
Cancer/Testis Antigens as Targets for RNA-Based Anticancer Therapy
Department of Biochemistry, College of Natural Sciences, Kangwon National University, Chuncheon 24341, Republic of Korea
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(19), 14679; https://doi.org/10.3390/ijms241914679
Submission received: 31 August 2023
/
Revised: 25 September 2023
/
Accepted: 27 September 2023
/
Published: 28 September 2023
(This article belongs to the Special Issue Advances in Targeted Immunotherapy in Cancers)
Abstract
:In the last few decades, RNA-based drugs have emerged as a promising candidate in the treatment of various diseases. The introduction of messenger RNA (mRNA) as a vaccine or therapeutic agent enables the production of almost any functional protein/peptide. The key to applying RNA therapy in clinical trials is developing safe and effective delivery systems. Exosomes and lipid nanoparticles (LNPs) have been exploited as promising vehicles for drug delivery. This review discusses the feasibility of exosomes and LNPs as vehicles for mRNA delivery. Cancer/testis antigens (CTAs) show restricted expression in normal tissues and widespread expression in cancer tissues. Many of these CTAs show expression in the sera of patients with cancers. These characteristics of CTAs make them excellent targets for cancer immunotherapy. This review summarizes the roles of CTAs in various life processes and current studies on mRNAs encoding CTAs. Clinical studies present the beneficial effects of mRNAs encoding CTAs in patients with cancers. This review highlight clinical studies employing mRNA-LNPs encoding CTAs.
1. Targets of Anticancer Immunotherapy: Tumor-Associated Antigens
Tumor-associated antigens (TAAs) exhibit abnormal expression in malignancies or are only produced during specific stages of differentiation, whereas their expression in normal tissues is restricted. TAAs include neoantigens and cancer/testis antigens (CTAs). Many clinical studies have examined the efficacy of immunotherapies targeting TAAs.
Neoantigens result from tumor-specific alterations, such as genomic mutation, abnormal RNA splicing, dysregulation of post-translational modification, and integration of viral open reading frames. Mutations include single nucleotide variations, insertions, deletions, and gene fusions. Next-generation sequencing has made the identification of neoantigens in individual patients easier [1]. Whole-exome sequencing and proteomic data from TCGA can also be employed to screen neoantigens.
Tumor-specific neoantigens are recognized as non-self and can elicit an immune response. Neoantigens can be presented by the major histocompatibility (MHC), alternatively known as human leukocyte antigen (HLA), molecules of cancer cells. The development of algorithms has made the prediction of immunogenic neoepitopes that can bind to MHC molecules possible [2]. These tumor-specific peptide-MHC complexes are recognized by T cells and trigger an anticancer immune response in cancer patients [3,4,5,6,7]. Highly antigenic neoantigens, through somatic mutations, make it possible for T cells specific for neoantigens to bypass negative selection in the thymus [8,9,10]. These characteristics make neoantigens ideal targets for developing anticancer immunotherapeutics.
CTAs, similar to neoantigens, are TAAs that play critical roles in various life processes, including angiogenesis [11,12], cancer progression [13], anti-apoptosis [14], epithelial to mesenchymal transition [15], metabolism [16], and anticancer drug resistance [17].
Sperm-associated antigen 9 promotes angiogenesis by activating vascular endothelial growth factor A signaling in an animal model of angiogenesis [18]. Prostate-associated gen 4 (PAGE4) protects prostate cancer (PCa) cells from apoptosis under oxidative stress by increasing the phosphorylation of ERK1/2 [19]. CTAs, such as sarcoma, synovial, X-chromosome-related (SSX), melanoma-associated gene-D4B (MAGE-D4B), cancer-associated gene (CAGE), piwil2, and CTA family 45A1 (CT45A1), promote epithelial to mesenchymal transition (EMT) and tumor progression [20]. SSX1 promotes EMT and stem cell-like properties by activating transforming growth factor- β1 signaling in synovial carcinoma [21]. SSX2 interacts with Rab3IP to promote EMT [22]. MAGEC2, which is highly expressed in non-small cell lung cancer cells, promotes angiogenesis and EMT [23]. MAGE-A3 shows high expression in cervical cancer cells and promotes cancer cell metastasis by activating Wingless-related integration site (Wnt) signaling [24]. PAGE4 specifically inhibits tankyrase 1 to activate canonical Wnt/β-catenin signaling in human cells [25]. The overexpression of CT45A enhances the proliferation and invasion of cancer cells by activating β-catenin [26]. High MAGE-A9 expression contributes to cancer stemness in hepatocellular carcinoma cells [27]. A high level of placental enriched 1 (PLAC1) promotes tumor invasion and is necessary for interaction between dendritic cells and macrophages, which suppresses antitumor immunity in head and neck squamous cell carcinoma [28]. CAGE, a CTA, confers resistance to anticancer drugs by directly regulating the expression of p53 in melanoma cells [29]. High expression of preferentially expressed antigen of melanoma (PRAME) is necessary for TNF-related apoptosis-inducing ligand (TRAIL) resistance in human lymphoid leukemia cells [30]. Chemotherapy-resistant Hodgkin lymphoma cells show high expression levels of CTAs, including MAGEA4, SSX2, survivin, and New York esophageal aquamous cell carcinoma-1 (NY-ESO-1) [31]. Semenogelin 1 (SEMG1) and SEMG2 interact with pyruvate kinase M2 and lactate dehydrogenase A to promote glycolysis and respiration in various cancer cell lines [32]. These reports suggest that CTAs can be targets for developing anticancer therapeutics. Table 1 shows the functions and locations of various TAAs. Figure 1 shows the roles of CTAs in various life processes.
Many CTAs are tissue-specific and are frequently expressed in cancer tissues, but not expressed under normal physiological conditions, rendering them promising candidates for cancer detection [33,34]. Autoantibodies against ubiquitin carboxyl-terminal esterase L1, SRY-box transcription factor 2, ATP-binding RNA helicase, and CAGE were higher in the advanced stage (IV) compared to the early stages (I-II) of lung cancer [35]. Spermatogenesis-associated 17 (SP17) can be employed as a diagnostic marker of Merkel cell carcinoma [36]. SP17-specific autoantibodies were detectable in the serum of HNSCC patients [37]. The presence of autoantibodies against CTAs indicates that these CTAs can be targets for anticancer immunotherapy.
CTAs have been considered as potential prognostic tumor markers [38] and targets for the immunotherapy of malignant tumors [39]. F-box only protein 39 is upregulated in glioma and is associated with a poor prognosis [40]. High expression of PRAME can predict poor prognosis in uveal melanoma [41]. PRAME promotes EMT and its presence in high levels is correlated with low overall survival in hepatocellular carcinoma [42]. High expression of PLAC1 is associated with low overall survival of patients with gastric cancers [43]. Taken together, these reports suggest that CTAs can be employed for developing anticancer immunotherapeutic agents.
2. Anticancer Immunotherapy Employing TAAs: Peptides, TCR-T and CAR-T
Cancer vaccines function as both prophylactics and therapeutics. CTAs are tumor-associated antigens and can be targets for developing therapeutic cancer vaccines [44]. Some CTAs, such as MAGE proteins, are present on the cell surface, making them targets for developing anticancer immunotherapeutic agents [45]. Cancer vaccines targeting mucin (MUC) and programmed death-ligand 1 (PD-L1) induce antitumor immunity in immunized mice by increasing the surface expression of MUC1 and PD-L1 [46].
CTAs induce humoral or cellular immune responses, providing anticancer immunotherapeutic opportunities for T-cell receptors (TCRs), chimeric antigen receptor-T (CAR-T) cells, antibody therapy, peptides, or mRNA-based vaccines. Cancer vaccines include whole-cell, DNA, RNA, and peptide-based vaccines [47]. Therapeutic cancer vaccines employ TAAs to induce cellular and humoral immunity to eliminate tumors. Anticancer immunotherapy eliminates cancer cells via the recognition of T cells of TAAs [48,49]. In other words, the recognition of TAA-derived peptides by T cells may eliminate cancer cells.
Dendritic cell (DC) vaccines loaded with different TAA-derived peptides have been widely used to study their therapeutic effects on cancer [50]. The peptides are derived from various TAAs, including MAGE A1, A3, A10, NY-ESO-1, and MUC1 [51,52,53]. DCs pulsed by the antigenic peptides derived from CTAs, such as MAGEA1 and human telomerase reverse transcriptase (hTERT), enhance cytolytic T lymphocyte (CTL) activity to inhibit the progression of acute myeloid leukemia [54]. HLA-A2 melanoma patients vaccinated with DCs pulsed with melanoma antigen-derived peptides (gp100 and tyrosinase) show an increased number of vaccine-specific T cells [55]. Peptide vaccines use short peptide fragments and usually do not include allergenic and/or reactogenic sequences such as lipopolysaccharides, lipids, and toxins. Peptide vaccines are generally weakly immunogenic and require particulate carriers for delivery and adjuvants.
TCR-based adoptive cell therapy employs genetically modified lymphocytes. TCR-T cell therapy (Figure 2) enables T cells to express antigen-specific TCR via retroviral or lentiviral vectors. The transfer of these engineered T cells into cancer patients can enhance the specificity and affinity of T cells to TAAs. Frequently employed TAAs for TCR-T cell therapy include NY-ESO-1, MAGE-A1/A4, PRAME, survivin, and SSX2 (NCT03192462 and NCT03093350). Adverse event data collection is still ongoing (NCT03192462). NCT03093350 measures the overall response (OR) and progression-free survival (PFS) in breast cancer patients who receive TAA-specific CTLs. A phase I clinical trial of TCR-T cells recognizing MAGE-A1 is underway in patients with refractory solid cancers (NCT03441100). The purpose of this trial is to determine safety and tolerability. In another phase I clinical study (NCT02111850), researchers constructed HLA-DPB1*0401-restricted TCRs to enable T cells to specifically recognize the MAGE-A3 antigen [56]. Among the seventeen patients with metastatic cancers, one had complete remission, and three had partial remission [56]. Adoptive transfer of CD4+ T cells transduced with MAGE-A3 TCR does not cause treatment-related deaths [56]. This suggests that TCR-T cell therapy targeting TAAs is safe and may warrant further investigations. Furthermore, T cell product ADP-A2 M4 targets MAGE-A4 peptide and has shown beneficial effects in a phase I trial of patients with synovial cell sarcomas (NCT04044768) [57]. ADP-A2M4 demonstrated potent anticancer activity in the absence of major off-target cross-reactivity against a range of human primary cells and cell lines [57]. A phase II clinical trial of ADP-A2M4 in combination with pembrolizumab is underway in patients with head and neck cancers (NCT04408898). A phase I clinical trial of TCR-T cells recognizing MAGEA4/8 is underway in patients with refractory solid cancers (NCT03247309). The purpose of this trial is to examine OS and PFS.
A clinical trial involving the transfer of MAGE-A10-specific TCR was performed [58]. Eleven patients with NSCLCs were treated with T cells (ADP-A2M10) transduced with a lentiviral vector containing the TCR targeting MAGE-A10. ADP-A2M10 showed trafficking to the tumor site and displayed an acceptable safety profile with no evidence of toxicity [58]. T cell lines targeting PRAME, SSX2, MAGEA4, NY-ESO-1, and survivin have shown safety with no systemic or neurotoxic effects in patients with metastatic pancreatic cancers (NCT03192462). A clinical trial of multi-antigen-targeted T cells (multiTAA-T) targeting survivin, NY-ESO-1, MAGE-A4, SSX2, and PRAME was performed in patients with refractory metastatic breast cancer (NCT03093350). In this trial, the multiTAA-T cells did not induce systemic toxicity and were well tolerated (NCT03093350). MultiTAA-T cells mostly induce disease stabilization in metastatic breast cancer (NCT03093350). Taken together, these reports suggest that TCR-T cell therapy that targets TAAs merits further investigations. Table 2 provides detailed information regarding clinical trials for multi-TAA-T cells.
Some CTAs are membrane-bound, making them potential targets for CAR-T therapy [59]. CAR-T cells recognize MHC and peptide complexes presented on the cell surface [59]. CAR-T cells can also recognize CTAs that are present on the surface of cancer cells (Figure 2). CARs are generated by fusing the antigen-binding domain to membrane-spanning and intracellular signaling domains, which enables T cells to recognize and eliminate cancer cells with CTAs. Although CAR-T therapy been effective for leukemia, it has not been successful in solid tumors [60]. Current applications of cancer/testis antigen-X (CT-X) in the CAR-T strategy are mainly limited to preclinical studies or early-phase clinical trials. CT-X antigen coding genes are located on the X chromosome. First-generation CARs combine a single-chain variable fragment (ScFv) with a cytoplasmic domain (CD3 zeta domain). Krebs et al. constructed CAR-T cells with interleukin-13 (IL-13) E13Y mutein for selective binding to IL-13Rα2 and reducing affinity to IL-13Rα1/IL-4Rα [61]. First-generation CAR-T cells recognize and attack both glioma stem cells and differentiated cells expressing the IL-13Rα antigen [62]. Second-generation CARs combine ScFv with CD3 zeta domain and CD28 (Figure 2). CD28 is a receptor for co-stimulatory molecules such as CD80. Third-generation CARs combine ScFv with the CD3 zeta domain, CD28, and 4-1BB, a co-stimulatory glycoprotein receptor (Figure 2). The added complexity of CARs may increase the number of active CAR-T cells. Cancer stem cells may lead to drug resistance and tumor relapse; hence, it might be a feasible strategy to generate CAR-T cells to target cancer stem cells expressing CT-X antigens.
CAR-T cells targeting MAGE A1 exhibit an anticancer effect in lung adenocarcinoma cells and xenografts [59]. A phase I clinical trial of CAR-T cells (MU-MA402C) that target MAGE-A4 is underway to determine safety, tolerability, and the anticancer effect [63]. CAR-T cells targeting the HLA-A2/NY-ESO-1 complex show an anticancer effect against HLA-A2+/NY-ESO-1+breast cancer cells [64]. CAR-expressing T cells targeting HLA-A-0201/SSX2 peptide eliminated acute myeloid leukemia cells [65]. These reports suggest that CAR-T cells targeting CTAs merit further studies for developing anticancer immunotherapeutic agents.
