
An Elusive Target: Inhibitors of JC Polyomavirus Infection and Their Development as Therapeutics for the Treatment of Progressive Multifocal Leukoencephalopathy


Abstract
:1. Introduction
2. Progressive Multifocal Leukoencephalopathy
2.1. History
2.2. Clinical Presentation
2.3. Management
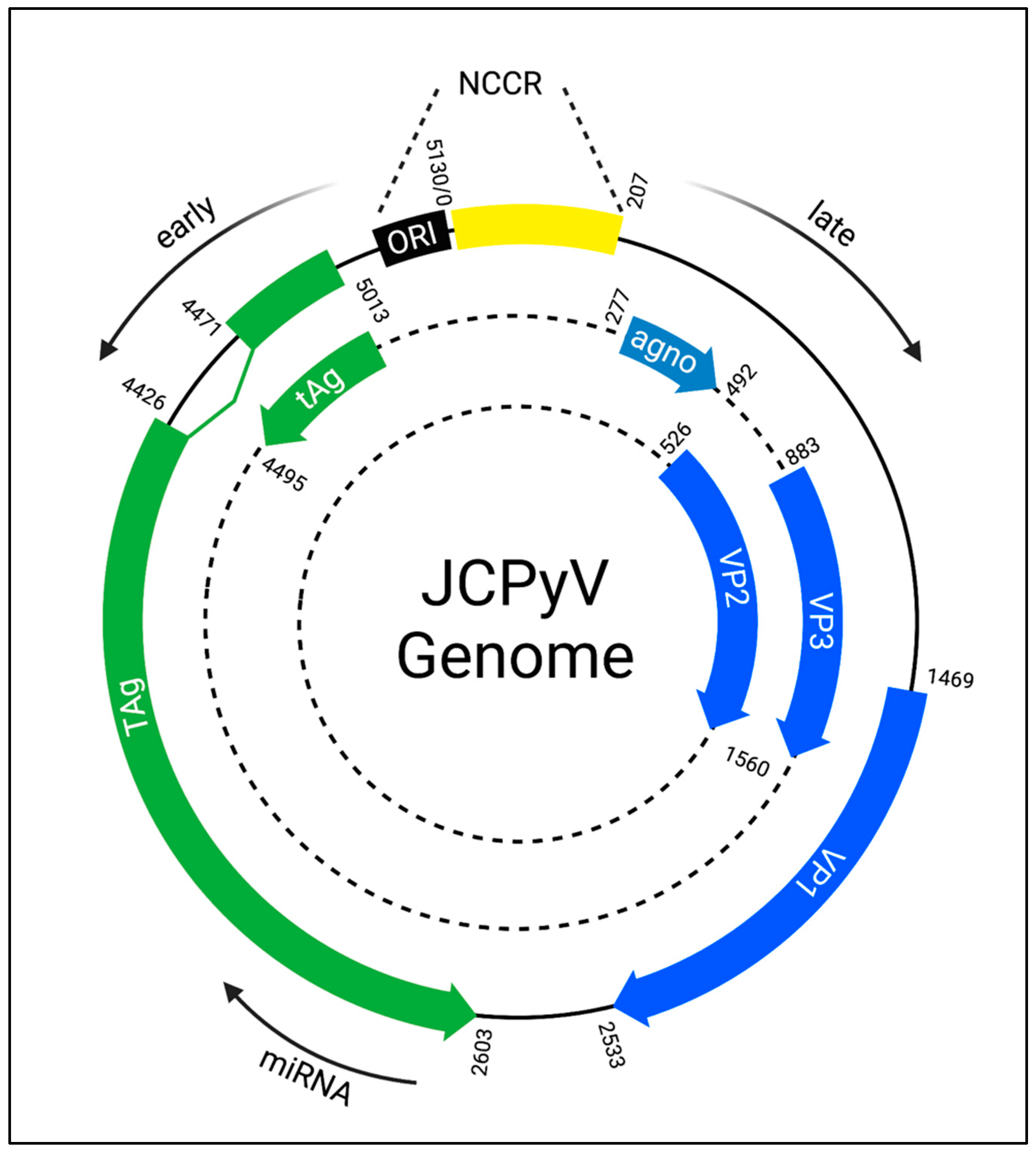
3. JCPyV Genome
4. Inhibitors of JCPyV Infection and Spread
4.1. Attachment Inhibitors
4.2. Entry Inhibitors
4.3. Endosome Acidification Inhibitors
4.4. Signaling Inhibitors
4.5. Trafficking Inhibitors
4.6. Uncoating Inhibitors
4.7. Nuclear Transport Inhibitors
4.8. Large T-Antigen Inhibitors
4.9. Replication Inhibitors
4.10. Extracellular Vesicle Association Inhibitors
4.11. Inhibitors of Unknown Mechanisms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism | Target | Compound Name | Year | Reference |
|---|---|---|---|---|
| Attachment inhibitor | LSTc binding pocket | AY4 | 2015 | [65] |
| Soluble LSTc | 2013 | [64] | ||
| Entry inhibitor | 5-HT2R | Clozapine | 2003 | [72] |
| Mirtazapine | 2008 | [71] | ||
| Cyproheptadine | ||||
| Risperidone | ||||
| Ketanserin | 2004 | [66] | ||
| Ritanserin | ||||
| Mianserin | ||||
| Volinsanserin | ||||
| Metoclopramide | ||||
| 5-HT2R | Chlorpromazine | 2000 | [83] | |
| Clathrin-mediated endocytosis | ||||
| Clathrin terminal domain | Pitstop2 | 2019 | [68] | |
| Dynamin | Dynole 34-2 | |||
| Macropinocytotis | EIPA | 2020 | [85] | |
| Endosomal acidification inhibitor | Unknown | Mefloquine | 2009 | [92] |
| Na+/H+ ATPase | Monensin | 2013 | [91] | |
| Vacuolar H+ ATPase | Bafilomycin A1 | |||
| Uncoating inhibitor | SERCA | Thapsigargin | ||
| Receptor tyrosine kinase inhibitor | Receptor tyrosine kinase | Genistein | 2004 | [70] |
| MAPK-ERK signaling inhibitor | c-Raf | GW-5074 | 2023 | [150] |
| Sorafenib | 2019 | [101] | ||
| MEK1/2 | U0126 | 2018 | [67] | |
| PD98059 | ||||
| PI3K-AKT-mTOR signaling inhibitor | AKT | MK2206 | 2021 | [97] |
| PI3K | Wortmannin | |||
| mTOR | PP242 | |||
| Rapamycin | ||||
| Ca2+ signaling | Calcineurin | Cyclosporine | 2012 | [108] |
| L-type Ca2+ channels | Topiramate | 2020 | [107] | |
| Phospholipase C | U73122 | 2018 | [109] | |
| IP3R | 2-APB | |||
| Xestospongin C | ||||
| Cyclin-dependent kinase inhibitor | CDK2, CDK7, CDK9 | Roscovitine | 2008 | [110] |
| Cytoskeleton disruptors | -tubulin | Nocodazole | 2003 | [90] |
| F-actin | Cytochalasin D | |||
| Retrograde transport inhibitors | ASNA1 | Retro-2cycl | 2013 | [111] |
| DHQZ36 | ||||
| Retro-2.1 | 2020 | [112] | ||
| ADP-ribosylation factor | Brefeldin A | 2006 | [89] | |
| TAg inhibitor | TAg ATPase | SMAL | 2016 | [120] |
| AMT580-043 | ||||
| LDN-0012754 | 2014 | [117] | ||
| Nucleoside analog | DNA Polymerase | Cytarabine | 1974 | [121,122] |
| Nucleotide analog | DNA Polymerase | Cidofovir | ||
| Brincidofovir | 2010 | [129,130,132] | ||
| Pyrimidine synthesis inhibitor | Dihydroorate dehydrogenase | Teriflunomide | 2006 | [133] |
| Topoisomerase inhibitor | Topoisomerase | -lapachone | 2016 | [138] |
| Topotecan | ||||
| CPT11 | 2017 | [143] | ||
| DNA repair inhibitor | PARP-1 | 3-AB | 2013 | [145] |
| Nuclear export inhibitor | SINE | Verdinexor | 2018 | [115] |
| Extracellular vesicle association inhibitor | nSMase2 | Cambinol | 2022 | [87] |
| Unknown | Unknown | Artesunate | 2014 | [146] |
| Ellagic acid | 2009 | [149] | ||
| Spiperone |
5. GW-5074: A Novel JCPyV Inhibitor That Antagonizes Signaling Events
6. Current Obstacles in PML Drug Development
6.1. Obstacle 1: PML Is a Rare Disease
6.2. Obstacle 2: No Reliable Animal Model for PML Exists
6.3. Obstacle 3: Few Compounds Accumulate in the CNS
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Polyomaviridae Study Group of the International Committee on Taxonomy of Viruses; Calvignac-Spencer, S.; Feltkamp, M.C.; Daugherty, M.D.; Moens, U.; Ramqvist, T.; Johne, R.; Ehlers, B. A taxonomy update for the family Polyomaviridae. Arch. Virol. 2016, 161, 1739–1750. [Google Scholar] [CrossRef]
- Moens, U.; Calvignac-Spencer, S.; Lauber, C.; Ramqvist, T.; Feltkamp, M.C.W.; Daugherty, M.D.; Verschoor, E.J.; Ehlers, B.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Polyomaviridae. J. Gen. Virol. 2017, 98, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Schmidlin, K.; Kraberger, S.; Cook, C.; DeNardo, D.F.; Fontenele, R.S.; Van Doorslaer, K.; Martin, D.P.; Buck, C.B.; Varsani, A. A novel lineage of polyomaviruses identified in bark scorpions. Virology 2021, 563, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Muller, H. Polyomaviruses of birds: Etiologic agents of inflammatory diseases in a tumor virus family. J. Virol. 2007, 81, 11554–11559. [Google Scholar] [CrossRef]
- Anthony, S.J.; St Leger, J.A.; Navarrete-Macias, I.; Nilson, E.; Sanchez-Leon, M.; Liang, E.; Seimon, T.; Jain, K.; Karesh, W.; Daszak, P.; et al. Identification of a novel cetacean polyomavirus from a common dolphin (Delphinus delphis) with Tracheobronchitis. PLoS ONE 2013, 8, e68239. [Google Scholar] [CrossRef] [PubMed]
- Sweet, B.H.; Hilleman, M.R. The vacuolating virus, S.V. 40. Proc. Soc. Exp. Biol. Med. 1960, 105, 420–427. [Google Scholar] [CrossRef]
- Wu, Z.; Graf, F.E.; Hirsch, H.H. Antivirals against human polyomaviruses: Leaving no stone unturned. Rev. Med. Virol. 2021, 31, e2220. [Google Scholar] [CrossRef]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef]
- Egli, A.; Infanti, L.; Dumoulin, A.; Buser, A.; Samaridis, J.; Stebler, C.; Gosert, R.; Hirsch, H.H. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J. Infect. Dis. 2009, 199, 837–846. [Google Scholar] [CrossRef]
- Kean, J.M.; Rao, S.; Wang, M.; Garcea, R.L. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009, 5, e1000363. [Google Scholar] [CrossRef] [PubMed]
