vasodilatory and vasoconstrictive agents, characterized by decreased vasodilatory signals, such as NO and prostacyclin, and increased vasoconstrictive signals, such as endothelin and reactive oxygen species (ROS). Aged SMCs have perturbed mechanosensing and compromised ECM-remodeling capacity, which causes adverse remodeling of elastin and collagen in large elastic arteries. The fragmentation and degradation of elastin occur in the arterial media, driven by the upregulation of matrix metalloproteinases (MMPs) from SMCs and infiltrated leukocytes. The deposition of collagen, potentially as a mechanical compensation for elastin loss, accelerates advanced glycation end product formation, in turn promoting the cross-linking of structural proteins, exacerbating arterial stiffening, and compromising arterial compliance. Moreover, reduced proliferative potential, chronic low-grade inflammation, and non-permissive mechanics act synergistically to impair the endogenous repair capacities of ECs and SMCs so as to counteract age-related “wear and tear” damage.
2.1.2. Morphological, Biochemical, Mechanical, and Metabolic Features of Senescent ECs
At the molecular level, cellular senescence, as an integral process of cellular aging, reflects a progressive failure of molecular mechanisms that leads to disturbances in DNA and cell nuclear and cytosolic environment [12]. The concept of cellular senescence, initially proposed in 1965 by Hayflick, suggests that it results from telomere shortening due to problems with the replication of evolutionarily conserved telomere ends. Subsequent studies identified cellular senescence as a permanent state of cell cycle arrest.
Senescent cells, including ECs, exhibit enlarged and flattened cell bodies. Such morphological features are closely associated with an aberrant hyper-adhesive phenotype [13]. Cho et al. reported hyperactivated focal adhesion kinase (FAK) and Rho GTPases Rac1, Cdc42, resulting in a larger focal adhesion complex and reduced cell motility [13]. Furthermore, multiple kinase pathways, such as mitogen-activated protein kinase (MAPK), mammalian target of rapamycin (mTOR), and Src, have been reported to play roles in senescent morphology [14,15]. Consequently, senescence alters the cell mechanics [16,17,18,19]. For example, Brauer et al. used traction force microscopy to demonstrate the increase in both adhesive and contractile force of senescent human skin fibroblasts at a single-cell resolution [16]. Such cellular biomechanical changes translate into the stiffening and contraction of an ex vivo 3D fibroblast-collagen construct, with a shifted capacity for collagenous matrix remodeling [16]. Most importantly, the morphological–mechanical coupling of senescent ECs has recently been described. Chala et al., with FluidFM-based single-cell force spectroscopy, reported that senescence of ECs leads to an enlarged spread area, enhanced EC-substratum adhesion, and loss of the ability to adapt their shapes in response to flow [20]. However, it must be noted that most morphological and biomechanical features of senescence are characterized in cultured cells. This warrants further study on the in vivo morphology and mechanics of EC senescence in aged or diseased vascular walls.
Intriguingly, senescent cells are metabolically active, albeit with dysregulated profiles. A wide spectrum of essential cellular metabolic processes is impacted by senescence [21,22], including in ECs [22,23]. Importantly, many senescence-related metabolic processes of ECs, including glycolysis, oxidative phosphorylation, fatty acid oxidation, and NAD+ metabolism, have been indicated for their functional involvement in vascular aging and/or atherosclerosis [22,23,24]. One particularly notable, but not yet explored process is glutamine metabolism. Specifically, glutamine metabolism in ECs maintains eNOS activity, suppresses inflammatory signaling, and, notably, plays an anti-senescent role in ECs [25]. The pharmacological inhibition of glutaminolysis, a process that converts glutamine into glutamate and ammonia, accelerates the replicative senescence of cultured ECs [26]. Moreover, deprivation of glutamine from the culture medium, or inhibition of glutaminase 1 (GLS1), a key glutaminolysis enzyme, in cultured ECs causes cell cycle arrest at the G0/G1 phase and sensitizes cells to cytotoxicity by H2O2 [27]. To date, the in vivo link between EC glutaminolysis and senescence in the context of atherosclerosis remains to be explored, although a pathogenic role of excess glutaminolysis in ECs and SMCs has been reported in human and rodent pulmonary hypertension [28], where EC senescence has a disease-driving role [29].
In addition to cell-autonomous effects, one critical outcome of cellular senescence is the modification of the cellular secretome, termed the senescence-associated secretory profile (SASP), which has been recognized as a crucial factor in age-related CVD, including atherosclerosis, and will be discussed in detail in the following sections.
A plethora of molecular mechanisms underlie senescence, including oxidative stress, inflammation, genomic instability, metabolite sensing longevity pathways (sirtuins, AMP-activated protein kinase/AMPK, mTOR, Klotho, fibroblast growth factor 21/FGF21), the activated renin angiotensin–aldosterone system (RAAS), uric acid and polyploidy which are the main topics of this review [30,31].
Currently, clinical assessments of aging are mostly made according to chronologic age, but natural aging does not assume a simple linear rate. The acceleration of organismal aging occurs around the last quarter of the lifespan, when senescent cells accumulate [32,33]. Critically, the rate of cellular senescence varies in the same tissue among different individuals, and among different tissues in the same individual [34,35]. To quantitatively assess the extent of cellular senescence in vivo, multiple methods have been developed, although a clinical assay has not yet been established. Specific advantages and considerations of each senescence assay are summarized in Table 1.
2.1.3. Cellular Senescence in Atherosclerosis
Atherosclerosis has a complex pathogenic course that involves multiple cell types, including ECs, SMCs, fibroblasts, perivascular adipocytes, and immune cells. Here, we briefly discuss the evidence of senescence of these cell types in the context of atherosclerosis. Senescent ECs have reduced NO production and compromised barrier functions attributed to disorganization intercellular adherens junctions and tight junctions [50]. The endothelial coverage of atherosclerotic lesions is one critical protective factor for plaque stability, and attrition of the endothelium leads to atherothrombosis at the affected lesion surface [51,52]. Moreover, senescent ECs have a heightened ability to attract leukocytes via increased expression of adhesive molecules such as VCAM-1 and ICAM-1. The heightened endothelial inflammation is considered as a detrimental process that maintains the chronic inflammatory environment of the atherosclerotic wall.
SMCs are the major cellular origin of atherosclerotic lesion. Lineage-tracing studies have demonstrated that up to 50% of lesional cells, including lipid-laden foam cells, are derived from pre-existing medial SMCs. SMC senescence is considered to be a major disease-driving force of atherogenesis, as senescent SMCs not only produce inflammatory cytokines such as tumor necrosis factor α (TNFα), MCP-1, and IL-6, but also express matrix-degrading enzymes such as MMP9 [53]. These complex senescence-associated dysfunctions of SMCs lead to reduced fibrous cap thickness and increased necrotic core size, which are features of plaque instability [54].
Macrophages have a plethora of pro- and anti-atherogenic functions. Once they have infiltrated into the arterial wall, macrophages can take up lipids/cholesterol and convert into foam cells, constituting the cellular lipid storage of the lesion. Notably, the relative contribution to foam cells by macrophages and SMCs is still a matter of active debate. Macrophages can undergo phenotypic polarization. While M1-polarized macrophage amplify atherosclerosis via the production of inflammatory cytokines, M2 macrophage can play a protective role [55]. Importantly, p16INK4a-induced cellular senescence promotes macrophage differentiation into M1-type macrophages [56,57]. In addition, the senescence of T cells, B cells, and dendritic cells also makes significant contributions to vascular aging and atherosclerosis [57].
Perivascular adipocytes have been reported to support the formation of vascular injury-induced neointima, an early event in atherogenesis [58]. Furthermore, inflammatory activation, as assessed by histological examination, of perivascular adipocytes was reported with an atherogenic diet, suggesting an active role in atherosclerotic vascular wall remodeling [59]. However, in situ evidence for the senescence of perivascular adipocytes is currently lacking.
2.2. Mechanism of Endothelial Cell Senescence
ECs maintain homeostasis mainly by producing and releasing the vasodilator NO and the vasoconstrictor endothelin-1 (ET-1). NO is one of the most important EC-derived anti-atherogenic molecules; it maintains vasorelaxation and prevents arterial stiffening [60]. Aging leads to decreased NO bioavailability [61], tipping the balance to favor vasoconstriction caused by ET-1 or other vasoactive stimuli (ROS, angiotensin II (Ang II), etc.), thereby contributing to age-related decline in vasoreactivity and end-organ perfusion [62]. Importantly, aged ECs display elevated expression of ET-1 and synthesize prostanoids, which in turn enhances vasodilation [63]. Notably, endothelial dysfunction is not only a product of aging and senescence, but also a reciprocal contributory factor in vascular senescence. Inactivated endothelial nitric oxide synthase (eNOS) and increased eNOS uncoupling in ECs enables the production of ROS or reactive nitrogen species (RNS) instead of NO, which disseminate prosenescent signals into the vascular wall [64]. Moreover, an aged or senescent endothelium has an elevated expression of adhesive molecules, such as VCAM-1, ICAM-1, and E-selectin, that attract leukocytes, thereby contributing to chronic vascular wall inflammation [7,65]. In the following sections, we will discuss the triggering of and regulatory signaling for endothelial senescence, including oxidative stress, inflammation, genomic instability, metabolite-sensing longevity pathways (sirtuins, AMPK, mTOR, Klotho, and FGF21), activated RAAS, uric acid, and polyploidization.
2.2.1. Oxidative Stress
Senescent ECs produce high levels of ROS, mainly including superoxide, H2O2, hydroxyl radical, and peroxynitrite, which hinder the vasodilator activity of NO. The redox imbalance is the main cause of endothelial dysfunction. ROS, at normal physiological levels, act as signaling molecules to regulate cell signaling, growth, and differentiation [66]. However, ROS at pathological levels result in oxidative stress, cell dysfunction, senescence, and even cell death [67]. Primary sources of endothelial ROS include mitochondria, NADPH oxidases, uncoupled eNOS, and xanthine oxidase (XO). Vascular stress signals, such as inflammation, Ang II, high glucose, oxidized low density lipoprotein (oxLDL), or even ROS itself, can serve as triggers of ROS production [68].
Mitochondria are the main ATP-producing organelle, and act as a primary source of ROS via the electron transport chain (ETC) [69]. Superoxide anion is produced by mitochondrial complex I and III via electron leakage from the ETC. Mitochondrial ROS (mtROS) further damages ETC complexes, in turn exacerbating ROS production [68]. Senescent ECs exhibit mitochondrial dysfunction, marked by a reduction in mitochondrial mass and changes in mitochondrial composition and ETC function. Aging is associated with reduced activities of complex IV and cytochrome c oxidase, both components of the mitochondrial ETC, leading to increased mitochondrial oxidative stress [70].
NOX4 is a subunit of the NADPH oxidase complex, which is arguably the most robust source of vascular ROS during atherogenesis. NOX4 can be activated by Ang II to generate superoxide, which reacts with NO to form peroxynitrite. NADPH-derived ROS and RNS, when present in excess, adversely impact endothelial biology, including endothelial inflammation, ischemic responses, and even NO production [71]. Specifically, Park and Datla et al. showed that the siRNA-mediated knockdown of NOX4 in human aortic or microvascular ECs significantly reduced ROS production and subsequent cell proliferation [72,73]. Furthermore, NOX4 downregulation inhibited endothelial inflammatory responses evoked by ROS, including ICAM-1, interleukin-8 (IL-8), and monocyte chemoattractant protein-1 (MCP-1) [72,73]. Importantly, NOX4 restricts the replicative lifespan of cultured human umbilical vein endothelial cells (HUVECs), and shRNA-mediated NOX4 knockdown delays the onset of replicative senescence [74]. Furthermore, downregulating NOX4 in ECs effectively inhibits replicative senescence by reducing DNA damage, which occurs independently of telomere attrition. The underlying mechanism by which NOX4 promotes senescence in ECs is associated with mitochondrial dysfunction [75]. On the other hand, NOX4 also has protective effects on vascular health. Mice with Nox4 knockout (KO) have been shown to exhibit intensified Ang II-mediated inflammation, EC dysfunction, and vascular wall remodeling [76]. Furthermore, mice with NOX4 overexpression showed enhanced vasodilation and reduced blood pressure by producing H2O2 [77]. These dual roles of NOX4 in EC functions are consistent with the dose-dependent effects of ROS in vascular functions [78,79,80].
eNOS uncoupling occurs under the conditions of insufficient substrates and/or cofactors such as L-arginine or tetrahydrobiopterin (BH4). Uncoupled eNOS mainly generates superoxide, which reacts with NO to generate peroxynitrite. Notably, peroxynitrite is a highly reactive, toxic metabolite, not only oxidizing nucleic acids, proteins, and lipids, but also oxidizing BH4 to further deplete NO production and accelerate vascular aging [81,82,83,84]. Although peroxynitrite induces the senescence and death of human red blood cells [85], there has yet to be a direct report on the cause–effect relationship between peroxynitrite and senescence in ECs. In this regard, it is important to note that NO can inhibit EC senescence at baseline and at high glucose levels, contributing to the anti-aging actions of estrogen [86]. Under normal physiological conditions, inducible nitric oxide synthase (iNOS) is undetectable in ECs, but abundantly expressed when stimulated by endotoxin or cytokines [87]. Both eNOS and iNOS produce NO, but mainly in the endothelium and intima, respectively [88,89,90,91]. Moreover, the downregulation of eNOS activity and concomitant upregulation of iNOS activity in the arterial wall are prominent molecular features of vascular aging [88,89,90,91]. Notably, selective inhibition of iNOS can partially reverse senescence-related endothelial dysfunction [87].
XO produces ROS during the oxidative conversion of hypoxanthine into uric acid, serving as a main source of ROS during tissue inflammation and ischemic injury [92]. Notably, both ROS and uric acid can induce EC senescence, and the prosenescent role of uric acid will be discussed in the following section. Hypoxia was shown to increase the abundance and activity of XO in the serum and lungs in a neonatal rat model of hypoxic pulmonary hypertension with chronic exposure to 13% O2 for 14 days after birth. Intraperitoneal administration of allopurinol, an XO inhibitor, ameliorated oxidative stress in the lungs and adverse pulmonary vascular remodeling [93]. Although the roles of XO-derived ROS in EC senescence have yet to be reported, accumulating evidence has revealed a close relation between XO, EC dysfunction, and atherosclerosis. For example, when exposed to atheroprone oscillatory shear stress, bovine aortic ECs showed an increase in superoxide production, mediated by NADPH oxidase-dependent XO activation [94]. The administration of oxypurinol and allopurinol improved flow-mediated endothelial-dependent vasodilation of radial arteries in nine patients with coronary artery disease [95]. Recently, the PRIZE study, a multi-center randomized trial involving 514 patients with carotid atherosclerosis demonstrated that febuxostat, an XO inhibitor, can improve arterial stiffness over 24 months, albeit without significant benefits to the atherosclerotic lesion burden [96].
ROS is an active inducer of genomic instability [97]. Unrepaired DNA lesions, especially those that occur on telomeric DNA segments, can cause robust DNA damage responses (DDRs) [98]. Persistent DDR is one of the leading signaling triggers of senescence in many cell types, including ECs [99]. In this regard, it is particularly important to note the well-documented relationship between vascular cells, vascular aging, and atherosclerosis. This topic will be discussed in the following section. In addition to DNA damage, ROS can activate a variety of intracellular signaling pathways, such as NFE2L2-Kelch-like ECH-associated protein 1 (Nrf2-Keap1) [100], peroxisome proliferator-activated receptor gamma (PPAR-γ) [101], sirtuin 1 (SIRT1) [102], nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [103], and Forkhead box (FOXO) [104], which in turn modulate EC senescence [105,106,107,108]. Intriguingly, ROS-activated signaling cascades also frequently elevate the expression of antioxidant genes, including superoxide dismutase (SOD) [109], catalase [110], glutathione peroxidase [111], peroxiredoxins [112], and thioredoxin [113], likely as a compensatory feedback mechanism. Taken together, ROS, when present in excess, reduce NO bioavailability, impair endothelium-dependent vasodilation, and drive EC senescence, all of which contribute to vascular aging and atherogenesis. Oxidative stress and EC senescence are summarized in Figure 1.
2.2.2. Inflammation
Chronic, low-grade inflammation is a key change that occurs in aging arteries [65]. Inflammation is argued to function as a link between the aging process and age-related diseases [114]. Compared with young people, the elderly have significantly increased plasma inflammatory markers associated with vascular diseases such as atherosclerosis, including TNF-α, interleukin-1β (IL-1β), interleukin-6 (IL-6), C-reactive protein (CRP), interferon γ (IFN-γ), MCP-1, and MMPs [115]. Such increases are proportional to age and independent of other cardiovascular risk factors [116]. Importantly, inflammation directly causes endothelial dysfunction and senescence, which constitute integral aspects of atherogenesis [117].
Transcription factor NF-κB has been established as the master regulator of inflammatory molecules, including TNF-α, interleukins (IL-1β, IL-2 and IL-6), chemokines (IL-8 and Regulation of Activation, Expression, and Secretion by Normal T Cells (RANTES)), adhesion molecules (ICAM-1 and VCAM-1), and enzymes (iNOS and COX-2) [118]. Thus, targeting the inhibition of these molecules could be a powerful strategy to prevent vascular aging and atherosclerosis. The recent CANTOS trial demonstrated that canakinumab, an IL-1β-targeting antibody, reduces the recurrence of cardiovascular events in patients with a previous myocardial infarction [119]. However, different types of vascular walls and circulating cells can contribute to local inflammation and atherosclerosis, but their relative contributions remain incompletely understood. Regarding endothelial inflammation specifically, Walker and Lesniewski et al. showed that inhibition of the NF-κB pathway significantly reduces inflammatory cytokine expression and enhances endothelium-dependent vasodilation in both aged mice and humans [120,121].
Senescence of vascular cells, including ECs, SMCs, and macrophages has been observed in atherosclerotic lesions [37]. Importantly, the elimination of p16Ink4a+ senescent cells alleviates atherogenesis and stabilizes established plaques in p16-3MR mice harboring a p16Ink4a promoter-based apoptosis executioner cassette, in which cellular apoptosis is activated by exogenous ganciclovir [122]. Specifically, EC senescence plays causal roles in the progression of atherosclerosis. Telomeric repeat binding factor 2 (TRF2) is a shelterin complex protein that maintains the structural integrity of telomeres [123]. Cellular expression of a dominant negative mutant of TRF2 (TRF2 DN) causes the uncapping of telomeres and, consequently, telomeric DNA damage and senescence [54,124]. Honda et al. reported that mice with forced EC senescence, via Tie2-based expression of TRF2 DN, exhibited an increased burden of atherosclerotic lesions in apolipoprotein E (Apoe-)−/− mice, with NF-κB-dependent VCAM-1 expression and endothelial hyper-inflammation [125]. However, given the fact that Tie2 is also expressed by hematopoietic cells [126], the contributions of the senescent endothelium and senescent immune cells to atherogenesis need to be further specified. This concern has recently been addressed in a mouse model with TRF2 KO under the control of a more specific endothelial promoter (Cdh5-Cre). These mice with endothelial-restricted senescence exhibit enhanced endothelial production of ROS and inflammatory cytokines MCP-1 and IL-1β, impaired endothelial-dependent vasodilation, and perturbed glucose metabolism due to reduced microvascular perfusion and increased endothelial inflammation in metabolically active organs (white adipose tissue and liver) [127]. Senescent ECs exhibit impaired NO production, elevated adhesive molecule expression, and pro-inflammatory cytokine secretion. Consequently, inflammation-mediated endothelial dysfunction has been observed in aged rat carotid arteries and the adipose tissue of the resistant arteries of diet-induced obese mice, with dysfunction arising downstream of TNF-α elevation [128,129]. TNF-α can induce oxidative stress by activating NOX4 or inducing mitochondrial dysfunction, causing endothelial dysfunction, apoptosis, iNOS induction, leukocyte adhesion, and even senescence [130,131,132]. The inhibition of TNF-α improves flow-mediated arterial dilation in aged rats and reduces the expression of inflammatory markers ICAM-1 and iNOS [129,133].
Another inflammatory senescence-signaling cascade is the Notch pathway, which has been reported to inhibit EC growth, promote vascular hyperpermeability, and induce EC senescence in a context-specific manner [134,135]. Notch signaling is enhanced in atherosclerotic regions of aortas from mice and humans, and is activated in the ECs of older adults [134]. Notch activity is also increased in accelerated senescence in response to progerin expression [136]. Progerin is an alternatively spliced prelamin A protein that accelerates telomere shortening and exacerbates aging-related transcription [137]. The LMNA gene encodes prelamin A protein, which normally splices into mature lamin A, a major structural constituent of the nuclear envelope. A mutation of the LMNA gene that favors the accumulation of progerin’s spliced form is the genetic cause of Hutchison Gilford progeroid syndrome (HGPS), with early-onset endothelial dysfunction and extensive SMC loss in elastic arteries. Patients with HGPS frequently die from atherosclerosis and related complications in their first or second decades of life. Importantly, the expression of the progerin protein is also elevated in vascular tissues and cells from otherwise healthy aged humans [138]. Importantly, endothelial progerin accumulation not only causes downregulation of eNOS and endothelial dysfunction, but also induces EC oxidative stress, inflammation, and senescence [139,140].
In conclusion, targeting signaling pathways such as NF-κB and Notch, along with the specific neutralization of inflammatory cytokines like IL-1β or TNF-α, may provide novel therapeutic strategies to combat inflammation-mediated endothelial dysfunction and EC senescence in the context of atherosclerosis. Inflammation and EC senescence are summarized in Figure 2.
2.2.3. Genomic Instability
Genomic instability, acquired via DNA damage as single- or double-strand DNA breaks or adducts over time, is an important mechanism that underlies the age-related accumulation of senescent cells in multiple organs, including elastic arteries [98,141]. Both external genotoxic stressors (oxidative stress and mechanical stress) [142,143] and telomere attrition are important triggers of DNA damage. Genomic instability in vascular ECs and SMCs has been recognized as a causal factor in senescence, vascular aging, and atherosclerosis [30,144,145]. Moreover, microsatellite instability and loss of heterozygosity, which is thought to be a consequence of non-productive repair of DNA breaks, has also been linked to atherosclerosis [146].
Telomere damage, including shortening or length-independent DNA lesions, is one of the best-established determinants of vascular cell senescence. Critically short telomeres trigger cellular senescence and limit EC proliferation, leading to impaired vascular repair and increased susceptibility to atherosclerosis [147,148]. Shelterin is a six-protein complex that protects telomere integrity at the end of chromosomes. Loss of the shelterin components with aging, inflammation, or oxidative stress causes telomere uncapping and excess DNA damage response, in turn eliciting prosenescent signals in ECs [8,149]. This mechanism, particularly via the loss of telomere-bound TRF2, a key shelterin protein, has been recently linked to in vivo EC shear response and senescence in a hyperlipidemic low-density lipoprotein receptor (Ldlr)−/− model of mouse atherosclerosis [150].
Multiple non-telomeric DNA damage/repair defects have been demonstrated as triggering the signaling of EC senescence and aging. One of the best examples is nucleotide excision repair (NER). NER defects have been associated with endothelial dysfunction and the pathogenesis of atherosclerosis. The Excision Repair Cross-Complementation group 1 (ERCC1) and xeroderma pigmentosum group D (XPD), which play essential roles in NER, have been found to protect against EC senescence in mice, suggesting a causal relationship between defective NER and vascular aging [151]. Importantly, the single-nucleotide polymorphism (SNP) of DNA damage binding Protein 2 (DDB2), another NER component, has been linked to carotid–femoral pulse wave velocity, a hallmark of arterial aging, in over 20,000 participants of the AortaGen Consortium [151]. Nevertheless, a causal link between EC-specific DNA repair machinery defects and atherosclerosis has yet to be established. Genomic instability and EC senescence are summarized in Figure 3.
2.2.4. Metabolite-Sensing Longevity Pathways
Metabolite-sensing pathways play a pivotal role in cellular energy homeostasis, nutrient sensing, and overall metabolic regulation, with strong roles in the development and progression of atherosclerosis. The three key players in these pathways are SIRT, AMPK, and mTOR. Notably, strong connections have been documented between these three metabolite-sensing signaling nodes, vascular cell senescence, and atherosclerosis [152,153,154].
Sirtuins
Sirtuins are a family of class III NAD+-dependent deacetylases with diverse roles in aging, inflammation, and stress responses [155]. Among the seven mammalian sirtuins (SIRT1–SIRT7), SIRT1 and SIRT6 have been widely studied in the context of EC senescence and atherosclerosis.
SIRT1 has been reported to play an endothelial-protective role, maintaining endothelial function, preventing endothelial senescence, and ameliorating atherosclerosis. The earliest experimental evidence has shown that chemical or siRNA inhibition of SIRT1 leads to senescence in HUVECs via enhancing the acetylation and activity of p53 [156]. Overexpression of SIRT1 prevents H2O2-induced EC senescence and downregulation of eNOS [156]. Subsequent studies have revealed that SIRT1 expression and activity decline with age in human vasculature. Such age-related SIRT1 repression is associated with human endothelial dysfunction, as determined by impaired flow-mediated vasodilation in the forearms [157]. Importantly, SIRT1 directly regulates NO expression and endothelial function in the arteries by deacetylating and subsequently activating eNOS [157]. Mice with endothelium-specific overexpression of SIRT1 are protected against diet-induced atherosclerosis in hyperlipidemic Apoe−/− mice, with improved aortic NO production independent of blood lipid and glucose levels [158]. Interestingly, global Sirt1 heterozygous KO enhances endothelial inflammation, without affecting eNOS activity, in atherosclerotic Apoe−/− mice [159]. It has been proposed that the NF-κB subunit RelA/p65 is a direct target of the deacetylase of SIRT1, and its deacetylation suppresses the transcriptional activity of NF-κB on proinflammatory adhesive molecules ICAM-1 and VCAM-1 in ECs [159,160]. In addition to eNOS, p53, and NF-κB, SIRT1 can function through many other target proteins, including FOXOs [161] and hypoxia-inducible factor-1α (HIF1α) [162], via its deacetylase activity to improve cardiovascular health. A pharmacological activator of SIRT1, SRT1720, has been shown to extend mouse lifespan and healthspan [163] and to reverse endothelial inflammation, ROS production, and endothelial dysfunction in aged mice [164]. Multiple small-molecule SIRT1 activators have entered clinical trials, but their cardiovascular benefits in humans have not been clearly established [165,166]. However, hope remains based on strong evidence from human genetics. Several SNPs of the SIRT1 gene have been associated with features of atherosclerotic CVDs, including carotid intima–media thickness and risk of coronary artery disease [167,168,169]. Notably, rs3758391 T→C, located in the SIRT1 gene promoter, is associated with elevated SIRT1 mRNA levels in patients with acute coronary syndromes [170].
SIRT6 is another well-documented sirtuin that can protect against EC senescence and atherosclerosis by promoting genomic stability, reducing inflammation, and epigenetically modulating age-related gene expression [171,172]. Global SIRT6 heterozygous KO (Sirt6+/−) exacerbates atherogenesis in Apoe−/− mice fed a high-fat diet, likely via derepressing the expression of proinflammatory adhesive molecules, such as VCAM-1, and promoting monocyte infiltration into the arterial wall [173]. Importantly, the endothelium-specific Sirt6 KO impairs endothelium-dependent vasodilation and causes hypertension [173,174]. Multiple mouse models with endothelial Sirt6 deficiency have generated critical evidence to support an anti-atherosclerotic role of SIRT6, although the in vivo senescence in these mice is not always clearly indicated [175,176]. At the cellular level, SIRT6 protects cultured human ECs from senescence via maintaining telomere integrity and reducing DNA damage [177], or by upregulating Forkhead box protein M1 (FOXM1) and promoting cell cycle progression [178].
In addition to SIRT1 and SIRT6, SIRT2 [179], SIRT3 [180], and SIRT7 [181,182] have also been reported to play anti-senescent roles in ECs. Taken together, emerging evidence suggests that sirtuin isoforms, particularly SIRT1 and SIRT6, play a protective role against EC senescence and atherosclerosis. They achieve this by modulating the oxidative stress response, inflammation, genomic stability, and epigenetics in vascular cells. Considering the complex clinical trial experience of SIRT1 activators, further research is needed to explore the potential therapeutic implications of these findings in the context of atherosclerosis and vascular aging.