There are few ideal CTAs expressed on the cell surface. Therefore, the development of CAR-T therapy for solid tumor treatment has been slow. In the context of solid tumors, there remain some obstacles in T cell therapy: (1) it is difficult to identify TAAs that are expressed ubiquitously in tumors and not in normal tissues. Cancer cells not expressing the target antigen may outgrow those with the target antigen, which may lead to immune escape; (2) the immunosuppressive microenvironment prevents CAR-T cells from being recruited into tumor tissues [66]. Chemotherapy may enhance CAR-T cell infiltration into the tumor microenvironment [67], and immune checkpoint inhibitors could improve antitumor efficacy [68].
3. mRNA Vaccines as Anti-Cancer Therapeutics
RNA-based drugs are mainly divided into two major classes: (1) oligonucleotide drugs, such as antisense oligonucleotides (ASOs), small-interfering RNAs (siRNAs), microRNAs (miRNAs), and RNA aptamers; (2) in vitro-transcribed (IVT) mRNA drugs. Due to their large molecular structure and negative charge, oligonucleotide drugs are easily degraded by RNases and have difficulty penetrating cell membranes. The goal of a vaccine is to stimulate the production of antibodies that target a pathogen (prophylactic). Traditional vaccines stimulate an antibody response by injecting either antigens, an attenuated (weakened) virus, an inactivated virus, or a recombinant antigen-encoding viral vector into the body.
RNA therapeutics include the use of mRNA, miRNA, siRNA, circular RNAs, and long non-coding RNA. miRNA targets multiple genes, and one gene can be targeted by many different miRNAs. Therefore, it is difficult to design a miRNA to regulate a specific gene, which can result in unexpected side effects. miRNA drugs have mostly been terminated due to safety issues, with only a few candidates continuing into clinical development, and none have entered Phase 3 clinical trials.
mRNA drugs have emerged as a safe and efficacious strategy for protecting patients from infectious diseases and cancers owing to their advantages, including high efficiency, low side efficacy, and ease of manufacture. mRNAs display high therapeutic efficacy due to their continuous translation into encoded proteins/peptides compared to transient traditional protein/peptide drugs [69]. mRNA has been successfully transfected and produced an immune response in a dose-dependent manner [70]. This indicates that mRNA can induce the activation of CD4+ T cells and /or CD8+ T cells. Unlike DNA-based drugs, mRNA transcripts have a relatively high transfection efficiency and low toxicity because they do not need to enter the nucleus [71]. In addition, mRNAs do not cause insertional mutagenesis [72,73]. A BNT162b2 mRNA vaccine against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) did not induce immune-related adverse events in patients with NSCLCs [74]. Taken together, these reports suggest that the feasibility of mRNA vaccines as anticancer therapeutics merits further investigation.
4. CTA-Based Vaccines for Anticancer Immunotherapy
CTAs are normally restricted to the male testis; however, these proteins are aberrantly overexpressed in cancer stem-like cells and a variety of cancers, suggesting their potential as a target for cancer immunotherapy. The increased expression of CTAs, such as MAGE-A2, enhances tumor recognition by T cells to eliminate esophageal cancer cells [75]. MAGE-A-derived peptides are presented on the cell surface by MHC class I molecules, which enables CD8+ T cells to recognize cancer cells. Thus, CTAs can be employed as targets for anticancer immunotherapy.
MAGE, NY-ESO-1, SSX, cancer-testis antigen SP-1, sperm lysozyme-like protein 1, PLAC1, SP17, and PRAME are the most widely employed CTAs for anticancer immunotherapy.
CTA-based tumor vaccines (peptides, DNA, or RNA) have been known to induce an anticancer response. M. smegmatis transfected with recombinant plasmids expressing MAGEA3 and SSX2 display anticancer effects in a xenograft of esophageal cancer cells [76]. In addition, a MAGE-A DNA vaccine induced interferon γ and tumor necrosis factor α CD8+ T cell responses and an anticancer effect in a mouse model of melanoma [77]. This indicates that recombinant CTAs can induce anticancer immunity to suppress tumor growth.
MAGE-As-derived peptides induce CTL to kill breast cancer cells [18]. In addition, DCs treated with SP17-derived peptides induced CTL activity, which led to the killing of autologous cancer cells [37]. Thus, CTA-derived peptides can stimulate immune responses to eliminate cancer cells. A clinical trial (phase I/II, NCT00243529) was conducted to determine the in vivo responses of DC vaccines presenting HLA Class I and II restricted tumor epitopes either via peptide-pulsing or mRNA transfection in melanoma patients (stage III or IV). In this study, dendritic cells were transfected with RNA encoding tumor antigens gp100 and tyrosinase. DC vaccines have some limits in that only patients with a certain HLA type can be treated. However, this problem did not occur when DCs were transfected with RNA encoding tumor antigens.
5. Cancer Vaccines Employing mRNA
The therapeutic avenues of mRNA can be categorized into three classes: prophylactic vaccines, therapeutic vaccines, and therapeutic drugs (ASOs, siRNAs, miRNAs, RNA aptamers). Prophylactic mRNAs can encode for specific foreign antigens to evoke protective immunity against infectious diseases [78] or prime the immune system to stimulate cell-mediated responses to target tumors as a therapeutic vaccine [79]. Delivery of IVT mRNA also allows host cells to produce encoded proteins, which can act as additional proteins for replacements.
Compared to DNA vaccines, mRNA vaccines display a higher protein expression rate. In addition, they exhibit a lower level of toxicity due to the bacteria-free manufacturing procedures used for their production [80]. mRNA vaccines introduce a fragment of the RNA sequence into the individual being vaccinated. IVT mRNA has been successfully introduced into animals [81]. An mRNA vaccine delivers antigen-encoding mRNA into immune cells, such as dendritic cells, to produce foreign proteins. These proteins induce adaptive immune responses to eliminate cancer cells. The advantages of mRNA vaccines over traditional vaccines include the induction of both cellular and humoral immunity and the lack of insertional mutagenesis of genomic DNA [82]. The manufacture of mRNA vaccines is easier and less time-consuming when compared with plasmid DNA [83,84]. Unlike subunit vaccines, mRNA vaccines do not require adjuvants [84]. An mRNA vaccine is expressed in the cytosol, but not in the nucleus. Neoantigen-specific mRNA cancer vaccines stimulate adaptive immune responses by delivering tumor antigens into antigen-presenting cells (APCs) and induce vigorous anticancer immunity in pancreatic cancer [85].
6. mRNA Vaccines Encoding TAAs Induce an Adaptive Immune Response
During vaccination, naked or vehicle-loaded mRNA vaccines efficiently express tumor antigens in APCs and induce innate/adaptive immune stimulation [86]. mRNA-based vaccines can induce both humoral and cellular immunity through the induction of the CD8+ T cell response [87,88]. Translated proteins can then activate the immune system, primarily in two ways (Figure 3): (1) proteins are degraded by the proteasome into peptides. These peptides are then presented on the cell surface by MHC class I molecules which bind to the TCR to activate CD8+ T cells. This results in the subsequent elimination of cancer cells through the secretion of perforin and granzyme [89]; (2) proteins secreted extracellularly are engulfed by APCs and degraded into peptides. These peptides are then presented on the cell surface by MHC class II molecules for recognition by CD4+ T cells, which can activate both the cellular immune responses by secreting cytokines and the humoral immune responses by co-activating B cells. MAGE-A4 mRNA enables CD4+ phytohemagglutinin (PHA) blasts to induce CD8+ T cell responses [90].
A study was conducted to determine the in vivo immunological response in patients with uveal melanomas. HLA-A2.1 patients with uveal melanomas were injected with autologous DCs electroporated with mRNA encoding melanoma-associated antigens tyrosinase and/or gp100. (NCT00929019). No results concerning safety have been reported. Intradermal injection of the mRNAs encoding melan-A, tyrosinase, gp100, MAGE-A1, MAGE-A3, and survivin decreases the frequency of forkhead Box P3+ (Foxp3+)/CD4+ regulatory T cells and Myeloid-derived suppressor cells (MDSCs) in metastatic melanoma patients [91]. mRNAs encoding melan-A, tyrosinase, gp100, MAGE-A1, MAGE-A3, and survivin increase vaccine-specific T cells in two of four immunologically evaluable patients [91]. No adverse events greater than grade II have been observed in patients receiving these mRNAs [91]. DCs containing mRNAs encoding TAAs, such as gp100, MAGE-A3, or MAGE-C2, induce a strong CTL response in many patients with advanced melanomas [92]. Melanoma patients receiving autologous DCs electroporated with mRNAs encoding MAGE-A1, -A3, -C2, tyrosinase, MelanA/MART-1, or gp100, show DC-related adverse events but no appreciable toxicity [93]. DCs stimulated with an mRNA vaccine targeting NY-ESO-1, MAGE-C2, and MUC1 induce strong T cell responses in patients with castration-resistant prostate cancer [94]. Autologous Langerhans-type dendritic cells (LCs) electroporated with mRNAs encoding CTA 7 (CT7), MAGE-A3, and Wilms tumor 1 (WT1) increase the number of antigen -specific CD4+ T cells and CD8+T cells in patients with multiple myelomas (MM) [95]. Patients with MM display mild delayed-type hypersensitivity but not appreciable toxicity [95]. These antigen-specific T cells show increased secretion of inflammatory cytokines such as IFN-γ, IL-2, and TNF-α [95]. Thus, triple antigen-bearing mRNA-electroporated autologous LC vaccination can induce antigen-specific immune reactivity and is safe. An mRNA vaccine (CV9201) that targets NY-ESO-1, MAGE-C1, MAGE-C2, survivin, and a trophoblast glycoprotein is well tolerated in patients with NSCLCs [96]. CV9201 induces antigen-specific T cell responses in 63% of patients and increases the number of activated IgD+CD38hi B cells in 60% (18/30) of evaluable patients [96]. The BI 1361849 mRNA vaccine, comprising MUC1, survivin, NY-ESO-1, 5T4, MAGE-C1, and MAGE-C2, was intradermally injected into metastatic NSCLC patients (NCT03164772, phase I/II). NSCLC patients also received an intravenous injection of anti-PD-L1 antibody or anti-cytotoxic T lymphocyte-associated protein 4 (CTL4) antibody (NCT03164772). Each mRNA was administered separately. The study aimed to measure objective response rate (OR), PFS, duration of response (DoR), and OS when administered with durvalumab and tremelimumab (NCT03164772) [97]. Taken together, these reports suggest that mRNA vaccines targeting CTAs merit further investigations. Table 3 provides detailed information regarding clinical trials of mRNAs encoding TAAs in combination with immune checkpoint inhibitors such as durvalumab.
7. mRNA Delivery System: Exosomes and LNPs
The clinical applications of RNA drugs are primarily limited by the delivery issue: the lack of efficient carriers for delivering RNA molecules to target cells and tissues [98]. Several studies have shown the unsatisfactory efficacy of delivering naked mRNA [99,100], which is caused by the high rates of RNA degradation during its circulation, RNA-induced innate immunity, and poor cellular infiltration [35,86,101]. To achieve the ideal mRNA potency, it is necessary to provide mRNA with protection and facilitate its cellular uptake as well as endosome escape. Since mRNA cannot pass through the membrane via passive diffusion, RNA-based drugs are usually taken up via endocytosis. Most of the mRNA is trapped in endosomes after entry and is unable to escape into the cytoplasm. In other words, enhanced endosomal escape of mRNA is required for developing mRNA-based therapeutics.
Exosomes can help mRNA overcome these problems. They are small (~30–150 nm) lipid bilayer-coated extracellular vesicles, which are released into the microenvironment after the fusion of multivesicular bodies with the plasma membranes [102,103]. Exosomes contain a variety of proteins and nucleic acids such as mRNA, noncoding RNA, and DNA [104,105].
Exosomes mediate cellular interactions and play crucial roles in various cellular processes, including cancer initiation and progression and immune regulation, in addition to anticancer drug resistance [106,107,108]. Macrophage-derived exosomal miR-342-3p binds to neural precursor cell expressed, developmentally down-regulated gene 4-like (NEDD4L), and consequently elevates centrosomal protein 55 (CEP55) expression, thereby exerting tumor-promoting effects [109]. Exosomes promote aerobic glycolysis and bladder cancer cell proliferation [110]. Exosomal PD-L1 binds to programmed death-1 (PD-1) on T cells to inhibit CTL activity and promote melanoma metastasis [108]. Exosomes secreted by regulatory T cells induce immune suppression and inhibit Th1 responses [111]. Exosomal B7H4 increases the expression of FOXP3, which induces immune evasion in glioblastoma cells [112]. Tumor-derived exosomal miR-183-5p increases the number of PD-L1-expressing macrophages to promote immune suppression and intrahepatic cholangiocarcinoma progression [113]. Exosomal Ring finger protein 157 mRNA from prostate cancer cells induces M2 macrophage polarization by binding to Histone deacetylase 1, which results in its ubiquitination [114]. These reports suggest that exosomes modified with mRNAs may induce adaptive immunity to eliminate cancers.
Exosomes have been increasingly utilized as a promising targeted delivery system for RNA therapeutics against cancers and other diseases [115,116]. Compared to other traditional delivery systems, exosomes have special advantages: (1) exosomes have relatively low cytotoxicity and immunogenicity, and they have better biocompatibility due to membrane proteins such as tetraspanin and fibronectin [117,118]; (2) exosomes display high stability in plasma [119] and can escape clearance by macrophages through CD47 [120].
Since mRNAs are too large to cross cell membranes, they need to be encapsulated in special nanoparticles to improve their stability, to cross cell membranes, and to limit their immunogenicity [116]. To enhance mRNA delivery, various vectors have been designed and synthesized, including LNPs, polymeric nanoparticles, cationic nanoemulsions, and other delivery systems [121,122,123,124]. Recently, LNPs have become a leading mRNA delivery vehicle [125,126,127]. LNPs consist of four components (Figure 4): an ionizable or cationic lipid, cholesterol, phospholipids, and lipid-linked polyethylene glycol (PEG) [128]. mRNA loading mechanisms include electrostatic interactions, hydrogen bonds, coordination interactions, nanoprecipitation, and microfluidic mixing. Figure 4 also shows the advantages and disadvantages of mRNA vaccines.