- Gossai, A.; Waterboer, T.; Nelson, H.H.; Michel, A.; Willhauck-Fleckenstein, M.; Farzan, S.F.; Hoen, A.G.; Christensen, B.C.; Kelsey, K.T.; Marsit, C.J.; et al. Seroepidemiology of Human Polyomaviruses in a US Population. Am. J. Epidemiol. 2016, 183, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Haley, S.A.; Atwood, W.J. Progressive Multifocal Leukoencephalopathy: Endemic Viruses and Lethal Brain Disease. Annu. Rev. Virol. 2017, 4, 349–367. [Google Scholar] [CrossRef] [PubMed]
- Assetta, B.; Atwood, W.J. The biology of JC polyomavirus. Biol. Chem. 2017, 398, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, A.L.; Atwood, W.J. Fifty Years of JC Polyomavirus: A Brief Overview and Remaining Questions. Viruses 2020, 12, 969. [Google Scholar] [CrossRef]
- Berger, J.R.; Aksamit, A.J.; Clifford, D.B.; Davis, L.; Koralnik, I.J.; Sejvar, J.J.; Bartt, R.; Major, E.O.; Nath, A. PML diagnostic criteria: Consensus statement from the AAN Neuroinfectious Disease Section. Neurology 2013, 80, 1430–1438. [Google Scholar] [CrossRef]
- Astrom, K.E.; Mancall, E.L.; Richardson, E.P., Jr. Progressive multifocal leuko-encephalopathy: A hitherto unrecognized complication of chronic lymphatic leukaemia and Hodgkin’s disease. Brain 1958, 81, 93–111. [Google Scholar] [CrossRef]
- Padgett, B.L.; Walker, D.L.; Zurhein, G.M.; Eckroade, R.J.; Dessel, B.H. Cultivation of Papova-Like Virus from Human Brain with Progressive Multifocal Leucoencephalopathy. Lancet 1971, 1, 1257–1260. [Google Scholar] [CrossRef]
- Padgett, B.L.; Walker, D.L.; ZuRhein, G.M.; Hodach, A.E.; Chou, S.M. JC Papovavirus in progressive multifocal leukoencephalopathy. J. Infect. Dis. 1976, 133, 686–690. [Google Scholar] [CrossRef]
- Zurhein, G.; Chou, S.M. Particles Resembling Papova Viruses in Human Cerebral Demyelinating Disease. Science 1965, 148, 1477–1479. [Google Scholar] [CrossRef]
- Brooks, B.R.; Walker, D.L. Progressive multifocal leukoencephalopathy. Neurol. Clin. 1984, 2, 299–313. [Google Scholar] [CrossRef]
- Berger, J.R.; Major, E.O. Progressive multifocal leukoencephalopathy. Semin. Neurol. 1999, 19, 193–200. [Google Scholar] [CrossRef]
- Miller, J.R.; Barrett, R.E.; Britton, C.B.; Tapper, M.L.; Bahr, G.S.; Bruno, P.J.; Marquardt, M.D.; Hays, A.P.; McMurtry, J.G., 3rd; Weissman, J.B.; et al. Progressive multifocal leukoencephalopathy in a male homosexual with T-cell immune deficiency. N. Engl. J. Med. 1982, 307, 1436–1438. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.L.Y.; Holman, R.C.; Hammett, T.A.; Belay, E.D.; Schonberger, L.B. Progressive Multifocal Leukoencephalopathy Deaths in the USA, 1979–2005. Neuroepidemiology 2010, 35, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Sacktor, N. The epidemiology of human immunodeficiency virus-associated neurological disease in the era of highly active antiretroviral therapy. J. Neurovirol. 2002, 8 (Suppl. S2), 115–121. [Google Scholar] [CrossRef] [PubMed]
- Aksamit, A.J.; Gendelman, H.E.; Orenstein, J.M.; Pezeshkpour, G.H. AIDS-associated progressive multifocal leukoencephalopathy (PML): Comparison to non-AIDS PML with in situ hybridization and immunohistochemistry. Neurology 1990, 40, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, R.H.; Ward, J.M.; Walker, D.L.; Ross, A.A. Progressive multifocal leukoencephalopathy and retroviral encephalitis in acquired immunodeficiency syndrome. Arch. Pathol. Lab. Med. 1988, 112, 1207–1213. [Google Scholar]
- Berger, J.R.; Kaszovitz, B.; Post, M.J.; Dickinson, G. Progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. A review of the literature with a report of sixteen cases. Ann. Intern. Med. 1987, 107, 78–87. [Google Scholar] [CrossRef]
- Van Assche, G.; Van Ranst, M.; Sciot, R.; Dubois, B.; Vermeire, S.; Noman, M.; Verbeeck, J.; Geboes, K.; Robberecht, W.; Rutgeerts, P. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N. Engl. J. Med. 2005, 353, 362–368. [Google Scholar] [CrossRef]
- Kleinschmidt-DeMasters, B.K.; Tyler, K.L. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N. Engl. J. Med. 2005, 353, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Langer-Gould, A.; Atlas, S.W.; Green, A.J.; Bollen, A.W.; Pelletier, D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 2005, 353, 375–381. [Google Scholar] [CrossRef]
- Williamson, E.M.L.; Berger, J.R. Diagnosis and Treatment of Progressive Multifocal Leukoencephalopathy Associated with Multiple Sclerosis Therapies. Neurotherapeutics 2017, 14, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.M.; Berger, J.R. Infection risk in patients on multiple sclerosis therapeutics. CNS Drugs 2015, 29, 229–244. [Google Scholar] [CrossRef]
- Berger, J.R.; Fox, R.J. Reassessing the risk of natalizumab-associated PML. J. Neurovirol. 2016, 22, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Major, E.O.; Nath, A. A link between long-term natalizumab dosing in MS and PML: Putting the puzzle together. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e235. [Google Scholar] [CrossRef]
- Zhovtis Ryerson, L.; Frohman, T.C.; Foley, J.; Kister, I.; Weinstock-Guttman, B.; Tornatore, C.; Pandey, K.; Donnelly, S.; Pawate, S.; Bomprezzi, R.; et al. Extended interval dosing of natalizumab in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Setien, S.; Sanchez de la Torre, J.R.; Torres-Barquin, M.; Misiego, M.; Perez, J.L.; Castillo-Trivino, T.; Menendez-Garcia, C.; Delgado-Alvarado, M. Does Extended Interval Dosing Natalizumab Preserve Effectiveness in Multiple Sclerosis? A 7 Year-Retrospective Observational Study. Front. Immunol. 2021, 12, 614715. [Google Scholar] [CrossRef]
- Chang, I.; Muralidharan, K.K.; Campbell, N.; Ho, P.R. Modeling the Efficacy of Natalizumab in Multiple Sclerosis Patients Who Switch from Every-4-Week Dosing to Extended-Interval Dosing. J. Clin. Pharmacol. 2021, 61, 339–348. [Google Scholar] [CrossRef]