AMPK
AMPK, a highly conserved heterotrimeric serine/threonine kinase, is an important energy-sensing signaling protein that integrates energy homeostasis, metabolism, and stress resistance [183]. In general, AMPK delays cellular senescence and ameliorates age-related signaling perturbations [184] in autophagy and mTOR [185,186] cascades. The signaling relationships between AMPK and autophagy or mTOR have been extensively studied and have proven to be very complex. AMPK activates autophagy via phosphorylating Unc-51-like kinase 1 (ULK1) or modulating autophagy-related transcription factors such as FOXOs. Ge et al. conducted an extensive review on the significant role of AMPK in the intricate connection between cellular autophagy and aging [184]. Moreover, AMPK can directly phosphorylate and inactivate mTOR, or do so indirectly via phosphorylating tuberous sclerosis complex 2 (TSC2) [184]. Interestingly, blunting age-related physiological dysfunction after mTOR inactivation also results in increased AMPK activity [187]. AMPK activation can also directly inhibit mTOR by phosphorylating Raptor, a crucial mTOR adaptor protein [188]. Additionally, AMPK signaling has been shown to be upregulated in ribosomal S6 protein kinase (S6K1) KO mice that have reduced mTOR signaling [187]. In ECs specifically, AMPK limits endothelial dysfunction by stimulating NO production. Mechanistically, AMPK enhances eNOS activity through Ras-related C3 botulinum toxin substrate 1 (Rac1)-AKT-dependent phosphorylation of eNOS [189]. Age is associated with declines in AMPK activity in mouse aortas and cerebral arteries [190]. Sustained activation of AMPK using AICAR, an AMP analog and AMPK agonist, improves the endothelium-dependent vasodilation of isolated carotid arteries of aged mice, and this effect is independent of NO bioavailability, suggesting a context-dependent vascular benefit of AMPK [190]. Furthermore, the amelioration of oxidative stress has been reproducibly reported as a convergent mechanism for the anti-aging and anti-senescence effects of AMPK in the endothelium [189,191,192,193]. On the other hand, it should be noted that hyperactivation of AMPK, through overexpression of liver kinase B1 (LKB1), is reported to induce senescence in cultured porcine aortic ECs [107]. In hyperlipidemic mouse models, activation of AMPK by AICAR or metformin has been reported to have an anti-atherogenic benefit, with the involvement of direct effects on the arterial wall, non-vascular organs, and immune cells [194,195]. For instance, metformin activation of AMPK improves mitochondrial biogenesis through interplay with SIRT1 and SIRT3 to antagonize replicative senescence of cultured ECs, and delays arterial senescence, inflammation, and spontaneous atherogenesis in Apoe−/− mice [196]. AMPK has a vaso-protective effect on endothelial aging and senescence via its pleiotropic signaling capacity involving autophagy, mTOR, sirtuins, eNOS, and ROS. However, in vivo evidence on the direct effects of AMPK on endothelial senescence in the context of atherosclerosis is still lacking.
mTOR
mTOR is a serine/threonine kinase which responds to nutrients or growth factors to modulate mRNA translation; protein synthesis; and cellular growth, proliferation, inflammation, and senescence [197]. Mounting studies have shown that decreasing mTOR signaling, either genetically or chemically with rapamycin or rapamycin-like drugs, can slow senescence and vascular aging and extend the lifespans of multiple organisms, including rodents [198,199,200]. Evidence has demonstrated that mTOR signaling is augmented in arteries from older mice which display endothelial dysfunction. Lifelong caloric restriction prevents augmented arterial mTOR signaling and endothelial dysfunction [201]. Activation of mTOR/S6K1 signaling has been demonstrated to play a causal role in endothelial senescence and endothelial dysfunction related to eNOS uncoupling in both a cell culture model and a rat aging model [202]. Furthermore, pilot studies suggested that dietary supplementation of rapamycin for 6 to 8 weeks improves NO and endothelial function in aged mice [203].
mTOR is a core component of two distinct multi-protein complexes: mTORC1, which contains mTOR, mammalian lethal with SEC13 protein 8 (mLST8), proline-rich AKT substrate of 40 kD (PRAS40), and DEP domain-containing mTOR-interacting protein (DEPTOR), and mTORC2, which contains mTOR, rapamycin-insensitive companion of mTOR (RICTOR), mammalian stress-activated protein kinase-interacting protein 1 (MSIN1), and protein observed with RICTOR 1 (PROTOR-1). These two complexes take part in endothelial senescence through different signaling cascades. mTORC1 relays the phosphoinositide 3-kinase (PI3K)-AKT signals evoked by replicative stress to NF-κB in cultured ECs to promote senescence-associated inflammatory secretion, such as IL-6, and paracrine senescence to impact proliferating neighbor cells [204]. Notably, such proinflammatory effects of mTOR on SASP have also been documented for tumor cells, involving diverse signaling cascades [205,206]. mTORC2 is also a prosenescent mediator in ECs when challenged with H2O2 [207]. Upon oxidative stress, mTORC2 acts upstream of AKT, which in turn suppresses CCAAT/enhancer binding protein alpha (CEBPα)-dependent expression of Nrf2 and its downstream antioxidant defense system [207]. siRNA knockdown of RICTOR diminishes the replicative and oxidative stress-induced senescence of HUVECs [207].
Attributable to the pleiotropic effects of mTOR on vascular cell activation and foam cell lipid metabolism, mTOR inhibition has been proposed as a promising therapeutic approach to atherosclerosis. Despite the established prosenescent and pro-aging roles of mTOR at the organ/tissue level, mTOR-mediated EC senescence has not been specifically investigated in the context of age-related vascular diseases. Recently, in a mouse model with tamoxifen-induced EC-specific KO of PRAS40, a negative regulatory component of mTORC1, endothelial inflammation and atherogenesis were exacerbated, and this was correlated with augmented mTORC1 signals [208]. Although the relationship between mTOR activity and endothelial senescence has not been specifically examined in these mouse atherosclerotic lesions, this study provides important evidence of mTOR modulation of endothelial function as a pathogenic determinant of atherosclerosis.
Klotho and FGF21
Klotho is a prominent anti-aging glycoprotein [209] serving as a coreceptor to facilitate the high-affinity binding of endocrine FGF19 family ligands (FGF19, FGF21, and FGF23) to their cognate FGF receptors. The main organ that Klotho directly targets is the kidney, where its protein expression is most abundant. As a comparison, conflicting data show that the vascular expression of Klotho mRNA and protein ranges from non-detectable to detectable levels. Nevertheless, Klotho has a wide spectrum of cardiovascular benefits, and genetic deficiency of Klotho in mice causes arterial medial calcification, intimal hyperplasia, hypertension, endothelial dysfunction, and even reduced lifespan [210]. Notably, low serum Klotho levels are associated with subclinical atherosclerosis or carotid atherosclerotic plaque burden in patients with type 1 diabetes or chronic kidney disease [211,212,213,214]. Furthermore, the serum and aortic levels of Klotho negatively correlate with coronary artery disease severity, independent of other established CVD risk factors [215]. At the cellular level, Klotho is reported to prevent Ang II- or IL-1β-induced senescence of HUVECs via upregulating the cytoprotective Nrf2-heme oxygenase-1 pathway [216]. In addition, Klotho can inhibit the transcriptional activity of NF-κB on cytokine genes, including IL-6 and TNF-α, in HUVECs [217,218]. Recently, Miao et al. have demonstrated that Klotho suppresses Ang II-induced endothelial senescence via inhibiting the wingless-type MMTV integration site family member 3A (Wnt3a)-glycogen synthase kinase-3β (GSK3β)-mTOR pathway, and established a strong association between low serum Klotho levels and an increased risk of major adverse cardiovascular events in 295 patients who underwent a percutaneous coronary intervention with 12 months of follow-up [219].
The anti-aging benefits of Klotho largely depend on endocrine FGFs, among which FGF21 is reported to play an anti-senescent role in ECs. FGF21 is a starvation hormone that induces stress responses by activating the sympathetic nervous system and the hypothalamus–pituitary–adrenal axis [220]. Importantly, growing evidence indicates that exogenous FGF21 has anti-atherosclerotic effects via its actions on lipid metabolism, oxidative stress, inflammation, and cell vitality (senescence and death) [221]. Importantly, FGF21 deficiency exacerbates the development of atherosclerotic plaque formation in Apoe−/− mice. This athero-protective effect of FGF21 is proposed to act via the inhibition of sterol regulatory element-binding protein-2 (SREBP2) and cholesterol biosynthesis in mouse liver [222]. FGF21 administration reverses endothelial dysfunction under oxidative stress in Apoe−/− mice and improves endothelial progenitor cell functions via the AKT/eNOS/NO pathway [223]. Similarly, FGF21 can improve endothelial dysfunction in type 1 diabetes, and the molecular mechanism involves AMPK activation [224]. Furthermore, FGF21 alleviates EC death in the form of apoptosis or pyroptosis, constituting another cytoprotective mechanism to curb atherogenesis [225,226,227]. Despite the accumulating evidence on the endothelial benefits of FGF21, the direct relationship between FGF21 and endothelial senescence remains poorly defined. Yan et al. reported that FGF21 protects HUVECs from H2O2-induced DNA damage, inflammation, and senescence in a SIRT1-dependent manner [228]. In addition, FGF21 can normalize the high-glucose-compromised vitality, migration, and eNOS activity of HUVECs, also lending support to an anti-senescent role of FGF21 [229]. Taken together, these data support the notion that Klotho and FGF21 have powerful vaso-protective effects during aging and under metabolic stress, and some of these benefits may act through the amelioration of endothelial senescence. Metabolite-sensing longevity pathways and EC senescence are summarized in Figure 4.
2.2.5. Activated Renin-Angiotensin System (RAS)
RAS is an essential regulatory component of blood pressure and vascular tone. Chronic activation of RAS contributes to vascular aging via directly modifying vascular cell functions, including enhancing endothelial senescence. Ang II acts through the angiotensin type 1 and type 2 receptors (AT1R and AT2R). Age is associated with elevated expression of Ang II, angiotensin-converting enzyme 1 (ACE1), and AT1R in multiple tissues and cell types, including arterial EC and SMCs [230]. AT1R activation on ECs and SMCs results in NAPDH oxidase-mediated ROS production, inflammation, fibrotic matrix remodeling, and vasoconstriction, whereas the activation of AT2R inhibits cell proliferation, inflammation, and fibrosis [9]. Moreover, Ang II can trigger mitochondrial dysfunction and enhance the production of mitochondrial ROS (mitoROS) [231], which is likely related to the mitochondrial adaptor protein p66Shc [232]. Both cytoplasmic and mitochondrial ROS decrease endothelial NO formation. The reduced NO bioavailability, in synergy with Ang II/AT1R/Rho-associated protein kinase (ROCK)-induced SMC contraction, compromises the endothelium-dependent vasodilation of arteries [233]. Importantly, global KO of AT1A, the closest murine homolog to human AT1R, prolongs mouse lifespan; limits oxidative damage in the heart, kidneys, and aorta, and diminishes age-related aortic damage, such as elastin fragmentation and atherosclerotic foam cell formation [234]. A meta-analysis of 2366 cases and 2414 controls demonstrated that the AT1R A1166C (rs5186) polymorphism is associated with a risk of coronary heart disease in the East Asian population, with an odds ratio of 1.50 for C versus A [235]. This SNP has also been linked with essential hypertension [236,237,238,239]. Pharmacological inhibition of RAS, especially AT1R and ACE1, which serve as rate-limiting steps of Ang II production, has been routinely prescribed as an anti-hypertensive therapy and proposed as a potential approach by which to delay vascular aging [240,241]. In this regard, mounting evidence shows that losartan, an AT1R blocker, reduces arterial stiffness, as measured by pulse wave velocity or ex vivo myography, in hypertensive patients [242,243,244]. In particular, a recent clinical study including 308 elderly, hypertensive subjects and with a 10-year follow-up demonstrated that long-term use of losartan is associated with a reduced risk of acute coronary syndrome [245]. More importantly, such cardiovascular benefits of RAS inhibition can be generalized to normotensive atherosclerotic patients, as established by a meta-analysis of 13 trials and 80,594 patients [245]. Animal experiments also provide striking data to support the life-extending effects of RAS inhibition. Treatment with ACE inhibitors or losartan significantly increases the lifespan of male Wistar rats by ~20% [246], partially via sustaining endothelial NO production during aging [247,248].
ROS, derived from NADPH oxidase or mitochondria, are recognized as major prosenescent signal mediators by Ang II. Furthermore, ROS-activated NF-κB is proposed to mediate surface presentation of adhesive molecules and secretion of proinflammatory cytokines by senescent ECs [249,250]. Beyond ROS and RNS, in recent years, more molecular targets have been identified to regulate Ang II-induced endothelial senescence: sodium-glucose cotransporter (SGLT) 1 and 2 are among the most intriguing, given the recent breakthrough of using a SGLT2 inhibitor as treatment for type 2 diabetes and its promising cardiovascular benefits [251,252,253]. Ang II can elevate the expression of SGLT1 and SGLT2 in cultured porcine coronary arterial ECs and rat aortic arch [254]. SGLT inhibitors, sotagliflozin or empagliflozin, almost completely block Ang II-induced EC senescence in vitro and normalize Ang II-induced endothelial inflammation and eNOS inactivation [254]. Interestingly, empagliflozin can also restore endothelial NO production and antagonize endothelial senescence induced by H2O2 or high glucose, with a potential suppressive effect on the protein levels of ACE1 and AT1R [255], suggesting a critical RAS-SGLT interplay in endothelial senescence. In summary, RAS, especially Ang II, is an important determinant of endothelial aging. Through provoking endothelial ROS, inflammation, death, and senescence, Ang II-mediated endothelial dysfunction confers a substantial risk of atherogenesis. Activated RAS system and EC senescence are summarized in Figure 5.
2.2.6. Uric Acid
An elevated uric acid level in serum is recognized as a risk factor for endothelial dysfunction and CVD [256,257]. As discussed in previous sections, uric acid is an enzymatic product of XO, and ROS are considered as integral components of hyperuricemia-associated vascular stress. The concomitant generation of superoxide and uric acid reacts with NO directly to decrease the bioavailability of NO and the production of peroxynitrite. High uric acid levels in serum are observed in aged individuals [258], and are associated with compromised endothelial function in patients with obesity, chronic kidney disease, and hypertension [259,260,261,262,263]. Remarkably, elevated uric acid levels in serum were found to be associated with CVD in a cohort of 675 patients, and this association was attributed to reduced vascular NO production [264]. A causal link between aortic XO activity and endothelial dysfunction or arterial stiffening has been established in mice with diet-induced cardiometabolic stress [265,266]. Importantly, uric acid-induced oxidative stress can activate RAS, in turn mediating endothelial dysfunction and EC senescence [267]. Uric acid also blocks the promitogenic effects of epidermal growth factor (EGF), thereby contributing to EC senescence [268]. It should be noted that the prosenescent effects of uric acid have only been observed in cultured HUVECs so far. It remains to be determined whether uric acid impacts endothelial senescence in atherosclerotic lesions. On the other hand, uric acid has also been reported to function as a scavenger of peroxynitrite to protect endothelium-dependent vasodilation. For example, the preincubation of mesenteric microvessels from healthy elderly subjects (>60 years of age) with uric acid partially restored endothelium-dependent vasodilation, to a similar extent as SOD or TEMPOL (an SOD mimetic) [269]. Uric acid is an important stress signal for the endothelium with vascular aging and diseases. However, whether uric acid functionally modifies endothelial senescence in the context of atherosclerosis warrants further study. Uric acid and EC senescence are summarized in Figure 6.
2.2.7. Polyploidization
Polyploidy is a cell state with more than two sets of chromosomes as a consequence of DNA replication without cell division, such as endoreduplication, cytokinesis failure, or cell fusion [270]. Polyploidization of vascular cells has been associated with a spectrum of pathological conditions, such as hypertension, stroke, atherosclerosis, and vascular aging [271,272,273,274,275]. Importantly, polyploidization of both SMCs and ECs is recognized as a precursor to cellular senescence [272,276,277]. Polyploid SMCs and ECs accumulate in aged arteries [271,272,273]. Moreover, electron microscopic studies have shown that the proportion of polyploid ECs correlates with the severity of atherosclerosis in the human aorta and iliac artery, as reported by Dr. Smirnov’s group [271] and Dr. Tokunaga’s group [273]. In particular, Dr. Borradaile and Dr. Pickering performed a series of careful studies on the relationship between polyploidy and senescence in human arterial ECs [276,277]. Polyploid ECs have defective cell cycle profiles featuring both G1 and mitotic arrest [277]. Between the cumulative population doubling 15 and 40, the prevalence of EC polyploidy is higher than that of senescence [276,277]. Although the exact cell cycle mechanism for polyploid ECs entering senescent status remains elusive, several critical aspects of endothelial dysfunction have been associated with polyploidization. Polyploid ECs have elevated ROS levels and reduced eNOS activity, correlated with increased expression of inflammatory adhesive protein ICAM-1. Moreover, transcriptomic analysis reveals a perturbed ECM profile and enhanced inflammation. Intriguingly, the overexpression of nicotinamide phosphoribosyltransferase (Nampt) and activation of SIRT1 rescues polyploidy-associated endothelial dysfunction, which is reminiscent of the anti-senescent role of this cascade [277]. In brief, age-driven EC polyploidization precedes senescence, and the polyploid phenotypes including cell cycle arrest, elevated ROS, and inflammation, very likely contribute to senescence and adverse vascular remodeling. Does this polyploidy–senescence link hold true in vivo? What are the relative contributions to vascular aging and atherosclerosis? These important questions remain unanswered and necessitate the development of an animal model that enables the genetic manipulation of endothelial cell ploidy, as in a hepatocyte study [278]. Polyploidization and EC senescence are summarized in Figure 7.
2.3. Senolysis—Therapeutic Targeting of Senescent ECs
2.3.1. Pharmacological Senolytic Approaches
Normally, senescent cells can be cleared by immune cells, such as natural killer cells and T lymphocytes [279,280,281]. However, with age or under atherosclerotic conditions, senescent ECs, SMCs, fibroblasts, and immune cells accumulate in the arterial walls [34,37,282,283]. The increase in senescence burden not only impairs the replicative and reparative capacity of SMCs and ECs, but also adversely impacts non-senescent neighbors and even distant organs via SASP [284,285]. Persistent cellular senescence and SASP are recognized as driving forces for chronic inflammation and adverse arterial remodeling in atherosclerosis. Therefore, senolysis, the selective elimination of senescent cells, is considered to be a novel therapeutic approach in addition to canonical lipid-lowering therapy and recently developed anti-inflammatory therapy.
The conceptual framework of senolysis was first proposed by Dr. Kirkland’s group in 2015 [286]. Through in-depth transcriptomic analysis of senescent and non-senescent cells, an upregulated anti-apoptotic gene set was identified as a common transcript signature of senescent preadipocytes and HUVECs via radiation-induced genomic instability or replicative aging. A targeted drug screening revealed that a combination of dasatinib and quercetin preferentially reduces the viability of cultured senescent preadipocytes and HUVECs, as compared with their non-senescent counterparts. This dasatinb/quercetin combination is able to reduce the senescent cell burden in white adipose tissue and liver of aged, 24-month-old mice, improve the physical performance of mouse limb muscles injured by radiation exposure, and, amazingly, demonstrate notable cardiovascular benefits, including improved left ventricular ejection fraction and endothelium-dependent vasorelaxation [286]. Since, this first-generation senolytic drug has shown considerable benefits in animal models of a wide spectrum of age-related diseases, such as cardiometabolic syndromes, including diabetes [287], obesity [288] and atherosclerosis [289]; cardiac dysfunction [290]; neurological deficits [291,292]; sarcopenia [293]; joint and skeletal disorders [294,295]; and even COVID-19 [296,297]. Importantly, this senolytic regimen has been evaluated in multiple phase 1 and 2 trials (NCT 02874989, 04785300, 02848131, 02652052, and 04685590).
In the past several years, the following studies have greatly expanded the realm of senolytic reagents. An increasing number of chemicals targeting anti-apoptotic BCL-2 signaling cascade have been identified or repurposed as senolytic drugs, including navitoclax (also known as ABT-263), Fisetin, UBX0101 (also known as nutlin-3a), A1331852, and UBX1967 [298]. Among these senolytics, navitoclax has demonstrated extensive cardiovascular benefits in animal models of chemotherapy-induced cardiomyopathy [299], angiotensin II-induced heart failure [300], and myocardial infarction [301,302]. Importantly, navitoclax has been carefully tested in mouse models of atherosclerosis [37,303,304]. In 2016, Dr. van Deursen’s group demonstrated that intraperitoneal injection with 100 mg/kg navitoclax drastically reduced Sudan IV+ early atherosclerotic lesions in the entire aorta by 70% in Ldlr−/− mice fed a high-fat diet [37]. In 2021, Dr. Martin Bennett’s group employed single-cell RNA sequencing to demonstrate that navitoclax (oral gavage, 50 mg/kg) could significantly reduce atherogenesis at the aortic root and in the descending thoracic aorta in Apoe−/− mice fed a high-fat diet [303]. The anti-atherosclerotic benefit of navitoclax was argued to act mainly through selective killing of dedifferentiated SMCs in the lesions. Furthermore, a very recent preprint from Dr. Gary Owens’ group has raised important concerns regarding the plaque-destabilizing effect of navitoclax [304]. Intraperitoneal injection with 100 mg/kg navitoclax (the same dose as the 2016 study by Dr. van Deursen’s group) in Apoe−/− mice fed a Western diet caused >50% mortality by 40 days after the onset of senolytic treatment. This is likely due to reduced plaque stability in the brachiocephalic artery. Single-cell RNA sequencing and SMC and EC lineage tracing revealed a 90% reduction in SMC content and increased endothelial contributions to the lesion via endothelial-to-mesenchymal transition. Such a dramatic alteration in the cellular origin of atherosclerotic plaques likely underlies the observed lethal plaque instability. Different drug sources and delivery methods, or different mouse lines and diets, could all contribute to the discrepancy among these navitoclax and atherosclerosis studies. Nevertheless, the Owens group’s study indicates the necessity of careful testing of senolytics across different mouse models and by multiple independent groups.
Other molecules have also been explored as senolytic targets, such as heat shock protein 90 (HSP90) [305]. Mechanistically, HSP90 protects AKT from proteasomal degradation to maintain survival signals in senescent cells. Inhibitors of HSP90, such as 17-DMAG, a geldanamycin derivative, lead to the selective induction of apoptosis in senescent cells, including HUVECs [305]. However, this has not been tested in animals with vascular aging or diseases yet. Another intriguing class of senolytic drugs is cardiac glycosides, a class of medicines to treat congestive heart failure and atrial fibrillation, that targets the Na+/K+ ATPase pump on the cell surface [306,307]. Although its efficacy on senescent ECs is yet to be determined, the clinical safety profiles of digoxin and ouabain make cardiac glycosides a tantalizing novel approach to reduce the senescent burden in diseased vessel walls.
Senescent cells have increased lysosomal biomass and β-galactosidase activity. One senolytic approach takes advantage of this feature to encapsulate cytotoxic drugs with galacto-oligosaccharides, or to covalently modify drugs with a galactose moiety [308,309,310]. This selectively enhances drug delivery and cytotoxicity to senescent cells. Notably, a preferential targeting of senescent lung cells, including ECs, in healthy and fibrotic mouse lungs by galacto-oligosaccharide nanoparticles has been demonstrated [308]. It will be intriguing to study the functional performance of this drug delivery system in an animal model of atherosclerosis.
Recent studies have demonstrated that lysosome dysfunction in senescent cells is associated with enhanced lysosomal permeability and cytosolic acidification. As a countermeasure, senescent cells upregulate GLS1 expression and glutaminolysis, which leads to increased ammonia production. This process can neutralize pH value lowering to improve senescent cell survival. Notably, the pharmacological inhibition of glutaminolysis by BPTES accelerates the replicative senescence of cultured ECs. However, the functional role of EC glutaminolysis is uncharted to date. Meaningful knowledge might be borrowed from vascular remodeling in pulmonary hypertension, another disease featuring EC senescence [311,312]. Bertero et al. reported that ECM stiffening increases GLS1 expression and enhances glutaminolytic flux and proliferation in cultured pulmonary arterial ECs [28]. Pharmacological inhibitors of GLS1, i.e., C968 and CB-839, reduces the proliferation of pulmonary arteriolar ECs and SMCs, thereby reducing pulmonary-hypertension-associated right ventricular dysfunction [28]. Similar matrix stiffening has been observed in hypertensive and atherosclerotic arterial walls, but no systematic study on EC glutaminolysis has been published to date. Therefore, the role of endothelial glutaminolysis in vascular aging and atherosclerosis remains a tantalizing candidate target for senolysis.
Senolysis is a fast-evolving field of research and innovation. The list of senolytic strategies has been growing to include the development of engineered chimeric antigen receptor/CAR T cells [313] that recognize uPAR-expressing senescent cells, as well as senolytic vaccination [314,315] that targets glycoprotein nonmetastatic melanoma protein B or CD153. Further studies are warranted to examine and optimize the efficacy of these novel senolytic methods in the vascular system.
2.3.2. Genetical Senolytic Approaches
Many high-quality studies have performed comparative studies between pharmacological agents and non-pharmacological, genetic approaches that enable the specific ablation of senescent cells in mouse organs. INK-ATTAC is the earliest mouse model, created by Dr. van Deursen’s group, that allows for the visualization and targeted ablation of senescent cells [316]. The INK-ATTAC transgene contains a p16Ink4a promoter and an expression cassette for FK509 binding protein (FKBP)-caspase-8 fusion protein, followed by an internal ribosome entry site (IRES)-segregated sequence encoding eGFP protein. In this mouse model, senescent cells, as marked by high p16 expression, express the FKBP-caspase-8 protein. These proteins remain inactive until the addition of AP20187, a synthetic drug that induces the dimerization and activation of FKBP-caspase-8, leading to apoptosis of senescent cells. This mouse model was first employed to study the dynamics and function of senescent cells in BubR1 progeroid mice. p16-3MR is another similar senolytic mouse model, created by Dr. Judi Campisi’s group [122]. This mouse model was first applied in a study on senescent ECs and mesenchymal cells in skin wound healing. Both mouse models have been widely employed to investigate the causal roles of cellular senescence in cardiovascular diseases, including vascular aging and atherosclerosis [37,289,303]. For example, Roos et al. demonstrated both a dasatinib/quercetin cocktail and the ablation of p16-high senescent cells using INK-ATTAC mice show a comparable benefit in reverting the vasomotor functions of aged mice [289]. In another seminal study, p16-3MR mice crossbred with Ldlr−/− mice were used to eliminate >90% of the senescent ECs, SMCs, and macrophages in atherosclerotic lesions [37]. This efficient senolytic approach led to a reduction of around 50% in the atherosclerotic burden induced by a high-fat diet, supporting a detrimental role of senescent cells in atherogenesis.
Further, more senolytic mouse models have been recently used, including a particularly notable p21-driven senolytic mouse model which was created and used to study the p21-high senescent cell population in a mouse model of obesity, including microvascular ECs [317,318]. As p16 and p21 may represent distinct senescent cell populations [319], a comparative study between p16- and p21-high senescent cell ablation in the context of atherosclerosis would be highly intriguing.
2.3.3. Challenges of Senolysis
Despite the ongoing promising progress in senolytic research, there are still many considerations from bench to bed. First, most senolytics are systemically administrated, and the potential side effects of senolytics in different vascular beds and organ systems need to be carefully examined. For example, navitoclax is well-known to cause thrombocytopenia, a condition characterized by a decrease in platelet count. Based on the registry of ClinicalTrials.gov, there are over 20 clinical studies regarding senolytic therapy, both in progress and completed. There are even more trials of drugs that possess senolytic property, although they do not aim to eliminate senescent cells. Knowledge from these trials will be critical to understanding and managing any adverse effects of senolytics. Notably, there are no registered trials of senolytics for atherosclerotic cardiovascular diseases yet. Second, the cell-type-specific effects of senolytic treatment remain to be defined. Atherosclerosis has a complex pathogenic course involving multiple cell types. Although senescent ECs are generally considered pro-atherogenic, the in vivo outcomes of senescent EC elimination in the context of atherosclerosis are far from clear. In this regard, the genetic ablation of p16-high senescent liver sinusoidal ECs in aged mice raises critical concerns for senolytics [320]. Recently, preclinical studies of atherosclerosis have begun to integrate senolytic treatment with single-cell omics. As mentioned earlier, Garrido et al. showed that navitoclax reduces the atherosclerotic lesion burden and necrotic core size in hyperlipidemic Apoe−/− mice [303]. This benefit is argued to act through the selective killing of SMCs, as the authors detected few ECs that strongly expressed p16 transcripts in single-cell RNA sequencing of mouse atherosclerotic plaques. However, this study alone is not sufficient to exclude the therapeutic value of targeting senescent ECs in atherosclerosis, as the p16 transcript level can be difficult to detect in single-cell RNA sequencing assays and may not acutely represent p16 protein abundance [321,322]. More experiments are needed to directly compare the effects of selective ablation of ECs versus other cell types in the same atherosclerotic animals. Understanding such cell-type-specific impacts of senolytics will be essential to advancing this promising regimen into the management of atherosclerotic cardiovascular diseases safely and efficiently.
3. Vascular EC Death in Atherosclerosis
3.1. EC Death and Atherosclerosis
The roles of various forms of regulated EC death, including apoptosis, autophagic cell death, NETosis-related cell death, ferroptosis, pyroptosis, and necroptosis, in the development of atherosclerosis have been investigated extensively, as summarized in Figure 8 and Table 2. Various proatherogenic signals, such as low-density lipoprotein (LDL), hyperglycemia, oxidative stress, and low shear stress, have been shown to induce EC death. Conversely, several athero-protective factors, like estrogen and NO, have been demonstrated to prevent EC death. The long-term influence of death-inducing factors causes a substantial loss of EC, leading to not only systemic endothelial dysfunction, but also plaque instability or surface erosion in atherosclerotic lesions locally. However, each mode of cell death is associated with the specific cellular signaling cascades and unique impacts on atherosclerosis, constituting a plethora of potential therapeutic targets. Before focusing on the death of ECs, we must emphasize the fact that multiple cell types have been reported to undergo cell death in atherosclerotic lesions, and the deaths of different cell types may yield various outcomes. For example, apoptosis of macrophages and foam cells precipitates cholesterol crystals, which enlarge the necrotic core and destabilize plaques [323]. Apoptosis of SMCs is frequently associated with the weakening of collagen fibrils, thinning of the fibrous cap, and calcification of lesions [324]. Non-apoptotic death has also been extensively characterized for macrophages and SMCs in atherosclerosis [325]. Herein, we will specifically discuss several well-studied forms of EC death in the context of atherosclerosis.