Many clinical trials are underway to evaluate the efficacy and safety of mRNA vaccines in combination with checkpoint inhibitor therapy or chemotherapy. LNP-based mRNA vaccines offer several advantages: (1) their efficacy, safety, and productive efficiency; (2) LNP-based mRNA vaccines can improve the stability of mRNA vaccines and reduce the dose and frequency of drugs; (3) compared with other therapies, LNP-based mRNA vaccines have few side effects and will not cause other adverse effects in patients. However, LNPs can be removed by macrophages or reticuloendothelial cells [129].
Liposomes, an early version of LNPs, are a versatile nanomedicine delivery platform. A clinical trial of a liposomal RNA vaccine that encodes four non-mutated TAAs (NY-ESO-1, tyrosinase, MAGE-A3, and trans-membrane phosphatase with tensin homology) was performed (NCT02410733). This liposomal RNA vaccine induces a durable objective response in combination with anti-PD-L1 antibodies in patients with metastatic melanoma [130]. This vaccine induces both CD4+ and CD8+ T cell responses. In addition, a clinical trial of a patient-specific liposome-based RNA was performed (NCT02316457). This trial determined the number of adverse events and the maximum tolerated dose as well as the changes in the induced T cell responses of patients with breast cancers.
mRNA-4157 (LNP) encoding neoantigens shows an acceptable safety profile and displays beneficial clinical responses in combination with pembrolizumab in patients with NSCLCs and melanomas (NCT03313778, Table 4). mRNA-LNPs encoding neoantigens show an acceptable safety profile. However, this personalized cancer vaccine does not induce any clinical responses in patients with gastrointestinal cancers (NCT03480152) [131]. A clinical trial of mRNA-LNPs encoding KRAS mutations in combination with pembrolizumab was performed (NCT03948763). This clinical trial determined dose-limiting toxicities, objective response rates, and the presence of mutant KRAS-specific T cells, and examined adverse events. Table 4 provides detailed information regarding clinical trials of mRNAs-LNPs encoding TAAs in cancer patients. Taken together, these reports suggest that mRNA-LNP vaccines merit further investigation.
8. Discussion and Perspectives
mRNA drugs exploit cells for functional protein production with efficacy and safety. They can also target multiple genetic molecules and induce long-term effects. Rapid production is another advantage of mRNA cancer vaccines, and the maturity of mRNA manufacturing techniques allows the production of novel vaccines in a short time. This advantage is especially important due to the frequent rapid disease progression in cancer patients. The promising results of IVT mRNA vaccines in preclinical studies demonstrate their great potential as immunotherapies for treating various cancers.
However, instability, insufficient translation potency in vivo, and high innate immunogenicity make it difficult to achieve success in the development of mRNA cancer vaccines. LNPs can protect RNAs against nuclease-mediated degradation. However, the LNP itself can be toxic and may induce immunostimulatory side effects [132]. Many mRNA vaccines tend to induce a Th1-biased immune response through interferon signals [132]. Therefore, it is necessary to reduce immunogenicity associated with LNPs. IIVT mRNA-based therapeutics are generally thought not to integrate into the genome and therefore do not induce insertional mutagenesis, although this has not been unequivocally demonstrated. The broad delivery of sufficient mRNA to target cell populations has relied to date on complex lipid nanoparticle formulations and is yet unresolved. The targeted delivery of mRNA vaccines to various tumor types would enhance the therapeutic values of these vaccines. mRNA vaccines can be a key feature of future personalized medicine. Sequencing of the genes from a patient’s tumor is necessary to identify driver mutations and/or drug-resistance mutations. These mutations can be selective targeted with mRNA vaccines. The mRNA vaccines of choice can be different for individual cancer patients depending on the mutations identified.
CT antigens are promising targets for anticancer immunotherapies such as mRNA-LNP, TCR-T, and CAR-T. The clinical translation of CTAs is still limited, despite promising results at the preclinical stage. The underlying reasons may be attributed to their heterogeneous expression in tumors and the restricted expression of certain CTAs in normal tissues. Thus, the co-expression pattern and structural homology of CTA subfamily members should be considered when designing CTA-based therapies. Since CTAs do not have mutations, CTA-based vaccines generally show low immunogenicity. The identification of mutant CTAs is necessary for developing efficient anticancer immunotherapy. Inefficient delivery of CTAs and immunosuppressive TME makes it difficult to develop CTA-based vaccines for clinical trials. Nanomaterial-based cancer vaccines, such as LNPs, may enhance the delivery of multiple CTA antigens and immunogenicity.
Most TAAs are also expressed in normal tissue or originate as oncofetal antigens. Therefore, peripheral or thymic tolerance to TAAs often exists and results in weak immunogenic response. It will be necessary to identify immunomodulators that can regulate TAA-specific immunity.
Therapeutic anticancer vaccines require multiple doses. Therefore, safety standards are high for mRNA production [133]. mRNA cancer vaccines encode multiple tumor antigens, which raises safety concerns for mRNA cancer vaccines. To be effective as a therapeutic cancer vaccine, mRNA modifications are necessary. It has been shown that N1-methylpseudouridine base modification can improve mRNA translation efficiency [134].
To further improve the anticancer efficacy of mRNA vaccines, specific adjuvants, immune checkpoint inhibitors, T cell-activated monoclonal antibodies, modulation of the TME, or combination with radiation therapy or chemotherapy should be used to avoid immune escape and thus improve vaccine efficacy.
LNPs are primarily transported to the liver or kidney. One of the problems regarding RNA-based drugs is the difficulty of delivering such drugs to target organs and tissues, except for the liver. It is therefore necessary to develop RNA vaccines that can be delivered to various organs and tissues.
mRNA vaccines are promising therapeutic candidates for future cancer treatments, especially in combination with additional immunotherapies. Over 20 mRNA-based vaccines have undergone clinical trials for solid tumors, such as melanoma and non-small cell lung cancer. In these clinical trials, mRNA-based vaccines were mostly combined with immune checkpoint regulators, such as PD-1 and CTLA-4. Based on the successful results obtained from the combination of cancer vaccines with immune checkpoint inhibitors, combination therapies should be considered as prominent approaches for cancer treatments. Most mRNA vaccines are known to be well tolerated and usually do not induce injection site-specific immune reactions [135].
Virus-like particles (VLPs) are employed for therapeutic developments such as vaccines and drug carriers. VLPs can encapsulate and protect mRNAs against degradation and maintain their shapes against serum [136]. The VLPs display low toxicity and minimal hemolytic activity [136]. The transfection efficiency of VLPs is low compared with lipoplex agents [136]. In addition, VLPs can boost the production of mRNA-induced antibodies [137]. VLPs encapsulating mRNAs encoding TAAs can be employed for tumor eradication via enhancing antitumor immunity [138].
With increasing numbers of clinical studies, especially regarding personalized vaccines, there is a growing possibility of developing mRNA vaccines against different cancers. Regarding clinical research, further clinical trials for different tumors are required. The recent discovery and identification of new antigens have facilitated the development of personalized therapeutic mRNA cancer vaccines. Therefore, with the advancement of RNA therapy technology, the development of more diverse RNA-based drugs and drug delivery methods is expected in the future. Several clinical studies performed by BioNTech and Moderna have demonstrated potent anticancer immunity using personalized vaccines for the treatment of multiple solid tumors, initiating a new era of therapeutic oncology vaccines.
A large number of vaccines are discarded before their application owing to exposure to sub-optimum temperatures [139]. The limited thermostability and need for ultracold storage conditions are the major drawbacks of the currently used LNP-formulated mRNA vaccines, which hamper the distribution of these vaccines in low-resource regions [140]. Therefore, there is a need for the development of a thermostable vaccine with a long shelf life at ambient temperature. Vaccine efficacy, thermostability, and other important properties can be modulated through RNA sequence and structure, both of which can be optimized. Lyophilization, encapsulation, modification in liquid formulation, or introduction of mutations can be employed for preparing thermostable vaccines [139].
Author Contributions
K.S., H.J. and D.J. contributed to the original draft preparation, review, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Research Foundation grants, (2020R1A2C1006996, 2022R1F1A1060031, and 2017M3A9G7072417) and a grant from the BK21 Four Program.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Song, Y.; Zhang, Y. Research progress of neoantigens in gynecologic cancers. Int. Immunopharmacol. 2022, 112, 109236. [Google Scholar] [CrossRef]
- Zhang, Q.; Jia, Q.; Zhang, J.; Zhu, B. Neoantigens in precision cancer immunotherapy: From identification to clinical applications. Chin. Med. J. 2022, 135, 1285–1298. [Google Scholar] [CrossRef]
- Apavaloaei, A.; Hardy, M.P.; Thibault, P.; Perreault, C. The origin and immune recognition of tumor-specific antigens. Cancers 2020, 12, 2607. [Google Scholar] [CrossRef]
- Gubin, M.M.; Artyomov, M.N.; Mardis, E.R.; Schreiber, R.D. Tumor neoantigens: Building a framework for personalized cancer immunotherapy. J. Clin. Investig. 2015, 125, 3413–3421. [Google Scholar] [CrossRef]
- Wu, Y.; Sang, M.; Liu, F.; Zhang, J.; Li, W.; Li, Z.; Gu, L.; Zheng, Y.; Li, J.; Shan, B. Epigenetic modulation combined with PD-1/PD-L1 blockade enhances immunotherapy based on MAGE-A11 antigen-specific CD8+T cells against esophageal carcinoma. Carcinogenesis 2020, 41, 894–903. [Google Scholar] [CrossRef]
- Li, Z.; Guo, P.; Guo, P.; Dong, K.; Liu, F.; Wu, Y.; Li, J.; Shan, B.; Sang, M. 5-aza-2′-deoxycytidine (DAC) treatment induces the MAGE-A10 expression and improves the cytotoxicity of MAGE-A10-specific CTLs in lung cancer cells. Transl. Cancer Res. 2020, 9, 1235–1245. [Google Scholar] [CrossRef]
- Fujii, S.I.; Yamasaki, S.; Hanada, K.; Ueda, S.; Kawamura, M.; Shimizu, K. Cancer immunotherapy using artificial adjuvant vector cells to deliver NY-ESO-1 antigen to dendritic cells in situ. Cancer Sci. 2022, 113, 864–874. [Google Scholar] [CrossRef]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef]
- Callebaut, A.; Bruggeman, Y.; Zamit, C.; Sodré, F.M.C.; Irla, M.; Mathieu, C.; Buitinga, M.; Overbergh, L. Aberrant expression of transglutaminase 2 in pancreas and thymus of NOD mice underscores the importance of deamidation in neoantigen generation. Front. Endocrinol. 2022, 3, 908248. [Google Scholar] [CrossRef]