- Berger, J.R.; Hartung, H.P. Commentary: Progressive multifocal leukoencephalopathy genetic risk variants for pharmacovigilance of immunosuppressant therapies. Front. Neurol. 2023, 14, 1146027. [Google Scholar] [CrossRef]
- Hatchwell, E.; Smith, E.B., 3rd; Jalilzadeh, S.; Bruno, C.D.; Taoufik, Y.; Hendel-Chavez, H.; Liblau, R.; Brassat, D.; Martin-Blondel, G.; Wiendl, H.; et al. Progressive multifocal leukoencephalopathy genetic risk variants for pharmacovigilance of immunosuppressant therapies. Front. Neurol. 2022, 13, 1016377. [Google Scholar] [CrossRef]
- Kartau, M.; Verkkoniemi-Ahola, A.; Paetau, A.; Palomaki, M.; Janes, R.; Ristola, M.; Lappalainen, M.; Anttila, V.J. The Incidence and Predisposing Factors of John Cunningham Virus-Induced Progressive Multifocal Leukoencephalopathy in Southern Finland: A Population-Based Study. Open Forum Infect. Dis. 2019, 6, ofz024. [Google Scholar] [CrossRef]
- Ferenczy, M.W.; Marshall, L.J.; Nelson, C.D.; Atwood, W.J.; Nath, A.; Khalili, K.; Major, E.O. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin. Microbiol. Rev. 2012, 25, 471–506. [Google Scholar] [CrossRef] [PubMed]
- Richardson, E.P., Jr. Progressive multifocal leukoencephalopathy. N. Engl. J. Med. 1961, 265, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Joly, M.; Conte, C.; Cazanave, C.; Le Moing, V.; Tattevin, P.; Delobel, P.; Sommet, A.; Martin-Blondel, G. Progressive multifocal leukoencephalopathy: Epidemiology and spectrum of predisposing conditions. Brain 2023, 146, 349–358. [Google Scholar] [CrossRef]
- Antinori, A.; Cingolani, A.; Lorenzini, P.; Giancola, M.L.; Uccella, I.; Bossolasco, S.; Grisetti, S.; Moretti, F.; Vigo, B.; Bongiovanni, M.; et al. Clinical epidemiology and survival of progressive multifocal leukoencephalopathy in the era of highly active antiretroviral therapy: Data from the Italian Registry Investigative Neuro AIDS (IRINA). J. Neurovirol. 2003, 9 (Suppl. S1), 47–53. [Google Scholar] [CrossRef]
- Engsig, F.N.; Hansen, A.B.; Omland, L.H.; Kronborg, G.; Gerstoft, J.; Laursen, A.L.; Pedersen, C.; Mogensen, C.B.; Nielsen, L.; Obel, N. Incidence, clinical presentation, and outcome of progressive multifocal leukoencephalopathy in HIV-infected patients during the highly active antiretroviral therapy era: A nationwide cohort study. J. Infect. Dis. 2009, 199, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Dahlhaus, S.; Hoepner, R.; Chan, A.; Kleiter, I.; Adams, O.; Lukas, C.; Hellwig, K.; Gold, R. Disease course and outcome of 15 monocentrically treated natalizumab-associated progressive multifocal leukoencephalopathy patients. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Kartau, M.; Sipila, J.O.T.; Auvineri, E.; Palomaki, M.; Verkkoniemi-Ahola, A. Progressive Multifocal Leukoencephalopathy: Current Insights. Degener. Neurol. Neuromuscul. Dis. 2019, 9, 109–121. [Google Scholar] [CrossRef]
- Himedan, M.; Camelo-Piragua, S.; Mills, E.A.; Gupta, A.; Aburashed, R.; Mao-Draayer, Y. Pathologic Findings of Chronic PML-IRIS in a Patient with Prolonged PML Survival Following Natalizumab Treatment. J. Investig. Med. High Impact Case Rep. 2017, 5, 2324709617734248. [Google Scholar] [CrossRef]
- Tan, K.; Roda, R.; Ostrow, L.; McArthur, J.; Nath, A. PML-IRIS in patients with HIV infection: Clinical manifestations and treatment with steroids. Neurology 2009, 72, 1458–1464. [Google Scholar] [CrossRef]
- Vidal, L.; Gafter-Gvili, A.; Salles, G.; Dreyling, M.H.; Ghielmini, M.; Schmitz, S.F.H.; Pettengell, R.; Witzens-Harig, M.; Shpilberg, O. Rituximab Maintenance for the Treatment of Patients With Follicular Lymphoma: An Updated Systematic Review and Meta-analysis of Randomized Trials. JNCI J. Natl. Cancer Inst. 2011, 103, 1799–1806. [Google Scholar] [CrossRef]
- Berger, J.R.; Cree, B.A.; Greenberg, B.; Hemmer, B.; Ward, B.J.; Dong, V.M.; Merschhemke, M. Progressive multifocal leukoencephalopathy after fingolimod treatment. Neurology 2018, 90, E1815–E1821. [Google Scholar] [CrossRef]
- Reddy, V.B.; Thimmappaya, B.; Dhar, R.; Subramanian, K.N.; Zain, B.S.; Pan, J.; Ghosh, P.K.; Celma, M.L.; Weissman, S.M. The genome of simian virus 40. Science 1978, 200, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Frisque, R.J.; Bream, G.L.; Cannella, M.T. Human polyomavirus JC virus genome. J. Virol. 1984, 51, 458–469. [Google Scholar] [CrossRef] [PubMed]
- White, M.K.; Safak, M.; Khalili, K. Regulation of gene expression in primate polyomaviruses. J. Virol. 2009, 83, 10846–10856. [Google Scholar] [CrossRef] [PubMed]
- Agostini, H.T.; Ryschkewitsch, C.F.; Singer, E.J.; Stoner, G.L. JC virus regulatory region rearrangements and genotypes in progressive multifocal leukoencephalopathy: Two independent aspects of virus variation. J. Gen. Virol. 1997, 78 Pt 3, 659–664. [Google Scholar] [CrossRef]
- Takahashi, K.; Sato, Y.; Sekizuka, T.; Kuroda, M.; Suzuki, T.; Hasegawa, H.; Katano, H. High expression of JC polyomavirus-encoded microRNAs in progressive multifocal leukoencephalopathy tissues and its repressive role in virus replication. PLoS Pathog. 2020, 16, e1008523. [Google Scholar] [CrossRef]
- Kumar, N.; Sharma, S.; Kumar, R.; Tripathi, B.N.; Barua, S.; Ly, H.; Rouse, B.T. Host-Directed Antiviral Therapy. Clin. Microbiol. Rev. 2020, 33, e00168-19. [Google Scholar] [CrossRef]
- Chitalia, V.C.; Munawar, A.H. A painful lesson from the COVID-19 pandemic: The need for broad-spectrum, host-directed antivirals. J. Transl. Med. 2020, 18, 390. [Google Scholar] [CrossRef]
- Mayberry, C.L.; Bond, A.C.; Wilczek, M.P.; Mehmood, K.; Maginnis, M.S. Sending mixed signals: Polyomavirus entry and trafficking. Curr. Opin. Virol. 2021, 47, 95–105. [Google Scholar] [CrossRef]
- Matrosovich, M.; Herrler, G.; Klenk, H.D. Sialic Acid Receptors of Viruses. Top. Curr. Chem. 2015, 367, 1–28. [Google Scholar] [CrossRef]