3.2. Autophagy—Protective and Detrimental
Autophagy is an essential cytoprotective process that degrades intracellular organelles and recycles the protein and lipids [340]. Genetic activation of autophagy by overexpression of ATG5, a key regulator of autophagosome formation, can even extend the lifespan of mice, partially through increasing cellular resistance to oxidative stress [341]. Notably, autophagic flux is critical to maintaining vascular cell homeostasis, and the decline in EC autophagy contributes to arterial aging [342]. Autophagy-enhancing agents, such as trehalose, spermidine, and rapamycin, revert the phenotypes of aged arteries in mice, including reduced vascular NO levels, elevated endothelial inflammation, and impaired endothelium-dependent vasodilation [199,342,343].
At homeostasis or in response to mild-to-moderate vascular stress, EC autophagy is generally considered to be vasoprotective [344,345]. This is attributed to its ability to balance the status of cellular redox and bioenergetics, which is central to proper EC functions and survival [346]. Multiple triggering signals for autophagy are present during aging, such as ROS, oxLDL, advanced glycation end-products (AGEs), and shear stress [347,348,349,350,351]. Specifically, autophagy can protect EC metabolism, NO production, and survival to restrict chronic vascular inflammation and atherogenesis, with control of the level of cellular ROS as a main mechanism [351,352,353]. In an inflammatory environment, intense autophagy in ECs has been found to reduce the expression of endothelial adhesion molecules, such as CD31 and VE-cadherin [354]. This process limits the migration of neutrophils across the endothelium, ultimately disrupting the cycle of inflammation. Moreover, EC autophagy mediates the vasoprotective effects of laminar shear stress, suppresses EC inflammation, apoptosis, and senescence, and alleviates the atherosclerotic burden in hyperlipidemic mice [355].
At the molecular level, SIRT1 has been recognized as a main signaling mediator of protective autophagy in ECs. Physiological, arterial-type laminar flow induces ROS in ECs, which stimulates EC autophagy through flow-activated SIRT1 and TFEB, a master transcriptional regulator of lysosomal biogenesis [356]. SIRT1 takes part in different stages of autophagic flux, ranging from autophagosome initiation to lysosomal degradation, which has been extensively reviewed elsewhere [357,358]. Herein, we briefly focus on the steps that have been experimentally validated in ECs. In HUVECs, SIRT1-dependent AMPK activation, likely via deacetylating/activating LKB1, inhibits mTORC1, in turn stimulating autophagy [359]. As mTOR is a master negative regulator of all main stages of autophagy [360,361], further studies are warranted to dissect the specific signaling role of the SIRT1-mTOR axis in EC autophagy. In addition to regulating the LKB1-AMPK-mTOR pathway, the deacetylase activity of SIRT1 works on a diverse range of pro-autophagic proteins. For example, upon nutrient starvation, SIRT1 deacetylates ATG5, ATG7, and ATG12 to facilitate the elongation of autophagic vesicles [362]. Importantly, the SIRT1-ATG5 link has been studied in oxLDL- and shear-stress-induced EC autophagy [363,364]. Another well-established pro-autophagic role of SIRT1 is that SIRT1-deactylated FOXO1 transcriptionally activates Rab7, which mediates autophagosome–lysosome fusion. The importance of the SIRT1-FOXO1 cascade in protective autophagy in ECs has been experimentally validated by several independent groups [365,366]. Interestingly, SIRT1 itself is susceptible to autophagic degradation, a process that contributes to the loss of SIRT1 during senescence and aging [367]. However, whether this bidirectional control between SIRT1 and autophagy functions in the aged endothelium is unknown.
On the other hand, autophagy also has broad adverse impacts on atherosclerosis via its profound detrimental effects on ECs, macrophages, and arterial SMCs, as it affects pathogenesis from the early to the advanced stages [368]. Critically, overactivation of autophagy can induce cell death, including death of EC, in a form distinct from other modes of death like apoptosis, necroptosis, and necrosis [369,370]. In 2003, Chau et al. demonstrated that endostatin, a collagen VIII cleaved fragment, can induce autophagic EC death in a ROS- and caspase-independent manner [371]. Importantly, serum endostatin levels correlate with subclinical atherosclerosis, as assessed by carotid intima–media thickness in a cohort of 648 healthy Japanese subjects [372], cardiovascular mortality in two Swedish community-based cohorts of 1689 elderly subjects [373], and mortality and severe disability after ischemic stroke in 3463 Chinese subjects [374]. However, the in vivo roles of endostatin in autophagic EC death remain undefined, although endostatin is reported to slow down mouse atherogenesis due to its anti-angiogenic effect or its capacity to interfere with LDL retention in intima [375,376]. Subsequent studies have identified multiple stress signals that trigger autophagic cell death, including ROS, cigarette smoke extract, glucose deprivation, and bisphosphonates [377,378,379,380]. Despite these advancements, in vivo evidence on autophagic EC death, especially its relevance to atherosclerosis, remains to be firmly investigated and established.
3.3. Apoptosis
Apoptosis is one of the most widely studied forms of programmed cell death, and it has diverse, context-dependent roles during development, homeostasis, and disease [381]. Apoptosis can be initiated via two main pathways: the extrinsic and intrinsic pathways. The extrinsic pathway is triggered by extracellular stress signals. Notably, many pro-atherogenic signals are also pro-apoptotic for ECs, including oxLDL, Ang II, TNF-α, homocysteine, and disturbed blood flow [382]. External stress triggers the assembly of a death-inducing signaling complex that activates caspase-8, which in turn cleaves and activates caspase-3, a major executor of apoptosis featuring chromatin condensation and DNA fragmentation [383]. In vivo, the remnants of apoptotic cells can be quickly cleared by phagocytic cells [381]. The intrinsic pathway is activated by perturbed intracellular signals, such as genomic instability due to DNA damage or mitotic defects, oxidative stress, or mitochondrial damage. This pathway mainly involves the release of pro-apoptotic proteins, typically cytochrome c, from the mitochondria into the cytoplasm. This then promotes the assembly of an apoptosome comprising APAF-1 and procaspase-9 for the downstream activation of caspase-3 and apoptotic cell death [384]. It needs to be noted that, despite being initially proposed as distinctive, the extrinsic and intrinsic apoptosis pathways have significant crosstalk and overlap. For instance, TNF-α not only activates tumor necrosis factor receptor 1 (TNFR1) to recruit tumor necrosis factor receptor type 1-associated DEATH domain (TRADD) for the assembly of the death-inducing signaling complex, but also causes mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c [385].
Near half a century ago, increased apoptosis of ECs was found at the pig aortic arch [386], the athero-prone aortic zone. Importantly, accumulating evidence has revealed that EC apoptosis is an early event in the development of atherosclerosis. Tricot et al. observed increased EC apoptosis in 42 human carotid atherosclerotic plaques, with a preferential localization of apoptotic EC in the downstream parts of the lesions, where the blood flow becomes slow and complex [387]. Redox status is one of the main determinants of EC apoptosis during atherogenesis [388], with excess ROS derived from NADPH oxidase and dysfunctional mitochondria as a major driver of EC apoptosis [389,390]. Moreover, pro-atherogenic stressors, such as oxLDL, Ang II, hyperglycemia, pro-inflammatory cytokine TNFα, and low shear stress, are shown to induce EC apoptosis [381]. In contrast, athero-protective factors, such as estrogen, NO, and arterial-type laminar shear stress, prevent EC apoptosis [391,392].
OxLDL is a key biomarker for atherosclerosis, and it induces chronic endothelial inflammation, endothelial dysfunction, and apoptosis [393]. In 1997, Dimmeler et al. identified that the oxLDL-induced death process in HUVECs is a mode of caspase-3-dependent apoptosis [394]. Subsequently, diverse molecular cascades have been identified for oxLDL-mediated apoptosis. Harada-Shiba et al. reported that oxLDL induces a rapid accumulation of ceramide and superoxide to promote the apoptosis of HUVECs [395]. This pro-apoptotic effect is mediated by the oxysterol, but not phospholipid, fraction of oxLDL [395]. At the same time, Sata et al. showed that oxLDL sensitizes Fas-mediated EC apoptosis [396]. Ca2+ activity is also an important intracellular mediator for cytotoxic signals of oxLDL [397]. OxLDL-induced Ca2+ mobilization and subsequent cell death are inhibited by athero-protective high-density lipoprotein (HDL) or delipidated apolipoprotein A (apoA) [397]. Notably, lipoxygenase-1 (LOX-1), a lectin-like endothelial receptor for oxLDL, is transcriptionally upregulated by oxLDL itself, and causes EC apoptosis in an NF-κB-dependent manner [398]. Subsequent studies greatly expanded the spectrum of oxLDL-elicited apoptotic cascades, ranging from surface receptors, stress-sensing kinases, and transcription factors to non-coding RNAs. One intriguing transcription factor is Krüppel-like Factor 2 (KLF2), a well-documented mechanotransducer for shear stress in ECs [399]. KLF2, whose expression is elevated by laminar flow [400], executes its athero-protective effects via transcriptional activation of eNOS and thrombomodulin, or repression of inflammatory mediators including E-selectin, MCP-1, VCAM-1, and plasminogen activator inhibitor-1 (PAI1) [399]. Notably, an anti-apoptotic role of KLF2 has been reported for ECs. Zhang et al. reported that protein tyrosine phosphatase 1B (PTB1B) knockdown can prevent oxLDL-induced inflammatory injury and dysfunction in ECs, which is regulated at least in part by the AMPK/SIRT1 signaling pathway through KLF2 [401]. Expression of the E3 ubiquitin ligase 3-hydroxy-3-methylglutaryl reductase degradation (HRD1) was significantly decreased in atherosclerotic intima. Mechanistically, decreases in HRD1 levels in ECs can be caused by oxLDL. Elevated expression of HRD1 inhibits the EC apoptosis induced by oxLDL, potentially via promoting the ubiquitination–proteasomal degradation of LOX-1, the oxLDL receptor [402]. Importantly, KLF2 has been identified as a transcriptional activator of HRD1 [402]. These studies support the prevention of EC apoptosis as a potential athero-protective mechanism of KLF2.
While laminar shear stress stabilizes the endothelial barrier, sustains eNOS activation, and promotes endothelial quiescence and survival, disturbed flow, an atherogenic risk factor, evokes endothelial inflammation, permeability, and apoptosis [403,404,405,406]. Several mechanisms have been discovered for disturbed flow-induced endothelial apoptosis. Disturbed flow leads to sustained X-box binding protein 1 (XBP1) activation and splicing, one branch of ER stress responses, thereby inducing apoptosis of ECs [406]. Adenovirus-mediated XBP1 overexpression results in the development of in situ EC apoptosis and atherosclerotic lesions in an Apoe−/− mouse model of aortic graft [407]. Moreover, disturbed flow increases endothelial peroxynitrite production, which in turns activates a cascade of the PKCζ/PIASy/p53 SUMOylation/BCL-2 pathway to execute apoptosis in vitro and in vivo [408]. Compared with the athero-protective greater curvature of the mouse aortic arch, the athero-susceptible lesser curvature exhibits higher expression and activity of PKCζ, the expression of p53, and the incidence of p53-dependent endothelial apoptosis [408].
In addition to shear stress, the endothelium in vivo is also impacted by cyclic mechanical stretch in a magnitude-dependent manner. Physiological levels of cyclic stretch (5–10% strain) are reported to protect endothelial survival, while excess stretch (15–20% strain) leads to a compromised endothelial barrier and cell apoptosis [408,409]. Importantly, such mechanosensitive apoptosis is associated with enhanced transcription of genes involved in endothelial inflammation, including IL-6, IL-8, MCP-1, ICAM-1 [410], and TGFβ [411]. Several intracellular signaling pathways have been shown to modulate stretch-dependent endothelial apoptosis. The PI3K-AKT cascade is considered to be a well-established pathway to sustain endothelial survival under mechanical stretch [412,413,414], while the production of ROS and activation of stress-responsive p38 MAPK or JNK is pro-apoptotic upon pathological stretch [411]. Recently, Zhuang et al. reported that cyclic stretch increases microparticle production in cultured ECs, with the endothelial microparticle proteomes dependent on the magnitude of stretch (5% versus 15% strain) [415]. Intriguingly, physiological stretch (5% stretch)-induced microparticles suppress EC apoptosis, potentially via activation of Src [415].
A continual endothelium is critical to suppressing atherogenesis and stabilizing established atherosclerotic plaques [415]. Superficial erosion of plaques, as a consequence of EC apoptosis, has been recognized as a potential cellular event triggering atherothrombosis. This mechanism involves neutrophil and NETosis, which will be discussed in a later section. A plethora of biochemical and biophysical pro-apoptotic stimuli, as discussed above, stress the endothelium of atherosclerosis lesions. However, a challenging question remains: whether the prevention of EC apoptosis constitutes a viable therapeutic invention to treat atherosclerosis. Several important and clinically approved anti-atherosclerotic therapies, including statins, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibition, and SGLT2 inhibitors, have been reported to protect against endothelial apoptosis [416,417,418,419,420,421]. However, given the fact that these medicines have pleiotropic benefits over endothelial functions, the role of endothelial apoptosis in the anti-atherosclerotic benefits are difficult to dissect. In fact, evidence from animal models shows that the genetic ablation of EC apoptotic machinery is largely inconclusive. For example, endothelial overexpression of the Fas ligand in hyperlipidemic Apoe−/− mice retards atherogenesis and is associated with dampened endothelial inflammation and reduced T cell and monocyte infiltration, but is indistinguishable from endothelial apoptosis, as assessed by an in situ TUNEL assay [422]. Further, global caspase-3 KO promotes, instead of inhibits, the growth and necrosis of atherosclerotic lesions in Apoe−/− mice without alteration of the plasma levels of cholesterol or triglyceride. Though EC apoptosis was not examined in the lesions, this study indicates that the prevention of cell apoptosis may not constitute an effective strategy to treat atherosclerosis. This can likely be attributed to the accumulated dysfunctional ECs upon failure in apoptosis execution [423]. Moreover, two alternative possibilities exist, either non-death functions of the apoptosis machinery [424,425] or the switch to other modes of cell death when the apoptosis cascade is inhibited. For the former, both caspase-3 and caspase-9 have a direct, non-death-inducing role in modulating endothelial barrier functions [424,425]. For the latter, pharmacological or genetic inactivation of caspase was reported to shunt the cells under lethal inflammatory stress into necroptosis, a mode of regulated cell death mediated by the formation of a necrosome, a death-inducing protein complex comprising receptor-interacting protein kinase (RIPK) and mixed-lineage kinase domain-like protein (MLKL). Notably, this apoptosis-to-necroptosis switch has been documented for ECs. For instance, induced deletion of endothelial caspase-8 in 6-week-old mice leads to diminished EC apoptosis and fatal hemorrhagic lesions in the small intestine [426]. Importantly, caspase-8-independent EC death and small intestinal hemorrhage are abolished by the genetic inactivation of necroptotic executioner MLKL [426]. We will discuss necroptosis in the following sections.
3.4. Necroptosis
Vulnerable atherosclerotic lesions are characterized by a large necrotic core, which comprises debris of dead cells and cholesterol crystals. The necrotic core is surrounded by layers of SMCs and ECM, termed the fibrous cap. The integrity and thickness of the fibrous cap are critical determinants of plaque stability. Notably, the fibrous cap is also covered by ECs, and endothelial denudation is considered one important contributory factor to atherothrombosis. Recent evidence has suggested that a majority of cell death events in atherosclerotic lesions are due to regulated necrosis. Regulated necrosis has three main modes: necroptosis, pyroptosis, and ferroptosis. These three modes of regulated cell death are triggered by different external stress signals, and have distinct signaling cascades and cellular outcomes. Their relevance to the endothelium in the context of atherosclerosis is the topic of the following sections.
Necroptosis is initiated by extracellular death signals, including TNFα, TNF-related apoptosis-inducing ligand (TRAIL), toll-like receptor (TLR) ligands, interferons, and viruses, which stimulate the formation of a death-inducing necrosome complex containing receptor-interacting serine/threonine-protein kinase 1 (RIPK1), RIPK3, and MLKL [427]. The activation of RIPK1 results in autophosphorylation and interaction with RIPK3, leading to RIPK3 oligomerization and necrosome formation [428,429]. Within the necrosome, activated RIPK3 recruits and phosphorylates MLKL, leading to its oligomerization. The phosphorylated MLKL oligomers interact with plasma membrane phospholipids to form pores [430], leading to membrane permeabilization, release of DAMPs, and necroptotic cell death [429]. Notably, necroptotic cells feature robust release of DAMPs and cytokines, in turn aggravating tissue inflammation [429]. Necroptotic inflammation can also be enhanced by crosstalk with pyroptosis, another form of regulated necrosis. For instance, RIPK3 activates the NLRP3 (NOD-like receptor family, pyrin domain-containing 3) inflammasome, which activates caspase-1 to cleave IL-1β into their mature forms [431]. Inflammasome activation is a key step of pyroptosis, which will be discussed later.
Human atherosclerotic plaques are associated with a noticeable increase in the expression of necroptosis mediators RIPK3 and MLKL, at both the mRNA and protein level [432], with their expression being even higher in individuals with unstable plaques compared to those with stable ones [432]. In hyperlipidemic Ldlr−/− mice, advanced plaques show a greater presence of RIPK3, primarily in macrophages [433]. When RIPK3 is absent, the development of advanced atherosclerotic lesions is reduced in Apoe−/− and Ldlr−/− mice, without affecting early atherogenesis [433]. Decisively, bone marrow transplantation experiments demonstrate that the anti-atherosclerotic effects of RIPK3 KO are mediated by bone marrow-derived cells [433]. Moreover, suppression of MLKL using antisense oligonucleotides or genetic deletion diminishes the necrotic cores of advanced plaques of Apoe−/− mice without impacting the total atherosclerotic plaque burden [434]. Interestingly, although cell death and necrotic cores are reduced in MLKL-deficient advanced lesions, the lesion lipid content is increased, which can mainly be attributed to exacerbated lipid accumulation in macrophage foam cells [434]. While RIPK1 is also predominantly found in macrophages within human carotid lesions, its deletion in LysM+ myeloid cells potentiates, instead of attenuates, macrophage necroptosis and necrotic core formation in atherosclerotic lesions in Apoe−/− mice [435]. This complex, regulatory role of RIPK1 in macrophage death can be at least partially attributed to its transactivation of the NF-κB survival pathway [435]. However, a comparative study using mice with different deficiencies in a multitude of necroptosis-related inflammatory disease models, ranging from systemic inflammation sepsis to localized ischemia-reperfusion injury of the kidney, showed that MLKL deficiency provides limited or no protection against adverse outcomes, whereas Ripk1D138N/D138N (a catalytically inactive mutant) and Ripk3−/− mice were protected [436].
While strong evidence supports the functional importance of macrophage necroptosis in atherosclerosis, the roles of EC necroptosis in the context of atherosclerosis remain elusive. Recently, Colijn et al. performed an important study to compare the effects of myeloid-, EC-, and SMC-specific KO of RIPK3 on atherosclerosis in Apoe−/− mice [437]. Mice with RIPK3-deficient ECs exhibit an increase in lesion burden to a similar extent as mice with RIPK3-deficient myeloid cells [437]. Mechanistically, RIPK3 deficiency in ECs enhances the expression of inflammatory mediators, including E-selectin and MCP-1, but had an inconclusive role in necroptotic cell death, as aortas with endothelial deletion of RIPK3 do not show significant changes in total or phosphorylated MLKL [437]. In the same regard, Karunakaran et al. reported that endothelial RIPK1 functions as a central driver of vascular inflammation in atherogenesis, instead of a regulator of cell survival [438]. In HUVECs, shRNA against RIPK1 reduces inflammatory gene expression (IL-1β, E-selectin, and ICAM-1) and monocyte attachment via promoting nuclear translocation of the NF-κB p65 subunit [438]. It must be noted that EC necroptosis has been investigated widely in microvasculature associated with a variety of diseases, including cardiac transplantation [439,440], lung injury [441,442], and tumor metastasis [443,444,445]. One typical example is that lung microvascular endothelial necroptosis serves as an essential event to facilitate the extravasation of circulating, metastatic tumor cells [443]. This intriguing intercellular crosstalk requires direct contact between tumor cells and ECs. EC expression of TNFR1 and death receptor 6 (DR6) mediates necroptotic cell death and opening of the endothelial barrier to facilitate tumor cell transmigration [443,444,445]. Does this intercellular communication also apply to the transendothelial migration of leukocytes into atherosclerotic lesions? Answers may come from more specific in situ analyses of leukocyte–endothelial contact with lesions of different stages and anatomical sites. With this evidence taken together, although the inhibition of necroptosis machinery, such as RIPK1/3 and MLKL, has demonstrated anti-atherosclerotic benefits in animals, the direct role of EC necroptosis warrants further study.
3.5. Pyroptosis
Pyroptosis is initiated through the activation of caspase-1 within a large complex called an inflammasome [332,446]. There are multiple forms of inflammasomes, including NLRP-1, NLRP-3, absent in melanoma 2 (AIM-2), NLR family CARD domain containing 4 (NLRC-4), and pyrin. The NLRP-3 inflammasome is the most widely studied in the vascular system, and consists of NLRP-3, apoptosis-associated speckle-like protein containing CARD (ASC), and procaspase-1 [447], all of which are expressed in the arterial wall [448,449]. Evidence suggests that atherosclerotic plaque components, such as oxLDL and cholesterol crystals, can activate NLRP-3 inflammasomes, partially involving lysosomal rupture and consequent cathepsin release [450,451,452]. Active caspase-1 promotes inflammation by converting pro-IL-1β and pro-IL-18 into bioactive forms, and by cleaving Gasdermin D (GSDMD). The cleaved GSDMD then forms pores in the cell membrane, leading to membrane disruption and the release of inflammatory factors to amplify tissue inflammation [332,446]. In addition to the canonical pyroptotic cascade, caspase-11 can directly sense and complex with lipopolysaccharide (LPS) in the absence of TLR4 or a canonical inflammasome in the context of sepsis [453]. Subsequently, active caspase-11 cleaves GSDMD, causing pore formation and pyroptotic cell death. Furthermore, caspase-11-induced GSDMD can also activate the NLRP-3 inflammasome, indirectly triggering caspase-1-dependent pyroptosis and the release of IL-1β and IL-18 [453]. However, the role of caspase-11-dependent non-canonical pyroptosis in atherosclerosis remains to be determined. Moreover, targeting different inflammasomes (NLRP-3, NLRP-1, AIM-2, or NLRC-4) has been considered as an anti-atherosclerotic strategy. Small molecule inhibitors selective for NLRP-3, such as MCC950, OLT1177, and CY-09 [454,455], have been shown to reduce IL-1β production and attenuate atherosclerosis progression in preclinical models. For example, van der Heijden et al. reported that intraperitoneal administration of MCC950 significantly ameliorates the development of atherosclerosis in Apoe−/− mice, and is associated with reduced plasma IL-1β levels and reduced macrophages in the lesions [456]. MCC950-mediated inactivation of the pyroptotic NLRP-3/ASC/caspase-1/GSDMD pathway in mouse aortas was clearly demonstrated in a subsequent study [457]. Importantly, the IL-1β-neutralizing antibody canakinumab has demonstrated considerable clinical benefits, reducing recurrent cardiovascular events by 17% in patients with a previous myocardial infarction (CANTOS trial), independent of the lipid level [119]. Mechanistically, although macrophages are currently recognized as the cell targets of anti-pyroptotic therapy [457,458], emerging evidence has established a role of EC pyroptosis in atherosclerosis. Many pro-atherogenic or pro-inflammatory factors activate the NLRP-3 inflammasome in ECs, including oxLDL, ROS, trimethylamine-N-oxide (TMAO), low shear stress, and nicotine [459,460]. These external stress signals likely converge on the redox-activated NF-κB-NLRP-3 axis [459,460] for the induction of endothelial pyroptosis. In particular, cholesterol crystals present in atherosclerotic plaques can induce NLRP-3 inflammasome activation and IL-1β secretion in cultured mouse carotid ECs [461], suggesting that the endothelial pyroptotic machine contributes to plaque vulnerability. Moreover, nicotine administration exacerbates the burden of atherosclerotic lesions of Apoe−/− mice fed a high-fat diet, with a functional involvement of ROS-dependent activation of NLRP3 inflammation, in addition to subsequent cytokine production and pyroptotic EC death [462].
While mice with EC-restricted KO of pyroptosis machinery have not been generated to study atherosclerosis, mechanistic insights can still be gained using mouse models with global deficiency of caspase-1 or NLRP-3. For example, Yin et al. reported that caspase-1 KO reduces early atherogenesis in Apoe−/− mice [463]. Caspase-1−/−; Apoe−/− mice fed a high-fat diet exhibit decreased endothelial activation, including reduced expression of leukocyte adhesion molecules (ICAM-1, VCAM-1, and E-selectin) and reduced secretion of cytokines/chemokines (chemokine (C-C motif) ligand 3 (CCL3), chemokine (C-X-C motif) ligand 2 (CXCL2), and CXCL10). Notably, oxLDL-induced pyroptosis of cultured mouse aortic ECs is abrogated by caspase-1 KO in a SIRT1-dependent manner [463]. Moreover, Zhuang et al. linked disturbed flow with endothelial NLRP3 inflammasome activation in atherosclerotic mice [464]. Pro-atherogenic oscillatory shear stress downregulates KLF2-dependent FoxP1 expression in ECs, in turn derepressing the activity of the NLRP3/caspase-1/IL-1β cascade to promote atherogenesis [464]. Although pyroptotic cell death was not examined in this specific scenario, a subsequent study found that low shear stress (5 dyn/cm2) stimulates pyroptosis of HUVECs via suppressing the expression of miR-181b-5p [465], which directly targets the signal transducer and activator of transcription 3 (STAT3) expression to restrain NLRP3 inflammasome activation and cell death [465]. Furthermore, genetic inactivation of GSDMD, the pyroptosis executioner, effectively blocks the development of atherosclerosis in hyperlipidemic mice. Recently, Puylaert et al. demonstrated that global GSDMD KO leads to reduced atherogenesis, which is associated with smaller necrotic cores and increased SMC-to-monocyte ratios in lesions, indicating that pyroptosis inhibition is a potential approach to stabilizing atherosclerotic plaques [466]. However, Puylaert et al. also noted that cleaved, pore-forming GSDMD species are not detected in human carotid lesions, in contrast with widely available evidence on the pyroptotic death of ECs in culture [467]. For now, it is not clear whether nondetectable expression of endothelial GSDMD is a biological observation or a technical issue. More studies are needed to ascertain the status of in vivo pyroptosis of ECs in human atherosclerosis. Notably, disulfiram, an FDA-approved drug, has been identified as a potent small molecule inhibitor of GSDMD pore formation [467], although its effects on atherosclerosis have yet to be tested [467]. Thus, the inhibition of caspase-1, NLRP3, or GSDMD in dyslipidemic and inflammatory contexts prevents endothelial pyroptosis and improves endothelial survival, thereby preserving vascular intima integrity, reducing vascular inflammation, and further attenuating the progression of atherosclerosis.
3.6. Ferroptosis
Ferroptosis was first discovered during anti-cancer small chemical screening as a non-apoptotic, MEK- and iron-dependent oxidative cell death process in tumor cells with oncogenic RAS [468]. The same study identified and characterized the first-generation ferroptosis inducers, erastin and Ras-selective lethal 3 (RSL3). Ferroptosis features excessive iron-dependent lipid peroxidation, associated with plasma membrane rupture with shrinkage of the nanopores and mitochondria [339]. The chemical processes of lipid peroxidation are complex and involve the dysregulation of several different pathways. First, iron overload contributes to the generation of hydroxyl radicals via the Fenton reaction to oxidize lipids. Normally, iron is buffered and stored by ferritin as Fe2+. Under the condition of iron overload, likely attributed to excessive hemoglobin, iron chloride, heme oxygenase-1 hyperactivation, or transferrin overabundance, the cytosolic labile/free ferrous iron pool grows to induce ferroptosis. In this regard, iron chelators like deferoxamine have anti-ferroptotic effects [469]. Second, enzymatic peroxidation of unsaturated fatty acid in the phospholipid bilayer can be mediated via the Achaete–Scute family BHLH transcription factor 4 (ACSL4)/lysophosphatidylcholine acyltransferase 3 (LPCAT3)/15-LOX cascade. Small chemical inhibitors of 15-LOX, such as PD146176 and ML351, have been used as ferroptosis inhibitors [470]. Third, and best characterized, is the inactivation of glutathione peroxidase (GPX)4 [471]. GPX4 catalyzes the reduction of lipid peroxides in the cellular membrane. Diminished gene expression of GPX4 or depletion of its substrate, glutathione, can permit the accumulation of lipid peroxides and subsequent membrane rupture and cell death. The depletion of glutathione can be caused by inhibition of the Xc-antiporter system, which is responsible for cellular uptake of cysteine in exchange for glutamate. Notably, the Xc-antiporter system is the molecular target of erastin [472], and GPX4 is inhibited by RSL3. In addition, ferrostatin-1, α-tocopherol, or liproxstatin, which directly trap lipid peroxides, can be used to inhibit the execution of ferroptosis.
