
Intratumor Heterogeneity and Treatment Resistance of Solid Tumors with a Focus on Polyploid/Senescent Giant Cancer Cells (PGCCs)

Abstract
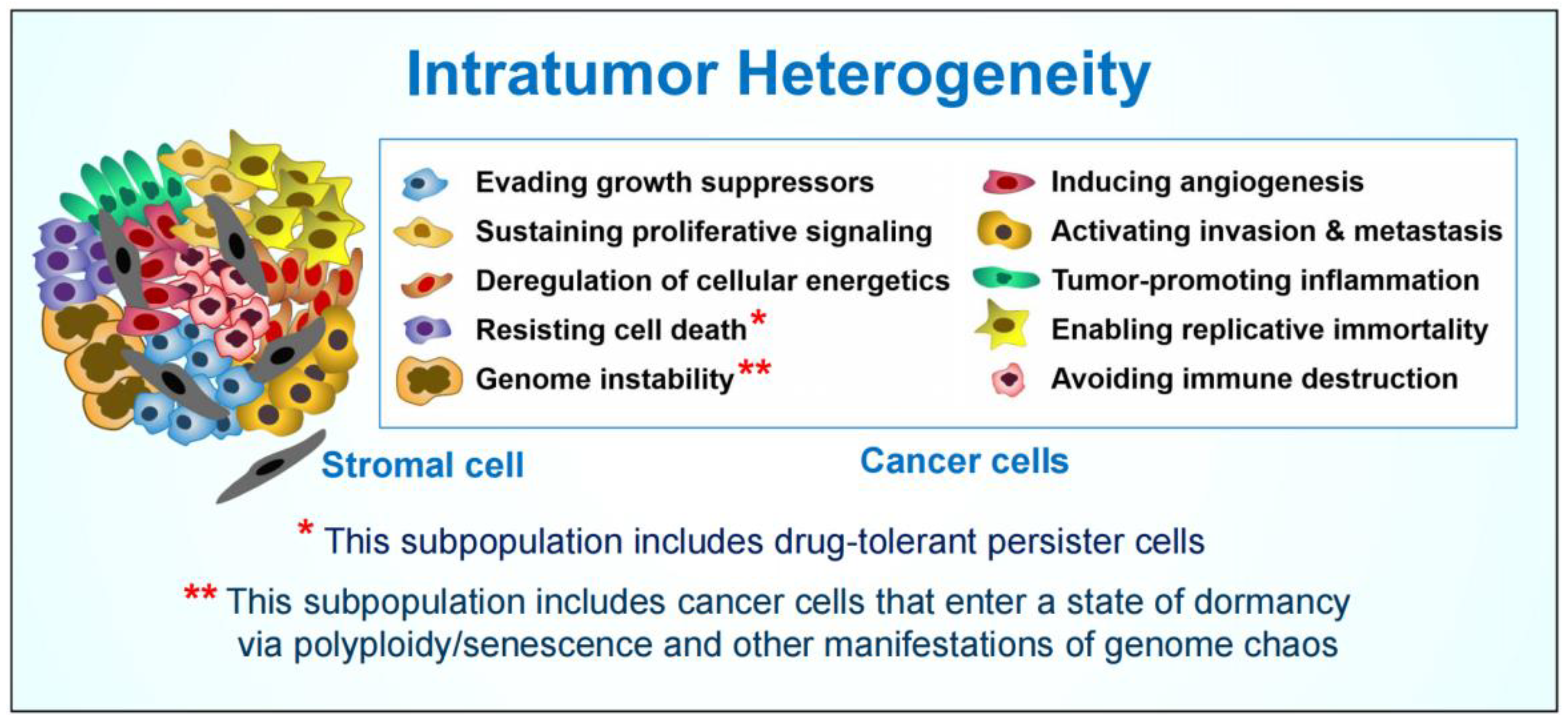
:1. Introduction
2. Therapy-Induced Cancer Cell Polyploidy/Senescence and Disease Recurrence
2.1. Prevalence and Prognostic Value of PGCCs
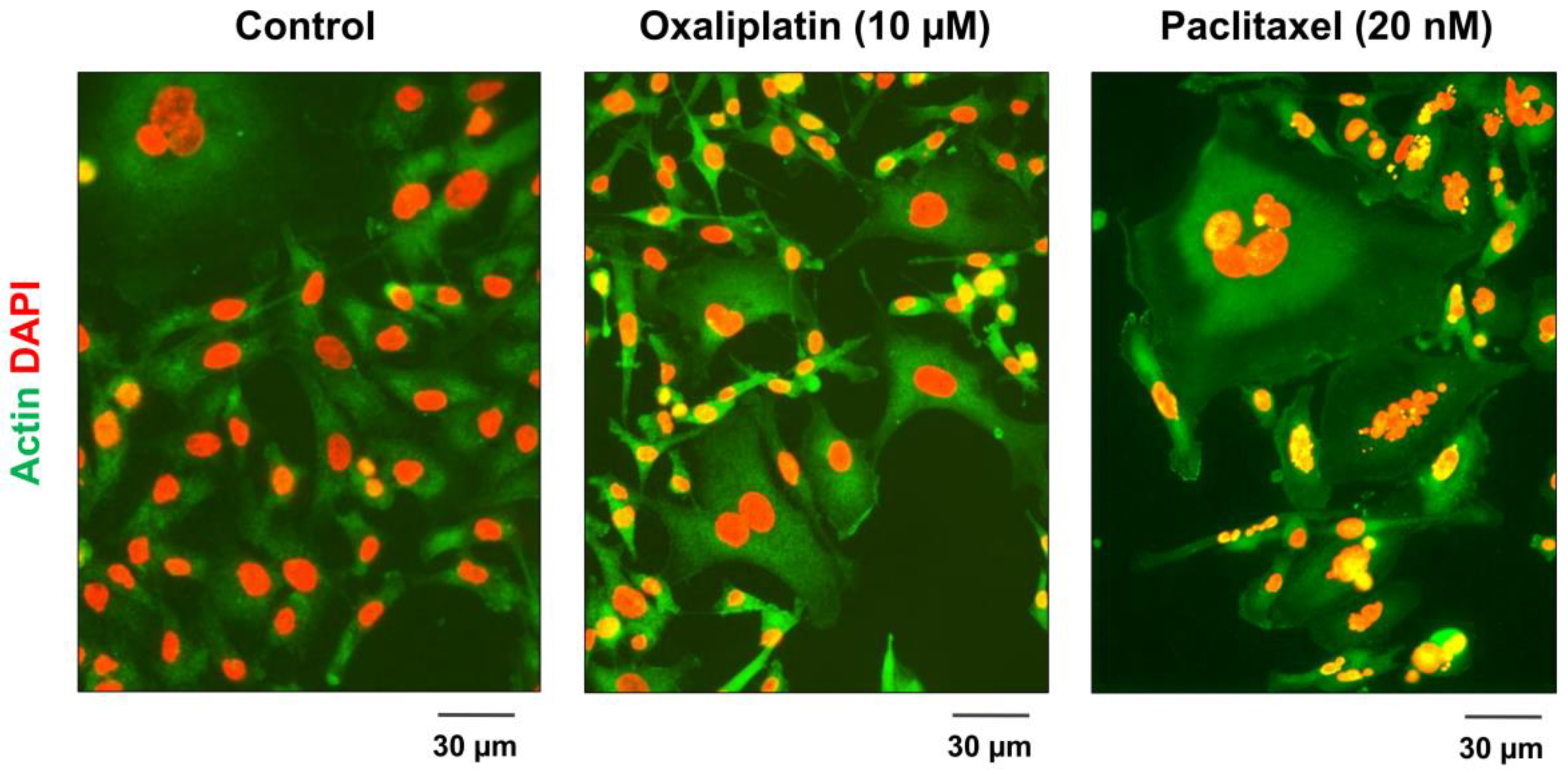
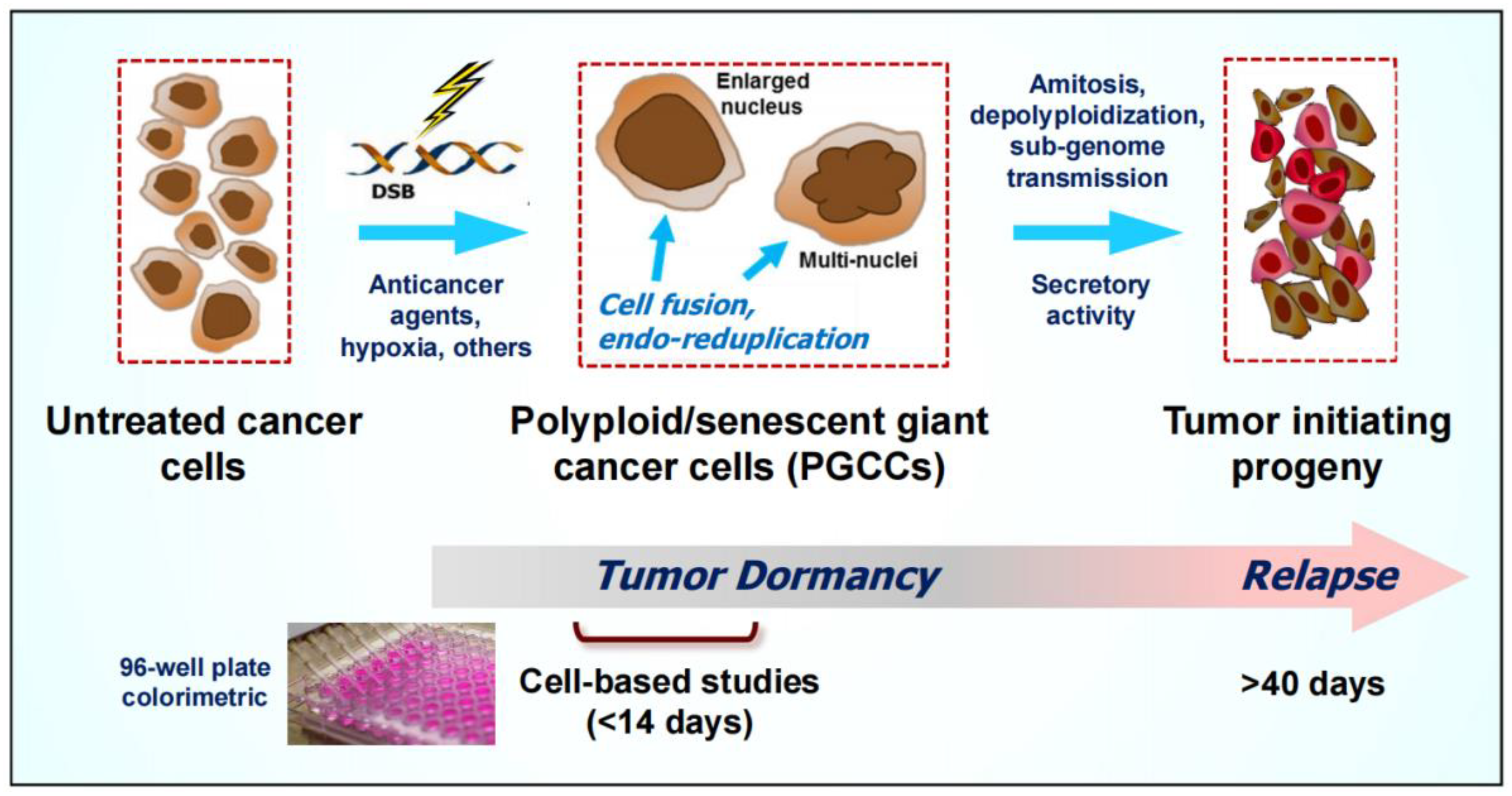