- Neu, U.; Maginnis, M.S.; Palma, A.S.; Stroh, L.J.; Nelson, C.D.; Feizi, T.; Atwood, W.J.; Stehle, T. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 2010, 8, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Dugan, A.S.; Gasparovic, M.L.; Atwood, W.J. Direct correlation between sialic acid binding and infection of cells by two human polyomaviruses (JC virus and BK virus). J. Virol. 2008, 82, 2560–2564. [Google Scholar] [CrossRef] [PubMed]
- Havranek, B.; Chan, K.K.; Wu, A.; Procko, E.; Islam, S.M. Computationally Designed ACE2 Decoy Receptor Binds SARS-CoV-2 Spike (S) Protein with Tight Nanomolar Affinity. J. Chem. Inf. Model. 2021, 61, 4656–4669. [Google Scholar] [CrossRef] [PubMed]
- Assetta, B.; Maginnis, M.S.; Gracia Ahufinger, I.; Haley, S.A.; Gee, G.V.; Nelson, C.D.; O’Hara, B.A.; Allen Ramdial, S.A.; Atwood, W.J. 5-HT2 receptors facilitate JC polyomavirus entry. J. Virol. 2013, 87, 13490–13498. [Google Scholar] [CrossRef] [PubMed]
- Yatawara, A.; Gaidos, G.; Rupasinghe, C.N.; O’Hara, B.A.; Pellegrini, M.; Atwood, W.J.; Mierke, D.F. Small-molecule inhibitors of JC polyomavirus infection. J. Pept. Sci. 2015, 21, 236–242. [Google Scholar] [CrossRef]
- Elphick, G.F.; Querbes, W.; Jordan, J.A.; Gee, G.V.; Eash, S.; Manley, K.; Dugan, A.; Stanifer, M.; Bhatnagar, A.; Kroeze, W.K.; et al. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science 2004, 306, 1380–1383. [Google Scholar] [CrossRef]
- DuShane, J.K.; Wilczek, M.P.; Mayberry, C.L.; Maginnis, M.S. ERK Is a Critical Regulator of JC Polyomavirus Infection. J. Virol. 2018, 92, e01529-17. [Google Scholar] [CrossRef]
- Mayberry, C.L.; Soucy, A.N.; Lajoie, C.R.; DuShane, J.K.; Maginnis, M.S. JC Polyomavirus Entry by Clathrin-Mediated Endocytosis Is Driven by beta-Arrestin. J. Virol. 2019, 93, e01948-18. [Google Scholar] [CrossRef]
- DuShane, J.K.; Mayberry, C.L.; Wilczek, M.P.; Nichols, S.L.; Maginnis, M.S. JCPyV-Induced MAPK Signaling Activates Transcription Factors during Infection. Int. J. Mol. Sci. 2019, 20, 4779. [Google Scholar] [CrossRef]
- Querbes, W.; Benmerah, A.; Tosoni, D.; Di Fiore, P.P.; Atwood, W.J. A JC virus-induced signal is required for infection of glial cells by a clathrin- and eps15-dependent pathway. J. Virol. 2004, 78, 250–256. [Google Scholar] [CrossRef]
- O’Hara, B.A.; Atwood, W.J. Interferon beta1-a and selective anti-5HT(2a) receptor antagonists inhibit infection of human glial cells by JC virus. Virus Res. 2008, 132, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Baum, S.; Ashok, A.; Gee, G.; Dimitrova, S.; Querbes, W.; Jordan, J.; Atwood, W.J. Early events in the life cycle of JC virus as potential therapeutic targets for the treatment of progressive multifocal leukoencephalopathy. J. Neurovirol. 2003, 9 (Suppl. S1), 32–37. [Google Scholar] [CrossRef]
- Goereci, Y.; Schweitzer, F.; Wellstein, A.; Silling, S.; Borchmann, S.; von Tresckow, B.; Adams, O.; Martin, R.; Schlamann, M.; Schroeter, M.; et al. Clearance of JC polyomavirus from cerebrospinal fluid following treatment with interleukin-2 and pembrolizumab in an individual with progressive multifocal leukoencephalopathy and no underlying immune deficiency syndrome. Eur. J. Neurol. 2020, 27, 2375–2377. [Google Scholar] [CrossRef] [PubMed]
- Trentalange, A.; Calcagno, A.; Ghisetti, V.; Atzori, C.; Busolli, P.; Bonora, S.; Imperiale, D. Clearance of cerebrospinal fluid JCV DNA with mirtazapine in a patient with progressive multifocal leukoencephalopathy and sarcoidosis. Antivir. Ther. 2016, 21, 633–635. [Google Scholar] [CrossRef] [PubMed]
- Cettomai, D.; McArthur, J.C. Mirtazapine use in human immunodeficiency virus-infected patients with progressive multifocal leukoencephalopathy. Arch. Neurol. 2009, 66, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Nambirajan, A.; Suri, V.; Kataria, V.; Sharma, M.C.; Goyal, V. Progressive multifocal leukoencephalopathy in a 44-year old male with idiopathic CD4+ T-lymphocytopenia treated with mirtazapine and mefloquine. Neurol. India 2017, 65, 1061–1064. [Google Scholar] [CrossRef]
- Cifci, B.; Yildiz, Y.; Altin, E.; Habibi, H.; Kocer, B.; Dizbay, M. Successful treatment of HIV-associated progressive multifocal leukoencephalopathy (PML) with mirtazapine, mefloquine, and IVIG combination therapy: A case report. J. Neurovirol. 2023, 29, 111–115. [Google Scholar] [CrossRef]
- Graf, L.M.; Rosenkranz, S.C.; Holzemer, A.; Hagel, C.; Goebell, E.; Jordan, S.; Friese, M.A.; Addo, M.M.; Schulze Zur Wiesch, J.; Beisel, C. Clinical Presentation and Disease Course of 37 Consecutive Cases of Progressive Multifocal Leukoencephalopathy (PML) at a German Tertiary-Care Hospital: A Retrospective Observational Study. Front. Neurol. 2021, 12, 632535. [Google Scholar] [CrossRef]
- Jamilloux, Y.; Kerever, S.; Ferry, T.; Broussolle, C.; Honnorat, J.; Seve, P. Treatment of Progressive Multifocal Leukoencephalopathy With Mirtazapine. Clin. Drug Investig. 2016, 36, 783–789. [Google Scholar] [CrossRef]
- Focosi, D.; Fazzi, R.; Montanaro, D.; Emdin, M.; Petrini, M. Progressive multifocal leukoencephalopathy in a haploidentical stem cell transplant recipient: A clinical, neuroradiological and virological response after treatment with risperidone. Antivir. Res. 2007, 74, 156–158. [Google Scholar] [CrossRef]
- Kast, R.E.; Focosi, D.; Petrini, M.; Altschuler, E.L. Treatment schedules for 5-HT2A blocking in progressive multifocal leukoencephalopathy using risperidone or ziprasidone. Bone Marrow Transplant. 2007, 39, 811–812. [Google Scholar] [CrossRef] [PubMed]