High iron levels have long been associated with an increased risk of atherosclerotic CVDs [473,474,475]. Martinet et al. suggested that intraplaque hemorrhage, iron deposition, and lipid peroxidation are common pathological features of advanced human atherosclerotic plaque [458]. In experimental animals, iron intake is a cause of atherogenesis [476,477,478,479]. Dietary iron aggravates atherosclerosis, while restricting iron intake via a low-iron diet or iron chelator ameliorates atherogenesis. Recently, Vinchi et al. showed that, in the presence of genetic iron overload caused by a heterozygous mutation of iron exporter Ferroportin C326S, Apoe−/− mice develop severe spontaneous atherosclerosis in the absence of a high-fat diet, which can be rescued by iron restriction (low-iron diet or iron chelator deferasirox) [477]. The iron-overloaded arterial wall displayed an elevated inflammation profile, including cytokine expression and endothelial dysfunction. In particular, a wide range of endothelial defects were observed, including increased aortic endothelial permeability, elevated expression of ICAM-1, VCAM-1, and E-selectin, reduced eNOS activity, and enhanced oxidative damage (as assessed by the nitrotyrosine content) [476]. Interestingly, iron overload, induced by ferric ammonium citrate, enhanced ROS and apoptosis in cultured human aortic ECs, but ferroptotic markers in dying ECs were not tested in this study [476]. Nevertheless, ferroptosis has been established as a mode of oxLDL-induced EC death [480,481,482]. The nature of endothelial ferroptosis is usually identified using ferroptosis inhibitors (e.g., ferrostatin-1) or molecular markers (iron content, GPX4 level, Solute Carrier Family 7 Member 11 (SLC7A11), the Xc-antiporter protein). For example, Bai et al. showed that intraperitoneal injection of ferrostatin-1 reduces the atherosclerotic lesion burden of Apoe−/− mice fed a high-fat diet, to a similar extent to oral administration of simvastatin [481]. Mechanistically, ferrostatin-1 treatment reduces serum and aortic iron content, restores the expression of GPX4 and SLC7A11, and reduces serum LDL-cholesterol levels. Notably, lesional endothelial expression of GPX4 appears to be increased with the treatment of ferrostatin-1 [481]. In addition, ferrostatin-1 rescues oxLDL-induced ferroptotic traits (mitochondria size, iron content, expression of GPX4 and SLC7A11) and the death of cultured mouse aortic ECs [481]. Prenyldiphosphate synthase subunit 2 (PDSS2) is a key enzyme involved in the biosynthesis of coenzyme Q10. Yang et al. found that serum PDSS2 levels are decreased in patients with coronary artery disease. Global PDSS2 KO increases the development of atherosclerosis induced by a Western high-fat diet. Overexpression of PDSS2 suppresses the oxLDL-induced ferroptosis in cultured human coronary arterial ECs, correlated with normalized levels of iron, glutathione, and ROS [480]. Important evidence also exists for the functional link between ferroptosis and atherosclerosis. Iron overloaded by FeSO4 induces both ferroptosis and apoptosis to promote the calcification of HUVECs, which can be alleviated by the application of ferrostatin and the iron chelator deferoxamine [483]. In addition to manipulation of the iron content, encouraging evidence also supports the therapeutic potential of targeting other ferroptotic pathways. Transgenic overexpression of GPX4 in Apoe−/− mice effectively suppresses atherogenesis, as well as necrotic core formation, in advanced plaque [483]. In cultured mouse aortic ECs, GPX4 overexpression reduces ROS production, the expression of ICAM-1 and VCAM-1, and monocyte adhesion induced by lysophosphatidylcholine or 7-ketocholesterol. Interestingly, both the necrotic and apoptotic modes of cell death are prevented by GPX4. Additionally, using the 15-LOX inhibitor PD146176 to hinder enzymatic lipid peroxidation leads to reduced plaque progression in atherosclerotic rabbits which already have established plaques [483]. Similarly, the genetic removal of 15-LOX contributes to a lower plaque load in Apoe−/− mice. Collectively, these investigations strongly imply that iron-dependent lipid peroxidation plays a significant role in the development of atherosclerosis.
3.7. NETosis
NETosis is a form of programmed cell death that is specific to neutrophils, involving the release of neutrophil extracellular traps (NETs) [335,484]. NETs are web-like structures composed of DNA, histones, and various antimicrobial proteins. They are released by neutrophils to trap and neutralize pathogens, such as bacteria, fungi, and parasites, in the extracellular space [335,484]. During NETosis, the neutrophil undergoes a sequence of morphological changes, including the decondensation of its nuclear material and the mixing of nuclear and cytoplasmic components. This leads to the rupture of the cell membrane and the release of NETs into the surrounding environment. NETs not only contain neutrophil materials, but also gather circulating elements in the blood, such as tissue factors, fibrin, and other procoagulants. While NETosis is essential for the immune response, excessive or uncontrolled NET formation can be harmful and has been implicated in the pathogenesis of various autoimmune and inflammatory diseases, including atherosclerosis [335,484]. Functionally, NET induces the activation of ECs, SMCs, antigen-presenting cells, and platelets, leading to localized tissue inflammation and thrombosis, thereby promoting atherogenesis and atherothrombosis [484,485,486]. Reciprocally, dysfunctional ECs enhance NETosis, amplifying the damage to neighboring cells and further injuring the arterial endothelial lining [487]. Coronary specimens from patients with acute myocardial infarction demonstrate the presence of NETs in both fresh and lytic, but not organized, thrombi, suggesting that NETosis occurs in the early stage of atherothrombosis [488]. Importantly, circulating NET components, including plasma levels of nucleosome and myeloperoxidase–DNA complexes, predict the extent of coronary stenosis, the number of atherosclerotic coronary vessels, and the occurrence of major adverse cardiac events, suggesting NET as a novel biomarker in atherosclerosis [489].
For our specific focus, NET not only induces endothelial dysfunction, but also results in EC death. Gupta et al. found that stressed ECs, conditioned by phorbol 12-myristate 13-acetate, TNF-α, or thapsigargin (ER stress inducer) enhance the NET formation of co-cultured neutrophils, which is partially dependent on endothelial release of IL-8 [490]. Reciprocally, NET causes EC death, as evidenced by the cellular uptake of SYTOX green, a membrane-impermeable nucleic acid dye. EC death can be prevented by DNase that disrupts the backbone of NETs [490]. Saffarzadeh et al. subsequently showed that extracellular histones are responsible for NET-induced cultured EC death, as assessed by lactate dehydrogenase release [491]. Notably, double KO of endogenous Dnase1 and Dnase1-like 3 causes severe NET clots and consequent lethal blood vessel occlusion in mouse models of chronic neutrophilia or sepsis [492]. While the specific mode of EC death caused by NET remains elusive, NET-associated citrullinated histone 4 has recently been reported to cause necrosis-like “lytic” cell death of cultured SMCs within 10 min [493]. Does this lytic cell death mechanism hold true for ECs within the proximal impact of NET? More studies are needed to answer this. However, apoptosis has been suggested as a mode of EC death in the context of NETosis, as is discussed in the following paragraph.
Peptidylarginine deiminase 4 (PAD4), a key enzyme of NETosis, mediates the citrullination of histones. Histone citrullination leads to the decondensation of chromatin, which is essential for the extrusion of DNA and the formation of NETs. PAD4 also takes part in ROS production and granule mobilization in neutrophils [335,484]. Given its central role in NETosis, research has been focusing on the impacts of PAD4 in atherosclerosis. Genetic inactivation of PAD4 in bone marrow abrogates NETosis, diminishes endothelial permeability, and reduces in situ thrombosis in an Ldlr−/− mouse model of flow-mediated superficial erosion of intima [484]. Notably, a decreased number of TUNEL+ apoptotic ECs were observed in mice with Pad4−/− bone marrow transplantation or DNase I treatment [494]. Importantly, neutrophils are found at the sites of superficially eroded human plaques, and NETs are localized near where apoptotic ECs reside [495,496]. Disturbed flow also plays a critical role in neutrophil recruitment to erosion-prone lesions and subsequent EC apoptosis and mural thrombosis in a TLR2-dependent manner [496]. Moreover, delivery of a PAD4 inhibitor GSK484 using a collagen IV-targeting nanoparticle reduces NET accumulation at sites of endothelial denudation, which is correlated with improved endothelial lining of superficially eroded arterial intima [52]. These findings indicate that NETosis-mediated apoptotic endothelial denudation is a critical cellular event that drives the superficial erosion of atherosclerotic lesions and atherothrombosis. Therefore, NETs represent a novel therapeutic target for the treatment and prevention of thrombotic complications of atherosclerosis.
3.8. Complexity and Limitations of Targeting EC Death in Atherosclerosis
The development and progression of atherosclerosis are closely related to various cell death modalities, including autophagy, apoptosis, necroptosis, pyroptosis, ferroptosis, and NETosis, as summarized in Figure 9. Cell death contributes to the development of atherosclerosis mainly by loss of endothelial lining, promoting endothelial dysfunction, and inflammatory monocyte recruitment. However, there are few registered trials on ClinicalTrials.gov that primarily evaluate a cell-death-targeting treatment in the context of atherosclerosis, although the mainstream medicines (statin, PCSK9 inhibitors, and SGLT2 inhibitors) have observable effects on vascular cell viability/death. Additionally, as was discussed above, a large collection of inhibitors of specific modes of EC death have been designed and have shown preclinical benefits. Why is there limited clinical translatability? There are several potential considerations. First, multiple modes of cell death can co-exist in the complex pro-atherogenic environment, with a composite stress input of hyperlipidemia, hyperglycemia, oxidative stress, and inflammatory stress. On some occasions, even one stress signal, such as H2O2 or oxidized LDL, can induce multiple modes of EC death. Moreover, different modes of cell death may compensate for each other. A typical example is that when caspase is inhibited in ECs and other cells, necroptosis is activated. This intricate death network could confound the trial of an inhibitor for a specific mode of death. In future, integrated analysis of two or more related modes of cell death should be compared in one atherosclerosis study. This kind of study will pave the way for a feasible strategy to target EC death. Second, pharmacological inhibitors of the death of ECs may impact the death of SMCs and immune cells in a similar or opposite manner, which would make EC-specific targeting impractical, and the outcomes difficult to predict. Recent advancement in single-cell omics at the transcript and epigenetic levels will allow for comprehensive analysis of the responses of different lesion cell types and their communications to a single death-modulating treatment.
4. Summary and Future Directions
In this review, we discussed EC senescence and death, with a particular focus on the context of vascular aging and atherosclerosis. The main points related to EC senescence include (1) morphological, biochemical, mechanical, and metabolic features of senescent EC; (2) molecular mechanisms of EC senescence with different stress signals and related contributions to aging and atherosclerosis; and (3) senolytic treatment for atherosclerosis. The main points regarding EC death include (1) molecular mechanisms of different modes of EC death, including apoptosis, autophagic cell death, necroptosis, pyroptosis, ferroptosis, and NETosis; and (2) the complexity and limitations of cell- death-targeting strategies for atherosclerosis.
Understanding how EC senescence and death influence atherosclerosis can determine which senescence and death modalities are most relevant to the design of effective therapeutics. Since the experimental observation of EC senescence and death in atherosclerosis, several milestones have been achieved in this area. First, EC senescence and death are now recognized as causal factors in endothelial dysfunction and key steps in the development and progression of atherosclerosis. Second, the establishment of pharmacological and genetic models of the manipulation of EC senescence and death has improved our understanding of the role of the endothelium in atherosclerosis. Finally, decades of evidence have clearly established that the adequate protection of ECs from oxidative stress- or inflammation-mediated cellular damage appears to be a promising strategy for maintaining endothelial functions in the context of an established atherosclerotic environment.
Herein, we proposed several potential future research directions that will enable us to gain an integrated understanding of EC senescence and death in atherosclerosis and to design novel therapeutics to treat atherosclerosis and related complications. First, atherosclerosis is a complex pathological process involving multiple cell types, and the senescence and death of EC could have localized and long-distance impacts on neighboring ECs and other cell types. Studies utilizing single-cell and spatial omics to understand intercellular communication between senescent or dying/dead or senescent ECs and other cell types, especially in human atherosclerotic lesions, need to be performed. Second, although senescent ECs are reported to be resistant to apoptosis, the relationship between senescence and other modes of death at the level of single ECs remains unresolved, even in culture. Tracking the fates of senescent ECs challenged with different death signals would be helpful. Third, dissecting the relative contributions of different modes of EC death across different stages of atherosclerosis is also critical for the design of endothelial-targeting therapies.
Author Contributions
All authors contributed to the development of this review article. The critical analysis and review of the literature were performed by L.-L.B. The manuscript was written by L.-L.B. and X.-L.Z., with revisions provided by H.-H.Y., L.-L.X., M.-H.G. and D.-F.L. All authors have read and agreed to the published version of the manuscript.
Funding
This review was supported by the Canadian Institutes of Health Research (CIHR, PJT178010 to X.-L. Zheng, PJT-165941 to X.-L. Zheng), the Natural Sciences and Engineering Research Council of Canada (RGPIN-2020-04592 to X.-L. Zheng), Key Discipline of Basic Medicine, Hunan University of Chinese Medicine (202302), the Science and Technology Innovation Program of Hunan Province (grant no. 2021RC4064), Key Laboratory of Vascular Biology and Translational Medicine, Hunan University of Chinese Medicine, and Basic Research Center of Integrated Chinese and Western Medicine for Prevention and Treatment of Vascular Diseases, Medical School, Hunan University of Chinese Medicine.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Rossman, M.J.; Kaplon, R.E.; Hill, S.D.; McNamara, M.N.; Santos-Parker, J.R.; Pierce, G.L.; Seals, D.R.; Donato, A.J. Endothelial cell senescence with aging in healthy humans: Prevention by habitual exercise and relation to vascular endothelial function. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H890–H895. [Google Scholar] [CrossRef] [PubMed]
- Lesniewski, L.A.; Connell, M.L.; Durrant, J.R.; Folian, B.J.; Anderson, M.C.; Donato, A.J.; Seals, D.R. B6D2F1 Mice are a suitable model of oxidative stress-mediated impaired endothelium-dependent dilation with aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2009, 64, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Takeshita, K.; Shimokawa, T.; Yi, H.; Isobe, K.; Loskutoff, D.J.; Saito, H. Plasminogen activator inhibitor-1 is a major stress-regulated gene: Implications for stress-induced thrombosis in aged individuals. Proc. Natl. Acad. Sci. USA 2002, 99, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Pelegrí, C.; Canudas, A.M.; del Valle, J.; Casadesus, G.; Smith, M.A.; Camins, A.; Pallàs, M.; Vilaplana, J. Increased permeability of blood-brain barrier on the hippocampus of a murine model of senescence. Mech. Ageing Dev. 2007, 128, 522–528. [Google Scholar] [CrossRef]
- Zhuo, Y.; Li, S.H.; Chen, M.S.; Wu, J.; Kinkaid, H.Y.; Fazel, S.; Weisel, R.D.; Li, R.K. Aging impairs the angiogenic response to ischemic injury and the activity of implanted cells: Combined consequences for cell therapy in older recipients. J. Thorac. Cardiovasc. Surg. 2010, 139, 1286–1294.e2. [Google Scholar] [CrossRef]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef]
- Hohensinner, P.J.; Kaun, C.; Buchberger, E.; Ebenbauer, B.; Demyanets, S.; Huk, I.; Eppel, W.; Maurer, G.; Huber, K.; Wojta, J. Age intrinsic loss of telomere protection via TRF1 reduction in endothelial cells. Biochim. Et Biophys. Acta 2016, 1863, 360–367. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; DeMarco, V.G.; Martinez-Lemus, L.A.; Meininger, G.A.; Sowers, J.R. Vascular stiffness in insulin resistance and obesity. Front. Physiol. 2015, 6, 231. [Google Scholar] [CrossRef]
- Martinet, W.; Schrijvers, D.M.; De Meyer, G.R. Pharmacological modulation of cell death in atherosclerosis: A promising approach towards plaque stabilization? Br. J. Pharmacol. 2011, 164, 1–13. [Google Scholar] [CrossRef]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular calcification: An update on mechanisms and challenges in treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. So! What’s aging? Is cardiovascular aging a disease? J. Mol. Cell. Cardiol. 2015, 83, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.A.; Ryu, S.J.; Oh, Y.S.; Park, J.H.; Lee, J.W.; Kim, H.P.; Kim, K.T.; Jang, I.S.; Park, S.C. Morphological adjustment of senescent cells by modulating caveolin-1 status. J. Biol. Chem. 2004, 279, 42270–42278. [Google Scholar] [CrossRef] [PubMed]
- Nishio, K.; Inoue, A. Senescence-associated alterations of cytoskeleton: Extraordinary production of vimentin that anchors cytoplasmic p53 in senescent human fibroblasts. Histochem. Cell Biol. 2005, 123, 263–273. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: Terminology for TOR-driven aging. Aging 2012, 4, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Brauer, E.; Lange, T.; Keller, D.; Görlitz, S.; Cho, S.; Keye, J.; Gossen, M.; Petersen, A.; Kornak, U. Dissecting the influence of cellular senescence on cell mechanics and extracellular matrix formation in vitro. Aging Cell 2023, 22, e13744. [Google Scholar] [CrossRef]
- Grandy, C.; Port, F.; Radzinski, M.; Singh, K.; Erz, D.; Pfeil, J.; Reichmann, D.; Gottschalk, K.E. Remodeling of the focal adhesion complex by hydrogen-peroxide-induced senescence. Sci. Rep. 2023, 13, 9735. [Google Scholar] [CrossRef]
- Shin, E.Y.; Park, J.H.; You, S.T.; Lee, C.S.; Won, S.Y.; Park, J.J.; Kim, H.B.; Shim, J.; Soung, N.K.; Lee, O.J.; et al. Integrin-mediated adhesions in regulation of cellular senescence. Sci. Adv. 2020, 6, eaay3909. [Google Scholar] [CrossRef]
- Phillip, J.M.; Aifuwa, I.; Walston, J.; Wirtz, D. The Mechanobiology of Aging. Annu. Rev. Biomed. Eng. 2015, 17, 113–141. [Google Scholar] [CrossRef]
- Chala, N.; Moimas, S.; Giampietro, C.; Zhang, X.; Zambelli, T.; Exarchos, V.; Nazari-Shafti, T.Z.; Poulikakos, D.; Ferrari, A. Mechanical Fingerprint of Senescence in Endothelial Cells. Nano Lett. 2021, 21, 4911–4920. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: Mechanisms and opportunities for intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Sabbatinelli, J.; Prattichizzo, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Giuliani, A. Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front. Physiol. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Le, N.T. Metabolic regulation of endothelial senescence. Front. Cardiovasc. Med. 2023, 10, 1232681. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef]
- Unterluggauer, H.; Mazurek, S.; Lener, B.; Hütter, E.; Eigenbrodt, E.; Zwerschke, W.; Jansen-Dürr, P. Premature senescence of human endothelial cells induced by inhibition of glutaminase. Biogerontology 2008, 9, 247–259. [Google Scholar] [CrossRef]
- Peyton, K.J.; Liu, X.M.; Yu, Y.; Yates, B.; Behnammanesh, G.; Durante, W. Glutaminase-1 stimulates the proliferation, migration, and survival of human endothelial cells. Biochem. Pharmacol. 2018, 156, 204–214. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef]
- Culley, M.K.; Chan, S.Y. Endothelial Senescence: A New Age in Pulmonary Hypertension. Circ. Res. 2022, 130, 928–941. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Sorond, F.; Merkely, B.; Csiszar, A. Mechanisms of Vascular Aging, A Geroscience Perspective: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Passos, J.F.; Kirkwood, T.B.L. On the evolution of cellular senescence. Aging Cell 2020, 19, e13270. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular senescence in aging primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhao, J.; Bukata, C.; Wade, E.A.; McGowan, S.J.; Angelini, L.A.; Bank, M.P.; Gurkar, A.U.; McGuckian, C.A.; Calubag, M.F.; et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 2020, 19, e13094. [Google Scholar] [CrossRef]
- Idda, M.L.; McClusky, W.G.; Lodde, V.; Munk, R.; Abdelmohsen, K.; Rossi, M.; Gorospe, M. Survey of senescent cell markers with age in human tissues. Aging 2020, 12, 4052–4066. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Herrmann, J.; Babic, M.; Tölle, M.; Eckardt, K.U.; van der Giet, M.; Schuchardt, M. A Novel Protocol for Detection of Senescence and Calcification Markers by Fluorescence Microscopy. Int. J. Mol. Sci. 2020, 21, 3475. [Google Scholar] [CrossRef]
- Evangelou, K.; Gorgoulis, V.G. Sudan Black B, The Specific Histochemical Stain for Lipofuscin: A Novel Method to Detect Senescent Cells. Methods Mol. Biol. 2017, 1534, 111–119. [Google Scholar]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef]
- Couchie, D.; Vaisman, B.; Abderrazak, A.; Mahmood, D.F.D.; Hamza, M.M.; Canesi, F.; Diderot, V.; El Hadri, K.; Nègre-Salvayre, A.; Le Page, A.; et al. Human Plasma Thioredoxin-80 Increases With Age and in Apoe(-/-) Mice Induces Inflammation, Angiogenesis, and Atherosclerosis. Circulation 2017, 136, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Geng, Q.; Gao, H.; Yang, R.; Guo, K.; Miao, D. Pyrroloquinoline Quinone Prevents Estrogen Deficiency-Induced Osteoporosis by Inhibiting Oxidative Stress and Osteocyte Senescence. Int. J. Biol. Sci. 2019, 15, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Viehmann, M.; Adams, V.; Rabald, K.; Mangner, N.; Höllriegel, R.; Lurz, P.; Erbs, S.; Linke, A.; Kirsch, K.; et al. Chronic heart failure and aging—Effects of exercise training on endothelial function and mechanisms of endothelial regeneration: Results from the Leipzig Exercise Intervention in Chronic heart failure and Aging (LEICA) study. Eur. J. Prev. Cardiol. 2016, 23, 349–358. [Google Scholar] [CrossRef]
- Mwasongwe, S.; Gao, Y.; Griswold, M.; Wilson, J.G.; Aviv, A.; Reiner, A.P.; Raffield, L.M. Leukocyte telomere length and cardiovascular disease in African Americans: The Jackson Heart Study. Atherosclerosis 2017, 266, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Tranah, G.J.; Katzman, S.M.; Lauterjung, K.; Yaffe, K.; Manini, T.M.; Kritchevsky, S.; Newman, A.B.; Harris, T.B.; Cummings, S.R. Mitochondrial DNA m.3243A > G heteroplasmy affects multiple aging phenotypes and risk of mortality. Sci. Rep. 2018, 8, 11887. [Google Scholar] [CrossRef]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- O’Neill, E.; Mosley, M.; Cornelissen, B. Imaging DNA damage response by γH2AX in vivo predicts treatment response to Lutetium-177 radioligand therapy and suggests senescence as a therapeutically desirable outcome. Theranostics 2023, 13, 1302–1310. [Google Scholar] [CrossRef]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Tréguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef]
- Krouwer, V.J.; Hekking, L.H.; Langelaar-Makkinje, M.; Regan-Klapisz, E.; Post, J.A. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc. Cell 2012, 4, 12. [Google Scholar] [CrossRef]
- Libby, P. Seeing and Sampling the Surface of the Atherosclerotic Plaque: Red or White Can Make Blue. Arter. Thromb. Vasc. Biol. 2016, 36, 2275–2277. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, R.; Yu, M.; Sausen, G.; Bichsel, C.A.; Corbo, C.; Folco, E.J.; Lee, G.Y.; Liu, Y.; Tesmenitsky, Y.; Shvartz, E.; et al. Targeted delivery of protein arginine deiminase-4 inhibitors to limit arterial intimal NETosis and preserve endothelial integrity. Cardiovasc. Res. 2021, 117, 2652–2663. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arter. Thromb. Vasc. Biol. 2015, 35, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Susser, L.I.; Rayner, K.J. Through the layers: How macrophages drive atherosclerosis across the vessel wall. J. Clin. Investig. 2022, 132, e157011. [Google Scholar] [CrossRef]
- Eshghjoo, S.; Kim, D.M.; Jayaraman, A.; Sun, Y.; Alaniz, R.C. Macrophage Polarization in Atherosclerosis. Genes 2022, 13, 756. [Google Scholar] [CrossRef]
- Vellasamy, D.M.; Lee, S.J.; Goh, K.W.; Goh, B.H.; Tang, Y.Q.; Ming, L.C.; Yap, W.H. Targeting Immune Senescence in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 13059. [Google Scholar] [CrossRef]
- Adachi, Y.; Ueda, K.; Takimoto, E. Perivascular adipose tissue in vascular pathologies-a novel therapeutic target for atherosclerotic disease? Front. Cardiovasc. Med. 2023, 10, 1151717. [Google Scholar] [CrossRef]
- Takaoka, M.; Nagata, D.; Kihara, S.; Shimomura, I.; Kimura, Y.; Tabata, Y.; Saito, Y.; Nagai, R.; Sata, M. Periadventitial adipose tissue plays a critical role in vascular remodeling. Circ. Res. 2009, 105, 906–911. [Google Scholar] [CrossRef]
- Alp, N.J.; Channon, K.M. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 413–420. [Google Scholar] [CrossRef]
- Lüscher, T.F.; Barton, M. Biology of the endothelium. Clin. Cardiol. 1997, 20 (Suppl. 2), Ii-3–Ii-10. [Google Scholar] [CrossRef]
- Novella, S.; Dantas, A.P.; Segarra, G.; Novensa, L.; Heras, M.; Hermenegildo, C.; Medina, P. Aging enhances contraction to thromboxane A2 in aorta from female senescence-accelerated mice. Age 2013, 35, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Westby, C.M.; Weil, B.R.; Greiner, J.J.; Stauffer, B.L.; DeSouza, C.A. Endothelin-1 vasoconstriction and the age-related decline in endothelium-dependent vasodilatation in men. Clin. Sci. 2011, 120, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.E.; Kim, E.N.; Kim, M.Y.; Lim, J.H.; Jang, I.A.; Ban, T.H.; Shin, S.J.; Park, C.W.; Chang, Y.S.; Choi, B.S. Age-Associated Changes in the Vascular Renin-Angiotensin System in Mice. Oxidative Med. Cell. Longev. 2016, 2016, 6731093. [Google Scholar] [CrossRef]
- Csiszar, A.; Wang, M.; Lakatta, E.G.; Ungvari, Z. Inflammation and endothelial dysfunction during aging: Role of NF-kappaB. J. Appl. Physiol. 2008, 105, 1333–1341. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Szarka, A.; Bánhegyi, G.; Sümegi, B. Mitochondria, oxidative stress and aging. Orvosi Hetil. 2014, 155, 447–452. [Google Scholar] [CrossRef]