2.2. Formation and Fate of PGCCs
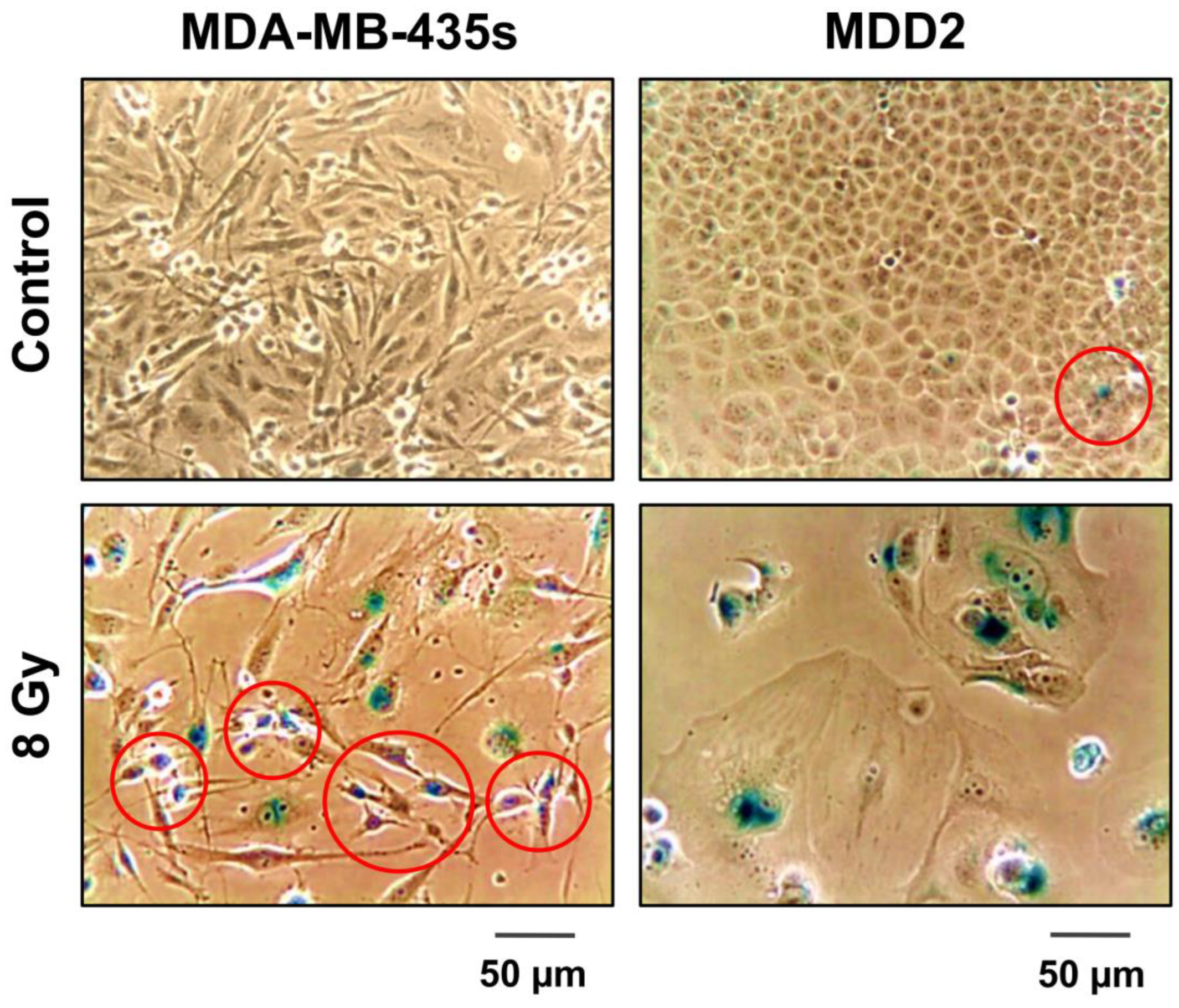
2.3. Contributions of Our Group to the Understanding of the Creation and Fate of PGCCs following Anticancer Treatment
3. Important Considerations When Assessing Cancer Cell Radiosensitivity and Chemosensitivity? What Does “Sensitivity” Actually Refer to?
3.1. Significance of p53-p21-WIP1 signaling in Suppressing Cancer Cell Death and Triggering (Reversible) Senescence