- Akagawa, Y.; Ueno, A.; Ikeda, J.; Ishii, W.; Shishido-Hara, Y.; Sekijima, Y. Two patients with progressive multifocal leukoencephalopathy with immune response against JC virus showing good long-term outcome by combination therapy of mefloquine, mirtazapine, and risperidone. Rinsho Shinkeigaku 2018, 58, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Pho, M.T.; Ashok, A.; Atwood, W.J. JC virus enters human glial cells by clathrin-dependent receptor-mediated endocytosis. J. Virol. 2000, 74, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, C.; Hochauf, K.; Rollig, C.; Schetelig, J.; Wunderlich, O.; Bandt, D.; Ehninger, G.; Jacobs, E.; Rohayem, J. Chlorpromazine combined with cidofovir for treatment of a patient suffering from progressive multifocal leukoencephalopathy. Intervirology 2007, 50, 412–417. [Google Scholar] [CrossRef]
- O’Hara, B.A.; Morris-Love, J.; Gee, G.V.; Haley, S.A.; Atwood, W.J. JC Virus infected choroid plexus epithelial cells produce extracellular vesicles that infect glial cells independently of the virus attachment receptor. PLoS Pathog. 2020, 16, e1008371. [Google Scholar] [CrossRef]
- Morris-Love, J.; Atwood, W.J. Complexities of JC Polyomavirus Receptor-Dependent and -Independent Mechanisms of Infection. Viruses 2022, 14, 1130. [Google Scholar] [CrossRef]
- Morris-Love, J.; O’Hara, B.A.; Gee, G.V.; Dugan, A.S.; O’Rourke, R.S.; Armstead, B.E.; Assetta, B.; Haley, S.A.; Atwood, W.J. Biogenesis of JC polyomavirus associated extracellular vesicles. J. Extracell. Biol. 2022, 1, e43. [Google Scholar] [CrossRef]
- Morris-Love, J.; Gee, G.V.; O’Hara, B.A.; Assetta, B.; Atkinson, A.L.; Dugan, A.S.; Haley, S.A.; Atwood, W.J.; Estes, M.K.; Imperiale, M.; et al. JC Polyomavirus Uses Extracellular Vesicles To Infect Target Cells. mBio 2019, 10, e00379-19. [Google Scholar] [CrossRef]
- Querbes, W.; O’Hara, B.A.; Williams, G.; Atwood, W.J. Invasion of host cells by JC virus identifies a novel role for caveolae in endosomal sorting of noncaveolar ligands. J. Virol. 2006, 80, 9402–9413. [Google Scholar] [CrossRef]
- Ashok, A.; Atwood, W.J. Contrasting roles of endosomal pH and the cytoskeleton in infection of human glial cells by JC virus and simian virus 40. J. Virol. 2003, 77, 1347–1356. [Google Scholar] [CrossRef]
- Gee, G.V.; O’Hara, B.A.; Derdowski, A.; Atwood, W.J. Pseudovirus mimics cell entry and trafficking of the human polyomavirus JCPyV. Virus Res. 2013, 178, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Brickelmaier, M.; Lugovskoy, A.; Kartikeyan, R.; Reviriego-Mendoza, M.M.; Allaire, N.; Simon, K.; Frisque, R.J.; Gorelik, L. Identification and characterization of mefloquine efficacy against JC virus in vitro. Antimicrob. Agents Chemother. 2009, 53, 1840–1849. [Google Scholar] [CrossRef]
- Clifford, D.B.; Nath, A.; Cinque, P.; Brew, B.J.; Zivadinov, R.; Gorelik, L.; Zhao, Z.; Duda, P. A study of mefloquine treatment for progressive multifocal leukoencephalopathy: Results and exploration of predictors of PML outcomes. J. Neurovirol. 2013, 19, 351–358. [Google Scholar] [CrossRef]
- Kurmann, R.; Weisstanner, C.; Kardas, P.; Hirsch, H.H.; Wiest, R.; Lammle, B.; Furrer, H.; Du Pasquier, R.; Bassetti, C.L.; Sturzenegger, M.; et al. Progressive multifocal leukoencephalopathy in common variable immunodeficiency: Mitigated course under mirtazapine and mefloquine. J. Neurovirol. 2015, 21, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Epperla, N.; Medina-Flores, R.; Mazza, J.J.; Yale, S.H. Mirtazapine and mefloquine therapy for non-AIDS-related progressive multifocal leukoencephalopathy. WMJ 2014, 113, 242–245. [Google Scholar] [PubMed]
- Moenster, R.P.; Jett, R.A. Mirtazapine and mefloquine therapy for progressive multifocal leukoencephalopathy in a patient infected with human immunodeficiency virus. Am. J. Health Syst. Pharm. 2012, 69, 496–498. [Google Scholar] [CrossRef]
- Wilczek, M.P.; Armstrong, F.J.; Mayberry, C.L.; King, B.L.; Maginnis, M.S. PI3K/AKT/mTOR Signaling Pathway Is Required for JCPyV Infection in Primary Astrocytes. Cells 2021, 10, 3218. [Google Scholar] [CrossRef]
- Wilczek, M.P.; Armstrong, F.J.; Geohegan, R.P.; Mayberry, C.L.; DuShane, J.K.; King, B.L.; Maginnis, M.S. The MAPK/ERK Pathway and the Role of DUSP1 in JCPyV Infection of Primary Astrocytes. Viruses 2021, 13, 1834. [Google Scholar] [CrossRef] [PubMed]
- Major, E.O.; Miller, A.E.; Mourrain, P.; Traub, R.G.; de Widt, E.; Sever, J. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc. Natl. Acad. Sci. USA 1985, 82, 1257–1261. [Google Scholar] [CrossRef]
- DuShane, J.K.; Maginnis, M.S. Human DNA Virus Exploitation of the MAPK-ERK Cascade. Int. J. Mol. Sci. 2019, 20, 3427. [Google Scholar] [CrossRef]
- DuShane, J.K.; Wilczek, M.P.; Crocker, M.A.; Maginnis, M.S. High-Throughput Characterization of Viral and Cellular Protein Expression Patterns during JC Polyomavirus Infection. Front. Microbiol. 2019, 10, 783. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Manley, K.; O’Hara, B.A.; Gee, G.V.; Sirakevich, C.P.; Sedivy, J.M.; Atwood, W.J. NFAT4 is required for JC virus infection of glial cells. J. Virol. 2006, 80, 12079–12085. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, B.A.; Gee, G.V.; Atwood, W.J.; Haley, S.A. Susceptibility of Primary Human Choroid Plexus Epithelial Cells and Meningeal Cells to Infection by JC Virus. J. Virol. 2018, 92, e00105-18. [Google Scholar] [CrossRef]
- Crocker, M.A. Development of a High-throughput Platform for the Determination of Antiviral Therapeutics. Master’s Thesis, The University of Maine, Orono, ME, USA, 2020. [Google Scholar]
- Wollebo, H.S.; Melis, S.; Khalili, K.; Safak, M.; White, M.K. Cooperative roles of NF-kappaB and NFAT4 in polyomavirus JC regulation at the KB control element. Virology 2012, 432, 146–154. [Google Scholar] [CrossRef]
- Soucy, A.N. Defining the Role of IP3R-Medicated er Calcium Flux in JC Polyomavirus Infection. Bachelor’s Thesis, Biochemistry. University of Maine, Orono, ME, USA, 2018. Available online: https://digitalcommons.library.umaine.edu/honors/354 (accessed on 24 April 2023).