- Lee, H.Y.; Zeeshan, H.M.A.; Kim, H.R.; Chae, H.J. Nox4 regulates the eNOS uncoupling process in aging endothelial cells. Free Radic. Biol. Med. 2017, 113, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Chun, J.N.; Jung, H.Y.; Choi, C.; Bae, Y.S. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc. Res. 2006, 72, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Datla, S.R.; Peshavariya, H.; Dusting, G.J.; Mahadev, K.; Goldstein, B.J.; Jiang, F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Lener, B.; Kozieł, R.; Pircher, H.; Hütter, E.; Greussing, R.; Herndler-Brandstetter, D.; Hermann, M.; Unterluggauer, H.; Jansen-Dürr, P. The NADPH oxidase Nox4 restricts the replicative lifespan of human endothelial cells. Biochem. J. 2009, 423, 363–374. [Google Scholar] [CrossRef]
- Salazar, G. NADPH Oxidases and Mitochondria in Vascular Senescence. Int. J. Mol. Sci. 2018, 19, 1327. [Google Scholar] [CrossRef]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. CB 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Wolin, M.S.; Gupte, S.A.; Neo, B.H.; Gao, Q.; Ahmad, M. Oxidant-redox regulation of pulmonary vascular responses to hypoxia and nitric oxide-cGMP signaling. Cardiol. Rev. 2010, 18, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Park, H.C.; Patschan, S.; Brodsky, S.V.; Gealikman, O.; Kuo, M.C.; Li, H.; Addabbo, F.; Zhang, F.; Nasjletti, A.; et al. Premature vascular senescence in metabolic syndrome: Could it be prevented and reversed by a selenorganic antioxidant and peroxynitrite scavenger ebselen? Drug Discov. Today Ther. Strateg. 2007, 4, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef] [PubMed]
- Matarrese, P.; Straface, E.; Pietraforte, D.; Gambardella, L.; Vona, R.; Maccaglia, A.; Minetti, M.; Malorni, W. Peroxynitrite induces senescence and apoptosis of red blood cells through the activation of aspartyl and cysteinyl proteases. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 416–418. [Google Scholar] [CrossRef]
- Hayashi, T.; Matsui-Hirai, H.; Miyazaki-Akita, A.; Fukatsu, A.; Funami, J.; Ding, Q.F.; Kamalanathan, S.; Hattori, Y.; Ignarro, L.J.; Iguchi, A. Endothelial cellular senescence is inhibited by nitric oxide: Implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. USA 2006, 103, 17018–17023. [Google Scholar] [CrossRef]
- Csiszar, A.; Ungvari, Z.; Edwards, J.G.; Kaminski, P.; Wolin, M.S.; Koller, A.; Kaley, G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ. Res. 2002, 90, 1159–1166. [Google Scholar] [CrossRef]
- Cernadas, M.R.; Sánchez de Miguel, L.; García-Durán, M.; González-Fernández, F.; Millás, I.; Montón, M.; Rodrigo, J.; Rico, L.; Fernández, P.; de Frutos, T.; et al. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ. Res. 1998, 83, 279–286. [Google Scholar] [CrossRef]
- Cau, S.B.; Carneiro, F.S.; Tostes, R.C. Differential modulation of nitric oxide synthases in aging: Therapeutic opportunities. Front. Physiol. 2012, 3, 218. [Google Scholar] [CrossRef]
- Ungvari, Z.; Csiszar, A.; Kaley, G. Vascular inflammation in aging. Herz 2004, 29, 733–740. [Google Scholar] [CrossRef]
- Chou, T.C.; Yen, M.H.; Li, C.Y.; Ding, Y.A. Alterations of nitric oxide synthase expression with aging and hypertension in rats. Hypertension 1998, 31, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Bortolotti, M.; Bolognesi, A.; Polito, L. Pro-Aging Effects of Xanthine Oxidoreductase Products. Antioxidants 2020, 9, 839. [Google Scholar] [CrossRef] [PubMed]
- Jankov, R.P.; Kantores, C.; Pan, J.; Belik, J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L233–L245. [Google Scholar] [CrossRef] [PubMed]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2290–H2297. [Google Scholar] [CrossRef]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef]
- Shiina, K.; Tomiyama, H.; Tanaka, A.; Yoshida, H.; Eguchi, K.; Kario, K.; Kato, T.; Teragawa, H.; Toyoda, S.; Ohishi, M.; et al. Differential effect of a xanthine oxidase inhibitor on arterial stiffness and carotid atherosclerosis: A subanalysis of the PRIZE study. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2022, 45, 602–611. [Google Scholar] [CrossRef]
- Ameziane El Hassani, R.; Buffet, C.; Leboulleux, S.; Dupuy, C. Oxidative stress in thyroid carcinomas: Biological and clinical significance. Endocr. Relat. Cancer 2019, 26, R131–R143. [Google Scholar] [CrossRef]
- Bencivenga, L.; De Souto Barreto, P.; Rolland, Y.; Hanon, O.; Vidal, J.S.; Cestac, P.; Vellas, B.; Rouch, L. Blood pressure variability: A potential marker of aging. Ageing Res. Rev. 2022, 80, 101677. [Google Scholar] [CrossRef]
- Nagane, M.; Yasui, H.; Kuppusamy, P.; Yamashita, T.; Inanami, O. DNA damage response in vascular endothelial senescence: Implication for radiation-induced cardiovascular diseases. J. Radiat. Res. 2021, 62, 564–573. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Et Biophys. Acta. Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Xie, T.; Xu, Y.; Ji, L.; Sui, X.; Zhang, A.; Zhang, Y.; Chen, J. Heme Oxygenase 1/Peroxisome Proliferator-Activated Receptor Gamma Pathway Protects Intimal Hyperplasia and Mitigates Arteriovenous Fistula Dysfunction by Regulating Oxidative Stress and Inflammatory Response. Cardiovasc. Ther. 2022, 2022, 7576388. [Google Scholar] [CrossRef]
- Zhu, S.L.; Wang, M.L.; He, Y.T.; Guo, S.W.; Li, T.T.; Peng, W.J.; Luo, D. Capsaicin ameliorates intermittent high glucose-mediated endothelial senescence via the TRPV1/SIRT1 pathway. Phytomed. Int. J. Phytother. Phytopharm. 2022, 100, 154081. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Li, Q.; Kou, X.; Qin, X.; Li, Z.; Li, J.; Chen, C. BMP-4 impedes endothelial cell migration in neointimal hyperplasia via FoXO-3 specific modulation of reactive oxygen species. Atherosclerosis 2022, 351, 9–17. [Google Scholar] [CrossRef] [PubMed]
- De Silva, T.M.; Li, Y.; Kinzenbaw, D.A.; Sigmund, C.D.; Faraci, F.M. Endothelial PPARγ (Peroxisome Proliferator-Activated Receptor-γ) Is Essential for Preventing Endothelial Dysfunction with Aging. Hypertension 2018, 72, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.F.; Chen, Z.Y.; Xia, J.B.; Zheng, L.; Zhao, H.; Pi, L.Q.; Park, K.S.; Kim, S.K.; Lee, K.J.; Cai, D.Q. FoxO3a suppresses the senescence of cardiac microvascular endothelial cells by regulating the ROS-mediated cell cycle. J. Mol. Cell. Cardiol. 2015, 81, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Liu, L.; Lee, M.Y.; Xu, C.; Liang, Y.; Man, R.Y.; Vanhoutte, P.M.; Wang, Y. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ. Res. 2010, 106, 1384–1393. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Nyúl-Tóth, Á.; Kiss, T.; Yabluchanskiy, A.; Csipo, T.; Balasubramanian, P.; Lipecz, A.; Benyo, Z.; Csiszar, A. Nrf2 dysfunction and impaired cellular resilience to oxidative stressors in the aged vasculature: From increased cellular senescence to the pathogenesis of age-related vascular diseases. GeroScience 2019, 41, 727–738. [Google Scholar] [CrossRef]
- Bresciani, G.; da Cruz, I.B.; González-Gallego, J. Manganese superoxide dismutase and oxidative stress modulation. Adv. Clin. Chem. 2015, 68, 87–130. [Google Scholar]
- Gebicka, L.; Krych-Madej, J. The role of catalases in the prevention/promotion of oxidative stress. J. Inorg. Biochem. 2019, 197, 110699. [Google Scholar] [CrossRef]
- Chu, Y.; Lan, R.S.; Huang, R.; Feng, H.; Kumar, R.; Dayal, S.; Chan, K.S.; Dai, D.F. Glutathione peroxidase-1 overexpression reduces oxidative stress, and improves pathology and proteome remodeling in the kidneys of old mice. Aging Cell 2020, 19, e13154. [Google Scholar] [CrossRef]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef]
- Cunningham, G.M.; Roman, M.G.; Flores, L.C.; Hubbard, G.B.; Salmon, A.B.; Zhang, Y.; Gelfond, J.; Ikeno, Y. The paradoxical role of thioredoxin on oxidative stress and aging. Arch. Biochem. Biophys. 2015, 576, 32–38. [Google Scholar] [CrossRef]
- Chung, H.Y.; Sung, B.; Jung, K.J.; Zou, Y.; Yu, B.P. The molecular inflammatory process in aging. Antioxid. Redox Signal. 2006, 8, 572–581. [Google Scholar] [CrossRef]
- Scuteri, A.; Orru, M.; Morrell, C.; Piras, M.G.; Taub, D.; Schlessinger, D.; Uda, M.; Lakatta, E.G. Independent and additive effects of cytokine patterns and the metabolic syndrome on arterial aging in the SardiNIA Study. Atherosclerosis 2011, 215, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Miles, E.A.; Rees, D.; Banerjee, T.; Cazzola, R.; Lewis, S.; Wood, R.; Oates, R.; Tallant, A.; Cestaro, B.; Yaqoob, P.; et al. Age-related increases in circulating inflammatory markers in men are independent of BMI, blood pressure and blood lipid concentrations. Atherosclerosis 2008, 196, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Botts, S.R.; Fish, J.E.; Howe, K.L. Dysfunctional Vascular Endothelium as a Driver of Atherosclerosis: Emerging Insights into Pathogenesis and Treatment. Front. Pharmacol. 2021, 12, 787541. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Cesari, M.; Anton, S.; Marzetti, E.; Giovannini, S.; Seo, A.Y.; Carter, C.; Yu, B.P.; Leeuwenburgh, C. Molecular inflammation: Underpinnings of aging and age-related diseases. Ageing Res. Rev. 2009, 8, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Walker, A.E.; Kaplon, R.E.; Pierce, G.L.; Nowlan, M.J.; Seals, D.R. Prevention of age-related endothelial dysfunction by habitual aerobic exercise in healthy humans: Possible role of nuclear factor κB. Clin. Sci. 2014, 127, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Lesniewski, L.A.; Durrant, J.R.; Connell, M.L.; Folian, B.J.; Donato, A.J.; Seals, D.R. Salicylate treatment improves age-associated vascular endothelial dysfunction: Potential role of nuclear factor kappaB and forkhead Box O phosphorylation. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 66, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Lechel, A.; Satyanarayana, A.; Ju, Z.; Plentz, R.R.; Schaetzlein, S.; Rudolph, C.; Wilkens, L.; Wiemann, S.U.; Saretzki, G.; Malek, N.P.; et al. The cellular level of telomere dysfunction determines induction of senescence or apoptosis in vivo. EMBO Rep. 2005, 6, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Ikeda, K.; Urata, R.; Yamazaki, E.; Emoto, N.; Matoba, S. Cellular senescence promotes endothelial activation through epigenetic alteration, and consequently accelerates atherosclerosis. Sci. Rep. 2021, 11, 14608. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Harrington, A.; Yang, X.; Friesel, R.E.; Liaw, L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis 2010, 48, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.I.; Liu, Y.; Tucker, J.R.; Islam, M.T.; Machin, D.R.; Abdeahad, H.; Thomas, T.G.; Bramwell, R.C.; Lesniewski, L.A.; Donato, A.J. Endothelial cell telomere dysfunction induces senescence and results in vascular and metabolic impairments. Aging Cell 2023, 22, e13875. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Henson, G.D.; Morgan, R.G.; Enz, R.A.; Walker, A.E.; Lesniewski, L.A. TNF-α impairs endothelial function in adipose tissue resistance arteries of mice with diet-induced obesity. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H672–H679. [Google Scholar] [CrossRef]
- Csiszar, A.; Labinskyy, N.; Smith, K.; Rivera, A.; Orosz, Z.; Ungvari, Z. Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am. J. Pathol. 2007, 170, 388–398. [Google Scholar] [CrossRef]
- Csiszar, A.; Ungvari, Z.; Koller, A.; Edwards, J.G.; Kaley, G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol. Genom. 2004, 17, 21–30. [Google Scholar] [CrossRef]
- Busik, J.V.; Mohr, S.; Grant, M.B. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes 2008, 57, 1952–1965. [Google Scholar] [CrossRef] [PubMed]
- Kandhaya-Pillai, R.; Miro-Mur, F.; Alijotas-Reig, J.; Tchkonia, T.; Kirkland, J.L.; Schwartz, S. TNFα-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging 2017, 9, 2411–2435. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Ungvari, Z.; Koller, A.; Edwards, J.G.; Kaley, G. Aging-induced proinflammatory shift in cytokine expression profile in coronary arteries. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 1183–1185. [Google Scholar] [CrossRef]
- Liu, Z.J.; Tan, Y.; Beecham, G.W.; Seo, D.M.; Tian, R.; Li, Y.; Vazquez-Padron, R.I.; Pericak-Vance, M.; Vance, J.M.; Goldschmidt-Clermont, P.J.; et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: Implication of Notch signaling in atherosclerosis. Atherosclerosis 2012, 225, 296–303. [Google Scholar] [CrossRef]
- Liang, J.; Ke, X.; Yang, R.; Wang, X.; Du, Z.; Hu, C. Notch pathway activation mediated the senescence of endothelial progenitor cells in hypercholesterolemic mice. J. Bioenerg. Biomembr. 2020, 52, 431–440. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef]
- Ragnauth, C.D.; Warren, D.T.; Liu, Y.; McNair, R.; Tajsic, T.; Figg, N.; Shroff, R.; Skepper, J.; Shanahan, C.M. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 2010, 121, 2200–2210. [Google Scholar] [CrossRef]
- Xu, Q.; Mojiri, A.; Boulahouache, L.; Morales, E.; Walther, B.K.; Cooke, J.P. Vascular senescence in progeria: Role of endothelial dysfunction. Eur. Heart J. Open 2022, 2, oeac047. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Investig. 2019, 129, 531–545. [Google Scholar] [CrossRef]
- Park, J.W.; Park, J.E.; Kim, S.R.; Sim, M.K.; Kang, C.M.; Kim, K.S. Metformin alleviates ionizing radiation-induced senescence by restoring BARD1-mediated DNA repair in human aortic endothelial cells. Exp. Gerontol. 2022, 160, 111706. [Google Scholar] [CrossRef]
- Shah, P.; Hobson, C.M.; Cheng, S.; Colville, M.J.; Paszek, M.J.; Superfine, R.; Lammerding, J. Nuclear Deformation Causes DNA Damage by Increasing Replication Stress. Curr. Biol. CB 2021, 31, 753–765.e6. [Google Scholar] [CrossRef] [PubMed]
- Mayr, M.; Hu, Y.; Hainaut, H.; Xu, Q. Mechanical stress-induced DNA damage and rac-p38MAPK signal pathways mediate p53-dependent apoptosis in vascular smooth muscle cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1423–1425. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.R.; Mahmoudi, M. The role of DNA damage and repair in atherosclerosis: A review. J. Mol. Cell. Cardiol. 2015, 86, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Machin, D.R.; Lesniewski, L.A. Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ. Res. 2018, 123, 825–848. [Google Scholar] [CrossRef]
- Kurz, C.; Hakimi, M.; Kloor, M.; Grond-Ginsbach, C.; Gross-Weissmann, M.L.; Böckler, D.; von Knebel Doeberitz, M.; Dihlmann, S. Coding Microsatellite Frameshift Mutations Accumulate in Atherosclerotic Carotid Artery Lesions: Evaluation of 26 Cases and Literature Review. Mol. Med. 2015, 21, 479–486. [Google Scholar] [CrossRef]
- Chang, E.; Harley, C.B. Telomere length and replicative aging in human vascular tissues. Proc. Natl. Acad. Sci. USA 1995, 92, 11190–11194. [Google Scholar] [CrossRef]
- Fuster, J.J.; Andrés, V. Telomere biology and cardiovascular disease. Circ. Res. 2006, 99, 1167–1180. [Google Scholar] [CrossRef]
- Liu, Y.; Bloom, S.I.; Donato, A.J. The role of senescence, telomere dysfunction and shelterin in vascular aging. Microcirculation 2019, 26, e12487. [Google Scholar] [CrossRef]
- Kotla, S.; Vu, H.T.; Ko, K.A.; Wang, Y.; Imanishi, M.; Heo, K.S.; Fujii, Y.; Thomas, T.N.; Gi, Y.J.; Mazhar, H.; et al. Endothelial senescence is induced by phosphorylation and nuclear export of telomeric repeat binding factor 2-interacting protein. JCI Insight 2019, 4, e124867. [Google Scholar] [CrossRef]
- Durik, M.; Kavousi, M.; van der Pluijm, I.; Isaacs, A.; Cheng, C.; Verdonk, K.; Loot, A.E.; Oeseburg, H.; Bhaggoe, U.M.; Leijten, F.; et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation 2012, 126, 468–478. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Bennett, M.R. Sirtuins in atherosclerosis: Guardians of healthspan and therapeutic targets. Nat. Rev. Cardiol. 2022, 19, 668–683. [Google Scholar] [CrossRef] [PubMed]
- Kaldirim, M.; Lang, A.; Pfeiler, S.; Fiegenbaum, P.; Kelm, M.; Bönner, F.; Gerdes, N. Modulation of mTOR Signaling in Cardiovascular Disease to Target Acute and Chronic Inflammation. Front. Cardiovasc. Med. 2022, 9, 907348. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zou, M.H. AMPK, Mitochondrial Function, and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Kume, S.; Takeda-Watanabe, A.; Kanasaki, K.; Koya, D. Sirtuins and renal diseases: Relationship with aging and diabetic nephropathy. Clin. Sci. 2013, 124, 153–164. [Google Scholar] [CrossRef]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell. Cardiol. 2007, 43, 571–579. [Google Scholar] [CrossRef]
- Donato, A.J.; Magerko, K.A.; Lawson, B.R.; Durrant, J.R.; Lesniewski, L.A.; Seals, D.R. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol. 2011, 589, 4545–4554. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Wang, Z.; Chen, H.Z.; Zhou, S.; Zheng, W.; Liu, G.; Wei, Y.S.; Cai, H.; Liu, D.P.; Liang, C.C. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc. Res. 2008, 80, 191–199. [Google Scholar] [CrossRef]
- Stein, S.; Schäfer, N.; Breitenstein, A.; Besler, C.; Winnik, S.; Lohmann, C.; Heinrich, K.; Brokopp, C.E.; Handschin, C.; Landmesser, U.; et al. SIRT1 reduces endothelial activation without affecting vascular function in Apoe-/-mice. Aging 2010, 2, 353–360. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.Y.; Yun, M.; Jeong, J.; Park, E.R.; Shin, H.J.; Woo, S.R.; Jung, J.K.; Kim, Y.M.; Park, J.J.; Kim, J.; et al. SIRT1 deacetylates and stabilizes hypoxia-inducible factor-1α (HIF-1α) via direct interactions during hypoxia. Biochem. Biophys. Res. Commun. 2015, 462, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Martin-Montalvo, A.; Mercken, E.M.; Palacios, H.H.; Ward, T.M.; Abulwerdi, G.; Minor, R.K.; Vlasuk, G.P.; Ellis, J.L.; Sinclair, D.A.; et al. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 2014, 6, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Gano, L.B.; Donato, A.J.; Pasha, H.M.; Hearon, C.M., Jr.; Sindler, A.L.; Seals, D.R. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1754–H1763. [Google Scholar] [CrossRef]
- Curry, A.M.; White, D.S.; Donu, D.; Cen, Y. Human Sirtuin Regulators: The “Success” Stories. Front. Physiol. 2021, 12, 752117. [Google Scholar] [CrossRef]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef]
- Kilic, U.; Gok, O.; Bacaksiz, A.; Izmirli, M.; Elibol-Can, B.; Uysal, O. SIRT1 gene polymorphisms affect the protein expression in cardiovascular diseases. PLoS ONE 2014, 9, e90428. [Google Scholar] [CrossRef]
- Nasiri, M.; Rauf, M.; Kamfiroozie, H.; Zibaeenezhad, M.J.; Jamali, Z. SIRT1 gene polymorphisms associated with decreased risk of atherosclerotic coronary artery disease. Gene 2018, 672, 16–20. [Google Scholar] [CrossRef]
- Kedenko, L.; Lamina, C.; Kedenko, I.; Kollerits, B.; Kiesslich, T.; Iglseder, B.; Kronenberg, F.; Paulweber, B. Genetic polymorphisms at SIRT1 and FOXO1 are associated with carotid atherosclerosis in the SAPHIR cohort. BMC Med. Genet. 2014, 15, 112. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, L.; Chen, S.; Liu, X.; Li, H.; Lu, X.; Yang, X.; Huang, J.; Gu, D. Association between the SIRT1 mRNA expression and acute coronary syndrome. J. Atheroscler. Thromb. 2015, 22, 165–182. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Li, T.; Liu, M.; Wang, Y.; Ma, H.; Mu, N.; Wang, H. SIRT6 in Senescence and Aging-Related Cardiovascular Diseases. Front. Cell Dev. Biol. 2021, 9, 641315. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Li, P.; Ge, J.; Li, H. SIRT6 in Aging, Metabolism, Inflammation and Cardiovascular Diseases. Aging Dis. 2022, 13, 1787–1822. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yin, M.; Koroleva, M.; Mastrangelo, M.A.; Zhang, W.; Bai, P.; Little, P.J.; Jin, Z.G. SIRT6 protects against endothelial dysfunction and atherosclerosis in mice. Aging 2016, 8, 1064–1082. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, Z.; Wu, J.; Liu, M.; Li, M.; Sun, Y.; Huang, W.; Li, Y.; Zhang, Y.; Tang, W.; et al. Endothelial SIRT6 Is Vital to Prevent Hypertension and Associated Cardiorenal Injury Through Targeting Nkx3.2-GATA5 Signaling. Circ. Res. 2019, 124, 1448–1461. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, J.; Huang, X.; Li, Z.; Liu, P. Deletion of sirtuin 6 accelerates endothelial dysfunction and atherosclerosis in apolipoprotein E-deficient mice. Transl. Res. J. Lab. Clin. Med. 2016, 172, 18–29.e2. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sun, C.; Hu, L.; Gao, E.; Li, C.; Wang, H.; Sun, D. Sirt6 stabilizes atherosclerosis plaques by promoting macrophage autophagy and reducing contact with endothelial cells. Biochem. Cell Biol. = Biochim. Et Biol. Cell. 2020, 98, 120–129. [Google Scholar] [CrossRef]
- Cardus, A.; Uryga, A.K.; Walters, G.; Erusalimsky, J.D. SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence. Cardiovasc. Res. 2013, 97, 571–579. [Google Scholar] [CrossRef]
- Lee, O.H.; Woo, Y.M.; Moon, S.; Lee, J.; Park, H.; Jang, H.; Park, Y.Y.; Bae, S.K.; Park, K.H.; Heo, J.H.; et al. Sirtuin 6 deficiency induces endothelial cell senescence via downregulation of forkhead box M1 expression. Aging 2020, 12, 20946–20967. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, C.; Nie, H.; Wu, D.; Ying, W. SIRT2 plays a significant role in maintaining the survival and energy metabolism of PIEC endothelial cells. Int. J. Physiol. Pathophysiol. Pharmacol. 2016, 8, 120–127. [Google Scholar]
- Chen, T.; Ma, C.; Fan, G.; Liu, H.; Lin, X.; Li, J.; Li, N.; Wang, S.; Zeng, M.; Zhang, Y.; et al. SIRT3 protects endothelial cells from high glucose-induced senescence and dysfunction via the p53 pathway. Life Sci. 2021, 264, 118724. [Google Scholar] [CrossRef]
- Sun, L.; Dang, W. SIRT7 slows down stem cell aging by preserving heterochromatin: A perspective on the new discovery. Protein Cell 2020, 11, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, J.; Yang, X.; Lai, P.; Mou, Y.; Deng, J.; Li, X.; Wang, H.; Liu, X.; Zhou, L.; et al. H2O2 down-regulates SIRT7’s protective role of endothelial premature dysfunction via microRNA-335-5p. Biosci. Rep. 2022, 42, BSR20211775. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Du, K. DHHC17 Is a New Regulator of AMPK Signaling. Mol. Cell. Biol. 2022, 42, e0013122. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Zhou, M.; Chen, C.; Wu, X.; Wang, X. Role of AMPK mediated pathways in autophagy and aging. Biochimie 2022, 195, 100–113. [Google Scholar] [CrossRef]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef]
- van Vliet, T.; Varela-Eirin, M.; Wang, B.; Borghesan, M.; Brandenburg, S.M.; Franzin, R.; Evangelou, K.; Seelen, M.; Gorgoulis, V.; Demaria, M. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol. Cell 2021, 81, 2041–2052.e6. [Google Scholar] [CrossRef]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef]
- Mori, H.; Inoki, K.; Münzberg, H.; Opland, D.; Faouzi, M.; Villanueva, E.C.; Ikenoue, T.; Kwiatkowski, D.; MacDougald, O.A.; Myers, M.G., Jr.; et al. Critical role for hypothalamic mTOR activity in energy balance. Cell Metab. 2009, 9, 362–374. [Google Scholar] [CrossRef]
- Levine, Y.C.; Li, G.K.; Michel, T. Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK -> Rac1 -> Akt -> endothelial nitric-oxide synthase pathway. J. Biol. Chem. 2007, 282, 20351–20364. [Google Scholar] [CrossRef]
- Lesniewski, L.A.; Zigler, M.C.; Durrant, J.R.; Donato, A.J.; Seals, D.R. Sustained activation of AMPK ameliorates age-associated vascular endothelial dysfunction via a nitric oxide-independent mechanism. Mech. Ageing Dev. 2012, 133, 368–371. [Google Scholar] [CrossRef]
- Yang, R.; Fang, W.; Liang, J.; Lin, C.; Wu, S.; Yan, S.; Hu, C.; Ke, X. Apelin/APJ axis improves angiotensin II-induced endothelial cell senescence through AMPK/SIRT1 signaling pathway. Arch. Med. Sci. AMS 2018, 14, 725–734. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, J.; Fu, M.; Dong, R.; Yang, Y.; Luo, J.; Hu, S.; Li, W.; Xu, X.; Tu, L. Dipeptidyl peptidase-4 inhibition improves endothelial senescence by activating AMPK/SIRT1/Nrf2 signaling pathway. Biochem. Pharmacol. 2020, 177, 113951. [Google Scholar] [CrossRef]
- Lyu, T.J.; Zhang, Z.X.; Chen, J.; Liu, Z.J. Ginsenoside Rg1 ameliorates apoptosis, senescence and oxidative stress in ox-LDL-induced vascular endothelial cells via the AMPK/SIRT3/p53 signaling pathway. Exp. Ther. Med. 2022, 24, 545. [Google Scholar] [CrossRef]
- Motoshima, H.; Goldstein, B.J.; Igata, M.; Araki, E. AMPK and cell proliferation—AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 2006, 574, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. TEM 2017, 28, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Karnewar, S.; Neeli, P.K.; Panuganti, D.; Kotagiri, S.; Mallappa, S.; Jain, N.; Jerald, M.K.; Kotamraju, S. Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: Relevance in age-associated vascular dysfunction. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2018, 1864, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Sharp, Z.D.; Strong, R. The role of mTOR signaling in controlling mammalian life span: What a fungicide teaches us about longevity. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65, 580–589. [Google Scholar] [CrossRef]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Müller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef]
- Lesniewski, L.A.; Seals, D.R.; Walker, A.E.; Henson, G.D.; Blimline, M.W.; Trott, D.W.; Bosshardt, G.C.; LaRocca, T.J.; Lawson, B.R.; Zigler, M.C.; et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell 2017, 16, 17–26. [Google Scholar] [CrossRef]
- Lin, A.L.; Zheng, W.; Halloran, J.J.; Burbank, R.R.; Hussong, S.A.; Hart, M.J.; Javors, M.; Shih, Y.Y.; Muir, E.; Solano Fonseca, R.; et al. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2013, 33, 1412–1421. [Google Scholar] [CrossRef]
- Donato, A.J.; Walker, A.E.; Magerko, K.A.; Bramwell, R.C.; Black, A.D.; Henson, G.D.; Lawson, B.R.; Lesniewski, L.A.; Seals, D.R. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell 2013, 12, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, A.G.; Yepuri, G.; Carvas, J.M.; Stein, S.; Matter, C.M.; Scerri, I.; Ruffieux, J.; Montani, J.P.; Ming, X.F.; Yang, Z. Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: Inhibition by resveratrol. PLoS ONE 2011, 6, e19237. [Google Scholar] [CrossRef] [PubMed]
- Sabini, E.; O’Mahony, A.; Caturegli, P. MyMD-1 Improves Health Span and Prolongs Life Span in Old Mice: A Noninferiority Study to Rapamycin. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2023, 78, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Bent, E.H.; Gilbert, L.A.; Hemann, M.T. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 2016, 30, 1811–1821. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Yang, H.W.; Hong, H.L.; Luo, W.W.; Dai, C.M.; Chen, X.Y.; Wang, L.P.; Li, Q.; Li, Z.Q.; Liu, P.Q.; Li, Z.M. mTORC2 facilitates endothelial cell senescence by suppressing Nrf2 expression via the Akt/GSK-3β/C/EBPα signaling pathway. Acta Pharmacol. Sin. 2018, 39, 1837–1846. [Google Scholar] [CrossRef]
- Zhang, K.S.; Schecker, J.; Krull, A.; Riechert, E.; Jürgensen, L.; Kamuf-Schenk, V.; Burghaus, J.; Kiper, L.; Cao Ho, T.; Wöltje, K.; et al. PRAS40 suppresses atherogenesis through inhibition of mTORC1-dependent pro-inflammatory signaling in endothelial cells. Sci. Rep. 2019, 9, 16787. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; Larsson, T.E. Arterial klotho expression and FGF23 effects on vascular calcification and function. PLoS ONE 2013, 8, e60658. [Google Scholar] [CrossRef]
- Donate-Correa, J.; Ferri, C.M.; Martín-Núñez, E.; Pérez-Delgado, N.; González-Luis, A.; Mora-Fernández, C.; Navarro-González, J.F. Klotho as a biomarker of subclinical atherosclerosis in patients with moderate to severe chronic kidney disease. Sci. Rep. 2021, 11, 15877. [Google Scholar] [CrossRef] [PubMed]