- Kim, Y.; Park, D.; Kim, H.; Choi, M.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeoung, D. miR-200b and cancer/testis antigen CAGE form a feedback loop to regulate the invasion and tumorigenic and angiogenic responses of a cancer cell line to microtubule-targeting drugs. J. Biol. Chem. 2013, 288, 36502–36518. [Google Scholar] [CrossRef]
- Sasahira, T.; Kurihara-Shimomura, M.; Nishiguchi, Y.; Shimomura, H.; Kirita, T. Sushi Repeat Containing Protein X-linked 2 Is a Downstream Signal of LEM Domain Containing 1 and Acts as a Tumor-Promoting Factor in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2020, 21, 3655. [Google Scholar] [CrossRef]
- Yang, P.; Qiao, Y.; Liao, H.; Huang, Y.; Meng, M.; Chen, Y.; Zhou, Q. The Cancer/Testis Antigen CT45A1 Promotes Transcription of Oncogenic Sulfatase-2 Gene in Breast Cancer Cells and Is Sensible Targets for Cancer Therapy. J. Breast Cancer 2023, 26, 168–185. [Google Scholar] [CrossRef]
- Zeng, Y.; He, Y.; Yang, F.; Mooney, S.M.; Getzenberg, R.H.; Orban, J.; Kulkarni, P. The cancer/testis antigen prostate-associated gene 4 (PAGE4) is a highly intrinsically disordered protein. J. Biol. Chem. 2011, 286, 13985–13994. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, W.; Wang, Y.; Min, Q.; Zhang, H.; Dong, D.; Zhan, Q. MAGE-C3 promotes cancer metastasis by inducing epithelial-mesenchymal transition and immunosuppression in esophageal squamous cell carcinoma. Cancer Commun. 2021, 41, 1354–1372. [Google Scholar] [CrossRef]
- Mizushima, E.; Tsukahara, T.; Emori, M.; Murata, K.; Akamatsu, A.; Shibayama, Y.; Hamada, S.; Watanabe, Y.; Kaya, M.; Hirohashi, Y.; et al. Osteosarcoma-initiating cells show high aerobic glycolysis and attenuation of oxidative phosphorylation mediated by LIN28B. Cancer Sci. 2020, 111, 36–46. [Google Scholar] [CrossRef]
- Suzuki, I.; Yoshida, S.; Tabu, K.; Kusunoki, S.; Matsumura, Y.; Izumi, H.; Asanoma, K.; Yagi, H.; Onoyama, I.; Sonoda, K.; et al. YBX2 and cancer testis antigen 45 contribute to stemness, chemoresistance and a high degree of malignancy in human endometrial cancer. Sci. Rep. 2021, 11, 4220. [Google Scholar] [CrossRef]
- Li, W.; Wang, F.; Shi, J.; Feng, Q.; Chen, Y.; Qi, X.; Wang, C.; Lu, H.; Lu, Z.; Jia, X.; et al. Sperm associated antigen 9 promotes oncogenic KSHV-encoded interferon regulatory factor-induced cellular transformation and angiogenesis by activating the JNK/VEGFA pathway. PLoS Pathog. 2020, 16, e1008730. [Google Scholar] [CrossRef]
- Lv, C.; Fu, S.; Dong, Q.; Yu, Z.; Zhang, G.; Kong, C.; Fu, C.; Zeng, Y. PAGE4 promotes prostate cancer cells survive under oxidative stress through modulating MAPK/JNK/ERK pathway. J. Exp. Clin. Cancer Res. 2019, 38, 24. [Google Scholar] [CrossRef]
- Yang, P.; Huo, Z.; Liao, H.; Zhou, Q. Cancer/testis antigens trigger epithelial-mesenchymal transition and genesis of cancer stem-like cells. Curr. Pharm. Des. 2015, 21, 1292–1300. [Google Scholar] [CrossRef]
- Qi, Y.; Dong, S.S.; He, Y.L.; Liu, Z.H.; Huang, Y.L.; Wang, N.; Zhang, Z.; Li, Z.; Shi, M.E.T.H.T.M.; Feng, X.; et al. SYT-SSX1 enhances the invasiveness and maintains stem-like cell properties in synovial sarcoma via induction of TGF-β1/Smad signaling. BMC Cancer 2022, 22, 166. [Google Scholar] [CrossRef]
- Ren, H.; Xu, Z.; Guo, W.; Deng, Z.; Yu, X. Rab3IP interacts with SSX2 and enhances the invasiveness of gastric cancer cells. Biochem. Biophys. Res. Commun. 2018, 503, 2563–2568. [Google Scholar] [CrossRef]
- Jiang, S.; Liu, X.; Li, D.; Yan, M.; Ju, C.; Sun, J.; Jiang, F. Study on Attenuating Angiogenesis and Epithelial-Mesenchymal Transition (EMT) of Non-Small Cell Lung Carcinoma (NSCLC) by Regulating MAGEC2. Technol. Cancer Res. Treat. 2018, 17, 1533033818797587. [Google Scholar] [CrossRef]
- Gao, X.; Chen, G.; Cai, H.; Wang, X.; Song, K.; Liu, L.; Qiu, T.; He, Y. Aberrantly enhanced melanoma-associated antigen (MAGE)-A3 expression facilitates cervical cancer cell proliferation and metastasis via actuating Wnt signaling pathway. Biomed. Pharmacother. 2020, 122, 109710. [Google Scholar] [CrossRef]
- Koirala, S.; Klein, J.; Zheng, Y.; Glenn, N.O.; Eisemann, T.; Fon Tacer, K.; Miller, D.J.; Kulak, O.; Lu, M.; Finkelstein, D.B.; et al. Tissue-Specific Regulation of the Wnt/β-Catenin Pathway by PAGE4 Inhibition of Tankyrase. Cell Rep. 2020, 32, 107922. [Google Scholar] [CrossRef]
- Wen, M.; Ren, H.; Zhang, S.; Li, T.; Zhang, J.; Ren, P. CT45A1 promotes the metastasis of osteosarcoma cells in vitro and in vivo through β-catenin. Cell Death Dis. 2021, 12, 650. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, Y.; Gong, J.; Rao, L.; Wu, Z.; Nie, T.; Shi, D.; Zhang, L. High expression of MAGE-A9 contributes to stemness and malignancy of human hepatocellular carcinoma. Int. J. Oncol. 2018, 52, 219–230. [Google Scholar] [CrossRef]
- Meng, X.; Liu, Z.; Zhang, L.; He, Y. Plac1 Remodels the Tumor Immune Evasion Microenvironment and Predicts Therapeutic Response in Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2022, 12, 919436. [Google Scholar] [CrossRef]
- Kim, Y.; Park, H.; Park, D.; Lee, Y.S.; Choe, J.; Hahn, J.H.; Lee, H.; Kim, Y.M.; Jeoung, D. Cancer/testis antigen CAGE exerts negative regulation on p53 expression through HDAC2 and confers resistance to anti-cancer drugs. J. Biol. Chem. 2010, 285, 25957–25968. [Google Scholar] [CrossRef]
- Naimi, A.; Safaei, S.; Entezari, A.; Solali, S.; Hassanzadeh, A. Knockdown of Enhancer of Zeste Homolog 2 Affects mRNA Expression of Genes Involved in the Induction of Resistance to Apoptosis in MOLT-4 Cells. Anticancer Agents Med. Chem. 2020, 20, 571–579. [Google Scholar] [CrossRef]
- Shafer, J.A.; Cruz, C.R.; Leen, A.M.; Ku, S.; Lu, A.; Rousseau, A.; Heslop, H.E.; Rooney, C.M.; Bollard, C.M.; Foster, A.E. Antigen-specific cytotoxic T lymphocytes can target chemoresistant side-population tumor cells in Hodgkin lymphoma. Leuk. Lymphoma 2010, 51, 870–880. [Google Scholar] [CrossRef]
- Shuvalov, O.; Kizenko, A.; Petukhov, A.; Aksenov, N.; Fedorova, O.; Vorobev, M.; Daks, A.; Barlev, N. Cancer-testis antigens, semenogelins 1 and 2, exhibit different anti-proliferative effects on human lung adenocarcinoma cells. Cell Death Discov. 2020, 6, 108. [Google Scholar] [CrossRef]
- Tsang, Y.H.; Wang, Y.; Kong, K.; Grzeskowiak, C.; Zagorodna, O.; Dogruluk, T.; Lu, H.; Villafane, N.; Bhavana, V.H.; Moreno, D.; et al. Differential expression of MAGEA6 toggles autophagy to promote pancreatic cancer progression. eLife 2020, 9, e48963. [Google Scholar] [CrossRef]
- Fon Tacer, K.; Montoya, M.C.; Oatley, M.J.; Lord, T.; Oatley, J.M.; Klein, J.; Ravichandran, R.; Tillman, H.; Kim, M.; Connelly, J.P.; et al. MAGE cancer-testis antigens protect the mammalian germline under environmental stress. Sci. Adv. 2019, 5, eaav4832. [Google Scholar] [CrossRef]
- Wang, Y.; Jiao, Y.; Ding, C.M.; Sun, W.Z. The role of autoantibody detection in the diagnosis and staging of lung cancer. Ann. Transl. Med. 2021, 9, 1673. [Google Scholar] [CrossRef]
- Dasgeb, B.; Mehregan, D.; Ring, C.; Nartker, N.; Brownell, I. Cancer-testis antigens as biomarkers for Merkel cell carcinoma: Pitfalls and opportunities. J. Cutan. Pathol. 2019, 46, 748–752. [Google Scholar] [CrossRef]
- Schutt, C.A.; Mirandola, L.; Figueroa, J.A.; Nguyen, D.D.; Cordero, J.; Bumm, K.; Judson, B.L.; Chiriva-Internati, M. The cancer-testis antigen, sperm protein 17, a new biomarker and immunological target in head and neck squamous cell carcinoma. Oncotarget 2017, 8, 100280–100287. [Google Scholar] [CrossRef]
- Luo, W.Z.; Li, X.; Wu, X.X.; Shang, Y.W.; Meng, D.H.; Chen, Y.L.; Zhang, Q.S. MAGED4B is a Poor Prognostic Marker of Stomach Adenocarcinoma and a Potential Therapeutic Target for Stomach Adenocarcinoma Tumorigenesis. Int. J. Gen. Med. 2023, 16, 1681–1693. [Google Scholar] [CrossRef]
- Zhang, J.; Sang, M.; Gu, L.; Liu, F.; Li, W.; Yin, D.; Wu, Y.; Liu, S.; Huang, W.; Shan, B. Zebularine Treatment Induces MAGE-A11 Expression and Improves CTL Cytotoxicity Using a Novel Identified HLA-A2-restricted MAGE-A11 Peptide. J. Immunother. 2017, 40, 211–220. [Google Scholar] [CrossRef]
- Wu, J.; Yao, F.; Li, Y.; Zhao, Z.; Liu, J.; Xu, T.; Chai, J.; Yang, Y.; Song, J.; Tian, C.; et al. The cancer-testis antigen FBXO39 predicts poor prognosis and is associated with stemness and aggressiveness in glioma. Pathol. Res. Pract. 2022, 239, 154168. [Google Scholar] [CrossRef]
- Broggi, G.; Failla, M.; Russo, A.; Longo, A.; Palicelli, A.; Zanelli, M.; Lombardo, C.; Loreto, C.; Merolla, F.; Di Crescenzo, R.M.; et al. Immunohistochemical expression of PRAME is a marker of poor prognosis in uveal melanoma: A clinico-pathologic and immunohistochemical study on a series of 85 cases. Pathol. Res. Pract. 2023, 247, 154543. [Google Scholar] [CrossRef]
- Hedrich, V.; Breitenecker, K.; Ortmayr, G.; Pupp, F.; Huber, H.; Chen, D.; Sahoo, S.; Jolly, M.K.; Mikulits, W. PRAME Is a Novel Target of Tumor-Intrinsic Gas6/Axl Activation and Promotes Cancer Cell Invasion in Hepatocellular Carcinoma. Cancers 2023, 15, 2415. [Google Scholar]
- Ansari, S.; Nikpour, P. Identification of Cancer/Testis Antigens Related to Gastric Cancer Prognosis Based on Co-Expression Network and Integrated Transcriptome Analyses. Adv. Biomed. Res. 2023, 12, 52. [Google Scholar]
- Burn, O.K.; Farrand, K.; Pritchard, T.; Draper, S.; Tang, C.W.; Mooney, A.H.; Schmidt, A.J.; Yang, S.H.; Williams, G.M.; Brimble, M.A.; et al. Glycolipid-peptide conjugate vaccines elicit CD8+ T-cell responses and prevent breast cancer metastasis. Clin. Transl. Immunol. 2022, 11, e1401. [Google Scholar] [CrossRef]
- Alsalloum, A.; Shevchenko, J.A.; Sennikov, S. The Melanoma-Associated Antigen Family A (MAGE-A): A Promising Target for Cancer Immunotherapy? Cancers 2023, 15, 1779. [Google Scholar] [CrossRef]
- Pan, J.; Zeng, W.; Jia, J.; Shi, Y.; Wang, D.; Dong, J.; Fang, Z.; He, J.; Yang, X.; Zhang, R.; et al. A Novel Therapeutic Tumor Vaccine Targeting MUC1 in Combination with PD-L1 Elicits Specific Anti-Tumor Immunity in Mice. Vaccines 2022, 10, 1092. [Google Scholar] [CrossRef]
- Nicolás-Morales, M.L.; Luisa-Sanjuan, A.; Gutiérrez-Torres, M.; Vences-Velázquez, A.; Ortuño-Pineda, C.; Espinoza-Rojo, M.; Navarro-Tito, N.; Cortés-Sarabia, K. Peptide-Based Vaccines in Clinical Phases and New Potential Therapeutic Targets as a New Approach for Breast Cancer: A Review. Vaccines 2022, 10, 1249. [Google Scholar] [CrossRef]
- Leko, V.; Rosenberg, S.A. Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell 2020, 38, 454–472. [Google Scholar]
- Su, H.; Imai, K.; Jia, W.; Li, Z.; DiCioccio, R.A.; Serody, J.S.; Poe, J.C.; Chen, B.J.; Doan, P.L.; Sarantopoulos, S. Alphavirus Replicon Particle Vaccine Breaks B Cell Tolerance and Rapidly Induces IgG to Murine Hematolymphoid Tumor Associated Antigens. Front. Immunol. 2022, 13, 865486. [Google Scholar]
- Liu, X.; Li, J.; Liu, Y.; Ding, J.; Tong, Z.; Liu, Y.; Zhou, Y.; Liu, Y. Calreticulin acts as an adjuvant to promote dendritic cell maturation and enhances antigen-specific cytotoxic T lymphocyte responses against non-small cell lung cancer cells. Cell. Immunol. 2016, 300, 46–53. [Google Scholar] [CrossRef]
- Patel, S.P.; Petroni, G.R.; Roszik, J.; Olson, W.C.; Wages, N.A.; Chianese-Bullock, K.A.; Smolkin, M.; Varhegyi, N.; Gaughan, E.; Smith, K.T.; et al. Phase I/II trial of a long peptide vaccine (LPV7) plus toll-like receptor (TLR) agonists with or without incomplete Freund’s adjuvant (IFA) for resected high-risk melanoma. J. Immunother. Cancer 2021, 9, e003220. [Google Scholar] [CrossRef]
- Shenderov, E.; Kandasamy, M.; Gileadi, U.; Chen, J.; Shepherd, D.; Gibbs, J.; Prota, G.; Silk, J.D.; Yewdell, J.W.; Cerundolo, V. Generation and characterization of HLA-A2 transgenic mice expressing the human TCR 1G4 specific for the HLA-A2 restricted NY-ESO-1157-165 tumor-specific peptide. J. Immunother. Cancer 2021, 9, e002544. [Google Scholar] [CrossRef]