- Orba, Y.; Sunden, Y.; Suzuki, T.; Nagashima, K.; Kimura, T.; Tanaka, S.; Sawa, H. Pharmacological cdk inhibitor R-Roscovitine suppresses JC virus proliferation. Virology 2008, 370, 173–183. [Google Scholar] [CrossRef]
- Nelson, C.D.; Carney, D.W.; Derdowski, A.; Lipovsky, A.; Gee, G.V.; O’Hara, B.; Williard, P.; DiMaio, D.; Sello, J.K.; Atwood, W.J. A retrograde trafficking inhibitor of ricin and Shiga-like toxins inhibits infection of cells by human and monkey polyomaviruses. mBio 2013, 4, e00729-13. [Google Scholar] [CrossRef] [PubMed]
- Treasure, T.; Nelson, C.D.S. Inhibition of JC polyomavirus infectivity by the retrograde transport inhibitor Retro-2.1. Microbiol. Immunol. 2020, 64, 783–791. [Google Scholar] [CrossRef]
- Qu, Q.; Sawa, H.; Suzuki, T.; Semba, S.; Henmi, C.; Okada, Y.; Tsuda, M.; Tanaka, S.; Atwood, W.J.; Nagashima, K. Nuclear entry mechanism of the human polyomavirus JC virus-like particle: Role of importins and the nuclear pore complex. J. Biol. Chem. 2004, 279, 27735–27742. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.M.; Zhao, L.; Bosard, C.; Imperiale, M.J. Role of a nuclear localization signal on the minor capsid proteins VP2 and VP3 in BKPyV nuclear entry. Virology 2015, 474, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Widman, D.G.; Gornisiewicz, S.; Shacham, S.; Tamir, S. In vitro toxicity and efficacy of verdinexor, an exportin 1 inhibitor, on opportunistic viruses affecting immunocompromised individuals. PLoS ONE 2018, 13, e0200043. [Google Scholar] [CrossRef] [PubMed]
- Pickens, J.A.; Tripp, R.A. Verdinexor Targeting of CRM1 is a Promising Therapeutic Approach against RSV and Influenza Viruses. Viruses 2018, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, P.; Zeng, G.; Bueno, M.; Salgarkar, A.; Lesniak, A.; Isse, K.; Seyb, K.; Perry, A.; Charles, I.; Hustus, C.; et al. Inhibition of large T antigen ATPase activity as a potential strategy to develop anti-polyomavirus JC drugs. Antivir. Res. 2014, 112, 113–119. [Google Scholar] [CrossRef]
- Meinke, G.; Phelan, P.J.; Kalekar, R.; Shin, J.; Archambault, J.; Bohm, A.; Bullock, P.A. Insights into the initiation of JC virus DNA replication derived from the crystal structure of the T-antigen origin binding domain. PLoS Pathog. 2014, 10, e1003966. [Google Scholar] [CrossRef]
- Topalis, D.; Andrei, G.; Snoeck, R. The large tumor antigen: A “Swiss Army knife” protein possessing the functions required for the polyomavirus life cycle. Antivir. Res. 2013, 97, 122–136. [Google Scholar] [CrossRef]
- Manos-Turvey, A.; Al-Ashtal, H.A.; Needham, P.G.; Hartline, C.B.; Prichard, M.N.; Wipf, P.; Brodsky, J.L. Dihydropyrimidinones and -thiones with improved activity against human polyomavirus family members. Bioorg. Med. Chem. Lett. 2016, 26, 5087–5091. [Google Scholar] [CrossRef]
- Conomy, J.P.; Beard, N.S.; Matsumoto, H.; Roessmann, U. Cytarabine treatment of progressive multifocal leukoencephalopathy. Clinical course and detection of virus-like particles after antiviral chemotherapy. JAMA 1974, 229, 1313–1316. [Google Scholar] [CrossRef]
- Hou, J.; Major, E.O. The efficacy of nucleoside analogs against JC virus multiplication in a persistently infected human fetal brain cell line. J. Neurovirol. 1998, 4, 451–456. [Google Scholar] [CrossRef]
- De Luca, A.; Giancola, M.L.; Cingolani, A.; Ammassari, A.; Gillini, L.; Murri, R.; Antinori, A. Clinical and virological monitoring during treatment with intrathecal cytarabine in patients with AIDS-associated progressive multifocal leukoencephalopathy. Clin. Infect. Dis. 1999, 28, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.D.; Dafni, U.; Simpson, D.; Clifford, D.; Wetherill, P.E.; Cohen, B.; McArthur, J.; Hollander, H.; Yainnoutsos, C.; Major, E.; et al. Failure of cytarabine in progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. AIDS Clinical Trials Group 243 Team. N. Engl. J. Med. 1998, 338, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Baker, W.J.; Royer, G.L., Jr.; Weiss, R.B. Cytarabine and neurologic toxicity. J. Clin. Oncol. 1991, 9, 679–693. [Google Scholar] [CrossRef]
- De Luca, A.; Ammassari, A.; Pezzotti, P.; Cinque, P.; Gasnault, J.; Berenguer, J.; Di Giambenedetto, S.; Cingolani, A.; Taoufik, Y.; Miralles, P.; et al. Cidofovir in addition to antiretroviral treatment is not effective for AIDS-associated progressive multifocal leukoencephalopathy: A multicohort analysis. AIDS 2008, 22, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, C.; Evers, S.; Nolting, T.; Arendt, G.; Husstedt, I.W. Cidofovir in combination with HAART and survival in AIDS-associated progressive multifocal leukoencephalopathy. J. Neurol. 2008, 255, 526–531. [Google Scholar] [CrossRef]
- Quenelle, D.C.; Lampert, B.; Collins, D.J.; Rice, T.L.; Painter, G.R.; Kern, E.R. Efficacy of CMX001 against herpes simplex virus infections in mice and correlations with drug distribution studies. J. Infect. Dis. 2010, 202, 1492–1499. [Google Scholar] [CrossRef]
- Jiang, Z.G.; Cohen, J.; Marshall, L.J.; Major, E.O. Hexadecyloxypropyl-cidofovir (CMX001) suppresses JC virus replication in human fetal brain SVG cell cultures. Antimicrob. Agents Chemother. 2010, 54, 4723–4732. [Google Scholar] [CrossRef]
- Gosert, R.; Rinaldo, C.H.; Wernli, M.; Major, E.O.; Hirsch, H.H. CMX001 (1-O-hexadecyloxypropyl-cidofovir) inhibits polyomavirus JC replication in human brain progenitor-derived astrocytes. Antimicrob. Agents Chemother. 2011, 55, 2129–2136. [Google Scholar] [CrossRef]
- Patel, A.; Patel, J.; Ikwuagwu, J. A case of progressive multifocal leukoencephalopathy and idiopathic CD4+ lymphocytopenia. J. Antimicrob. Chemother. 2010, 65, 2697–2698. [Google Scholar] [CrossRef]
- El Chakhtoura, N.G.; Ghamrawi, R.; Cowan, R.; Richards, S.; Silver, S.A.; Tsigrelis, C. Progressive Multifocal Leukoencephalopathy Treated With CMX001 in a Non-Human Immunodeficiency Virus Patient After Rituximab Therapy for Lymphoma A Case Report. Infect. Dis. Clin. Prac. 2018, 26, 170–172. [Google Scholar] [CrossRef]
- O’Hara, B.A.; Gee, G.V.; Haley, S.A.; Morris-Love, J.; Nyblade, C.; Nieves, C.; Hanson, B.A.; Dang, X.; Turner, T.J.; Chavin, J.M.; et al. Teriflunomide Inhibits JCPyV Infection and Spread in Glial Cells and Choroid Plexus Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 9809. [Google Scholar] [CrossRef] [PubMed]