- Castelblanco, E.; Hernández, M.; Alonso, N.; Ribes-Betriu, A.; Real, J.; Granado-Casas, M.; Rossell, J.; Rojo-López, M.I.; Dusso, A.S.; Julve, J.; et al. Association of α-klotho with subclinical carotid atherosclerosis in subjects with type 1 diabetes mellitus. Cardiovasc. Diabetol. 2022, 21, 207. [Google Scholar] [CrossRef] [PubMed]
- Keles, N.; Dogan, B.; Kalcik, M.; Caliskan, M.; Keles, N.N.; Aksu, F.; Bulut, M.; Kostek, O.; Isbilen, B.; Yilmaz, Y.; et al. Is serum Klotho protective against atherosclerosis in patients with type 1 diabetes mellitus? J. Diabetes Complicat. 2016, 30, 126–132. [Google Scholar] [CrossRef]
- Yu, L.; Kang, L.; Ren, X.Z.; Diao, Z.L.; Liu, W.H. Circulating α-Klotho Levels in Hemodialysis Patients and Their Relationship to Atherosclerosis. Kidney Blood Press. Res. 2018, 43, 1174–1182. [Google Scholar] [CrossRef]
- Navarro-González, J.F.; Donate-Correa, J.; Muros de Fuentes, M.; Pérez-Hernández, H.; Martínez-Sanz, R.; Mora-Fernández, C. Reduced Klotho is associated with the presence and severity of coronary artery disease. Heart (Br. Card. Soc.) 2014, 100, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; San Hipólito-Luengo, Á.; Villalobos, L.A.; Vallejo, S.; Valencia, I.; Michalska, P.; Pajuelo-Lozano, N.; Sánchez-Pérez, I.; León, R.; Bartha, J.L.; et al. The angiotensin-(1-7)/Mas receptor axis protects from endothelial cell senescence via klotho and Nrf2 activation. Aging Cell 2019, 18, e12913. [Google Scholar] [CrossRef]
- Buendía, P.; Carracedo, J.; Soriano, S.; Madueño, J.A.; Ortiz, A.; Martín-Malo, A.; Aljama, P.; Ramírez, R. Klotho Prevents NFκB Translocation and Protects Endothelial Cell from Senescence Induced by Uremia. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 1198–1209. [Google Scholar] [CrossRef]
- Liu, F.; Wu, S.; Ren, H.; Gu, J. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat. Cell Biol. 2011, 13, 254–262. [Google Scholar] [CrossRef]
- Mao, Q.; Deng, M.; Zhao, J.; Zhou, D.; Tong, W.; Xu, S.; Zhao, X. Klotho ameliorates angiotension-II-induced endothelial senescence via restoration of autophagy by inhibiting Wnt3a/GSK-3β/mTOR signaling: A potential mechanism involved in prognostic performance of Klotho in coronary atherosclerotic disease. Mech. Ageing Dev. 2023, 211, 111789. [Google Scholar] [CrossRef]
- Kuro, O.M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef]
- Nar, G.; Cetin, S.S.; Nar, R.; Kilic, O.; Furkan, O.M.; Gunver, G.; Ilyas, S.C. Is serum fibroblast growth factor 21 associated with the severity or presence of coronary artery disease? J. Med. Biochem. 2022, 41, 162–167. [Google Scholar] [CrossRef]
- Lin, Z.; Pan, X.; Wu, F.; Ye, D.; Zhang, Y.; Wang, Y.; Jin, L.; Lian, Q.; Huang, Y.; Ding, H.; et al. Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation 2015, 131, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.P.; Chen, C.Y.; Lin, T.W.; Kuo, C.S.; Huang, H.L.; Huang, P.H.; Lin, S.J. Fibroblast growth factor 21 reverses high-fat diet-induced impairment of vascular function via the anti-oxidative pathway in Apoe knockout mice. J. Cell. Mol. Med. 2022, 26, 2451–2461. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Li, N.; He, Z.; Zeng, X.; Nan, Y.; Chen, J.; Miao, P.; Ying, Y.; Lin, W.; Zhao, X.; et al. Fibroblast growth factor 21 Ameliorates diabetes-induced endothelial dysfunction in mouse aorta via activation of the CaMKK2/AMPKα signaling pathway. Cell Death Dis. 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Zheng, Q.; Chen, J.; Tan, X.; Li, Q.; Ding, L.; Zhang, R.; Lin, X. FGF21 mitigates atherosclerosis via inhibition of NLRP3 inflammasome-mediated vascular endothelial cells pyroptosis. Exp. Cell Res. 2020, 393, 112108. [Google Scholar] [CrossRef]
- Chen, J.J.; Tao, J.; Zhang, X.L.; Xia, L.Z.; Zeng, J.F.; Zhang, H.; Wei, D.H.; Lv, Y.C.; Li, G.H.; Wang, Z. Inhibition of the ox-LDL-Induced Pyroptosis by FGF21 of Human Umbilical Vein Endothelial Cells Through the TET2-UQCRC1-ROS Pathway. DNA Cell Biol. 2020, 39, 661–670. [Google Scholar] [CrossRef]
- Yan, X.; Gou, Z.; Li, Y.; Wang, Y.; Zhu, J.; Xu, G.; Zhang, Q. Fibroblast growth factor 21 inhibits atherosclerosis in Apoe-/- mice by ameliorating Fas-mediated apoptosis. Lipids Health Dis. 2018, 17, 203. [Google Scholar] [CrossRef]
- Yan, J.; Wang, J.; Huang, H.; Huang, Y.; Mi, T.; Zhang, C.; Zhang, L. Fibroblast growth factor 21 delayed endothelial replicative senescence and protected cells from H(2)O(2)-induced premature senescence through SIRT1. Am. J. Transl. Res. 2017, 9, 4492–4501. [Google Scholar]
- Wang, X.M.; Song, S.S.; Xiao, H.; Gao, P.; Li, X.J.; Si, L.Y. Fibroblast growth factor 21 protects against high glucose induced cellular damage and dysfunction of endothelial nitric-oxide synthase in endothelial cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2014, 34, 658–671. [Google Scholar] [CrossRef]
- Lauder, L.; Mahfoud, F.; Azizi, M.; Bhatt, D.L.; Ewen, S.; Kario, K.; Parati, G.; Rossignol, P.; Schlaich, M.P.; Teo, K.K.; et al. Hypertension management in patients with cardiovascular comorbidities. Eur. Heart J. 2023, 44, 2066–2077. [Google Scholar] [CrossRef]
- de Cavanagh, E.M.; Inserra, F.; Ferder, M.; Ferder, L. From mitochondria to disease: Role of the renin-angiotensin system. Am. J. Nephrol. 2007, 27, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Haslem, L.; Hays, J.M.; Hays, F.A. p66Shc in Cardiovascular Pathology. Cells 2022, 11, 1855. [Google Scholar] [CrossRef] [PubMed]
- Widder, J.D.; Fraccarollo, D.; Galuppo, P.; Hansen, J.M.; Jones, D.P.; Ertl, G.; Bauersachs, J. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension 2009, 54, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Corna, D.; Zoja, C.; Sonzogni, A.; Latini, R.; Salio, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Cassis, P.; et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Investig. 2009, 119, 524–530. [Google Scholar] [CrossRef]
- Zhang, K.; Zhou, B.; Zhang, L. Association study of angiotensin II type 1 receptor: A1166C (rs5186) polymorphism with coronary heart disease using systematic meta-analysis. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2013, 14, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Mo, Y.P.; He, Y.S.; Yang, F.; Xu, Y.; Li, C.C.; Wang, J.; Reng, H.M.; Long, L. Correlation between renin-angiotensin system gene polymorphisms and essential hypertension in the Chinese Yi ethnic group. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2015, 16, 975–981. [Google Scholar] [CrossRef]
- Parchwani, D.N.; Patel, D.D.; Rawtani, J.; Yadav, D. Analysis of Association of Angiotensin II Type 1 Receptor Gene A1166C Gene Polymorphism with Essential Hypertension. Indian J. Clin. Biochem. IJCB 2018, 33, 53–60. [Google Scholar] [CrossRef]
- Xu, Y.; Bao, Q.; He, B.; Pan, Y.; Zhang, R.; Mao, X.; Tang, Z.; Qu, L.; Zhu, C.; Tian, F.; et al. Association of angiotensin I converting enzyme, angiotensin II type 1 receptor and angiotensin I converting enzyme 2 gene polymorphisms with the dyslipidemia in type 2 diabetic patients of Chinese Han origin. J. Endocrinol. Investig. 2012, 35, 378–383. [Google Scholar]
- Chandra, S.; Narang, R.; Sreenivas, V.; Bhatia, J.; Saluja, D.; Srivastava, K. Association of angiotensin II type 1 receptor (A1166C) gene polymorphism and its increased expression in essential hypertension: A case-control study. PLoS ONE 2014, 9, e101502. [Google Scholar] [CrossRef]
- Yi, W.; Chen, F.; Zhang, H.; Tang, P.; Yuan, M.; Wen, J.; Wang, S.; Cai, Z. Role of angiotensin II in aging. Front. Aging Neurosci. 2022, 14, 1002138. [Google Scholar] [CrossRef]
- Neves, M.F.; Cunha, A.R.; Cunha, M.R.; Gismondi, R.A.; Oigman, W. The Role of Renin-Angiotensin-Aldosterone System and Its New Components in Arterial Stiffness and Vascular Aging. High Blood Press. Cardiovasc. Prev. Off. J. Ital. Soc. Hypertens. 2018, 25, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, A.; Feely, J. Effect of angiotensin ii receptor blockade on arterial stiffness: Beyond blood pressure reduction. Am. J. Hypertens. 2002, 15, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Intengan, H.D.; Schiffrin, E.L. Reduction of resistance artery stiffness by treatment with the AT(1)-receptor antagonist losartan in essential hypertension. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2000, 1, 40–45. [Google Scholar] [CrossRef]
- Mahmud, A.; Feely, J. Arterial stiffness and the renin-angiotensin-aldosterone system. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2004, 5, 102–108. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Wang, B.X.; Ou, W.S.; Lv, Y.H.; Li, M.M.; Miao, Z.; Wang, S.X.; Fei, J.C.; Guo, T. Administration of losartan improves aortic arterial stiffness and reduces the occurrence of acute coronary syndrome in aged patients with essential hypertension. J. Cell. Biochem. 2019, 120, 5713–5721. [Google Scholar] [CrossRef] [PubMed]
- Basso, N.; Cini, R.; Pietrelli, A.; Ferder, L.; Terragno, N.A.; Inserra, F. Protective effect of long-term angiotensin II inhibition. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1351–H1358. [Google Scholar] [CrossRef] [PubMed]
- González Bosc, L.V.; Kurnjek, M.L.; Müller, A.; Terragno, N.A.; Basso, N. Effect of chronic angiotensin II inhibition on the nitric oxide synthase in the normal rat during aging. J. Hypertens. 2001, 19, 1403–1409. [Google Scholar] [CrossRef]
- González Bosc, L.; Kurnjek, M.L.; Müller, A.; Basso, N. Effect of chronic angiotensin II inhibition on the cardiovascular system of the normal rat. Am. J. Hypertens. 2000, 13, 1301–1307. [Google Scholar] [CrossRef]
- Xu, S.; Zhi, H.; Hou, X.; Jiang, B. Angiotensin II modulates interleukin-1β-induced inflammatory gene expression in vascular smooth muscle cells via interfering with ERK-NF-κB crosstalk. Biochem. Biophys. Res. Commun. 2011, 410, 543–548. [Google Scholar] [CrossRef]
- Zhang, G.H.; Chao, M.; Hui, L.H.; Xu, D.L.; Cai, W.L.; Zheng, J.; Gao, M.; Zhang, M.X.; Wang, J.; Lu, Q.H. Poly(ADP-ribose)polymerase 1 inhibition protects against age-dependent endothelial dysfunction. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1266–1274. [Google Scholar] [CrossRef]
- Nassif, M.E.; Windsor, S.L.; Borlaug, B.A.; Kitzman, D.W.; Shah, S.J.; Tang, F.; Khariton, Y.; Malik, A.O.; Khumri, T.; Umpierrez, G.; et al. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: A multicenter randomized trial. Nat. Med. 2021, 27, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef]
- Udell, J.A.; Jones, W.S.; Petrie, M.C.; Harrington, J.; Anker, S.D.; Bhatt, D.L.; Hernandez, A.F.; Butler, J. Sodium Glucose Cotransporter-2 Inhibition for Acute Myocardial Infarction: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2022, 79, 2058–2068. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Belcastro, E.; Hasan, H.; Matsushita, K.; Marchandot, B.; Abbas, M.; Toti, F.; Auger, C.; Jesel, L.; Ohlmann, P.; et al. Angiotensin II-induced upregulation of SGLT1 and 2 contributes to human microparticle-stimulated endothelial senescence and dysfunction: Protective effect of gliflozins. Cardiovasc. Diabetol. 2021, 20, 65. [Google Scholar] [CrossRef] [PubMed]
- Khemais-Benkhiat, S.; Belcastro, E.; Idris-Khodja, N.; Park, S.H.; Amoura, L.; Abbas, M.; Auger, C.; Kessler, L.; Mayoux, E.; Toti, F.; et al. Angiotensin II-induced redox-sensitive SGLT1 and 2 expression promotes high glucose-induced endothelial cell senescence. J. Cell. Mol. Med. 2020, 24, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.; Malhotra, K.; Sowers, J.; Aroor, A. Uric Acid—Key ingredient in the recipe for cardiorenal metabolic syndrome. Cardiorenal Med. 2013, 3, 208–220. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Whaley-Connell, A.T.; Sowers, J.R. Fructose and uric acid: Is there a role in endothelial function? Curr. Hypertens. Rep. 2014, 16, 434. [Google Scholar] [CrossRef]
- Canepa, M.; Viazzi, F.; Strait, J.B.; Ameri, P.; Pontremoli, R.; Brunelli, C.; Studenski, S.; Ferrucci, L.; Lakatta, E.G.; AlGhatrif, M. Longitudinal Association Between Serum Uric Acid and Arterial Stiffness: Results from the Baltimore Longitudinal Study of Aging. Hypertension 2017, 69, 228–235. [Google Scholar] [CrossRef]
- Puddu, P.; Puddu, G.M.; Cravero, E.; Vizioli, L.; Muscari, A. Relationships among hyperuricemia, endothelial dysfunction and cardiovascular disease: Molecular mechanisms and clinical implications. J. Cardiol. 2012, 59, 235–242. [Google Scholar] [CrossRef]
- Kanbay, M.; Yilmaz, M.I.; Sonmez, A.; Turgut, F.; Saglam, M.; Cakir, E.; Yenicesu, M.; Covic, A.; Jalal, D.; Johnson, R.J. Serum uric acid level and endothelial dysfunction in patients with nondiabetic chronic kidney disease. Am. J. Nephrol. 2011, 33, 298–304. [Google Scholar] [CrossRef]
- Tang, Z.; Cheng, L.T.; Li, H.Y.; Wang, T. Serum uric acid and endothelial dysfunction in continuous ambulatory peritoneal dialysis patients. Am. J. Nephrol. 2009, 29, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Maio, R.; Mallamaci, F.; Sesti, G.; Perticone, F. Uric acid and endothelial dysfunction in essential hypertension. J. Am. Soc. Nephrol. JASN 2006, 17, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Sincer, I.; Kurtoglu, E.; Calıskan, M.; Akkaya, E.; Vuruskan, E.; Küçükosmanoglu, M.; Çoşkun, F.Y.; Inci, M.F.; Zorlu, A. Significant correlation between uric acid levels and flow-mediated dilatation in patients with masked hypertension. Clin. Exp. Hypertens. 2014, 36, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, A.J.; Bruinsma, K.A. Uric acid is closely linked to vascular nitric oxide activity. Evidence for mechanism of association with cardiovascular disease. J. Am. Coll. Cardiol. 2001, 38, 1850–1858. [Google Scholar] [CrossRef]
- Aroor, A.R.; Jia, G.; Habibi, J.; Sun, Z.; Ramirez-Perez, F.I.; Brady, B.; Chen, D.; Martinez-Lemus, L.A.; Manrique, C.; Nistala, R.; et al. Uric acid promotes vascular stiffness, maladaptive inflammatory responses and proteinuria in western diet fed mice. Metab. Clin. Exp. 2017, 74, 32–40. [Google Scholar] [CrossRef]
- Lastra, G.; Manrique, C.; Jia, G.; Aroor, A.R.; Hayden, M.R.; Barron, B.J.; Niles, B.; Padilla, J.; Sowers, J.R. Xanthine oxidase inhibition protects against Western diet-induced aortic stiffness and impaired vasorelaxation in female mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R67–R77. [Google Scholar] [CrossRef]
- Yu, M.A.; Sánchez-Lozada, L.G.; Johnson, R.J.; Kang, D.H. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J. Hypertens. 2010, 28, 1234–1242. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Qi, W. Uric acid, as a double-edged sword, affects the activity of epidermal growth factor (EGF) on human umbilical vein endothelial cells by regulating aging process. Bioengineered 2022, 13, 3877–3895. [Google Scholar] [CrossRef]
- Rodríguez-Mañas, L.; El-Assar, M.; Vallejo, S.; López-Dóriga, P.; Solís, J.; Petidier, R.; Montes, M.; Nevado, J.; Castro, M.; Gómez-Guerrero, C.; et al. Endothelial dysfunction in aged humans is related with oxidative stress and vascular inflammation. Aging Cell 2009, 8, 226–238. [Google Scholar] [CrossRef]
- Davoli, T.; de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef]
- Repin, V.S.; Dolgov, V.V.; Zaikina, O.E.; Novikov, I.D.; Antonov, A.S.; Nikolaeva, M.A.; Smirnov, V.N. Heterogeneity of endothelium in human aorta. A quantitative analysis by scanning electron microscopy. Atherosclerosis 1984, 50, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; McCrann, D.J.; Nguyen, H.; St Hilaire, C.; DePinho, R.A.; Jones, M.R.; Ravid, K. Increased polyploidy in aortic vascular smooth muscle cells during aging is marked by cellular senescence. Aging Cell 2007, 6, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, O.; Fan, J.L.; Watanabe, T. Atherosclerosis- and age-related multinucleated variant endothelial cells in primary culture from human aorta. Am. J. Pathol. 1989, 135, 967–976. [Google Scholar] [PubMed]
- Devlin, A.M.; Davidson, A.O.; Gordon, J.F.; Campbell, A.M.; Morton, J.J.; Reid, J.L.; Dominiczak, A.F. Vascular smooth muscle polyploidy in genetic hypertension: The role of angiotensin II. J. Hum. Hypertens. 1995, 9, 497–500. [Google Scholar]
- Devlin, A.M.; Gordon, J.F.; Davidson, A.O.; Clark, J.S.; Hamilton, C.A.; Morton, J.J.; Campbell, A.M.; Reid, J.L.; Dominiczak, A.F. The effects of perindopril on vascular smooth muscle polyploidy in stroke-prone spontaneously hypertensive rats. J. Hypertens. 1995, 13, 211–218. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Pickering, J.G. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell 2009, 8, 100–112. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Pickering, J.G. Polyploidy impairs human aortic endothelial cell function and is prevented by nicotinamide phosphoribosyltransferase. Am. J. Physiol. Cell Physiol. 2010, 298, C66–C74. [Google Scholar] [CrossRef]
- Pandit, S.K.; Westendorp, B.; Nantasanti, S.; van Liere, E.; Tooten, P.C.; Cornelissen, P.W.; Toussaint, M.J.; Lamers, W.H.; de Bruin, A. E2F8 is essential for polyploidization in mammalian cells. Nat. Cell Biol. 2012, 14, 1181–1191. [Google Scholar] [CrossRef]
- Prata, L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat. Commun. 2019, 10, 2387. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef]
- Minamino, T.; Komuro, I. Vascular cell senescence: Contribution to atherosclerosis. Circ. Res. 2007, 100, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Macdonald, K.; Chan, S.W.; Boyle, J.J.; Weissberg, P.L. Cooperative interactions between RB and p53 regulate cell proliferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ. Res. 1998, 82, 704–712. [Google Scholar] [CrossRef]
- Suda, M.; Paul, K.H.; Minamino, T.; Miller, J.D.; Lerman, A.; Ellison-Hughes, G.M.; Tchkonia, T.; Kirkland, J.L. Senescent Cells: A Therapeutic Target in Cardiovascular Diseases. Cells 2023, 12, 1296. [Google Scholar] [CrossRef] [PubMed]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B., Jr.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab 2019, 30, 129–142.e4. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef]
- Wang, J.; Lu, Y.; Carr, C.; Dhandapani, K.M.; Brann, D.W. Senolytic therapy is neuroprotective and improves functional outcome long-term after traumatic brain injury in mice. Front. Neurosci. 2023, 17, 1227705. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Novais, E.J.; Tran, V.A.; Johnston, S.N.; Darris, K.R.; Roupas, A.J.; Sessions, G.A.; Shapiro, I.M.; Diekman, B.O.; Risbud, M.V. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 2021, 12, 5213. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Prata, L.; Huggins, M.A.; Pierson, M.; Zhang, L.; O’Kelly, R.D.; Pirtskhalava, T.; Xun, P.; et al. Senolytics reduce coronavirus-related mortality in old mice. Science 2021, 373, eabe4832. [Google Scholar] [CrossRef]
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M.; et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 2021, 599, 283–289. [Google Scholar] [CrossRef]
- Crespo-Garcia, S.; Tsuruda, P.R.; Dejda, A.; Ryan, R.D.; Fournier, F.; Chaney, S.Y.; Pilon, F.; Dogan, T.; Cagnone, G.; Patel, P.; et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab 2021, 33, 818–832.e7. [Google Scholar] [CrossRef]
- Lérida-Viso, A.; Estepa-Fernández, A.; Morellá-Aucejo, Á.; Lozano-Torres, B.; Alfonso, M.; Blandez, J.F.; Bisbal, V.; Sepúlveda, P.; García-Fernández, A.; Orzáez, M. Pharmacological senolysis reduces doxorubicin-induced cardiotoxicity and improves cardiac function in mice. Pharmacol. Res. 2022, 183, 106356. [Google Scholar] [CrossRef]
- Jia, K.; Dai, Y.; Liu, A.; Li, X.; Wu, L.; Lu, L.; Bao, Y.; Jin, Q. Senolytic Agent Navitoclax Inhibits Angiotensin II-Induced Heart Failure in Mice. J. Cardiovasc. Pharmacol. 2020, 76, 452–460. [Google Scholar] [CrossRef]
- Salerno, N.; Marino, F.; Scalise, M.; Salerno, L.; Molinaro, C.; Filardo, A.; Chiefalo, A.; Panuccio, G.; De Angelis, A.; Urbanek, K.; et al. Pharmacological clearance of senescent cells improves cardiac remodeling and function after myocardial infarction in female aged mice. Mech. Ageing Dev. 2022, 208, 111740. [Google Scholar] [PubMed]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.M.; Kaistha, A.; Uryga, A.K.; Oc, S.; Foote, K.; Shah, A.; Finigan, A.; Figg, N.; Dobnikar, L.; Jørgensen, H.; et al. Efficacy and limitations of senolysis in atherosclerosis. Cardiovasc. Res. 2022, 118, 1713–1727. [Google Scholar] [CrossRef] [PubMed]
- Karnewar, S.; Karnewar, V.; Shankman, L.S.; Owens, G.K. Treatment of advanced atherosclerotic mice with the senolytic agent ABT-263 is associated with reduced indices of plaque stability and increased mortality. bioRxiv 2023. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [PubMed]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [PubMed]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Rovira, M.; Galiana, I.; Giménez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Muñoz-Martín, M.; Ucero, A.C.; et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 2018, 10, e9355. [Google Scholar] [CrossRef]
- Guerrero, A.; Guiho, R.; Herranz, N.; Uren, A.; Withers, D.J.; Martínez-Barbera, J.P.; Tietze, L.F.; Gil, J. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell 2020, 19, e13133. [Google Scholar] [CrossRef]
- González-Gualda, E.; Pàez-Ribes, M.; Lozano-Torres, B.; Macias, D.; Wilson, J.R., 3rd; González-López, C.; Ou, H.L.; Mirón-Barroso, S.; Zhang, Z.; Lérida-Viso, A.; et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 2020, 19, e13142. [Google Scholar] [CrossRef]
- van der Feen, D.E.; Bossers, G.P.L.; Hagdorn, Q.A.J.; Moonen, J.R.; Kurakula, K.; Szulcek, R.; Chappell, J.; Vallania, F.; Donato, M.; Kok, K.; et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci. Transl. Med. 2020, 12, eaaw4974. [Google Scholar] [PubMed]
- Ramadhiani, R.; Ikeda, K.; Miyagawa, K.; Ryanto, G.R.T.; Tamada, N.; Suzuki, Y.; Kirita, Y.; Matoba, S.; Hirata, K.I.; Emoto, N. Endothelial cell senescence exacerbates pulmonary hypertension by inducing juxtacrine Notch signaling in smooth muscle cells. iScience 2023, 26, 106662. [Google Scholar]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [PubMed]
- Yoshida, S.; Nakagami, H.; Hayashi, H.; Ikeda, Y.; Sun, J.; Tenma, A.; Tomioka, H.; Kawano, T.; Shimamura, M.; Morishita, R.; et al. The CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat. Commun. 2020, 11, 2482. [Google Scholar] [CrossRef] [PubMed]
- Suda, M.; Shimizu, I.; Katsuumi, G.; Yoshida, Y.; Hayashi, Y.; Ikegami, R.; Matsumoto, N.; Yoshida, Y.; Mikawa, R.; Katayama, A.; et al. Senolytic vaccination improves normal and pathological age-related phenotypes and increases lifespan in progeroid mice. Nat. Aging 2021, 1, 1117–1126. [Google Scholar]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Wang, L.; Wang, B.; Gasek, N.S.; Zhou, Y.; Cohn, R.L.; Martin, D.E.; Zuo, W.; Flynn, W.F.; Guo, C.; Jellison, E.R.; et al. Targeting p21(Cip1) highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell Metab 2022, 34, 75–89.e8. [Google Scholar] [CrossRef]
- Wang, B.; Wang, L.; Gasek, N.S.; Zhou, Y.; Kim, T.; Guo, C.; Jellison, E.R.; Haynes, L.; Yadav, S.; Tchkonia, T.; et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat. Aging 2021, 1, 962–973. [Google Scholar]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar]
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.D.; Bulavin, D.V. Defined p16High Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020, 32, 87–99.e6. [Google Scholar]
- Kim, S.; Kim, C. Transcriptomic Analysis of Cellular Senescence: One Step Closer to Senescence Atlas. Mol. Cells 2021, 44, 136–145. [Google Scholar] [PubMed]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.L.; Huby, T.; Witztum, J.L.; Ouzilleau, B.; Miller, E.R.; Saint-Charles, F.; Aucouturier, P.; Chapman, M.J.; Lesnik, P. Macrophage apoptosis exerts divergent effects on atherogenesis as a function of lesion stage. Circulation 2009, 119, 1795–1804. [Google Scholar] [CrossRef]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.W.; Fang, L.J.; Cheng, S.Q.; Wang, X.; Liu, N.F. Programmed cell death in atherosclerosis and vascular calcification. Cell Death Dis. 2022, 13, 467. [Google Scholar]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [PubMed]
- Zhao, F.; Satyanarayana, G.; Zhang, Z.; Zhao, J.; Ma, X.L.; Wang, Y. Endothelial Autophagy in Coronary Microvascular Dysfunction and Cardiovascular Disease. Cells 2022, 11, 2081. [Google Scholar] [PubMed]
- Choy, J.C.; Granville, D.J.; Hunt, D.W.; McManus, B.M. Endothelial cell apoptosis: Biochemical characteristics and potential implications for atherosclerosis. J. Mol. Cell. Cardiol. 2001, 33, 1673–1690. [Google Scholar]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Michel, J.B. Anoikis in the cardiovascular system: Known and unknown extracellular mediators. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2146–2154. [Google Scholar] [CrossRef]
- Taddei, M.L.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: An emerging hallmark in health and diseases. J. Pathol. 2012, 226, 380–393. [Google Scholar] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar]
- Ju, J.; Liu, Y.; Liang, H.; Yang, B. The role of pyroptosis in endothelial dysfunction induced by diseases. Front. Immunol. 2022, 13, 1093985. [Google Scholar] [PubMed]
- Zdanyte, M.; Borst, O.; Münzer, P. NET-(works) in arterial and venous thrombo-occlusive diseases. Front. Cardiovasc. Med. 2023, 10, 1155512. [Google Scholar] [PubMed]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar]
- Zhang, X.; Ren, Z.; Xu, W.; Jiang, Z. Necroptosis in atherosclerosis. Clin. Chim. Acta Int. J. Clin. Chem. 2022, 534, 22–28. [Google Scholar]
- Zhang, H.; Zhou, S.; Sun, M.; Hua, M.; Liu, Z.; Mu, G.; Wang, Z.; Xiang, Q.; Cui, Y. Ferroptosis of Endothelial Cells in Vascular Diseases. Nutrients 2022, 14, 4506. [Google Scholar]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar]
- LaRocca, T.J.; Henson, G.D.; Thorburn, A.; Sindler, A.L.; Pierce, G.L.; Seals, D.R. Translational evidence that impaired autophagy contributes to arterial ageing. J. Physiol. 2012, 590, 3305–3316. [Google Scholar]
- LaRocca, T.J.; Gioscia-Ryan, R.A.; Hearon, C.M., Jr.; Seals, D.R. The autophagy enhancer spermidine reverses arterial aging. Mech. Ageing Dev. 2013, 134, 314–320. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.N.; Xu, S.B.; Wu, H.; Dai, B.; Jian, D.D.; Yang, M.; Wu, Y.T.; Feng, Q.; Zhu, J.H.; et al. MicroRNA-214-3p: A link between autophagy and endothelial cell dysfunction in atherosclerosis. Acta Physiol. 2018, 222, e12973. [Google Scholar] [CrossRef]
- Chen, Y.; Zeng, A.; He, S.; He, S.; Li, C.; Mei, W.; Lu, Q. Autophagy-Related Genes in Atherosclerosis. J. Healthc. Eng. 2021, 2021, 6402206. [Google Scholar]
- Patella, F.; Neilson, L.J.; Athineos, D.; Erami, Z.; Anderson, K.I.; Blyth, K.; Ryan, K.M.; Zanivan, S. In-Depth Proteomics Identifies a Role for Autophagy in Controlling Reactive Oxygen Species Mediated Endothelial Permeability. J. Proteome Res. 2016, 15, 2187–2197. [Google Scholar]
- Zhu, L.; Wu, G.; Yang, X.; Jia, X.; Li, J.; Bai, X.; Li, W.; Zhao, Y.; Li, Y.; Cheng, W.; et al. Low density lipoprotein mimics insulin action on autophagy and glucose uptake in endothelial cells. Sci. Rep. 2019, 9, 3020. [Google Scholar] [PubMed]
- Yuan, P.; Hu, Q.; He, X.; Long, Y.; Song, X.; Wu, F.; He, Y.; Zhou, X. Laminar flow inhibits the Hippo/YAP pathway via autophagy and SIRT1-mediated deacetylation against atherosclerosis. Cell Death Dis. 2020, 11, 141. [Google Scholar] [PubMed]
- Torisu, K.; Singh, K.K.; Torisu, T.; Lovren, F.; Liu, J.; Pan, Y.; Quan, A.; Ramadan, A.; Al-Omran, M.; Pankova, N.; et al. Intact endothelial autophagy is required to maintain vascular lipid homeostasis. Aging Cell 2016, 15, 187–191. [Google Scholar]