- Dillon, P.M.; Petroni, G.R.; Smolkin, M.E.; Brenin, D.R.; Chianese-Bullock, K.A.; Smith, K.T.; Olson, W.C.; Fanous, I.S.; Nail, C.J.; Brenin, C.M.; et al. A pilot study of the immunogenicity of a 9-peptide breast cancer vaccine plus poly-ICLC in early stage breast cancer. J. Immunother. Cancer 2017, 5, 92. [Google Scholar] [CrossRef]
- Zhong, G.; Zhao, W.; Li, Y.; Jin, G.; Zeng, W.; Yu, C.; Zhou, J.; Yu, L. MAGEA1 and hTERT Peptide Treatment Improves the Potency of The Dendritic Cell- Cytotoxic T Lymphocytes (DC-CTL) Immunotherapy in DAC Treated Acute Myeloid Leukemia. J. Cancer 2022, 13, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- De Vries, I.J.; Bernsen, M.R.; Lesterhuis, W.J.; Scharenborg, N.M.; Strijk, S.P.; Gerritsen, M.J.; Ruiter, D.J.; Figdor, C.G.; Punt, C.J.; Adema, G.J. Immunomonitoring tumor-specific T cells in delayed-type hypersensitivity skin biopsies after dendritic cell vaccination correlates with clinical outcome. J. Clin. Oncol. 2005, 23, 5779–5787. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Parker, L.L.; Lu, T.; Zheng, Z.; Toomey, M.A.; White, D.E.; Yao, X.; Li, Y.F.; Robbins, P.F.; Feldman, S.A.; et al. Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II–Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J. Clin. Oncol. 2017, 35, 3322–3329. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, J.P.; Crowley, D.J.; Wiedermann, G.E.; Quinn, L.L.; Crossland, K.L.; Tunbridge, H.M.; Cornforth, T.V.; Barnes, C.S.; Ahmed, T.; Howe, K.; et al. Preclinical evaluation of an affinity-enhanced MAGE-A4-specific T-cell receptor for adoptive T-cell therapy. Oncoimmunology 2019, 9, 1682381. [Google Scholar] [CrossRef]
- Blumenschein, G.R.; Devarakonda, S.; Johnson, M.; Moreno, V.; Gainor, J.; Edelman, M.J.; Heymach, J.V.; Govindan, R.; Bachier, C.; de Spéville, B.D.; et al. Phase I clinical trial evaluating the safety and efficacy of ADP-A2M10 SPEAR T cells in patients with MAGE-A10+ advanced non-small cell lung cancer. J. Immunother. Cancer 2022, 10, e003581. [Google Scholar] [CrossRef]
- Mao, Y.; Fan, W.; Hu, H.; Zhang, L.; Michel, J.; Wu, Y.; Wang, J.; Jia, L.; Tang, X.; Xu, L.; et al. MAGE-A1 in lung adenocarcinoma as a promising target of chimeric antigen receptor T cells. J. Hematol. Oncol. 2019, 12, 106. [Google Scholar] [CrossRef]
- Khawar, M.B.; Ge, F.; Afzal, A.; Sun, H. From barriers to novel strategies: Smarter CAR T therapy hits hard to tumors. Front. Immunol. 2023, 14, 1203230. [Google Scholar] [CrossRef]
- Krebs, S.; Chow, K.K.; Yi, Z.; Rodriguez-Cruz, T.; Hegde, M.; Gerken, C.; Ahmed, N.; Gottschalk, S. T cells redirected to interleukin-13Rα2 with interleukin-13 mutein--chimeric antigen receptors have anti-glioma activity but also recognize interleukin-13Rα1. Cytotherapy 2014, 16, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Ishihara, M.; Kiyota, N.; Yakushijin, K.; Takada, K.; Kobayashi, S.; Ikeda, H.; Endo, M.; Kato, K.; Kitano, S.; et al. Chimeric antigen receptor T-cell therapy targeting a MAGE A4 peptide and HLA-A*02:01 complex for unresectable advanced or recurrent solid cancer: Protocol for a multi-institutional phase 1 clinical trial. BMJ Open 2022, 12, e065109. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, Y.; Xiong, W.; Yin, B.; Huang, Y.; Chu, J.; Xing, C.; Qian, C.; Du, Y.; Duan, T.; et al. Development of a TCR-like antibody and chimeric antigen receptor against NY-ESO-1/HLA-A2 for cancer immunotherapy. J. Immunother. Cancer 2022, 10, e004035. [Google Scholar] [CrossRef]
- Raskin, S.; Van Pelt, S.; Toner, K.; Balakrishnan, P.B.; Dave, H.; Bollard, C.M.; Yvon, E. Novel TCR-like CAR-T cells targeting an HLA∗0201-restricted SSX2 epitope display strong activity against acute myeloid leukemia. Mol. Ther. Methods Clin. Dev. 2021, 23, 296–306. [Google Scholar] [CrossRef]
- Akbari, B.; Soltantoyeh, T.; Shahosseini, Z.; Jadidi-Niaragh, F.; Hadjati, J.; Brown, C.E.; Mirzaei, H.R. PGE2-EP2/EP4 signaling elicits mesoCAR T cell immunosuppression in pancreatic cancer. Front. Immunol. 2023, 14, 1209572. [Google Scholar] [CrossRef]
- Cowan, A.J.; Pont, M.J.; Sather, B.D.; Turtle, C.J.; Till, B.G.; Libby, E.N., 3rd; Coffey, D.G.; Tuazon, S.A.; Wood, B.; Gooley, T.; et al. γ-Secretase inhibitor in combination with BCMA chimeric antigen receptor T-cell immunotherapy for individuals with relapsed or refractory multiple myeloma: A phase 1, first-in-human trial. Lancet Oncol. 2023, 24, 811–822. [Google Scholar] [CrossRef]
- Pensato, U.; Guarino, M.; Muccioli, L. The role of neurologists in the era of cancer immunotherapy: Focus on CAR T-cell therapy and immune checkpoint inhibitors. Front. Neurol. 2022, 13, 936141. [Google Scholar] [CrossRef]
- Kashida, S.; Wang, D.O.; Saito, H.; Gueroui, Z. Nanoparticle-based local translation reveals mRNA as a translation-coupled scaffold with anchoring function. Proc. Natl. Acad. Sci. USA 2019, 116, 13346–13351. [Google Scholar] [CrossRef]
- Mancuso, K.; Zamagni, E.; Solli, V.; Gabrielli, L.; Leone, M.; Pantani, L.; Rocchi, S.; Rizzello, I.; Tacchetti, P.; Ghibellini, S.; et al. Long term follow-up of humoral and cellular response to mRNA-based vaccines for SARS-CoV-2 in patients with active multiple myeloma. Front. Oncol. 2023, 13, 1208741. [Google Scholar] [CrossRef]
- Duan, L.J.; Wang, Q.; Zhang, C.; Yang, D.X.; Zhang, X.Y. Potentialities and Challenges of mRNA Vaccine in Cancer Immunotherapy. Front. Immunol. 2022, 13, 923647. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H. Protein-Based Systems for Translational Regulation of Synthetic mRNAs in Mammalian Cells. Life 2021, 11, 1192. [Google Scholar] [CrossRef]
- Hirschberger, K.; Jarzebinska, A.; Kessel, E.; Kretzschmann, V.; Aneja, M.K.; Dohmen, C.; Herrmann-Janson, A.; Wagner, E.; Plank, C.; Rudolph, C. Exploring Cytotoxic mRNAs as a Novel Class of Anti-cancer Biotherapeutics. Mol. Ther. Methods Clin. Dev. 2017, 8, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Diamantopoulos, P.T.; Kontandreopoulou, C.N.; Gkoufa, A.; Solomou, E.; Anastasopoulou, A.; Palli, E.; Kouzis, P.; Bouros, S.; Samarkos, M.; Magiorkinis, G.; et al. Immunogenicity and Safety of the BNT162b2 mRNA COVID-19 Vaccine in Patients with Melanoma Treated with Immunotherapy. Cancers 2022, 14, 3791. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Chen, X.; Fang, B.; Ping, Y.; Qin, G.; Yue, D.; Li, F.; Yang, S.; Zhang, Y. Decitabine enhances tumor recognition by T cells through upregulating the MAGE-A3 expression in esophageal carcinoma. Biomed. Pharmacother. 2019, 112, 108632. [Google Scholar] [CrossRef] [PubMed]
- Jian, W.; Li, X.; Kang, J.; Lei, Y.; Bai, Y.; Xue, Y. Antitumor effect of recombinant Mycobacterium smegmatis expressing MAGEA3 and SSX2 fusion proteins. Exp. Ther. Med. 2018, 16, 2160–2166. [Google Scholar] [CrossRef]
- Duperret, E.K.; Liu, S.; Paik, M.; Trautz, A.; Stoltz, R.; Liu, X.; Ze, K.; Perales-Puchalt, A.; Reed, C.; Yan, J.; et al. A Designer Cross-reactive DNA Immunotherapeutic Vaccine that Targets Multiple MAGE-A Family Members Simultaneously for Cancer Therapy. Clin. Cancer Res. 2018, 24, 6015–6027. [Google Scholar] [CrossRef]
- Gu, Y.; Yang, J.; He, C.; Zhao, T.; Lu, R.; Liu, J.; Mo, X.; Wen, F.; Shi, H. Incorporation of a Toll-like receptor 2/6 agonist potentiates mRNA vaccines against cancer and infectious diseases. Signal Transduct. Target Ther. 2023, 8, 273. [Google Scholar]
- Touray, B.J.B.; Hanafy, M.; Phanse, Y.; Hildebrand, R.; Talaat, A.M. Protective RNA nanovaccines against Mycobacterium avium subspecies hominissuis. Front. Immunol. 2023, 14, 1188754. [Google Scholar] [CrossRef]
- Syama, K.; Jakubek, Z.J.; Chen, S.; Zaifman, J.; Tam, Y.Y.C.; Zou, S. Development of lipid nanoparticles and liposomes reference materials (II): Cytotoxic profiles. Sci. Rep. 2022, 12, 18071. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Qureischi, M.; Mohr, J.; Arellano-Viera, E.; Knudsen, S.E.; Vohidov, F.; Garitano-Trojaola, A. mRNA-based therapies: Preclinical and clinical applications. Int. Rev. Cell. Mol. Biol. 2022, 372, 1–54. [Google Scholar] [PubMed]
- Liu, C.; Shi, Q.; Huang, X.; Koo, S.; Kong, N.; Tao, W. mRNA-based cancer therapeutics. Nat. Rev. Cancer 2023. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Cao, H.; Luan, N.; Wang, Y.; Hu, J.; Liu, C. Comparison of the Immune Effects of an mRNA Vaccine and a Subunit Vaccine against Herpes Zoster Administered by Different Injection Methods. Vaccines 2023, 11, 1003. [Google Scholar] [CrossRef]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef]
- Oladipo, E.K.; Adeniyi, M.O.; Ogunlowo, M.T.; Irewolede, B.A.; Adekanola, V.O.; Oluseyi, G.S.; Omilola, J.A.; Udoh, A.F.; Olufemi, S.E.; Adediran, D.A.; et al. Bioinformatics Designing and Molecular Modelling of a Universal mRNA Vaccine for SARS-CoV-2 Infection. Vaccines 2022, 10, 2107. [Google Scholar] [CrossRef]
- Egri, N.; Calderón, H.; Martinez, R.; Vazquez, M.; Gómez-Caverzaschi, V.; Pascal, M.; Araújo, O.; Juan, M.; González-Navarro, E.A.; Hernández-Rodríguez, J. Cellular and humoral responses after second and third SARS-CoV-2 vaccinations in patients with autoimmune diseases treated with rituximab: Specific T cell immunity remains longer and plays a protective role against SARS-CoV-2 reinfections. Front. Immunol. 2023, 14, 1146841. [Google Scholar] [CrossRef]
- Poveda, C.; Leão, A.C.; Mancino, C.; Taraballi, F.; Chen, Y.L.; Adhikari, R.; Villar, M.J.; Kundu, R.; Nguyen, D.M.; Versteeg, L.; et al. Heterologous mRNA-protein vaccination with Tc24 induces a robust cellular immune response against Trypanosoma cruzi, characterized by an increased level of polyfunctional CD8+ T-cells. Curr. Res. Immunol. 2023, 4, 100066. [Google Scholar] [CrossRef]
- Naota, H.; Miyahara, Y.; Okumura, S.; Kuzushima, K.; Akatsuka, Y.; Hiasa, A.; Kitano, S.; Takahashi, T.; Yuta, A.; Majima, Y.; et al. Generation of peptide-specific CD8+ T cells by phytohemagglutinin-stimulated antigen-mRNA-transduced CD4+ T cells. J. Immunol. Methods 2006, 314, 54–66. [Google Scholar] [CrossRef]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Pawelec, G.; Hoerr, I.; Rammensee, H.G.; Garbe, C. Direct injection of protamine-protected mRNA: Results of a phase 1/2 vaccination trial in metastatic melanoma patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef] [PubMed]
- Wilgenhof, S.; Corthals, J.; Van Nuffel, A.M.; Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Neyns, B. Long-term clinical outcome of melanoma patients treated with messenger RNA-electroporated dendritic cell therapy following complete resection of metastases. Cancer Immunol. Immunother. 2015, 64, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Westdorp, H.; Creemers, J.H.A.; van Oort, I.M.; Schreibelt, G.; Gorris, M.A.J.; Mehra, N.; Simons, M.; de Goede, A.L.; van Rossum, M.M.; Croockewit, A.J.; et al. Blood-derived dendritic cell vaccinations induce immune responses that correlate with clinical outcome in patients with chemo-naive castration-resistant prostate cancer. J. Immunother. Cancer 2019, 7, 302. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.J.; Sharma, S.; Rangesa, M.; DeWolf, S.; Elhanati, Y.; Perica, K.; Young, J.W. Langerhans dendritic cell vaccine bearing mRNA-encoded tumor antigens induces antimyeloma immunity after autotransplant. Blood Adv. 2022, 6, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, M.; Schröder, A.; Scheel, B.; Hong, H.S.; Muth, A.; von Boehmer, L.; Zippelius, A.; Mayer, F.; Reck, M.; Atanackovic, D.; et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol. Immunother. 2019, 68, 799–812. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Gustà, M.F.; Edel, M.J.; Salazar, V.A.; Alvarez-Palomo, B.; Juan, M.; Broggini, M.; Damia, G.; Bigini, P.; Corbelli, A.; Fiordaliso, F.; et al. Exploiting endocytosis for transfection of mRNA for cytoplasmatic delivery using cationic gold nanoparticles. Front. Immunol. 2023, 14, 1128582. [Google Scholar] [CrossRef]