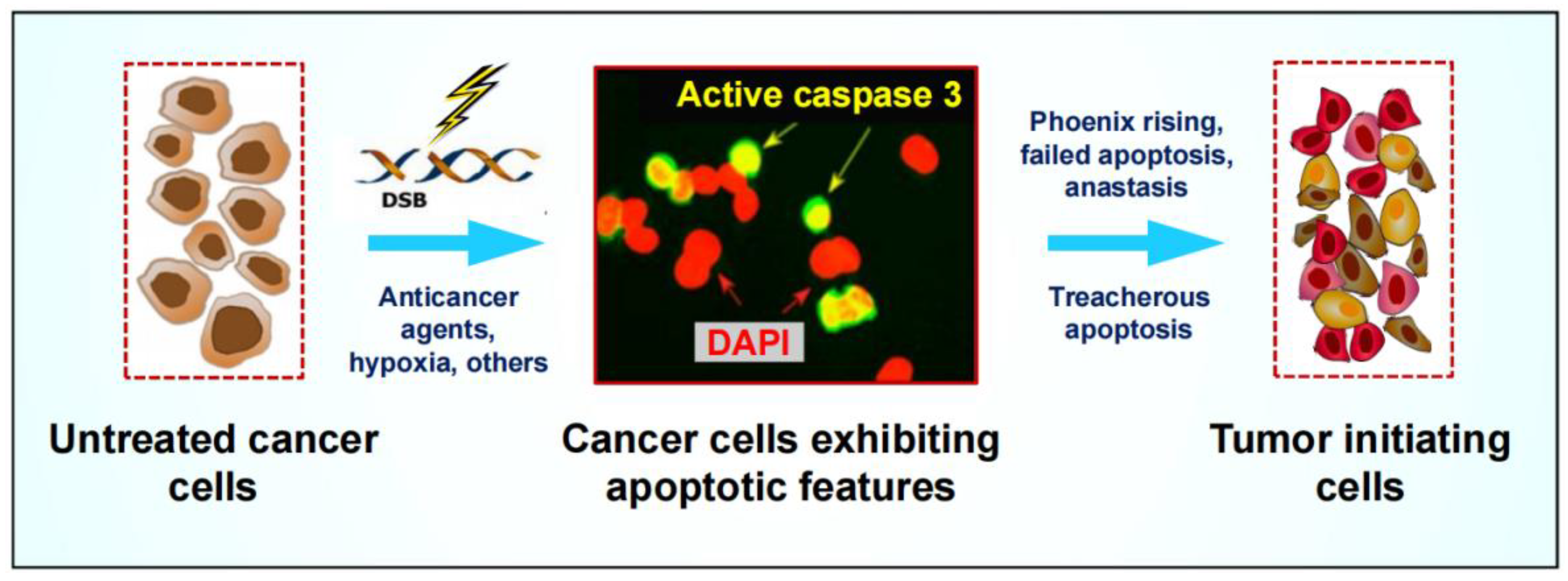
3.2. Pro-Survival Properties of Cancer Cells Triggered to Undergo Apoptosis
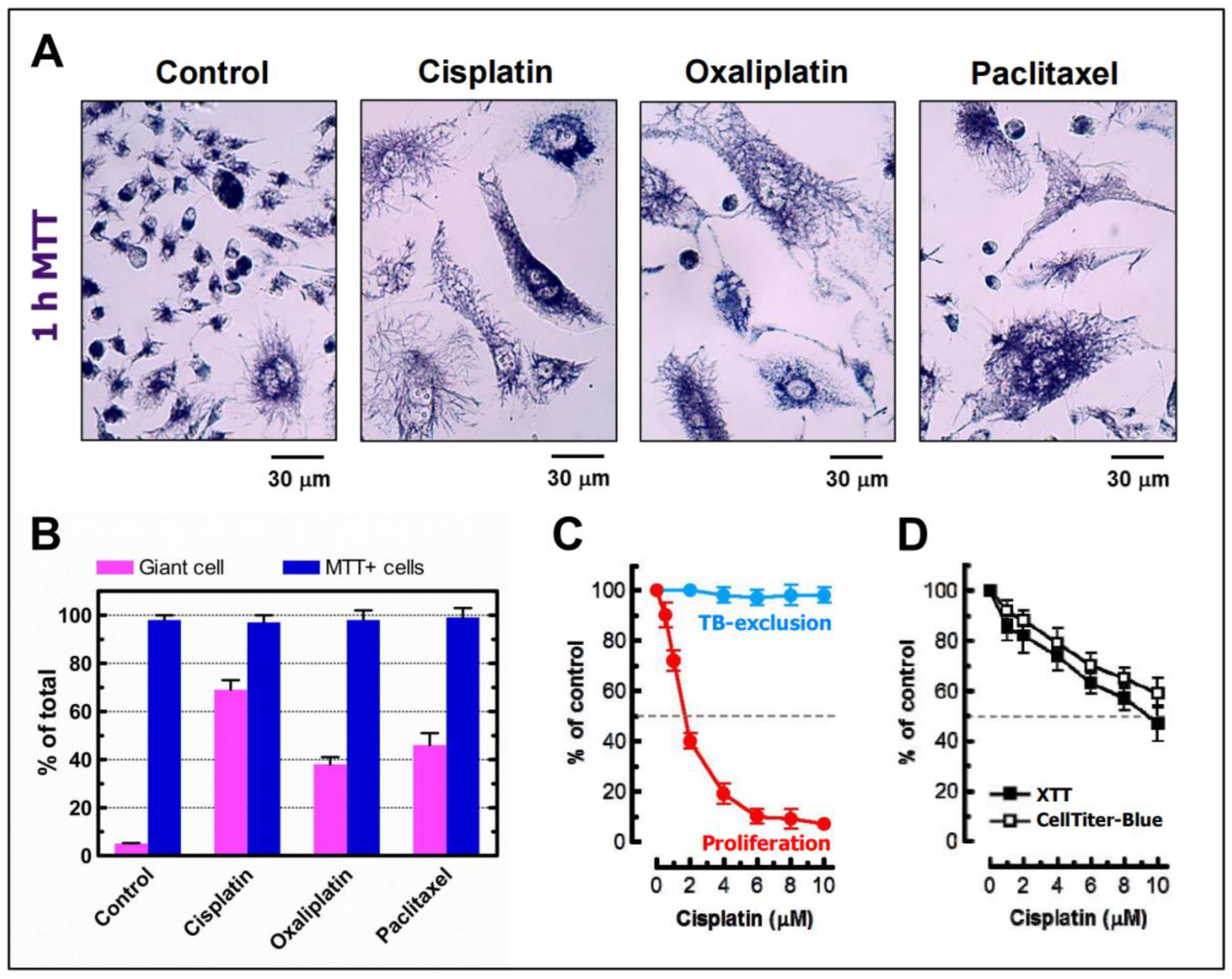
3.3. Danger of Relying on High Content Multiwell Plate Assays for Cancer Cell “Lethality” Assessment
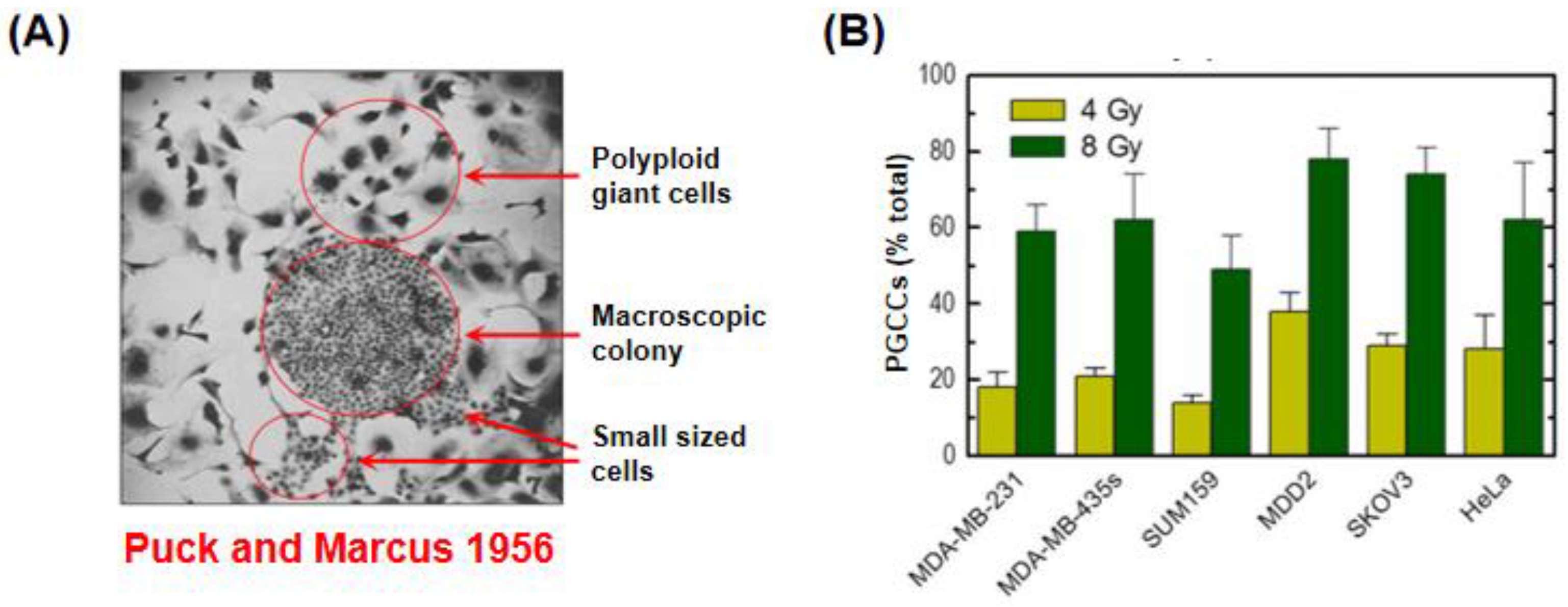
3.4. Danger of Relying on the Clonogenic “Survival” Assay for Assessment of Cancer Cell Death
3.5. Single Cell Biology: A Step towards Generating Clinically Relevant Information
4. Possible Reasons Why PGCCs and Their Tumor Repopulating Properties Continue to Be Overlooked in Most Preclinical Anticancer Studies
4.1. Misleading Assumption That PGCCs Represent Dead or Dying Cells That Will Be Eventually Eliminated via Apoptosis and Other Means