- Xie, Y.; You, S.J.; Zhang, Y.L.; Han, Q.; Cao, Y.J.; Xu, X.S.; Yang, Y.P.; Li, J.; Liu, C.F. Protective role of autophagy in AGE-induced early injury of human vascular endothelial cells. Mol. Med. Rep. 2011, 4, 459–464. [Google Scholar] [PubMed]
- Zhang, X.; Yu, L.; Xu, H. Lysosome calcium in ROS regulation of autophagy. Autophagy 2016, 12, 1954–1955. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Choi, S.H. ASK1 Mediates Apoptosis and Autophagy during oxLDL-CD36 Signaling in Senescent Endothelial Cells. Oxidative Med. Cell. Longev. 2019, 2019, 2840437. [Google Scholar]
- Meng, Q.; Pu, L.; Lu, Q.; Wang, B.; Li, S.; Liu, B.; Li, F. Morin hydrate inhibits atherosclerosis and LPS-induced endothelial cells inflammatory responses by modulating the NFκB signaling-mediated autophagy. Int. Immunopharmacol. 2021, 100, 108096. [Google Scholar] [PubMed]
- Reglero-Real, N.; Pérez-Gutiérrez, L.; Yoshimura, A.; Rolas, L.; Garrido-Mesa, J.; Barkaway, A.; Pickworth, C.; Saleeb, R.S.; Gonzalez-Nuñez, M.; Austin-Williams, S.N.; et al. Autophagy modulates endothelial junctions to restrain neutrophil diapedesis during inflammation. Immunity 2021, 54, 1989–2004.e9. [Google Scholar] [PubMed]
- Vion, A.C.; Kheloufi, M.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc. Natl. Acad. Sci. USA 2017, 114, E8675–E8684. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Fan, Y.; Qiao, C.; Liang, W.; Hu, W.; Zhu, T.; Zhang, J.; Chen, Y.E. TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Sci. Signal. 2017, 10, eaah4214. [Google Scholar]
- Kim, J.Y.; Mondaca-Ruff, D.; Singh, S.; Wang, Y. SIRT1 and Autophagy: Implications in Endocrine Disorders. Front. Endocrinol. 2022, 13, 930919. [Google Scholar]
- Xu, Y.; Wan, W. Acetylation in the regulation of autophagy. Autophagy 2023, 19, 379–387. [Google Scholar]
- Gu, S.; Zhou, C.; Pei, J.; Wu, Y.; Wan, S.; Zhao, X.; Hu, J.; Hua, X. Esomeprazole inhibits hypoxia/endothelial dysfunction-induced autophagy in preeclampsia. Cell Tissue Res. 2022, 388, 181–194. [Google Scholar]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422. [Google Scholar]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Hao, R.; Wang, W.; Gao, H.; Wang, C. SIRT1/Atg5/autophagy are involved in the antiatherosclerosis effects of ursolic acid. Mol. Cell. Biochem. 2016, 420, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bi, X.; Chen, T.; Zhang, Q.; Wang, S.X.; Chiu, J.J.; Liu, G.S.; Zhang, Y.; Bu, P.; Jiang, F. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015, 6, e1827. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Duan, W.; Wu, G.; Zhang, D.; Wang, L.; Chen, D.; Chen, Z.; Yang, B. Protective effect of hydrogen sulfide on endothelial cells through Sirt1-FoxO1-mediated autophagy. Ann. Transl. Med. 2020, 8, 1586. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, X.; Zhang, Z.; Liu, J.; Wu, C.; Chen, Y.; Zhou, J.; Zhang, J.; Zhang, X. Autophagy promotes directed migration of HUVEC in response to electric fields through the ROS/SIRT1/FOXO1 pathway. Free Radic. Biol. Med. 2022, 192, 213–223. [Google Scholar] [CrossRef]
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat. Cell Biol. 2020, 22, 1170–1179. [Google Scholar] [CrossRef]
- Hua, Y.; Zhang, J.; Liu, Q.; Su, J.; Zhao, Y.; Zheng, G.; Yang, Z.; Zhuo, D.; Ma, C.; Fan, G. The Induction of Endothelial Autophagy and Its Role in the Development of Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 831847. [Google Scholar] [CrossRef]
- De Meyer, G.R.; Grootaert, M.O.; Michiels, C.F.; Kurdi, A.; Schrijvers, D.M.; Martinet, W. Autophagy in vascular disease. Circ. Res. 2015, 116, 468–479. [Google Scholar] [CrossRef]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef]
- Chau, Y.P.; Lin, S.Y.; Chen, J.H.; Tai, M.H. Endostatin induces autophagic cell death in EAhy926 human endothelial cells. Histol. Histopathol. 2003, 18, 715–726. [Google Scholar] [PubMed]
- Kato, Y.; Furusyo, N.; Tanaka, Y.; Ueyama, T.; Yamasaki, S.; Murata, M.; Hayashi, J. The Relation between Serum Endostatin Level and Carotid Atherosclerosis in Healthy Residents of Japan: Results from the Kyushu and Okinawa Population Study (KOPS). J. Atheroscler. Thromb. 2017, 24, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Ärnlöv, J.; Ruge, T.; Ingelsson, E.; Larsson, A.; Sundström, J.; Lind, L. Serum endostatin and risk of mortality in the elderly: Findings from 2 community-based cohorts. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Qian, S.; Zhang, R.; Guo, D.; Wang, A.; Peng, Y.; Peng, H.; Li, Q.; Ju, Z.; Geng, D.; et al. Endostatin as a novel prognostic biomarker in acute ischemic stroke. Atherosclerosis 2020, 293, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S.; Heller, E.; Konerding, M.A.; Flynn, E.; Palinski, W.; Folkman, J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 1999, 99, 1726–1732. [Google Scholar] [CrossRef]
- Zeng, X.; Chen, J.; Miller, Y.I.; Javaherian, K.; Moulton, K.S. Endostatin binds biglycan and LDL and interferes with LDL retention to the subendothelial matrix during atherosclerosis. J. Lipid Res. 2005, 46, 1849–1859. [Google Scholar] [CrossRef]
- Zeng, M.; Wei, X.; Wu, Z.; Li, W.; Zheng, Y.; Li, B.; Meng, X.; Fu, X.; Fei, Y. Simulated ischemia/reperfusion-induced p65-Beclin 1-dependent autophagic cell death in human umbilical vein endothelial cells. Sci. Rep. 2016, 6, 37448. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Z.; Han, W.; Li, H. Zoledronate induces autophagic cell death in human umbilical vein endothelial cells via Beclin-1 dependent pathway activation. Mol. Med. Rep. 2016, 14, 4747–4754. [Google Scholar] [CrossRef]
- Csordas, A.; Kreutmayer, S.; Ploner, C.; Braun, P.R.; Karlas, A.; Backovic, A.; Wick, G.; Bernhard, D. Cigarette smoke extract induces prolonged endoplasmic reticulum stress and autophagic cell death in human umbilical vein endothelial cells. Cardiovasc. Res. 2011, 92, 141–148. [Google Scholar] [CrossRef]
- Zeng, M.; Wei, X.; Wu, Z.; Li, W.; Li, B.; Fei, Y.; He, Y.; Chen, J.; Wang, P.; Liu, X. Reactive oxygen species contribute to simulated ischemia/reperfusion-induced autophagic cell death in human umbilical vein endothelial cells. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2014, 20, 1017–1023. [Google Scholar]
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K.H. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell. Mol. Life Sci. CMLS 2019, 76, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Zhang, Q.; Liu, J.; Li, R.; Wang, D.; Peng, W.; Wu, C. Suppression of apoptosis in vascular endothelial cell, the promising way for natural medicines to treat atherosclerosis. Pharmacol. Res. 2021, 168, 105599. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Ketelut-Carneiro, N.; Fitzgerald, K.A. Apoptosis, Pyroptosis, and Necroptosis-Oh My! The Many Ways a Cell Can Die. J. Mol. Biol. 2022, 434, 167378. [Google Scholar] [CrossRef]
- Webster, J.D.; Vucic, D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell Dev. Biol. 2020, 8, 365. [Google Scholar] [CrossRef]
- Gerrity, R.G.; Richardson, M.; Somer, J.B.; Bell, F.P.; Schwartz, C.J. Endothelial cell morphology in areas of in vivo Evans blue uptake in the aorta of young pigs. II. Ultrastructure of the intima in areas of differing permeability to proteins. Am. J. Pathol. 1977, 89, 313–334. [Google Scholar]
- Tricot, O.; Mallat, Z.; Heymes, C.; Belmin, J.; Lesèche, G.; Tedgui, A. Relation between endothelial cell apoptosis and blood flow direction in human atherosclerotic plaques. Circulation 2000, 101, 2450–2453. [Google Scholar] [CrossRef]
- Dimmeler, S.; Haendeler, J.; Zeiher, A.M. Regulation of endothelial cell apoptosis in atherothrombosis. Curr. Opin. Lipidol. 2002, 13, 531–536. [Google Scholar] [CrossRef]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef]
- Qu, K.; Yan, F.; Qin, X.; Zhang, K.; He, W.; Dong, M.; Wu, G. Mitochondrial dysfunction in vascular endothelial cells and its role in atherosclerosis. Front. Physiol. 2022, 13, 1084604. [Google Scholar] [CrossRef]
- Su, Q.; Wang, Y.; Yang, X.; Li, X.D.; Qi, Y.F.; He, X.J.; Wang, Y.J. Inhibition of Endoplasmic Reticulum Stress Apoptosis by Estrogen Protects Human Umbilical Vein Endothelial Cells Through the PI3 Kinase-Akt Signaling Pathway. J. Cell. Biochem. 2017, 118, 4568–4574. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Momi, S.; Guglielmini, G. Nitric oxide-enhancing or -releasing agents as antithrombotic drugs. Biochem. Pharmacol. 2019, 166, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Haendeler, J.; Galle, J.; Zeiher, A.M. Oxidized low-density lipoprotein induces apoptosis of human endothelial cells by activation of CPP32-like proteases. A mechanistic clue to the ‘response to injury’ hypothesis. Circulation 1997, 95, 1760–1763. [Google Scholar] [CrossRef]
- Harada-Shiba, M.; Kinoshita, M.; Kamido, H.; Shimokado, K. Oxidized low density lipoprotein induces apoptosis in cultured human umbilical vein endothelial cells by common and unique mechanisms. J. Biol. Chem. 1998, 273, 9681–9687. [Google Scholar] [CrossRef]
- Sata, M.; Walsh, K. Oxidized LDL activates fas-mediated endothelial cell apoptosis. J. Clin. Investig. 1998, 102, 1682–1689. [Google Scholar] [CrossRef]
- Suc, I.; Escargueil-Blanc, I.; Troly, M.; Salvayre, R.; Nègre-Salvayre, A. HDL and ApoA prevent cell death of endothelial cells induced by oxidized LDL. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2158–2166. [Google Scholar] [CrossRef]
- Li, D.; Mehta, J.L. Upregulation of endothelial receptor for oxidized LDL (LOX-1) by oxidized LDL and implications in apoptosis of human coronary artery endothelial cells: Evidence from use of antisense LOX-1 mRNA and chemical inhibitors. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1116–1122. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Sukhorukov, V.N.; Kalmykov, V.A.; Grechko, A.V.; Shakhpazyan, N.K.; Orekhov, A.N. The Role of KLF2 in the Regulation of Atherosclerosis Development and Potential Use of KLF2-Targeted Therapy. Biomedicines 2022, 10, 254. [Google Scholar] [CrossRef]
- Dekker, R.J.; van Soest, S.; Fontijn, R.D.; Salamanca, S.; de Groot, P.G.; VanBavel, E.; Pannekoek, H.; Horrevoets, A.J. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2). Blood 2002, 100, 1689–1698. [Google Scholar] [CrossRef]
- Zhang, Y.; Guan, Q.; Wang, Z. PTP1B inhibition ameliorates inflammatory injury and dysfunction in ox-LDL-induced HUVECs by activating the AMPK/SIRT1 signaling pathway via negative regulation of KLF2. Exp. Ther. Med. 2022, 24, 467. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xuan, W.; Jia, Z.; Li, H.; Li, M.; Liang, X.; Su, D. HRD1 prevents atherosclerosis-mediated endothelial cell apoptosis by promoting LOX-1 degradation. Cell Cycle 2020, 19, 1466–1477. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Hsu, P.P.; Chen, B.P.; Yuan, S.; Usami, S.; Shyy, J.Y.; Li, Y.S.; Chien, S. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc. Natl. Acad. Sci. USA 2000, 97, 9385–9389. [Google Scholar] [CrossRef]
- Malek, A.M.; Jiang, L.; Lee, I.; Sessa, W.C.; Izumo, S.; Alper, S.L. Induction of nitric oxide synthase mRNA by shear stress requires intracellular calcium and G-protein signals and is modulated by PI 3 kinase. Biochem. Biophys. Res. Commun. 1999, 254, 231–242. [Google Scholar] [CrossRef]
- Tardy, Y.; Resnick, N.; Nagel, T.; Gimbrone, M.A., Jr.; Dewey, C.F., Jr. Shear stress gradients remodel endothelial monolayers in vitro via a cell proliferation-migration-loss cycle. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3102–3106. [Google Scholar] [CrossRef] [PubMed]
- Chien, S. Effects of disturbed flow on endothelial cells. Ann. Biomed. Eng. 2008, 36, 554–562. [Google Scholar] [CrossRef]
- Zeng, L.; Zampetaki, A.; Margariti, A.; Pepe, A.E.; Alam, S.; Martin, D.; Xiao, Q.; Wang, W.; Jin, Z.G.; Cockerill, G.; et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2009, 106, 8326–8331. [Google Scholar] [CrossRef] [PubMed]
- Heo, K.S.; Lee, H.; Nigro, P.; Thomas, T.; Le, N.T.; Chang, E.; McClain, C.; Reinhart-King, C.A.; King, M.R.; Berk, B.C.; et al. PKCζ mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J. Cell Biol. 2011, 193, 867–884. [Google Scholar] [CrossRef]
- Birukov, K.G.; Jacobson, J.R.; Flores, A.A.; Ye, S.Q.; Birukova, A.A.; Verin, A.D.; Garcia, J.G. Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L785–L797. [Google Scholar] [CrossRef]
- Wang, J.; Fan, B.; Wei, Y.; Suo, X.; Ding, Y. A simple multi-well stretching device to induce inflammatory responses of vascular endothelial cells. Lab A Chip 2016, 16, 360–367. [Google Scholar] [CrossRef]
- Dong, G.; Huang, X.; Jiang, S.; Ni, L.; Chen, S. Simvastatin Mitigates Apoptosis and Transforming Growth Factor-Beta Upregulation in Stretch-Induced Endothelial Cells. Oxidative Med. Cell. Longev. 2019, 2019, 6026051. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chiou, K.R.; Bugayenko, A.; Irani, K.; Kass, D.A. Reduced wall compliance suppresses Akt-dependent apoptosis protection stimulated by pulse perfusion. Circ. Res. 2005, 97, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.M.; Ensenat, D.; Wang, H.; Schafer, A.I.; Durante, W. Physiologic cyclic stretch inhibits apoptosis in vascular endothelium. FEBS Lett. 2003, 541, 52–56. [Google Scholar] [CrossRef]
- Raaz, U.; Kuhn, H.; Wirtz, H.; Hammerschmidt, S. Rapamycin reduces high-amplitude, mechanical stretch-induced apoptosis in pulmonary microvascular endothelial cells. Microvasc. Res. 2009, 77, 297–303. [Google Scholar] [CrossRef]
- Zhuang, F.; Bao, H.; Shi, Q.; Li, J.; Jiang, Z.L.; Wang, Y.; Qi, Y.X. Endothelial microparticles induced by cyclic stretch activate Src and modulate cell apoptosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 13586–13596. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, Y.; Fard, J.K.; Ghafoor, D.; Eid, A.H.; Sahebkar, A. Paradoxical effects of statins on endothelial and cancer cells: The impact of concentrations. Cancer Cell Int. 2023, 23, 43. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Nuki, T.; Gomita, K.; Weyand, C.M.; Hagiwara, N. Statins reduce endothelial cell apoptosis via inhibition of TRAIL expression on activated CD4 T cells in acute coronary syndrome. Atherosclerosis 2010, 213, 33–39. [Google Scholar] [CrossRef]
- Safaeian, L.; Mirian, M.; Bahrizadeh, S. Evolocumab, a PCSK9 inhibitor, protects human endothelial cells against H(2)O(2)-induced oxidative stress. Arch. Physiol. Biochem. 2022, 128, 1681–1686. [Google Scholar] [CrossRef]
- Zeng, J.; Tao, J.; Xi, L.; Wang, Z.; Liu, L. PCSK9 mediates the oxidative low-density lipoprotein-induced pyroptosis of vascular endothelial cells via the UQCRC1/ROS pathway. Int. J. Mol. Med. 2021, 47, 53. [Google Scholar] [CrossRef]
- Hu, Y.; Xu, Q.; Li, H.; Meng, Z.; Hao, M.; Ma, X.; Lin, W.; Kuang, H. Dapagliflozin Reduces Apoptosis of Diabetic Retina and Human Retinal Microvascular Endothelial Cells Through ERK1/2/cPLA2/AA/ROS Pathway Independent of Hypoglycemic. Front. Pharmacol. 2022, 13, 827896. [Google Scholar] [CrossRef]
- Faridvand, Y.; Kazemzadeh, H.; Vahedian, V.; Mirzajanzadeh, P.; Nejabati, H.R.; Safaie, N.; Maroufi, N.F.; Pezeshkian, M.; Nouri, M.; Jodati, A. Dapagliflozin attenuates high glucose-induced endothelial cell apoptosis and inflammation through AMPK/SIRT1 activation. Clin. Exp. Pharmacol. Physiol. 2022, 49, 643–651. [Google Scholar] [CrossRef]
- Yang, J.; Sato, K.; Aprahamian, T.; Brown, N.J.; Hutcheson, J.; Bialik, A.; Perlman, H.; Walsh, K. Endothelial overexpression of Fas ligand decreases atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Carino, K.; Johnston, L.; Servinsky, L.; Machamer, C.E.; Kolb, T.M.; Lam, H.; Dudek, S.M.; An, S.S.; Rane, M.J.; et al. A nonapoptotic endothelial barrier-protective role for caspase-3. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L1118–L1126. [Google Scholar] [CrossRef] [PubMed]
- Avrutsky, M.I.; Ortiz, C.C.; Johnson, K.V.; Potenski, A.M.; Chen, C.W.; Lawson, J.M.; White, A.J.; Yuen, S.K.; Morales, F.N.; Canepa, E.; et al. Endothelial activation of caspase-9 promotes neurovascular injury in retinal vein occlusion. Nat. Commun. 2020, 11, 3173. [Google Scholar] [CrossRef]
- Bader, S.M.; Preston, S.P.; Saliba, K.; Lipszyc, A.; Grant, Z.L.; Mackiewicz, L.; Baldi, A.; Hempel, A.; Clark, M.P.; Peiris, T.; et al. Endothelial Caspase-8 prevents fatal necroptotic hemorrhage caused by commensal bacteria. Cell Death Differ. 2023, 30, 27–36. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yin, J.; Starovasnik, M.A.; Fairbrother, W.J.; Dixit, V.M. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 2002, 277, 9505–9511. [Google Scholar] [CrossRef] [PubMed]
- Petrie, E.J.; Czabotar, P.E.; Murphy, J.M. The Structural Basis of Necroptotic Cell Death Signaling. Trends Biochem. Sci. 2019, 44, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL forms cation channels. Cell Res. 2016, 26, 517–528. [Google Scholar] [CrossRef]
- Vince, J.E.; Wong, W.W.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef]
- Karunakaran, D.; Geoffrion, M.; Wei, L.; Gan, W.; Richards, L.; Shangari, P.; DeKemp, E.M.; Beanlands, R.A.; Perisic, L.; Maegdefessel, L.; et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2016, 2, e1600224. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, H.; Yang, M.; Ren, J.; Huang, Z.; Han, F.; Huang, J.; Ma, J.; Zhang, D.; Zhang, Z.; et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013, 3, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, A.; Robichaud, S.; Nguyen, M.A.; Geoffrion, M.; Wyatt, H.; Cottee, M.L.; Dennison, T.; Pietrangelo, A.; Lee, R.; Lagace, T.A.; et al. Loss of MLKL (Mixed Lineage Kinase Domain-Like Protein) Decreases Necrotic Core but Increases Macrophage Lipid Accumulation in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1155–1167. [Google Scholar] [CrossRef]
- Coornaert, I.; Puylaert, P.; Marcasolli, G.; Grootaert, M.O.J.; Vandenabeele, P.; De Meyer, G.R.Y.; Martinet, W. Impact of myeloid RIPK1 gene deletion on atherogenesis in Apoe-deficient mice. Atherosclerosis 2021, 322, 51–60. [Google Scholar] [CrossRef]
- Newton, K.; Dugger, D.L.; Maltzman, A.; Greve, J.M.; Hedehus, M.; Martin-McNulty, B.; Carano, R.A.; Cao, T.C.; van Bruggen, N.; Bernstein, L.; et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016, 23, 1565–1576. [Google Scholar] [CrossRef]
- Colijn, S.; Muthukumar, V.; Xie, J.; Gao, S.; Griffin, C.T. Cell-specific and athero-protective roles for RIPK3 in a murine model of atherosclerosis. Dis. Models Mech. 2020, 13, dmm041962. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, D.; Nguyen, M.A.; Geoffrion, M.; Vreeken, D.; Lister, Z.; Cheng, H.S.; Otte, N.; Essebier, P.; Wyatt, H.; Kandiah, J.W.; et al. RIPK1 Expression Associates with Inflammation in Early Atherosclerosis in Humans and Can Be Therapeutically Silenced to Reduce NF-κB Activation and Atherogenesis in Mice. Circulation 2021, 143, 163–177. [Google Scholar] [CrossRef]
- Gan, I.; Jiang, J.; Lian, D.; Huang, X.; Fuhrmann, B.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.X. Mitochondrial permeability regulates cardiac endothelial cell necroptosis and cardiac allograft rejection. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2019, 19, 686–698. [Google Scholar] [CrossRef]
- Kwok, C.; Pavlosky, A.; Lian, D.; Jiang, J.; Huang, X.; Yin, Z.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.X. Necroptosis Is Involved in CD4+ T Cell-Mediated Microvascular Endothelial Cell Death and Chronic Cardiac Allograft Rejection. Transplantation 2017, 101, 2026–2037. [Google Scholar] [CrossRef]
- Zhu, P.; Wang, J.; Du, W.; Ren, J.; Zhang, Y.; Xie, F.; Xu, G. NR4A1 Promotes LPS-Induced Acute Lung Injury through Inhibition of Opa1-Mediated Mitochondrial Fusion and Activation of PGAM5-Related Necroptosis. Oxidative Med. Cell. Longev. 2022, 2022, 6638244. [Google Scholar] [CrossRef] [PubMed]
- Singla, S.; Sysol, J.R.; Dille, B.; Jones, N.; Chen, J.; Machado, R.F. Hemin Causes Lung Microvascular Endothelial Barrier Dysfunction by Necroptotic Cell Death. Am. J. Respir. Cell Mol. Biol. 2017, 57, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Paku, S.; Laszlo, V.; Dezso, K.; Nagy, P.; Hoda, M.A.; Klepetko, W.; Renyi-Vamos, F.; Timar, J.; Reynolds, A.R.; Dome, B. The evidence for and against different modes of tumour cell extravasation in the lung: Diapedesis, capillary destruction, necroptosis, and endothelialization. J. Pathol. 2017, 241, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef]
- Bolik, J.; Krause, F.; Stevanovic, M.; Gandraß, M.; Thomsen, I.; Schacht, S.S.; Rieser, E.; Müller, M.; Schumacher, N.; Fritsch, J.; et al. Inhibition of ADAM17 impairs endothelial cell necroptosis and blocks metastasis. J. Exp. Med. 2022, 219, e20201039. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Coll, R.C.; Schroder, K.; Pelegrín, P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol. Sci. 2022, 43, 653–668. [Google Scholar] [CrossRef]
- Shi, X.; Xie, W.L.; Kong, W.W.; Chen, D.; Qu, P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2015, 24, 2455–2466. [Google Scholar] [CrossRef]
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sørensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031. [Google Scholar] [CrossRef]
- Jin, X.; Fu, W.; Zhou, J.; Shuai, N.; Yang, Y.; Wang, B. Oxymatrine attenuates oxidized low-density lipoprotein-induced HUVEC injury by inhibiting NLRP3 inflammasome-mediated pyroptosis via the activation of the SIRT1/Nrf2 signaling pathway. Int. J. Mol. Med. 2021, 48, 187. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, L.; Guo, X.; Liu, J.; Xu, L.; Li, Y. Exogenous spermine inhibits high glucose/oxidized LDL-induced oxidative stress and macrophage pyroptosis by activating the Nrf2 pathway. Exp. Ther. Med. 2022, 23, 310. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Das, B.; Sarkar, C.; Rawat, V.S.; Kalita, D.; Deka, S.; Agnihotri, A. Promise of the NLRP3 Inflammasome Inhibitors in In Vivo Disease Models. Molecules 2021, 26, 4996. [Google Scholar] [CrossRef] [PubMed]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slütter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Wu, D.; Sun, Y.; Suo, Y.; Yu, Q.; Zeng, M.; Gao, Q.; Yu, B.; Jiang, X.; Wang, Y. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci. Rep. 2021, 11, 19305. [Google Scholar] [CrossRef]
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 2019, 10, 306. [Google Scholar] [CrossRef]
- Qian, Z.; Zhao, Y.; Wan, C.; Deng, Y.; Zhuang, Y.; Xu, Y.; Zhu, Y.; Lu, S.; Bao, Z. Pyroptosis in the Initiation and Progression of Atherosclerosis. Front. Pharmacol. 2021, 12, 652963. [Google Scholar] [CrossRef]
- Lin, X.; Ouyang, S.; Zhi, C.; Li, P.; Tan, X.; Ma, W.; Yu, J.; Peng, T.; Chen, X.; Li, L.; et al. Focus on ferroptosis, pyroptosis, apoptosis and autophagy of vascular endothelial cells to the strategic targets for the treatment of atherosclerosis. Arch. Biochem. Biophys. 2022, 715, 109098. [Google Scholar] [CrossRef]
- Koka, S.; Xia, M.; Chen, Y.; Bhat, O.M.; Yuan, X.; Boini, K.M.; Li, P.L. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. 2017, 13, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, X.; Sha, X.; Xi, H.; Li, Y.F.; Shao, Y.; Mai, J.; Virtue, A.; Lopez-Pastrana, J.; Meng, S.; et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, T.; Liu, J.; Chen, X.; Zhang, L.; Pi, J.; Sun, H.; Li, L.; Bauer, R.; Wang, H.; Yu, Z.; et al. Endothelial Foxp1 Suppresses Atherosclerosis via Modulation of Nlrp3 Inflammasome Activation. Circ. Res. 2019, 125, 590–605. [Google Scholar] [CrossRef]
- Xu, X.; Yang, Y.; Wang, G.; Yin, Y.; Han, S.; Zheng, D.; Zhou, S.; Zhao, Y.; Chen, Y.; Jin, Y. Low shear stress regulates vascular endothelial cell pyroptosis through miR-181b-5p/STAT-3 axis. J. Cell. Physiol. 2021, 236, 318–327. [Google Scholar] [CrossRef]
- Puylaert, P.; Van Praet, M.; Vaes, F.; Neutel, C.H.G.; Roth, L.; Guns, P.J.; De Meyer, G.R.Y.; Martinet, W. Gasdermin D Deficiency Limits the Transition of Atherosclerotic Plaques to an Inflammatory Phenotype in Apoe Knock-Out Mice. Biomedicines 2022, 10, 1171. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.L. Iron and the sex difference in heart disease risk. Lancet 1981, 1, 1293–1294. [Google Scholar] [CrossRef] [PubMed]
- Kiechl, S.; Willeit, J.; Egger, G.; Poewe, W.; Oberhollenzer, F. Body iron stores and the risk of carotid atherosclerosis: Prospective results from the Bruneck study. Circulation 1997, 96, 3300–3307. [Google Scholar] [CrossRef]
- Meyers, D.G.; Jensen, K.C.; Menitove, J.E. A historical cohort study of the effect of lowering body iron through blood donation on incident cardiac events. Transfusion 2002, 42, 1135–1139. [Google Scholar] [CrossRef]
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.N.; Spaich, S.; Seide, S.E.; Sparla, R.; et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Heart J. 2020, 41, 2681–2695. [Google Scholar] [CrossRef]
- Lee, T.S.; Shiao, M.S.; Pan, C.C.; Chau, L.Y. Iron-deficient diet reduces atherosclerotic lesions in Apoe-deficient mice. Circulation 1999, 99, 1222–1229. [Google Scholar] [CrossRef]
- Minqin, R.; Rajendran, R.; Pan, N.; Tan, B.K.; Ong, W.Y.; Watt, F.; Halliwell, B. The iron chelator desferrioxamine inhibits atherosclerotic lesion development and decreases lesion iron concentrations in the cholesterol-fed rabbit. Free Radic. Biol. Med. 2005, 38, 1206–1211. [Google Scholar] [CrossRef]
- Zhang, W.J.; Wei, H.; Frei, B. The iron chelator, desferrioxamine, reduces inflammation and atherosclerotic lesion development in experimental mice. Exp. Biol. Med. 2010, 235, 633–641. [Google Scholar] [CrossRef]
- Yang, K.; Song, H.; Yin, D. PDSS2 Inhibits the Ferroptosis of Vascular Endothelial Cells in Atherosclerosis by Activating Nrf2. J. Cardiovasc. Pharmacol. 2021, 77, 767–776. [Google Scholar] [CrossRef]
- Bai, T.; Li, M.; Liu, Y.; Qiao, Z.; Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Biol. Med. 2020, 160, 92–102. [Google Scholar] [CrossRef]
- Li, L.; Wang, H.; Zhang, J.; Chen, X.; Zhang, Z.; Li, Q. Effect of endothelial progenitor cell-derived extracellular vesicles on endothelial cell ferroptosis and atherosclerotic vascular endothelial injury. Cell Death Discov. 2021, 7, 235. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yang, N.; Song, Y.; Si, H.; Qin, Q.; Guo, Z. Effect of iron overload on endothelial cell calcification and its mechanism. Ann. Transl. Med. 2021, 9, 1658. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, M.N.; Osama, M. The implications of neutrophil extracellular traps in the pathophysiology of atherosclerosis and atherothrombosis. Exp. Biol. Med. 2020, 245, 1376–1384. [Google Scholar] [CrossRef]
- Döring, Y.; Libby, P.; Soehnlein, O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Nija, R.J.; Sanju, S.; Sidharthan, N.; Mony, U. Extracellular Trap by Blood Cells: Clinical Implications. Tissue Eng. Regen. Med. 2020, 17, 141–153. [Google Scholar] [CrossRef]
- de Boer, O.J.; Li, X.; Teeling, P.; Mackaay, C.; Ploegmakers, H.J.; van der Loos, C.M.; Daemen, M.J.; de Winter, R.J.; van der Wal, A.C. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb. Haemost. 2013, 109, 290–297. [Google Scholar]
- Döring, Y.; Weber, C.; Soehnlein, O. Footprints of neutrophil extracellular traps as predictors of cardiovascular risk. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1735–1736. [Google Scholar] [CrossRef]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef]
- Quillard, T.; Araújo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. Eur. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef]
- Franck, G.; Mawson, T.; Sausen, G.; Salinas, M.; Masson, G.S.; Cole, A.; Beltrami-Moreira, M.; Chatzizisis, Y.; Quillard, T.; Tesmenitsky, Y.; et al. Flow Perturbation Mediates Neutrophil Recruitment and Potentiates Endothelial Injury via TLR2 in Mice: Implications for Superficial Erosion. Circ. Res. 2017, 121, 31–42. [Google Scholar] [CrossRef]
Figure 1.