- Geng, L.; Kato, N.; Kodama, Y.; Mukai, H.; Kawakami, S. Influence of lipid composition of messenger RNA-loaded lipid nanoparticles on the protein expression via intratracheal administration in mice. Int. J. Pharm. 2023, 637, 122896. [Google Scholar] [CrossRef]
- Maruf, A.; Milewska, M.; Lalik, A.; Student, S.; Wandzik, I. A Simple Synthesis of Reduction-Responsive Acrylamide-Type Nanogels for miRNA Delivery. Molecules 2023, 28, 761. [Google Scholar] [CrossRef]
- Semple, S.L.; Au, S.K.W.; Jacob, R.A.; Mossman, K.L.; DeWitte-Orr, S.J. Discovery and Use of Long dsRNA Mediated RNA Interference to Stimulate Antiviral Protection in Interferon Competent Mammalian Cells. Front. Immunol. 2022, 13, 859749. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Duan, L.; Lu, J.; Xia, J. Engineering exosomes for targeted drug delivery. Theranostics 2021, 11, 3183–3195. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Shim, K.; Jeoung, D. Exosomes: Diagnostic and Therapeutic Implications in Cancer. Pharmaceutics 2023, 15, 1465. [Google Scholar] [CrossRef] [PubMed]
- Si, M.Y.; Rao, D.Y.; Xia, Y.; Sang, C.P.; Mao, K.Y.; Liu, X.J.; Zhang, Z.X.; Tang, Z.X. Role of exosomal noncoding RNA in esophageal carcinoma. Front. Oncol. 2023, 13, 1126890. [Google Scholar] [CrossRef]
- Alipoor, S.D.; Chang, H. Exosomal miRNAs in the Tumor Microenvironment of Multiple Myeloma. Cells 2023, 12, 1030. [Google Scholar] [CrossRef]
- Ni, Q.; Zhang, H.; Shi, X.; Li, X. Exosomal lncRNA HCG18 contributes to cholangiocarcinoma growth and metastasis through mediating miR-424-5p/SOX9 axis through PI3K/AKT pathway. Cancer Gene Ther. 2023, 30, 582–595. [Google Scholar] [CrossRef]
- Jiang, X.; Xu, Y.; Liu, R.; Guo, S. Exosomal lincROR Promotes Docetaxel Resistance in Prostate Cancer through a β-catenin/HIF1α Positive Feedback Loop. Mol. Cancer Res. 2023, 21, 472–482. [Google Scholar] [CrossRef]
- Chen, J.; Song, Y.; Miao, F.; Chen, G.; Zhu, Y.; Wu, N.; Pang, L.; Chen, Z.; Chen, X. PDL1-positive exosomes suppress antitumor immunity by inducing tumor-specific CD8+ T cell exhaustion during metastasis. Cancer Sci. 2021, 112, 3437–3454. [Google Scholar] [CrossRef]
- Feng, J.; Xu, B.; Dai, C.; Wang, Y.; Xie, G.; Yang, W.; Zhang, B.; Li, X.; Wang, J. Macrophage-derived exosomal miR-342-3p promotes the progression of renal cell carcinoma through the NEDD4L/CEP55 axis. Oncol. Res. 2022, 29, 331–349. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, Y.; Liu, J.; Fu, H.; Yang, Q.; Song, W.; Li, Y. Exosomes from PYCR1 knockdown bone marrow mesenchymal stem inhibits aerobic glycolysis and the growth of bladder cancer cells via regulation of the EGFR/PI3K/AKT pathway. Int. J. Oncol. 2023, 63, 84. [Google Scholar] [CrossRef]
- Smyth, L.A.; Ratnasothy, K.; Tsang, J.Y.; Boardman, D.; Warley, A.; Lechler, R.; Lombardi, G. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur. J. Immunol. 2013, 43, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, C.; Li, Z.; Ai, M.; Wang, B.; Du, K.; Liu, W.; Wang, H.; Yu, P.; Chen, C.; et al. Exosomal B7-H4 from irradiated glioblastoma cells contributes to increase FoxP3 expression of differentiating Th1 cells and promotes tumor growth. Redox Biol. 2022, 56, 102454. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Xin, H.; Zhou, Z.; Hu, Z.; Sun, R.; Yao, N.; Sun, Q.; Borjigin, U.; Wu, X.; Fan, J.; et al. Tumor-derived exosomes induce immunosuppressive macrophages to foster intrahepatic cholangiocarcinoma progression. Hepatology 2022, 76, 982–999. [Google Scholar] [CrossRef]
- Guan, H.; Mao, L.; Wang, J.; Wang, S.; Yang, S.; Wu, H.; Sun, W.; Chen, Z.; Chen, M. Exosomal RNF157 mRNA from prostate cancer cells contributes to M2 macrophage polarization through destabilizing HDAC1. Front. Oncol. 2022, 12, 1021270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, M.; Han, C.; Wen, Z.; Meng, X.; Qi, D.; Wang, N.; Du, H.; Wang, J.; Lu, L.; et al. Intraduodenal Delivery of Exosome-Loaded SARS-CoV-2 RBD mRNA Induces a Neutralizing Antibody Response in Mice. Vaccines 2023, 11, 673. [Google Scholar] [CrossRef]
- Haltom, A.R.; Hassen, W.E.; Hensel, J.; Kim, J.; Sugimoto, H.; Li, B.; McAndrews, K.M.; Conner, M.R.; Kirtley, M.L.; Luo, X.; et al. Engineered exosomes targeting MYC reverse the proneural-mesenchymal transition and extend survival of glioblastoma. Extracell. Vesicle 2022, 1, 100014. [Google Scholar] [CrossRef]
- Avgoulas, D.I.; Tasioulis, K.S.; Papi, R.M.; Pantazaki, A.A. Therapeutic and Diagnostic Potential of Exosomes as Drug Delivery Systems in Brain Cancer. Pharmaceutics 2023, 15, 1439. [Google Scholar] [CrossRef]
- Moholkar, D.N.; Kandimalla, R.; Gupta, R.C.; Aqil, F. Advances in lipid-based carriers for cancer therapeutics: Liposomes, exosomes and hybrid exosomes. Cancer Lett. 2023, 565, 216220. [Google Scholar] [CrossRef]
- Feng, X.; Iliuk, A.; Zhang, X.; Jia, S.; Shen, A.; Zhang, W.; Hu, L.; Tao, W.A. Supramolecular Exosome Array for Efficient Capture and In Situ Detection of Protein Biomarkers. Anal. Chem. 2023, 95, 2812–2821. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Y.; Federzoni, E.A.; Wang, X.; Dharmawan, A.; Hu, X.; Wang, H.; Hawley, R.J.; Stevens, S.; Sykes, M.; et al. CD47 cross-dressing by extracellular vesicles expressing CD47 inhibits phagocytosis without transmitting cell death signals. eLife 2022, 11, e73677. [Google Scholar] [CrossRef]
- Van Rijn, C.J.M.; Vlaming, K.E.; Bem, R.A.; Dekker, R.J.; Poortinga, A.; Breit, T.; van Leeuwen, S.; Ensink, W.A.; van Wijnbergen, K.; van Hamme, J.L.; et al. Low energy nebulization preserves integrity of SARS-CoV-2 mRNA vaccines for respiratory delivery. Sci. Rep. 2023, 13, 8851. [Google Scholar]
- Cai, X.; Dou, R.; Guo, C.; Tang, J.; Li, X.; Chen, J.; Zhang, J. Cationic Polymers as Transfection Reagents for Nucleic Acid Delivery. Pharmaceutics 2023, 15, 1502. [Google Scholar] [CrossRef]
- Donahue, D.A.; Ballesteros, C.; Maruggi, G.; Glover, C.; Ringenberg, M.A.; Marquis, M.; Ben Abdeljelil, N.; Ashraf, A.; Rodriguez, L.A.; Stokes, A.H. Nonclinical Safety Assessment of Lipid Nanoparticle-and Emulsion-Based Self-Amplifying mRNA Vaccines in Rats. Int. J. Toxicol. 2023, 42, 37–49. [Google Scholar] [CrossRef]
- Sharma, A.; Kontodimas, K.; Bosmann, M. Nanomedicine: A Diagnostic and Therapeutic Approach to COVID-19. Front. Med. 2021, 8, 648005. [Google Scholar] [CrossRef]
- Patel, N.; Davis, Z.; Hofmann, C.; Vlasak, J.; Loughney, J.W.; DePhillips, P.; Mukherjee, M. Development and Characterization of an In Vitro Cell-Based Assay to Predict Potency of mRNA-LNP-Based Vaccines. Vaccines 2023, 11, 1224. [Google Scholar] [CrossRef]
- Bernard, M.C.; Bazin, E.; Petiot, N.; Lemdani, K.; Commandeur, S.; Verdelet, C.; Margot, S.; Perkov, V.; Ripoll, M.; Garinot, M.; et al. The impact of nucleoside base modification in mRNA vaccine is influenced by the chemistry of its lipid nanoparticle delivery system. Mol. Ther. Nucleic Acids. 2023, 32, 794–806. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, J.; Hou, X.; Wang, C.; Kang, D.D.; Xue, Y.; Du, S.; Deng, B.; McComb, D.W.; Liu, S.L.; et al. STING Agonist-Derived LNP-mRNA Vaccine Enhances Protective Immunity Against SARS-CoV-2. Nano Lett. 2023, 23, 2593–2600. [Google Scholar] [CrossRef]
- Hajiaghapour Asr, M.; Dayani, F.; Saedi Segherloo, F.; Kamedi, A.; Neill, A.O.; MacLoughlin, R.; Doroudian, M. Lipid Nanoparticles as Promising Carriers for mRNA Vaccines for Viral Lung Infections. Pharmaceutics 2023, 15, 1127. [Google Scholar] [CrossRef]
- Estapé Senti, M.; de Jongh, C.A.; Dijkxhoorn, K.; Verhoef, J.J.F.; Szebeni, J.; Storm, G.; Hack, C.E.; Schiffelers, R.M.; Fens, M.H.; Boross, P. Anti-PEG antibodies compromise the integrity of PEGylated lipid-based nanoparticles via complement. J. Control. Release 2022, 341, 475–486. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Cafri, G.; Gartner, J.J.; Zaks, T.; Hopson, K.; Levin, N.; Paria, B.C.; Parkhurst, M.R.; Yossef, R.; Lowery, F.J.; Jafferji, M.S.; et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Investig. 2020, 130, 5976–5988. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Zhang, L.; Deng, W.; Lou, J.; Gao, X.; Lou, X.; Liu, Y.; Yao, X.; Sheng, Y.; Yan, Y.; et al. Spleen-selective co-delivery of mRNA and TLR4 agonist-loaded LNPs for synergistic immunostimulation and Th1 immune responses. J. Control. Release 2023, 357, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, A.; Kuter, B.; Pilon-Thomas, S.; Whiting, J.; Mo, Q.; Leav, B.; Sirak, B.; Cubitt, C.; Dukes, C.; Isaacs-Soriano, K.; et al. Safety and immunogenicity of a third dose of mRNA-1273 vaccine among cancer patients. Cancer Commun. 2023, 43, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Parr, C.J.C.; Wada, S.; Kotake, K.; Kameda, S.; Matsuura, S.; Sakashita, S.; Park, S.; Sugiyama, H.; Kuang, Y.; Saito, H. N 1-Methylpseudouridine substitution enhances the performance of synthetic mRNA switches in cells. Nucleic Acids Res. 2020, 48, e35. [Google Scholar] [CrossRef]
- Faghfuri, E.; Pourfarzi, F.; Faghfouri, A.H.; Abdoli Shadbad, M.; Hajiasgharzadeh, K.; Baradaran, B. Recent Developments of RNA-Based Vaccines in Cancer Immunotherapy. Expert Opin. Biol. Ther. 2021, 21, 201–218. [Google Scholar] [CrossRef]
- Jekhmane, S.; de Haas, R.; da Silva Filho, O.P.; van Asbeck, A.H.; Favretto, M.E.; Hernandez Garcia, A.; Brock, R.; de Vries, R. Virus-Like Particles of mRNA with Artificial Minimal Coat Proteins: Particle Formation, Stability, and Transfection Efficiency. Nucleic Acid Ther. 2017, 27, 159–167. [Google Scholar] [CrossRef]
- Vogt, A.S.; Jörg, L.; Martina, B.; Krenger, P.S.; Chang, X.; Zeltins, A.; Vogel, M.; Mohsen, M.O.; Bachmann, M.F. Virus-Like Particles Are Efficient Tools for Boosting mRNA-Induced Antibodies. Front. Immunol. 2022, 13, 864718. [Google Scholar] [CrossRef]
- Patel, J.M.; Vartabedian, V.F.; Kim, M.C.; He, S.; Kang, S.M.; Selvaraj, P. Influenza virus-like particles engineered by protein transfer with tumor-associated antigens induces protective antitumor immunity. Biotechnol. Bioeng. 2015, 112, 1102–1110. [Google Scholar] [CrossRef]
- Kumar, R.; Srivastava, V.; Baindara, P.; Ahmad, A. Thermostable vaccines: An innovative concept in vaccine development. Expert Rev. Vaccines 2022, 21, 811–824. [Google Scholar] [CrossRef]
- Lamoot, A.; Lammens, J.; De Lombaerde, E.; Zhong, Z.; Gontsarik, M.; Chen, Y.; De Beer, T.R.M.; De Geest, B.G. Successful batch and continuous lyophilization of mRNA LNP formulations depend on cryoprotectants and ionizable lipids. Biomater. Sci. 2023, 11, 4327–4334. [Google Scholar] [CrossRef]
Figure 1.
The roles of CTAs in various life processes. EMT denotes epithelial to mesenchymal transition. Hollow arrows indicate the roles of cancer/testis antigens.
Figure 1.
The roles of CTAs in various life processes. EMT denotes epithelial to mesenchymal transition. Hollow arrows indicate the roles of cancer/testis antigens.

Figure 2.
TCR-T cells therapy and CAR-T therapy. CAR-T cells employ chimeric antigen receptors which bind to tumor cell surface TAAs (left). TCR-T cell therapy employs engineered T cells that express exogenous TCR which binds to TAA-derived peptides (middle). TAAs include neoantigens and CTAs. CTAs are present on the cell surface or secreted (right), making CTAs targets of immunotherapy. ITAM denotes immunoreceptor tyrosine-based activation motifs. CTA denotes cancer/testis antigen. CD28 is a receptor of costimulatory molecules. ScFv denotes single-chain variable fragment. Black arrows denote the direction of reaction.
Figure 2.