4.2. Misleading/Inappropriate Preclinical Assays?
4.3. Dishonesty in Data Reporting?
5. Relevance to the Future Direction of Precision Oncology: A Personal Perspective
- ●
- Classical DNA-damaging cancer therapeutics (e.g., ionizing radiation, cisplatin) have been shown to induce significant PGCC formation (reviewed in [20]). Do more recent cancer mutation-targeted strategies such as exploiting synthetic–“lethal” partnerships also cause the generation of PGCCs?
- ●
- Can we develop reliable and high throughput imaging-based versions of the currently cumbersome and expertise-dependent assays for entities such as PGCCs that will be accepted/taken up by the scientific community such that screening these responses to therapy in tissue culture can be done in a time- and cost-effective manner?
- ●
- With such assays in hand, can we identify drugs/combinations (with or without radiation) that either circumvent the generation of these treacherous PGCCs or trigger their demise?
- ●
- Given that PGCCs are highly atypical in many regards, such as their size, shape, and ploidy, can we devise strategies that will harness the full power of the immune system to eradicate these potentially harmful aberrant cells?
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Levine, A.J.; Perry, M.E.; Chang, A.; Silver, A.; Dittmer, D.; Wu, M.; Welsh, D. The 1993 Walter Hubert Lecture: The role of the p53 tumour-suppressor gene in tumorigenesis. Br. J. Cancer 1994, 69, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Enoch, T.; Norbury, C. Cellular responses to DNA damage: Cell-cycle checkpoints, apoptosis and the roles of p53 and ATM. Trends Biochem. Sci. 1995, 20, 426–430. [Google Scholar] [CrossRef]
- Morgan, S.E.; Kastan, M.B. p53 and ATM: Cell cycle, cell death, and cancer. Adv. Cancer Res. 1997, 71, 1–25. [Google Scholar] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Rybinski, B.; Yun, K. Addressing intra-tumoral heterogeneity and therapy resistance. Oncotarget 2016, 7, 72322–72342. [Google Scholar] [CrossRef] [Green Version]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-H.; Altschuler, S.J.; Wu, L.F. Patterns of early p21 dynamics determine proliferation-senescence cell fate after chemotherapy. Cell 2019, 178, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Ramón y Cajal, S.; Sesé, M.; Capdevila, C.; Trond, A.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernández-Losa, J.; Castellví, J. Clinical implications of intratumor heterogeneity: Challenges and opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef] [Green Version]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor heterogeneity: The Rosetta Stone of therapy resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. Intratumor heterogeneity and therapy resistance: Contributions of dormancy, apoptosis reversal (anastasis) and cell fusion to disease recurrence. Int. J. Mol. Sci. 2020, 21, 1308. [Google Scholar] [CrossRef] [Green Version]
- Gilson, P.; Merlin, J.-L.; Harlé, A. Deciphering tumour heterogeneity: From tissue to liquid biopsy. Cancers 2022, 14, 1384. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R. Treacherous apoptosis—Cancer cells sacrifice themselves at the altar of heterogeneity. Hepatolog. 2022, 76, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Kalkavan, H.; Rühl, S.; Shaw, J.J.P.; Green, D.R. Non-lethal outcomes of engaging regulated cell death pathways in cancer. Nat. Cancer 2023, 4, 795–806. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Eastman, A. Improving anticancer drug development begins with cell culture: Misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget 2017, 8, 8854–8866. [Google Scholar] [CrossRef] [Green Version]
- Zaitceva, V.; Kopeina, G.S.; Zhivotovsky, B. Anastasis: Return journey from cell death. Cancers 2021, 13, 3671. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Murray, D. What are the reasons for continuing failures in cancer therapy? Are misleading/inappropriate preclinical assays to be blamed? Might some modern therapies cause more harm than benefit? Int. J. Mol. Sci. 2022, 23, 13217. [Google Scholar] [CrossRef]
- Borroni, E.M.; Grizzi, F. Cancer Immunoediting and beyond in 2021. Int. J. Mol. Sci. 2021, 22, 13275. [Google Scholar] [CrossRef]
- Brandmaier, A.; Formenti, S.C. The impact of radiation therapy on innate and adaptive tumor immunity. Semin. Radiat. Oncol. 2020, 30, 139–144. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of polyploid/multinucleated giant cancer cells in metastasis and disease relapse following anticancer treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy road to therapy-induced cellular senescence and escape. Int. J. Cancer 2012, 132, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Coward, J.; Harding, A. Size does matter: Why polyploid tumor cells are critical drug targets in the war on cancer. Front. Oncol. 2014, 4, 123. [Google Scholar] [CrossRef]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy formation in doxorubicin-treated cancer cells can favor escape from senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [Green Version]