Oxidative stress and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.
Figure 1.
Oxidative stress and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.

Figure 2.
Inflammation and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.
Figure 2.
Inflammation and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.

Figure 3.
Genomic instability and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.
Figure 3.
Genomic instability and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.

Figure 4.
Metabolite-sensing longevity pathways and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.
Figure 4.
Metabolite-sensing longevity pathways and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.

Figure 5.
Activated RAS and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.
Figure 5.
Activated RAS and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.

Figure 6.
Uric acid and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.
Figure 6.
Uric acid and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.

Figure 7.
Polyploidization and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.
Figure 7.
Polyploidization and EC senescence. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.

Figure 8.
Timeline of cell death research.

Figure 9.
EC death in atherosclerosis. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.
Figure 9.
EC death in atherosclerosis. Green pointed arrows indicate stimulation; red blunted arrows indicate inhibition.


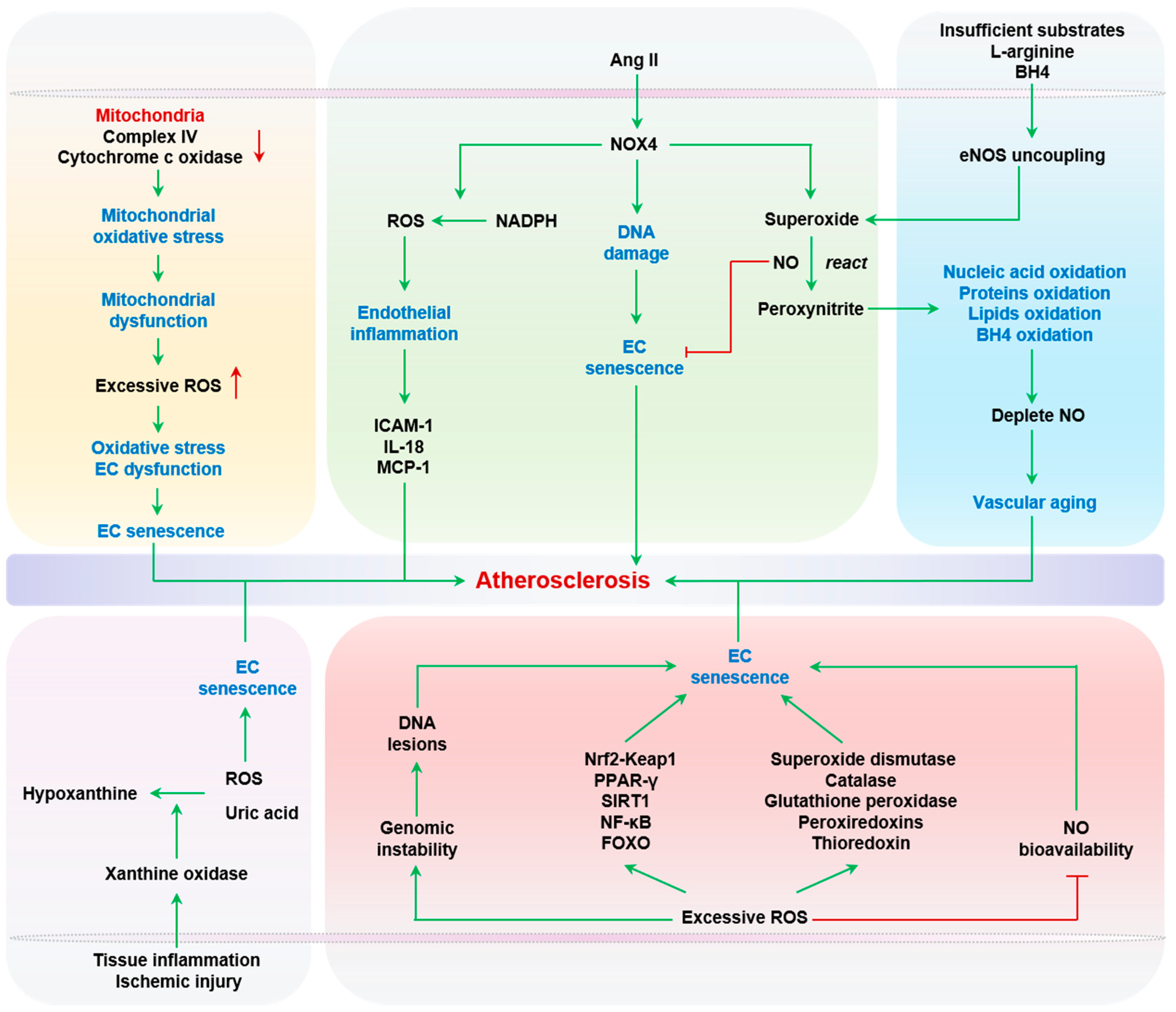{kind=link}
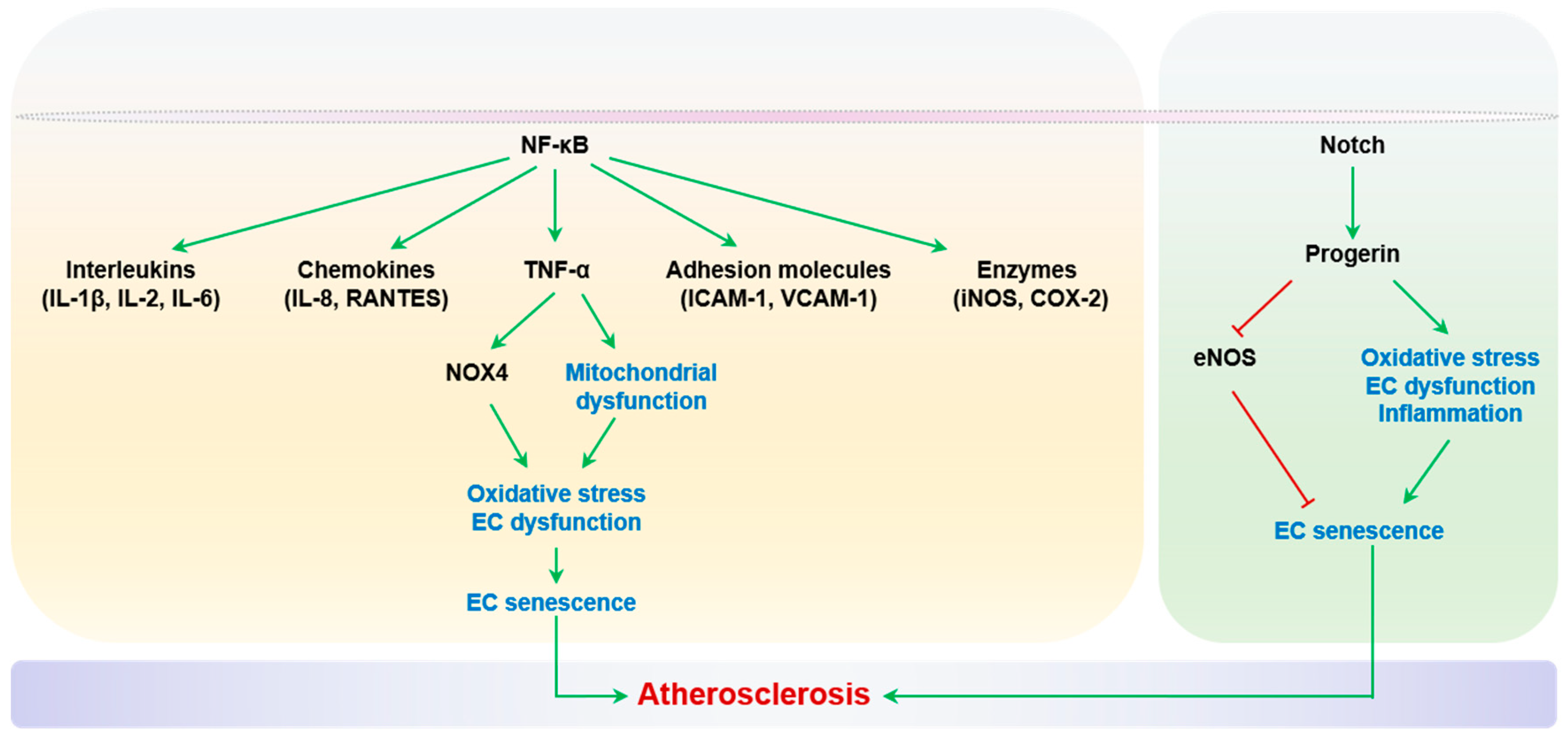{kind=link}
{kind=link}
{kind=link}
{kind=link}
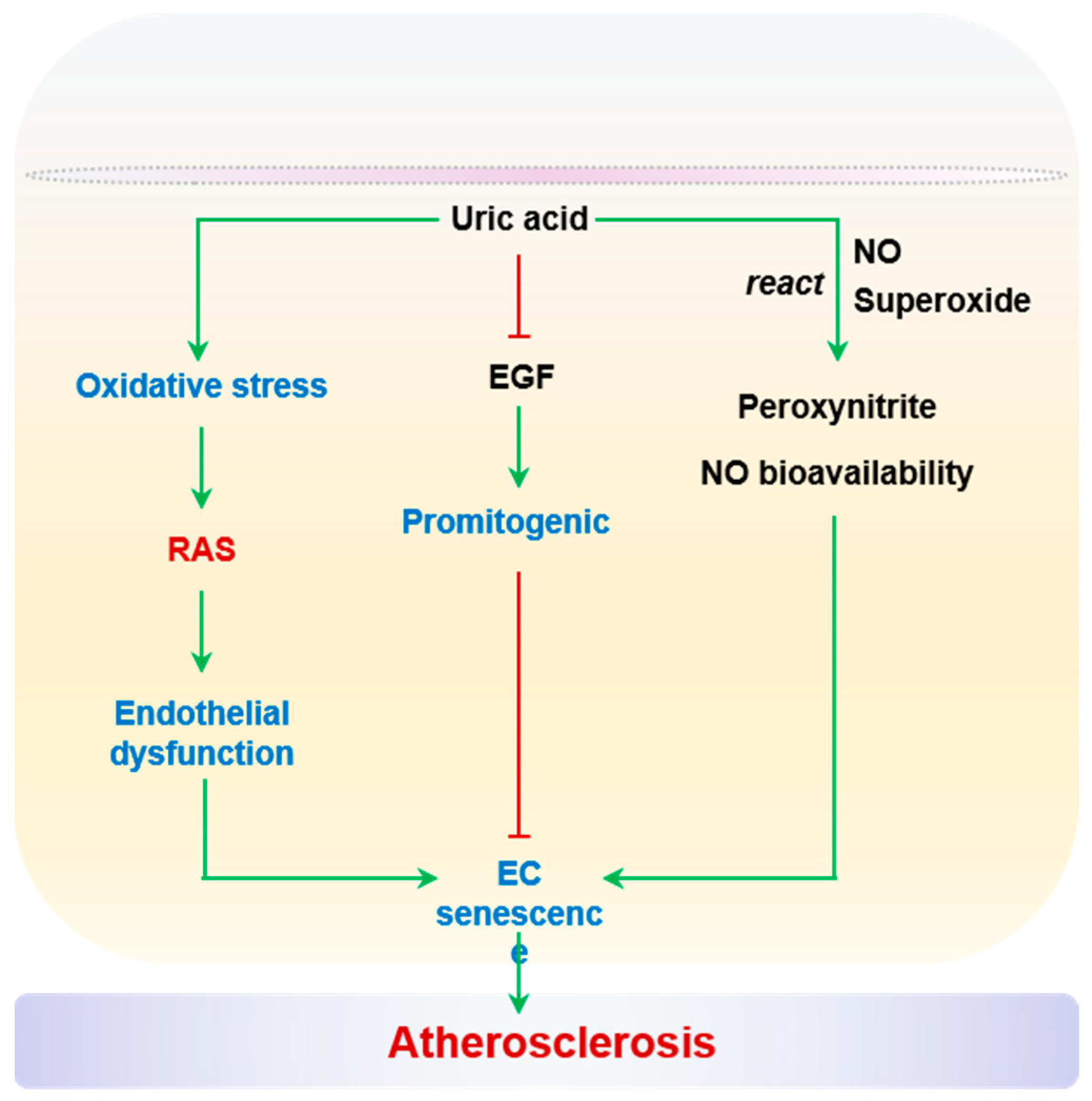{kind=link}
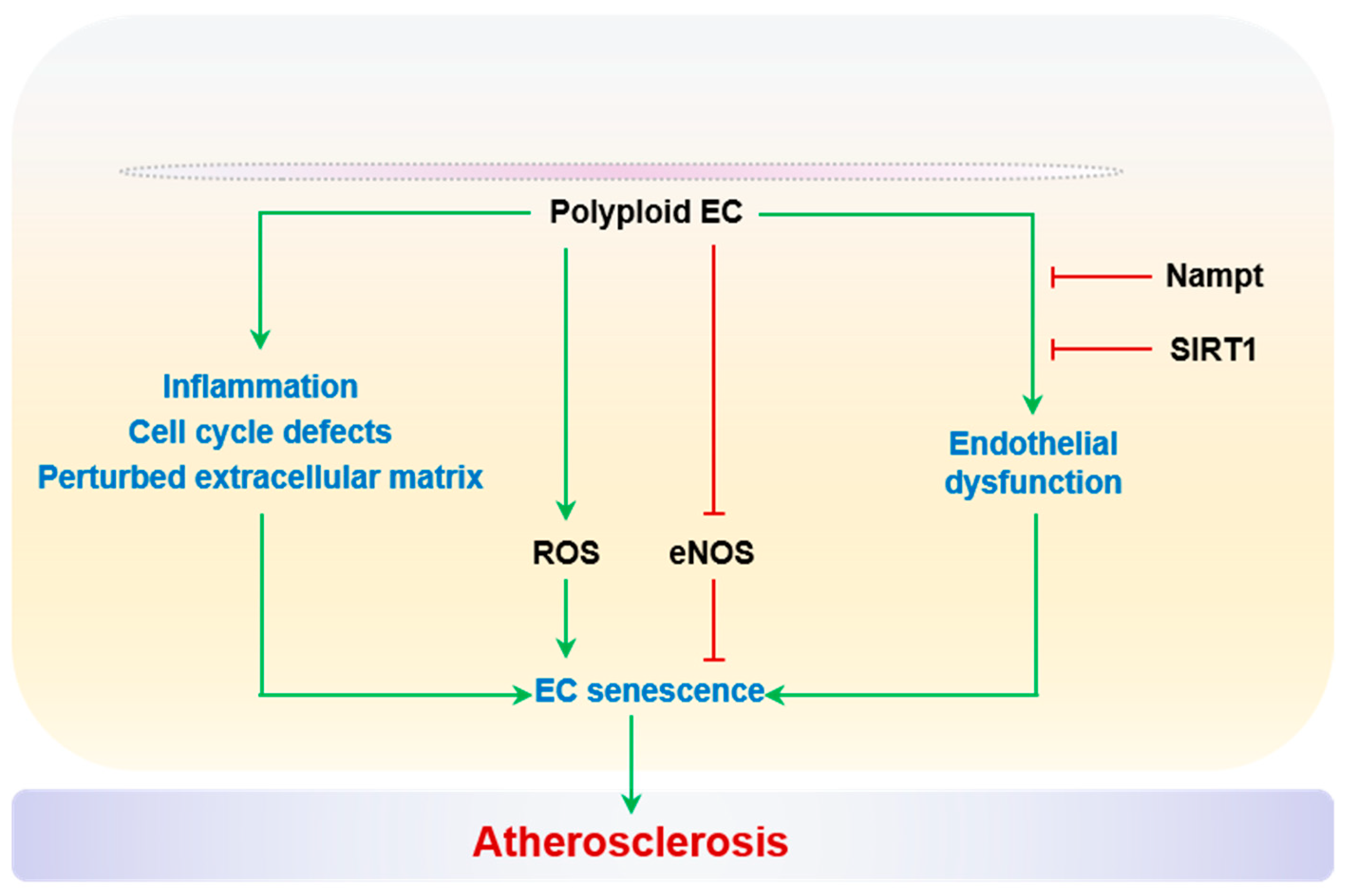{kind=link}
{kind=link}
{kind=link}
Table 1.
Methods to detect in vivo cellular senescence.
| Samples | Indexes | Methods | Advantage | Shortcoming | Ref |
|---|---|---|---|---|---|
| Protein (tissues, cells) | SA-β-gal | SA-β-gal test | Gold standard | Traumatically harvested, time-consuming | [36] |
| Gal-EM | Transmission electron microscopy | Gold standard | Traumatically harvested, expensive instruments | [37] | |
| p16, p21, SAHF | RNAscope Immunohistochemistry | High specificity, quantifiable | Operational complexity, time-consuming | [38] | |
| Lipofuscin | Sudan black B staining | High specificity | Time-consuming | [39] | |
| Serum/Plasma | SASP: IL8/CXCL1/CXCL2/CXCL3 /TNF/IL-1α/IL-1β/ICAM-1/GM-CSF | ELISA Western blot | Mature method, quantifiable | Low specificity | [40] |
| TRX80 | ELISA | Mature method, quantifiable | Low specificity | [41] | |
| TNF-α/IL-1β/IL-6 | ELISA | Mature method, quantifiable | Large sample sizes are required | [42] | |
| Endothelial progenitor cells | Flow cytometry | High specificity, quantifiable | Operational complexity | [43] | |
| DNA | Telomere loss | Southern blot qPCR | High specificity, mature method, quantifiable | Operational complexity, time-consuming | [44] |
| g (POLG) mutation | DNA sequencing | Mature method, quantifiable | Low specificity, expensive | [45] | |
| m.3243A > G | DNA sequencing | Mature method, quantifiable | Low specificity, expensive | [46] | |
| LINE-1 elements | RT-qPCR | Mature method, quantifiable | Low specificity | [47] | |
| γH2AX | Dual isotope SPECT imaging | Quantifiable | Low specificity, expensive | [48] | |
| Non-coding RNA | miR-183/miR-34a | PCR | Mature method, quantifiable | Low specificity | [49] |
Abbreviations: CXCL1/2/3: C-X-C motif chemokine ligand 1/2/3; Gal-EM: galactosidase-EM; GM-CSF: granulocyte monocyte colony-stimulating factor; ICAM-1: intercellular adhesion molecule 1; IL-1α: interleukin 1 alpha; IL-1β: interleukin 1 beta; IL-6/8: interleukin 6/8; p16: cyclin dependent kinase inhibitor 2A; p21: cyclin-dependent kinase inhibitor 1A; POLG: polymerase gamma; SA-β-gal: senescence-associated β-galactosidase; SAHF: senescence-associated heterochromatin aggregation; TNF: tumor necrosis factor; TRX80: thioredoxin 80; γH2AX: gamma histone variant H2AX.
Table 2.
The main features of cell death.
| Cell Death | Morphological Features | Biochemical Features | Key Biomarkers | Hallmarks | Related Signaling Molecules and Pathways | |
|---|---|---|---|---|---|---|
| Autophagy | Formation of double-membraned autolysosomes | Increased lysosomal activity | ATG5, ATG7, DRAM3, TFEB | LC3-II/I, autophagic vesicles, autophagic flow | PI3K-AKT-mTOR, MAPK-ERK1/2 pathway | [326,327] |
| Apoptosis | Cell membrane blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, DNA fragmentation, and apoptotic body formation | Activation of caspases, oligonucleosomal DNA fragmentation, modification of nuclear polypeptides | Caspase, p53, Fas, Bcl-2, Bax | Cell morphology, annexin V/PI, mitochondrial membrane potential, cytochrome C | Death receptor, mitochondrial, endoplasmic reticulum pathway, caspase, p53, Bcl-2-mediated pathway | [328,329] |
| Anoikis | Insufficient cell–matrix interactions | Integrins interact with ECM components to form adhesion complexes | Bcl-2, Bcl-XL, Mcl1, Bax, Bak, Bok, Bid, Bik, Bmf, Noxa, Bad, Bim, Puma | Caspases activation, DNA fragmentation | JNK, ERK, PI3K, PKB/AKT pathway, integrin, caveolin-1 | [330,331] |
| Pyroptosis | Apoptosis-like chromatin condensation and DNA fragmentation in the early stage, karyopyknosis, necrosis-like cell membrane pore formation, cellular swelling, membrane rupture | Dependent on caspase-1 and proinflammatory cytokine releases | Caspase-1, IL-1β, IL-18 | Gasdermin D, caspase-4, cell morphology | Caspase-1, NLRP3-mediated pathway | [332,333] |
| NETosis | Enzymes from granules translocate to the nucleus, chromatin de-condensation, internal membranes break down, cytolysis releases NETs | NETs generation | LPS, GM-CSF, IL8, PAD4, HMGB1, TF, MMP9, PMA, TGFβ, MPO, NE, RAGE, Histone, Anti-DsDNA | Promoting the transcription and translation of NE, MPO, and PAD4 | Raf-MEK-ERK, TLR2-fibronectin, caspase-4/5-GSDMD pathway | [334,335] |
| Necroptosis | Rupture of the cellular membrane, progressively translucent cytoplasm and swelling of organelles, moderate chromatin condensation | Drop in ATP levels, Activation of RIP1, RIP3, MLKL | LEF1, RIP1, RIP3 | RIPK1, RIPK3 | TNFα, TNFR1, TLR3, TRAIL, FasL, ROS, PKC-MAPK-AP1-mediated pathway | [336,337] |
| Ferroptosis | Small mitochondria with condensed mitochondrial membrane densities, reduction in or vanishing of mitochondria crista, outer mitochondrial membrane rupture | Inhibition of xCT and reduced GSH, inhibition of GPX4 Iron accumulation, lipid peroxidation | GPX4, Nrf2, LSH, TFR1, xCT | Fe2+, ROS, GSH, lipid peroxidation | MVA, HSF1-HSPB1, p62-Keap1-Nrf2 pathway; LSH pathway | [338,339] |
Abbreviations: ATG5/7: Autophagy-related 5/7, Bad: BCL-2 associated agonist of cell death, Bak: BCL-2 antagonist/killer 1, Bax: BCL-2-associated X, Bcl-2: BCL-2 apoptosis regulator, Bcl-XL: BCL-2-like 1, Bid: BH3-interacting domain death agonist, Bik: BCL-2-interacting killer, Bim: Bcl-2-like 11, Bmf: Bcl-2 modifying factor, Bok: BCL-2 family apoptosis regulator BOK, DRAM3: transmembrane protein 150B, Fas: Fas cell surface death receptor, GM-CSF: granulocyte–macrophage colony-stimulating factor, GPX4: glutathione peroxidase 4, Histone: hypothetical protein, HMGB1: high mobility group box 1, IL-18/1β/8: interleukin 18/1β/8, LEF1: lymphoid enhancer binding factor 1, LPS: lipopolysaccharide, LSH: solute carrier family 11 member 1, Mcl1: MCL1 apoptosis regulator, BCL-2 family member, MMP9: matrix metallopeptidase 9, MPO: myeloperoxidase, NE: Nelson’s mutant, Noxa: phorbol-12-myristate-13-acetate-induced protein 1, Nrf2: NFE2 like bZIP transcription factor 2, p53: tumor protein p53, PAD4: peptidyl arginine deiminase 4, PMA: Phorbol-12-myristate-13-acetate, Puma: BCL-2 binding component 3, RAGE: advanced glycosylation end-product-specific receptor, RIP1/3: ralA binding protein 1/3, TF: transferrin, TFEB: transcription factor EB, TFR1: transferrin receptor, TGFβ: transforming growth factor beta 1, xCT: solute carrier family 7 member 11.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bu, L.-L.; Yuan, H.-H.; Xie, L.-L.; Guo, M.-H.; Liao, D.-F.; Zheng, X.-L. New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death. Int. J. Mol. Sci. 2023, 24, 15160. https://doi.org/10.3390/ijms242015160
AMA Style
Bu L-L, Yuan H-H, Xie L-L, Guo M-H, Liao D-F, Zheng X-L. New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death. International Journal of Molecular Sciences. 2023; 24(20):15160. https://doi.org/10.3390/ijms242015160
Chicago/Turabian StyleBu, Lan-Lan, Huan-Huan Yuan, Ling-Li Xie, Min-Hua Guo, Duan-Fang Liao, and Xi-Long Zheng. 2023. "New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death" International Journal of Molecular Sciences 24, no. 20: 15160. https://doi.org/10.3390/ijms242015160
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.