- Rahmlow, M.; Shuster, E.A.; Dominik, J.; Deen, H.G., Jr.; Dickson, D.W.; Aksamit, A.J., Jr.; Robles, H.A.; Freeman, W.D. Leflunomide-associated progressive multifocal leukoencephalopathy. Arch. Neurol. 2008, 65, 1538–1539. [Google Scholar] [CrossRef]
- Miller, A.E.; Olsson, T.P.; Wolinsky, J.S.; Comi, G.; Kappos, L.; Hu, X.; Xu, X.; Lublin, A.L.; Truffinet, P.; Chavin, J.; et al. Long-term safety and efficacy of teriflunomide in patients with relapsing multiple sclerosis: Results from the TOWER extension study. Mult. Scler. Relat. Disord. 2020, 46, 102438. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.W.; Javaid, B.; Kadambi, P.V.; Gillen, D.; Harland, R.; Thistlewaite, J.R.; Garfinkel, M.; Foster, P.; Atwood, W.; Millis, J.M.; et al. Leflunomide for polyomavirus type BK nephropathy. N. Engl. J. Med. 2005, 352, 1157–1158. [Google Scholar] [CrossRef] [PubMed]
- Bernhoff, E.; Tylden, G.D.; Kjerpeseth, L.J.; Gutteberg, T.J.; Hirsch, H.H.; Rinaldo, C.H. Leflunomide inhibition of BK virus replication in renal tubular epithelial cells. J. Virol. 2010, 84, 2150–2156. [Google Scholar] [CrossRef]
- Nukuzuma, S.; Nakamichi, K.; Kameoka, M.; Sugiura, S.; Nukuzuma, C.; Tasaki, T.; Takegami, T. Suppressive effect of topoisomerase inhibitors on JC polyomavirus propagation in human neuroblastoma cells. Microbiol. Immunol. 2016, 60, 253–260. [Google Scholar] [CrossRef]
- Royal, W., 3rd; Dupont, B.; McGuire, D.; Chang, L.; Goodkin, K.; Ernst, T.; Post, M.J.; Fish, D.; Pailloux, G.; Poncelet, H.; et al. Topotecan in the treatment of acquired immunodeficiency syndrome-related progressive multifocal leukoencephalopathy. J. Neurovirol. 2003, 9, 411–419. [Google Scholar] [CrossRef]
- Gerber, D.E.; Beg, M.S.; Fattah, F.; Frankel, A.E.; Fatunde, O.; Arriaga, Y.; Dowell, J.E.; Bisen, A.; Leff, R.D.; Meek, C.C.; et al. Phase 1 study of ARQ 761, a beta-lapachone analogue that promotes NQO1-mediated programmed cancer cell necrosis. Br. J. Cancer 2018, 119, 928–936. [Google Scholar] [CrossRef]
- Lee, E.J.; Ko, H.M.; Jeong, Y.H.; Park, E.M.; Kim, H.S. beta-Lapachone suppresses neuroinflammation by modulating the expression of cytokines and matrix metalloproteinases in activated microglia. J. Neuroinflamm. 2015, 12, 133. [Google Scholar] [CrossRef]
- Wong, E.T.; Berkenblit, A. The role of topotecan in the treatment of brain metastases. Oncologist 2004, 9, 68–79. [Google Scholar] [CrossRef]
- Nukuzuma, S.; Nukuzuma, C.; Kameoka, M.; Sugiura, S.; Nakamichi, K.; Tasaki, T.; Takegami, T. CPT11 prevents virus replication in JCI cells persistently infected with JC polyomavirus. Microbiol. Immunol. 2017, 61, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Blaney, S.M.; Takimoto, C.; Murry, D.J.; Kuttesch, N.; McCully, C.; Cole, D.E.; Godwin, K.; Balis, F.M. Plasma and cerebrospinal fluid pharmacokinetics of 9-aminocamptothecin (9-AC), irinotecan (CPT-11), and SN-38 in nonhuman primates. Cancer Chemother. Pharmacol. 1998, 41, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Nukuzuma, S.; Kameoka, M.; Sugiura, S.; Nakamichi, K.; Nukuzuma, C.; Takegami, T. Suppressive effect of PARP-1 inhibitor on JC virus replication in vitro. J. Med. Virol. 2013, 85, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.N.; Marschall, M.; Rinaldo, C.H. Antiviral Effects of Artesunate on JC Polyomavirus Replication in COS-7 Cells. Antimicrob. Agents Chemother. 2014, 58, 6724–6734. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.; Al-Shaibani, Z.; Kumar, D.; Viswabandya, A.; Thyagu, S.; Michelis, F.V.; Kim, D.D.; Lipton, J.H.; Messner, H.A.; Deotare, U. Progressive multifocal leukoencephalopathy due to John Cunningham (JC) virus following allogeneic haematopoietic cell transplantation. Antivir. Ther. 2017, 22, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.M.; Binh, T.Q.; Ilett, K.F.; Batty, K.T.; Phuong, H.L.; Chiswell, G.M.; Phuong, V.D.; Agus, C. Penetration of dihydroartemisinin into cerebrospinal fluid after administration of intravenous artesunate in severe falciparum malaria. Antimicrob. Agents Chemother. 2003, 47, 368–370. [Google Scholar] [CrossRef]
- Goodwin, E.C.; Atwood, W.J.; DiMaio, D. High-Throughput Cell-Based Screen for Chemicals That Inhibit Infection by Simian Virus 40 and Human Polyomaviruses. J. Virol. 2009, 83, 5630–5639. [Google Scholar] [CrossRef]
- Kaiserman, J.; O’Hara, B.A.; Garabian, K.; Lukacher, A.; Haley, S.A.; Atwood, W.J. The Oxindole GW-5074 Inhibits JC Polyomavirus Infection and Spread by Antagonizing the MAPK-ERK Signaling Pathway. mBio 2023, 14, e03583-22. [Google Scholar] [CrossRef]
- Kim, A.; McCully, C.; Cruz, R.; Cole, D.E.; Fox, E.; Balis, F.M.; Widemann, B.C. The plasma and cerebrospinal fluid pharmacokinetics of sorafenib after intravenous administration in non-human primates. Investig. New Drugs 2012, 30, 524–528. [Google Scholar] [CrossRef]
- Lackey, K.; Cory, M.; Davis, R.; Frye, S.V.; Harris, P.A.; Hunter, R.N.; Jung, D.K.; McDonald, O.B.; McNutt, R.W.; Peel, M.R.; et al. The discovery of potent cRaf1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2000, 10, 223–226. [Google Scholar] [CrossRef]
- Laus, G.; Brossner, D.; Keplinger, K. Alkaloids of Peruvian Uncaria tomentosa. Phytochemistry 1997, 45, 855–860. [Google Scholar] [CrossRef]
- Khetmalis, Y.M.; Shivani, M.; Murugesan, S.; Sekhar, K.V.G.C. Oxindole and its derivatives: A review on recent progress in biological activities. Biomed. Pharmacother. 2021, 141, 111842. [Google Scholar] [CrossRef]
- Kaur, M.; Singh, M.; Chadha, N.; Silakari, O. Oxindole: A chemical prism carrying plethora of therapeutic benefits. Eur. J. Med. Chem. 2016, 123, 858–894. [Google Scholar] [CrossRef] [PubMed]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Nintedanib: A Review in Fibrotic Interstitial Lung Diseases. Drugs 2021, 81, 575–586. [Google Scholar] [CrossRef]
- Makino, S. Progressive fibrosing interstitial lung diseases: A new concept and indication of nintedanib. Mod. Rheumatol. 2021, 31, 13–19. [Google Scholar] [CrossRef]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef]
- Reck, M.; Kaiser, R.; Mellemgaard, A.; Douillard, J.Y.; Orlov, S.; Krzakowski, M.; von Pawel, J.; Gottfried, M.; Bondarenko, I.; Liao, M.; et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): A phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014, 15, 143–155. [Google Scholar] [CrossRef]
- Jin, J.; Xie, Y.; Zhang, J.S.; Wang, J.Q.; Dai, S.J.; He, W.F.; Li, S.Y.; Ashby, C.R., Jr.; Chen, Z.S.; He, Q. Sunitinib resistance in renal cell carcinoma: From molecular mechanisms to predictive biomarkers. Drug Resist. Updates 2023, 67, 100929. [Google Scholar] [CrossRef]