TCR-T cells therapy and CAR-T therapy. CAR-T cells employ chimeric antigen receptors which bind to tumor cell surface TAAs (left). TCR-T cell therapy employs engineered T cells that express exogenous TCR which binds to TAA-derived peptides (middle). TAAs include neoantigens and CTAs. CTAs are present on the cell surface or secreted (right), making CTAs targets of immunotherapy. ITAM denotes immunoreceptor tyrosine-based activation motifs. CTA denotes cancer/testis antigen. CD28 is a receptor of costimulatory molecules. ScFv denotes single-chain variable fragment. Black arrows denote the direction of reaction.

Figure 3.
Immune responses induced by an mRNA vaccine encoding CTA. (A) Following endocytosis of mRNA vaccine by antigen presenting cells such as dendritic cells (DCs), the CTA mRNA is translated into protein. After translation, the protein is degraded by proteasome complex to yield the antigenic peptide. The antigenic peptide can be presented on the cell surface via MHC class I. The MHC class I presented epitopes are recognized by CD8+ cytotoxic T cells for activation to kill infected cells or cancer cells. (B) After translation of CTA mRNA, the secreted CTA protein can be recognized by B cells and engulfed. The protein is then degraded into antigenic peptide, which is bound to MHC II. The MHC II-peptide complex on the cell surface can activate CD4+ T cells. The activated CD4+ T cells induce B cells to produce antibodies. ER denotes endoplasmic reticulum. BCR denotes B cell receptor. Hollow arrows denote the direction of reaction.
Figure 3.
Immune responses induced by an mRNA vaccine encoding CTA. (A) Following endocytosis of mRNA vaccine by antigen presenting cells such as dendritic cells (DCs), the CTA mRNA is translated into protein. After translation, the protein is degraded by proteasome complex to yield the antigenic peptide. The antigenic peptide can be presented on the cell surface via MHC class I. The MHC class I presented epitopes are recognized by CD8+ cytotoxic T cells for activation to kill infected cells or cancer cells. (B) After translation of CTA mRNA, the secreted CTA protein can be recognized by B cells and engulfed. The protein is then degraded into antigenic peptide, which is bound to MHC II. The MHC II-peptide complex on the cell surface can activate CD4+ T cells. The activated CD4+ T cells induce B cells to produce antibodies. ER denotes endoplasmic reticulum. BCR denotes B cell receptor. Hollow arrows denote the direction of reaction.

Figure 4.
Structures of lipid nanoparticles (LNPs). LNPs are made of four types of lipid molecules: phospholipid, ionizable lipid, cholesterol, and PEG-lipid. (A) shows mRNA organized in inverse lipid micelles inside the nanoparticle. (B) shows mRNA intercalated between the lipid bilayers. (C) shows advantages and disadvantages of mRNA-LNP vaccine.
Figure 4.
Structures of lipid nanoparticles (LNPs). LNPs are made of four types of lipid molecules: phospholipid, ionizable lipid, cholesterol, and PEG-lipid. (A) shows mRNA organized in inverse lipid micelles inside the nanoparticle. (B) shows mRNA intercalated between the lipid bilayers. (C) shows advantages and disadvantages of mRNA-LNP vaccine.


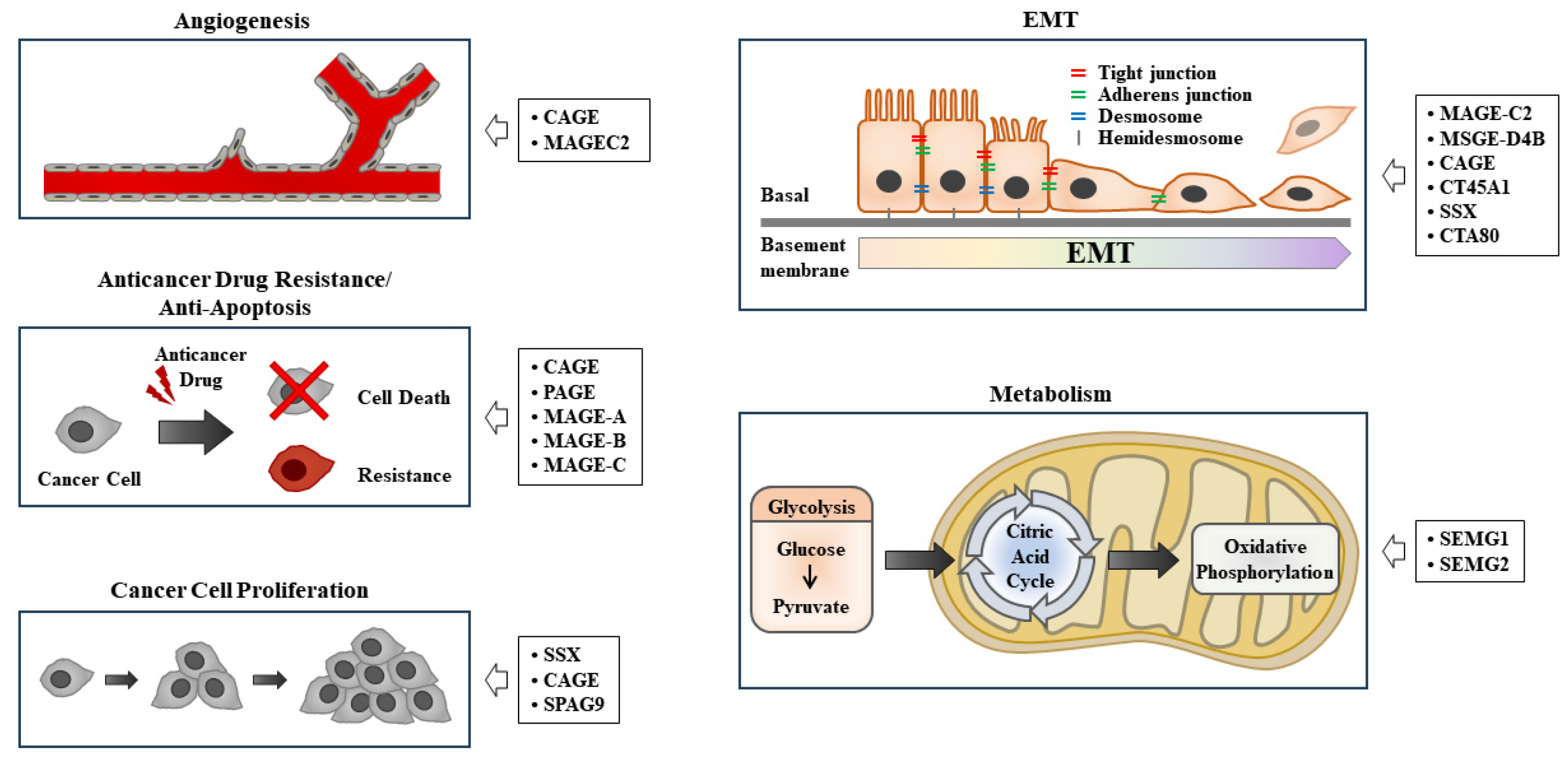{kind=link}
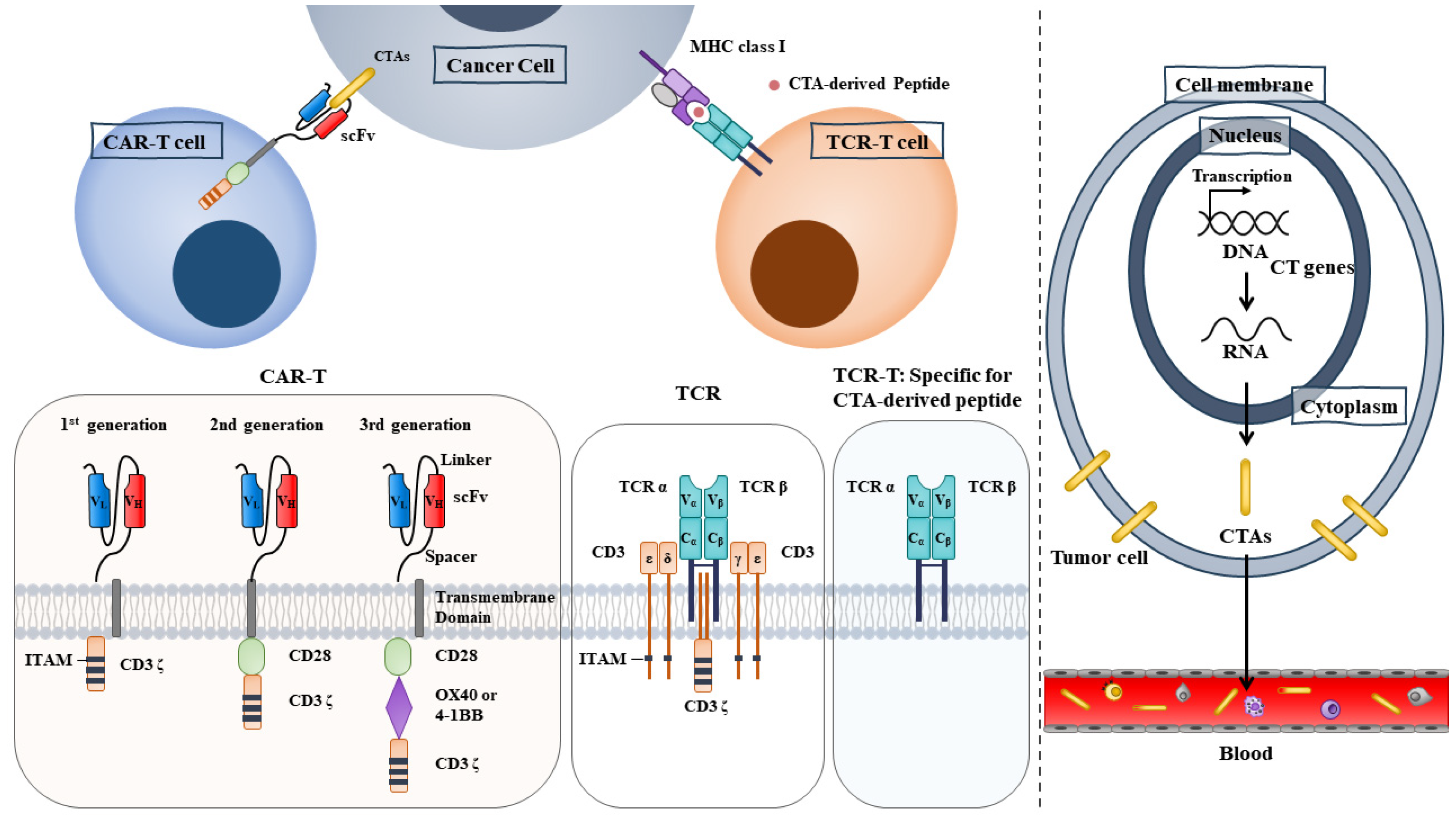{kind=link}
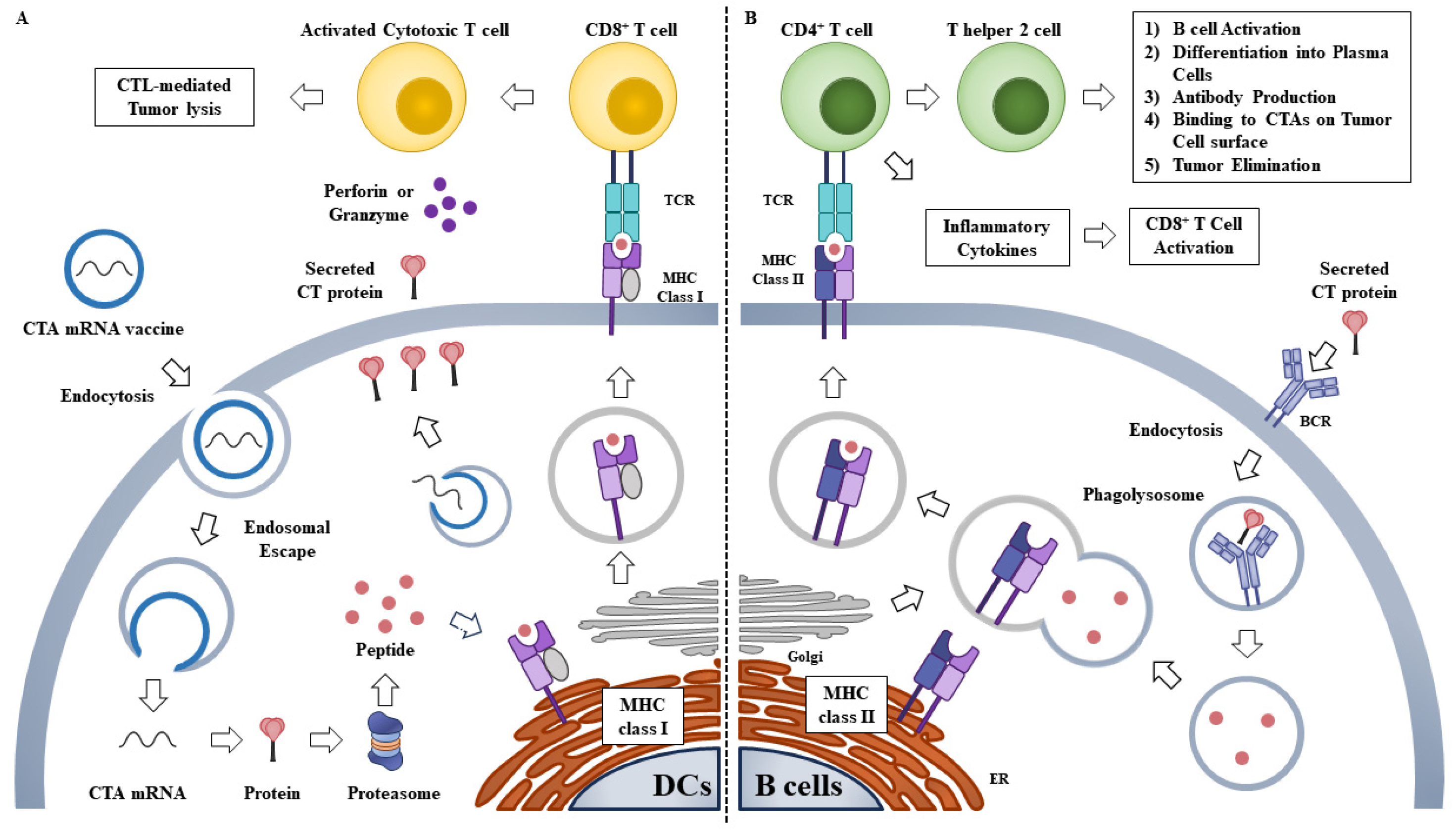{kind=link}
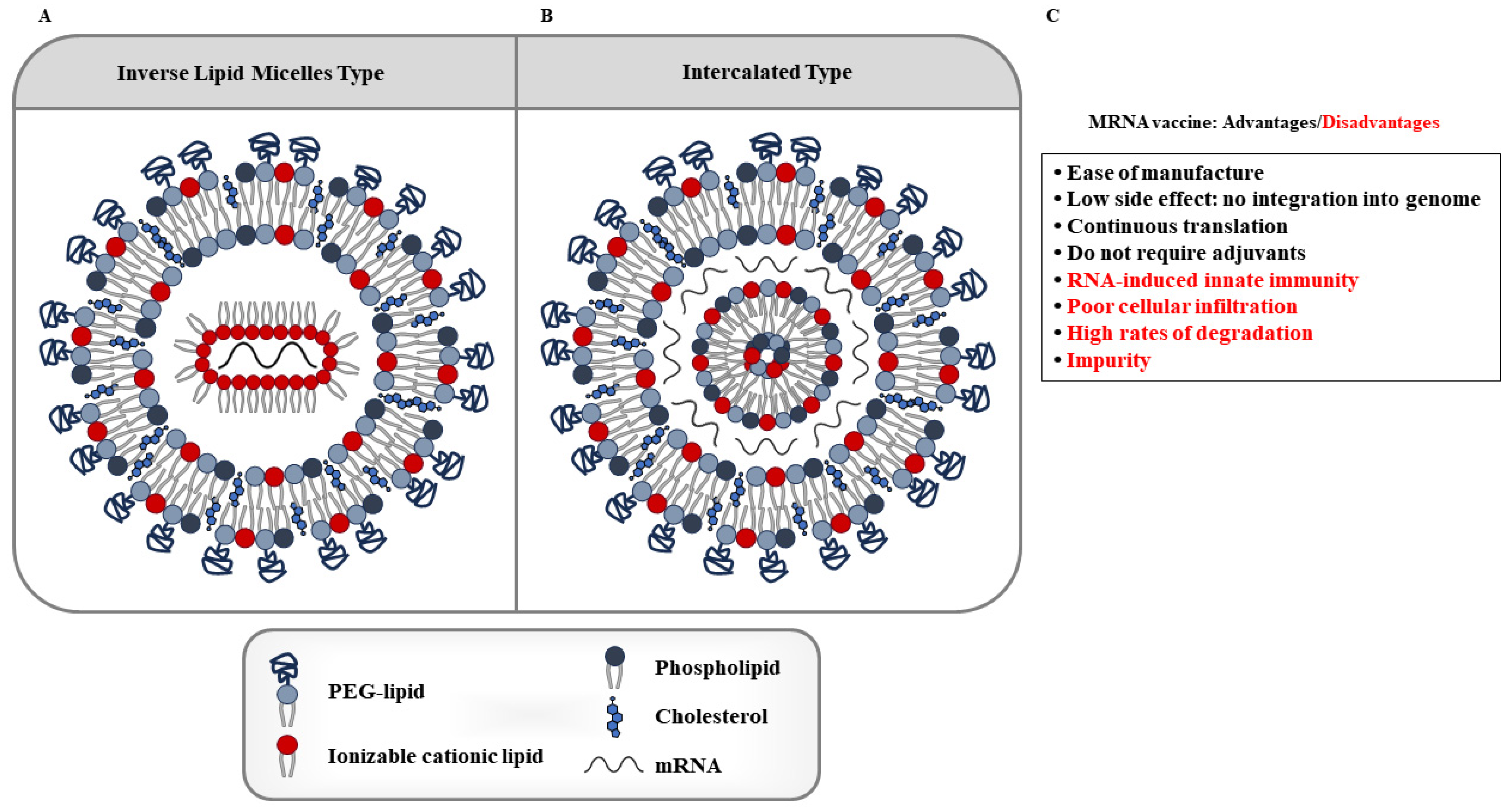{kind=link}
Table 1.
Functions and locations of TAAs.
| TAAs | Function | Location | Refs |
|---|---|---|---|
| SPAG9 | Angiogenesis | Cytosol, Perinuclear region | [18] |
| PAGE4 | Antiapoptosis | Cytosol, Nucleus, Mitochondria | [19] |
| CT45A1 | Promotes EMT | Nucleus, Cytosol | [20] |
| SSX1 | Promotes EMT and cancer stem cell-like properties | Nucleus, Cytosol, Extracellular | [21] |
| SSX2 | Promotes EMT | Nucleus, Cytosol, Extracellular | [22] |
| MAGEC2 | Angiogenesis, EMT | Nucleus, Cytosol, Extracellular, Mitochondria | [23] |
| MAGEA3 | Cancer metastasis | Nucleus, Cytosol, Extracellular, ER | [23] |
| MAGEA9 | Cancer stemness | Nucleus, Cytosol, Extracellular, Mitochondria | [27] |
| PLAC1 | Promotes tumor invasion and inhibit antitumor immunity | Extracellular, Cytosol, ER, Nucleus, Mitochondria | [28] |
| CAGE | Promotes anticancer drug resistance | Nucleus, Cytosol, Extracellular, Mitochondria | [29] |
| PRAME | Promotes resistance to TRAIL | Nucleus, Plasma membrane, Extracellular, Mitochondria | [30] |
| SEMG1 and SEMG2 | Promotes glycolysis and respiration | Nucleus, Extracellular | [32] |
Table 2.
Clinical trials of T cells targeting multiple TAAs.
| Study Title | Conditions | Interventions | Study Type | Phase | Number Enrolled | NCT Number | Study Start/ Completion Study Completion |
|---|---|---|---|---|---|---|---|
| TAA Specific Cytotoxic T Lymphocytes in Patients with Pancreatic Cancer (TACTOPS) | Pancreatic cancer |
| Interventional | Phase1/II | 37 | NCT03192462 | Jan 2018/May 2027 |
| Tumor-associated Antigen (TAA) Specific Cytotoxic T Lymphocytes Administered in Patients With Breast Cancer | Breast cancer |
| Interventional | Phase II | 12 | NCT03093350 | Oct 2017/May 2024 |
| A Phase I/II Study of the Treatment of Metastatic Cancer That Expresses MAGE-A3 Using Lymphodepleting Conditioning Followed by Infusion of HLA-DP0401/0402 Restricted Anti-MAGE-A3 TCR-Gene Engineered Lymphocytes and Aldesleukin | Metastatic cancers:
|
| Interventional | Phase I/II | 21 | NCT02111850 | Feb 2014/March 2021 |
| A Phase I Dose Escalation Open Label Clinical Trial Evaluating the Safety and Efficacy of MAGE A10c796T in Subjects With Stage IIIb or Stage IV Non-Small Cell Lung Cancer (NSCLC) |