- Bojko, A.; Staniak, K.; Czarnecka-Herok, J.; Sunderland, P.; Dudkowska, M.; Śliwińska, M.A.; Salmina, K.; Sikora, E. Improved autophagic flux in escapers from doxorubicin-induced senescence/polyploidy of breast cancer cells. Int. J. Mol. Sci. 2020, 21, 6084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qiao, Q.; Xu, H.; Zhou, R.; Liu, X. Human cell polyploidization: The good and the evil. Semin. Cancer Biol. 2022, 81, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.; Heng, H.H. Genome chaos, information creation, and cancer emergence: Searching for new frameworks on the 50th anniversary of the “war on cancer”. Genes 2022, 13, 101. [Google Scholar] [CrossRef]
- Ye, J.C.; Horne, S.; Zhang, J.Z.; Jackson, L.; Heng, H.H. Therapy induced genome chaos: A novel mechanism of rapid cancer drug resistance. Front. Cell Dev. Biol. 2021, 9, 676344. [Google Scholar] [CrossRef]
- Chen, J.; Niu, N.; Zhang, J.; Qi, L.; Shen, W.; Donkena, K.V.; Feng, Z.; Liu, J. Polyploid giant cancer cells (PGCCs): The evil roots of cancer. Curr. Cancer Drug Targets 2019, 19, 360–367. [Google Scholar] [CrossRef]
- Liu, J.; Erenpreisa, J.; Sikora, E. Polyploid giant cancer cells: An emerging new field of cancer biology. Semin. Cancer Biol. 2022, 81, 1–4. [Google Scholar] [CrossRef]
- Liu, J.; Niu, N.; Li, X.; Zhang, X.; Sood, A.K. The life cycle of polyploid giant cancer cells and dormancy in cancer: Opportunities for novel therapeutic interventions. Semin. Cancer Biol. 2022, 81, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Murray, D. Impact of chemotherapeutic drugs on cancer cell proliferation, morphology and metabolic activity. J. Cancer Biol. Res. 2018, 6, 1118. [Google Scholar]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Donovan, P.; Cato, K.; Legaie, R.; Jayalath, R.; Olsson, G.; Hall, B.; Olson, S.; Boros, S.; Reynolds, B.A.; Harding, A. Hyperdiploid tumor cells increase phenotypic heterogeneity within Glioblastoma tumors. Mol. Biosyst. 2014, 10, 741–758. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Trabzonlu, L.; Pienta, K.J.; Trock, B.J.; De Marzo, A.M.; Amend, S.R. Presence of cells in the polyaneuploid cancer cell (PACC) state predicts the risk of recurrence in prostate cancer. Prostate 2023, 83, 277–285. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, L.; Rong, Z.; He, T.; Zhang, S. Number of glioma polyploid giant cancer cells (PGCCs) associated with vasculogenic mimicry formation and tumor grade in human glioma. J. Exp. Clin. Cancer Res. 2013, 32, 75. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Wang, Y.; Fei, F.; Wang, X.; Li, C.; Liu, K.; Du, J.; Cao, Y.; Zhang, S. Clinical characteristics and preliminary morphological observation of 47 cases of primary anorectal malignant melanomas. Melanoma Res. 2018, 28, 592–599. [Google Scholar] [CrossRef]
- Liu, H.T.; Xia, T.; You, Y.W.; Zhang, Q.C.; Ni, H.S.; Liu, Y.F.; Liu, Y.R.; Xu, Y.Q.; You, B.; Zhang, Z.X. Characteristics and clinical significance of polyploid giant cancer cells in laryngeal carcinoma. Laryngoscope Investig. Otolaryngol. 2021, 6, 1228–1234. [Google Scholar] [CrossRef]
- Fei, F.; Zhang, D.; Yang, Z.; Wang, S.; Wang, X.; Wu, Z.; Wu, Q.; Zhang, S. The number of polyploid giant cancer cells and epithelial–mesenchymal transition-related proteins are associated with invasion and metastasis in human breast cancer. J. Exp. Clin. Cancer Res. 2015, 34, 158. [Google Scholar] [CrossRef] [Green Version]
- Gerashchenko, B.I.; Salmina, K.; Eglitis, J.; Huna, A.; Grjunberga, V.; Erenpreisa, J. Disentangling the aneuploidy and senescence paradoxes: A study of triploid breast cancers non-responsive to neoadjuvant therapy. Histochem. Cell Biol. 2016, 145, 497–508. [Google Scholar] [CrossRef]
- Lv, H.; Shi, Y.; Zhang, L.; Zhang, D.; Liu, G.; Yang, Z.; Li, Y.; Fei, F.; Zhang, S. Polyploid giant cancer cells with budding and the expression of cyclin E, S-phase kinase-associated protein 2, stathmin associated with the grading and metastasis in serous ovarian tumor. BMC Cancer 2014, 14, 576. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Yang, X.; Yang, Z.; Fei, F.; Li, S.; Qu, J.; Zhang, M.; Li, Y.; Zhang, X.; Zhang, S. Daughter cells and erythroid cells budding from PGCCs and their clinicopathological significances in colorectal cancer. J. Cancer 2017, 8, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Beltran, A.; Eble, J.N.; Bostwick, D.G. Pleomorphic giant cell carcinoma of the prostate. Arch. Pathol. Lab. Med. 2005, 129, 683–685. [Google Scholar] [CrossRef]
- Mannan, R.; Wang, X.; Bawa, P.S.; Spratt, D.E.; Wilson, A.; Jentzen, J.; Chinnaiyan, A.M.; Reichert, Z.R.; Mehra, R. Polypoidal giant cancer cells in metastatic castration-resistant prostate cancer: Observations from the Michigan Legacy Tissue Program. Med. Oncol. 2020, 37, 16. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, A.M.; De Marzo, A.M.; Hicks, J.L.; Lotan, T.L.; Epstein, J.I. Prostatic adenocarcinoma with focal pleomorphic giant cell features: A series of 30 cases. Am. J. Surg. Pathol. 2018, 42, 1286–1296. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [Green Version]
- Amend, S.R.; Torga, G.; Lin, K.C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef]
- Mirzayans, R.; Scott, A.; Cameron, M.; Murray, D. Induction of accelerated senescence by gamma radiation in human solid tumor-derived cell lines expressing wild-type TP53. Radiat. Res. 2005, 163, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Dhanyamraju, P.K.; Schell, T.D.; Amin, S.; Robertson, G.P. Drug-tolerant persister cells in cancer therapy resistance. Cancer Res. 2022, 82, 2503–2514. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Hansen, G.; Murray, D. Role of p16INK4A in replicative senescence and DNA damage-induced premature senescence in p53-deficient human cells. Biochem. Res. Int. 2012, 2012, 951574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Was, H.; Czarnecka, J.; Kominek, A.; Barszcz, K.; Bernas, T.; Piwocka, K.; Kaminska, B. Some chemotherapeutics-treated colon cancer cells display a specific phenotype being a combination of stem-like and senescent cell features. Cancer Biol. The. 2018, 19, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Czarnecka-Herok, J.; Sliwinska, M.A.; Herok, M.; Targonska, A.; Strzeszewska-Potyrala, A.; Bojko, A.; Wolny, A.; Mosieniak, G.; Sikora, E. Therapy-induced senescent/polyploid cancer cells undergo atypical divisions associated with altered expression of meiosis, spermatogenesis and EMT genes. Int. J. Mol. Sci. 2022, 23, 8288. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Kumar, P.; Murray, D. Multinucleated giant cancer cells produced in response to ionizing radiation retain viability and replicate their genome. Int. J. Mol. Sci. 2017, 18, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzayans, R.; Andrais, B.; Murray, D. Do multiwell plate high throughput assays measure loss of cell viability following exposure to genotoxic agents? Int. J. Mol. Sci. 2017, 18, 1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzayans, R.; Andrais, B.; Murray, D. Viability assessment following anticancer treatment requires single-cell visualization. Cancers 2018, 10, 255. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Scott, A.; Murray, D. New insights into p53 signaling and cancer-cell response to DNA damage: Implications for cancer therapy. J. Biomed. Biotechnol. 2012, 2012, 170325. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Murray, D. Ionizing radiation-induced responses in human cells with differing TP53 status. Int. J. Mol. Sci. 2013, 14, 22409–22435. [Google Scholar]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. The growing complexity of cancer cell response to DNA-damaging agents: Caspase 3 mediates cell death or survival? Int. J. Mol. Sci. 2016, 17, 708. [Google Scholar] [CrossRef] [Green Version]
- Murray, D.; Mirzayans, R. Cellular responses to platinum-based anticancer drugs and UVC: Role of p53 and implications for cancer therapy. Int. J. Mol. Sci. 2020, 21, 5766. [Google Scholar] [CrossRef]
- Jänicke, R.U.; Sohn, D.; Schulze-Osthoff, K. The dark side of a tumor suppressor: Anti-apoptotic p53. Cell Death Differ. 2008, 15, 959–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, D.; Mirzayans, R.; McBride, W.H. Defenses against pro-oxidant forces—Maintenance of cellular and genomic integrity and longevity. Radiat. Res. 2018, 190, 331–349. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of wild-type p53 signaling in suppressing apoptosis in response to chemical genotoxic agents: Impact on chemotherapy outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar] [PubMed]
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017, 3, 17049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shekhar, M.P.V. The dark side of apoptosis. In Molecular Mechanisms of Tumor Cell Resistance to Chemotherapy, Resistance to Targeted Anti-Cancer Therapeutics 1; Bonavida, B., Ed.; Springer: New York, NY, USA, 2013; pp. 245–258. [Google Scholar] [CrossRef]
- Corsi, F.; Capradossi, F.; Pelliccia, A.; Briganti, S.; Bruni, E.; Traversa, E.; Torino, F.; Reichle, A.; Ghibelli, L. Apoptosis as driver of therapy-induced cancer repopulation and acquired cell-resistance (CRAC): A simple in vitro model of Phoenix Rising in prostate cancer. Int. J. Mol. Sci. 2022, 23, 1152. [Google Scholar] [CrossRef]
- Berthenet, K.; Castillo Ferrer, C.; Fanfone, D.; Popgeorgiev, N.; Neves, D.; Bertolino, P.; Gibert, B.; Hernandez-Vargas, H.; Ichim, G. Failed apoptosis enhances melanoma cancer cell aggressiveness. Cell Rep. 2020, 31, 107731. [Google Scholar] [CrossRef]
- Khatib, S.A.; Ma, L.; Dang, H.; Forgues, M.; Chung, J.-Y.; Ylaya, K.; Hewitt, S.M.; Chaisaingmongkol, J.; Rucchirawat, M.; Wang, X.W. Single-cell biology uncovers apoptotic cell death and its spatial organization as a potential modifier of tumor diversity in HCC. Hepatology 2022, 76, 599–611. [Google Scholar] [CrossRef]
- Brix, N.; Samaga, D.; Belka, C.; Zitzelsberger, H.; Lauber, K. Analysis of clonogenic growth in vitro. Nat. Protoc. 2021, 16, 4963–4991. [Google Scholar] [CrossRef]
- Puck, T.T.; Marcus, P.I. Action of X-rays on mammalian cells. J. Exp. Med. 1956, 103, 653–666. [Google Scholar] [CrossRef]
- Weinberg, R.A. Coming full circle-from endless complexity to simplicity and back again. Cell 2014, 157, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Pienta, K.J.; Hammarlund, E.U.; Austin, R.H.; Axelrod, R.; Brown, J.S.; Amend, S.R. Cancer cells employ an evolutionarily conserved polyploidization program to resist therapy. Semin. Cancer Biol. 2022, 81, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Kailen, W.G. Publish houses of brick, not mansions of straw. Nature 2017, 5454, 387. [Google Scholar]
- Kailen, W.G. Preclinical Cancer Target Validation: How Not to Be Wrong. NIH Wednesday Afternoon Lectures (WELS) Series. Available online: https://videocast.nih.gov/watch=27066 (accessed on 19 June 2023).