|
| Interventional | Phase I | 28 | NCT02592577 | Nov 2015/Mar 2017 |
| A Phase 2 Single Arm Open-Label Clinical Trial of ADP-A2M4 SPEAR™ T Cells in Subjects With Advanced Synovial Sarcoma or Myxoid/Round Cell Liposarcoma |
|
| Interventional | Phase II | 90 | NCT04044768 | Aug 2019/Nov 2034 |
| A Phase 2 Pilot Study of ADP-A2M4 in Combination With Pembrolizumab in Subjects With Recurrent or Metastatic Head and Neck Cancer | Head and neck cancer |
| Interventional | Phase II | 10 | NCT04408898 | Jul 2020/Dec 2021 |
| Phase 1 Study Evaluating Genetically Modified Autologous T Cells Expressing a T-cell Receptor Recognizing a Cancer/Germline Antigen in Patients With Recurrent and/or Refractory Solid Tumors(ACTengine® IMA202-101) |
|
| Interventional | Phase I | 16 | NCT03441100 | May 2019/Mar 2023 |
| Phase 1 Study Evaluating Genetically Modified Autologous T Cells Expressing a T-cell Receptor Recognizing a Cancer/Germline Antigen in Patients With Recurrent and/or Refractory Solid Tumors (ACTengine® IMA201-101) |
|
| Interventional | Phase I | 22 | NCT03247309 | Dec 2018/Dec 2024 |
| Tumor-Associated Antigen (TAA) Specific Cytotoxic T Lymphocytes Administered in Patients With Pancreatic Cancer | Pancreatic cancer |
| Interventional | Phase II | 37 | NCT03192462 | Jan 2018/May 2027 |
TAAs denote tumor-associated antigens.
Table 3.
Clinical trial of mRNAs encoding CTAs.
| Study Title | Conditions | Interventions | Study Type | Phase | Number Enrolled | NCT Number | Study Start/Completion Study Completion |
|---|---|---|---|---|---|---|---|
| Intradermal Vaccination With Stabilized Tumor mRNA—a Clinical Phase I/II Trial in Melanoma Patients |
|
| Interventional | Phase I/II | 20 | NCT00204607 | July 2004 /Jan 2007 |
| Vaccination With Tumor mRNA in Metastatic Melanoma—Fixed Combination Versus Individual Selection of Targeted Antigens |
|
| Interventional | Phase I/II | 31 | NCT00204516 | Apr 2007 /December 2012 |
| CT7, MAGE-A3, and WT1 mRNA-electroporated Autologous Langerhans-type Dendritic Cells as Consolidation for Multiple Myeloma Patients Undergoing Autologous Stem Cell Transplantation |
|
| Interventional | Phase I | 28 | NCT01995708 | Jan 31 2014 /June 20 2022 |
| Phase I/II Study of Combination Immunotherapy and Messenger Ribonucleic Acid (mRNA) Vaccine in Subjects With NSCLC |
|
| Interventional | Phase I/II | 61 | NCT03164772 | December 20 2017 /October 29 2021 |
| Messenger Ribonucleic Acid (mRNA) Transfected Dendritic Cell Vaccination in High Risk Uveal Melanoma Patients |
|
| Interventional | Phase I/II | 23 | NCT00929019 | June 2009 /Apr 2016 |
| Peptide-pulsed vs. RNA-transfected Dendritic Cell Vaccines in Melanoma Patients |
|
| Interventional | Phase I/II | 64 | NCT00243529 | Apr 2004 |
| Study of MAGE-3/Melan-A/gp 100/NA17 and rhIL-12 With/Out Low Dose IL-2 in Metastatic Melanoma |
|
| Interventional | Phase II | 19 | NCT00203879 | Feb 2002/May 2007 |
Table 4.
Clinical trials of LNP-mRNAs encoding TAAs.
| Study Title | Conditions | Interventions | Study Type | Phase | Number Enrolled | NCT Number | Study Start/ Completion Study Completion |
|---|---|---|---|---|---|---|---|
| A Phase I, Open-Label, Multicenter Study to Assess the Safety, Tolerability, and Immunogenicity of mRNA-4157 Alone in Subjects With Resected Solid Tumors and in Combination With Pembrolizumab in Subjects With Unresectable Solid Tumors | NSCLC Melanoma | Biological: mRNA-4157 (LNP) encoding neoantigen Personalized cancer vaccine, IM injection Biological: Pembrolizumab Intravenous infusion | Interventional | Phase I | 108 | NCT03313778 | Aug 2017/June 2025 |
| A Phase I/II Trial to Evaluate the Safety and Immunogenicity of a Messenger RNA (mRNA)-Based, Personalized Cancer Vaccine Against Neoantigens Expressed by the Autologous Cancer |
|
| Interventional | Phase I/II | 5 | NCT03480152 | May 2018/Nov 2019 |
| A Phase I, Open-Label, Multicenter Study to Assess the Safety and Tolerability of mRNA-5671/V941 as a Monotherapy and in Combination With Pembrolizumab in Participants With KRAS Mutant Advanced or Metastatic Non-Small Cell Lung Cancer, Colorectal Cancer or Pancreatic Adenocarcinoma |
|
| Interventional | Phase I | 70 | NCT03948763 | June 2019/Aug 2022 |
| Clinical First-in-human Dose Escalation Study Evaluating the Safety and Tolerability of Intravenous Administration of a Tetravalent RNA-lipoplex Cancer Vaccine Targeting the Tumor-associated Antigens NY-ESO-1, Tyrosinase, MAGE-A3, and TPTE in Patients With Advanced Melanoma | Metastatic melanoma |
| Interventional | Phase I | 119 | NCT02410733 | Mar 2015/June 2023 |
| First-in-human Clinical Study With RNA-Immunotherapy Combination of IVAC_W_bre1_uID and IVAC_M_uID for Individualized Tumor Therapy in Triple Negative Breast Cancer Patients | Breast cancer |
| Interventional | Phase I | 42 | NCT02316457 | Oct 2016/May 2023 |
MERIT denotes Mutanom Engineered RNA Immunotherapy.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shim, K.; Jo, H.; Jeoung, D. Cancer/Testis Antigens as Targets for RNA-Based Anticancer Therapy. Int. J. Mol. Sci. 2023, 24, 14679. https://doi.org/10.3390/ijms241914679
AMA Style
Shim K, Jo H, Jeoung D. Cancer/Testis Antigens as Targets for RNA-Based Anticancer Therapy. International Journal of Molecular Sciences. 2023; 24(19):14679. https://doi.org/10.3390/ijms241914679
Chicago/Turabian StyleShim, Kyeonghee, Hyein Jo, and Dooil Jeoung. 2023. "Cancer/Testis Antigens as Targets for RNA-Based Anticancer Therapy" International Journal of Molecular Sciences 24, no. 19: 14679. https://doi.org/10.3390/ijms241914679
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.