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Holly, E. Top Journals Retract DNA-Repair Studies after Misconduct Probe: Investigation Found That Science and Nature Papers Contained Data Falsified by One Author. Nature News. 2019. Available online: https://www.nature.com/articles/d41586-019-00406-4 (accessed on 19 June 2023).
- Hill, R.; Madureira, P.A.; Waisman, D.M.; Lee, P.W.K. DNA-PKCS binding to p53 on the p21WAF1/CIP1 promoter blocks transcription resulting in cell death. Oncotarget 2019, 10, 5572. [Google Scholar] [CrossRef] [PubMed]
- Raj, L.; Ide, T.; Gurkar, A.U.; Foley, M.; Schenone, M.; Li, X.; Tolliday, N.J.; Golub, T.R.; Carr, S.A.; Shamji, A.F.; et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2018, 561, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Csibi, A.; Yang, S.; Hoffman, G.R.; Li, C.; Zhang, E.; Yu, J.J.; Blenis, J. Synthetic lethality of combined glutaminase and Hsp90 inhibition in mTORC1-driven tumor cells. Proc. Natl. Acad. Sci USA 2023, 120, e2220405120. [Google Scholar] [CrossRef]
- Williams, C.L.; Casadevall, A.; Jackson, S. Figure errors, sloppy science, and fraud: Keeping eyes on your data. J. Clin. Invest. 2019, 129, 1805–1807. [Google Scholar] [CrossRef]
- Available online: https://retractionwatch.com/2015/07/06/cancer-research-retraction-is-fifth-for-robert-weinberg-fourth-for-his-former-student/ (accessed on 19 June 2023).
- Setton, J.; Zinda, M.; Riaz, N.; Durocher, D.; Zimmermann, M.; Koehler, M.; Reis-Filho, J.S.; Powell, S.N. Synthetic lethality in cancer therapeutics: The next generation. Cancer Discov. 2021, 11, 1626–1635. [Google Scholar] [CrossRef]
- Bruin, M.A.C.; Sonke, G.S.; Beijnen, J.H.; Huitema, A.D.R. Pharmacokinetics and pharmacodynamics of PARP inhibitors in oncology. Clin. Pharmacokinet. 2022, 61, 1649–1675. [Google Scholar] [CrossRef]
- Ryan, C.J.; Mehta, I.; Kebabci, N.; Adams, D.J. Targeting synthetic lethal paralogs in cancer. Trends Cancer 2023, 9, 397–409. [Google Scholar] [CrossRef]
- Hass, R.; von der Ohe, J.; Dittmar, T. Hybrid Formation and Fusion of Cancer Cells In Vitro and In Vivo. Cancers 2021, 13, 4496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, H.; Yang, X.; Fan, L.; Tian, S.; Niu, R.; Yan, M.; Zheng, M.; Zhang, S. Cell fusion-related proteins and signaling pathways, and their roles in the development and progression of cancer. Front. Cell Dev. Biol. 2022, 9, 809668. [Google Scholar] [CrossRef]
- Laberge, G.S.; Duvall, E.; Haedicke, K.; Pawelek, J. Leukocyte–cancer cell fusion—Genesis of a deadly journey. Cells 2019, 8, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabo, I.; Svanvik, J.; Lindström, A.; Lechertier, T.; Trabulo, S.; Hulit, J.; Sparey, T.; Pawelek, J. Roles of cell fusion, hybridization and polyploid cell formation in cancer metastasis. World J. Clin. Oncol. 2020, 11, 121–135. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Date | Cancer Type | No of Patients | Outcome |
|---|---|---|---|---|
| Wang et al. [22] | 2012 | Non-small-cell lung cancer | 18 | Patients expressing markers of senescence following neoadjuvant therapy had a significantly worse prognosis than patients who did not express these markers. |
| Qu et al. [37] | 2013 | Glioma | 76 | The number of PGCCs increased with the grade of tumors. |
| Lv et al. [42] | 2014 | Serous ovarian cancer | 80 | The presence of PGCCs in the primary tumor correlated with metastasis. |
| Fei et al. [40] | 2015 | Primary breast tumors, lymph node metastases, and benign tissue | 167 | The number of PGCCs was the highest in patients with lymph node metastases. |
| Gerashchenko et al. [41] | 2016 | Breast cancer | 30 | Tumors with a higher proportion of PGCCs showed a poorer response to neoadjuvant chemotherapy. |
| Zhang et al. [43] | 2017 | Colon cancer | 169 | The presence of PGCCs with budding increased as tumors became more dedifferentiated. |
| Liu et al. [38] | 2018 | Anorectal melanoma | 47 | The proportion of PGCCs increased with tumor size. |
| Alharbi et al. [46] | 2018 | Prostate cancer | 30 | Pleomorphic giant cells were present in all 30 patients with a rare variant of prostate cancer. |
| Mannan et al. [45] | 2020 | Prostate cancer | 5 | Multiple cells with highly irregular polylobulated nuclei or multiple pleomorphic nuclei were present in autopsy samples of patients who had failed multiple lines of therapy. |
| Liu et al. [39] | 2021 | Laryngeal cancer | 102 | High numbers of PGCCs correlated with poor prognosis. |
| Trabzonlu et al. [36] | 2023 | Prostate cancer | 209 | PGCCs were significant prognostic factors for metastasis in patients who underwent radical prostatectomy with curative intent to treat their presumed localized cancer. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirzayans, R.; Murray, D. Intratumor Heterogeneity and Treatment Resistance of Solid Tumors with a Focus on Polyploid/Senescent Giant Cancer Cells (PGCCs). Int. J. Mol. Sci. 2023, 24, 11534. https://doi.org/10.3390/ijms241411534
Mirzayans R, Murray D. Intratumor Heterogeneity and Treatment Resistance of Solid Tumors with a Focus on Polyploid/Senescent Giant Cancer Cells (PGCCs). International Journal of Molecular Sciences. 2023; 24(14):11534. https://doi.org/10.3390/ijms241411534
Chicago/Turabian StyleMirzayans, Razmik, and David Murray. 2023. "Intratumor Heterogeneity and Treatment Resistance of Solid Tumors with a Focus on Polyploid/Senescent Giant Cancer Cells (PGCCs)" International Journal of Molecular Sciences 24, no. 14: 11534. https://doi.org/10.3390/ijms241411534