- Heng, D.Y.; Kollmannsberger, C. Sunitinib. Recent Results Cancer Res. 2010, 184, 71–82. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Sunitinib: A VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem. Biophys. Res. Commun. 2007, 356, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Matheson, A.J.; Spencer, C.M. Ropinirole: A review of its use in the management of Parkinson’s disease. Drugs 2000, 60, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Bogan, R.K. Ropinirole treatment for restless legs syndrome. Expert. Opin. Pharmacother. 2008, 9, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.R.; Franco, C.; Torrent, C.; Comes, M.; Cruz, N.; Horga, G.; Benabarre, A.; Vieta, E. Ziprasidone in the treatment of affective disorders: A review. CNS Neurosci. Ther. 2008, 14, 278–286. [Google Scholar] [CrossRef]
- Chander, S.; Tang, C.R.; Penta, A.; Wang, P.; Bhagwat, D.P.; Vanthuyne, N.; Albalat, M.; Patel, P.; Sankpal, S.; Zheng, Y.T.; et al. Hit optimization studies of 3-hydroxy-indolin-2-one analogs as potential anti-HIV-1 agents. Bioorg. Chem. 2018, 79, 212–222. [Google Scholar] [CrossRef]
- Conti, I.; Morigi, R.; Locatelli, A.; Rambaldi, M.; Bua, G.; Gallinella, G.; Leoni, A. Synthesis of 3-(Imidazo[2,1-b]thiazol-6-yl)-2H-chromen-2-one Derivatives and Study of Their Antiviral Activity against Parvovirus B19. Molecules 2019, 24, 1037. [Google Scholar] [CrossRef]
- Yang, S.N.Y.; Atkinson, S.C.; Fraser, J.E.; Wang, C.; Maher, B.; Roman, N.; Forwood, J.K.; Wagstaff, K.M.; Borg, N.A.; Jans, D.A. Novel Flavivirus Antiviral That Targets the Host Nuclear Transport Importin alpha/beta1 Heterodimer. Cells 2019, 8, 281. [Google Scholar] [CrossRef]
- Kao, C.C.; Ho, C.L.; Yang, M.H.; Tsai, Y.T.; Liu, S.Y.; Chang, P.Y.; Wu, Y.Y.; Chen, J.H.; Huang, T.C.; Yehn, R.H.; et al. Phase I Targeted Combination Trial of Sorafenib and GW5074 in Patients with Advanced Refractory Solid Tumors. J. Clin. Med. 2022, 11, 2183. [Google Scholar] [CrossRef]
- Clifford, D.B. Progressive multifocal leukoencephalopathy therapy. J. Neurovirol. 2015, 21, 632–636. [Google Scholar] [CrossRef]
- Bruckner, A.; Stadlbauer, F.; Guarino, L.A.; Brunahl, A.; Schneider, C.; Rehfuess, C.; Previes, C.; Fanning, E.; Nasheuer, H.P. The mouse DNA polymerase alpha-primase subunit p48 mediates species-specific replication of polyomavirus DNA in vitro. Mol. Cell/ Biol. 1995, 15, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Tikhanovich, I.; Nasheuer, H.P. Host-specific replication of BK virus DNA in mouse cell extracts is independently controlled by DNA polymerase alpha-primase and inhibitory activities. J. Virol. 2010, 84, 6636–6644. [Google Scholar] [CrossRef] [PubMed]
- London, W.T.; Houff, S.A.; McKeever, P.E.; Wallen, W.C.; Sever, J.L.; Padgett, B.L.; Walker, D.L. Viral-induced astrocytomas in squirrel monkeys. Prog. Clin. Biol. Res. 1983, 105, 227–237. [Google Scholar] [PubMed]
- Major, E.O.; Mourrain, P.; Cummins, C. JC virus-induced owl monkey glioblastoma cells in culture: Biological properties associated with the viral early gene product. Virology 1984, 136, 359–367. [Google Scholar] [CrossRef]
- Gordon, J.; Del Valle, L.; Otte, J.; Khalili, K. Pituitary neoplasia induced by expression of human neurotropic polyomavirus, JCV, early genome in transgenic mice. Oncogene 2000, 19, 4840–4846. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, K.; Yasui, K.; Kimura, J.; Washizu, M.; Yamaguchi, K.; Mori, W. Induction of brain tumors by a newly isolated JC virus (Tokyo-1 strain). Am. J. Pathol. 1984, 116, 455–463. [Google Scholar]
- Drake, D.R., 3rd; Lukacher, A.E. Beta 2-microglobulin knockout mice are highly susceptible to polyoma virus tumorigenesis. Virology 1998, 252, 275–284. [Google Scholar] [CrossRef]
- Frost, E.L.; Lukacher, A.E. The importance of mouse models to define immunovirologic determinants of progressive multifocal leukoencephalopathy. Front. Immunol. 2014, 5, 646. [Google Scholar] [CrossRef]
- Moser, J.M.; Altman, J.D.; Lukacher, A.E. Antiviral CD8+ T cell responses in neonatal mice: Susceptibility to polyoma virus-induced tumors is associated with lack of cytotoxic function by viral antigen-specific T cells. J. Exp. Med. 2001, 193, 595–606. [Google Scholar] [CrossRef]
- Swanson, P.A., 2nd; Lukacher, A.E.; Szomolanyi-Tsuda, E. Immunity to polyomavirus infection: The polyomavirus-mouse model. Semin. Cancer Biol. 2009, 19, 244–251. [Google Scholar] [CrossRef]
- Ayers, K.N.; Carey, S.N.; Lukacher, A.E. Understanding polyomavirus CNS disease—A perspective from mouse models. FEBS J. 2022, 289, 5744–5761. [Google Scholar] [CrossRef]
- Mockus, T.E.; Netherby-Winslow, C.S.; Atkins, H.M.; Lauver, M.D.; Jin, G.; Ren, H.M.; Lukacher, A.E. CD8 T Cells and STAT1 Signaling Are Essential Codeterminants in Protection from Polyomavirus Encephalopathy. J. Virol. 2020, 94, e02038-19. [Google Scholar] [CrossRef] [PubMed]
- Maru, S.; Jin, G.; Desai, D.; Amin, S.; Shwetank; Lauver, M.D.; Lukacher, A.E. Inhibition of Retrograde Transport Limits Polyomavirus Infection In Vivo. mSphere 2017, 2, e00494-17. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.Z.; Berg, S.; Blaney, S.M. Intrathecal chemotherapy. Crit. Rev. Oncol. Hematol. 2001, 37, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Argoff, C.E.; Dubin, A.; Pilitis, J.G. Pain Management Secrets, 4th ed.; Elsevier, Inc.: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef]
- Wager, T.T.; Chandrasekaran, R.Y.; Hou, X.; Troutman, M.D.; Verhoest, P.R.; Villalobos, A.; Will, Y. Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem. Neurosci. 2010, 1, 420–434. [Google Scholar] [CrossRef]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRX 2005, 2, 541–553. [Google Scholar] [CrossRef]
- Tanigawara, Y. Role of P-glycoprotein in drug disposition. Ther. Drug Monit. 2000, 22, 137–140. [Google Scholar] [CrossRef]
- Stanimirovic, D.B.; Bani-Yaghoub, M.; Perkins, M.; Haqqani, A.S. Blood-brain barrier models: In vitro to in vivo translation in preclinical development of CNS-targeting biotherapeutics. Expert. Opin. Drug Discov. 2015, 10, 141–155. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]




| Compound Structure | Commercial Name | Disease Target(s) | Year Licensed | Reference |
|---|---|---|---|---|
 | Nintedanib | Idiopathic pulmonary fibrosis | 2014 | [156,157,158,159,160] |
| Non-small-cell lung cancer | ||||
 | Sunitinib | Renal cell carcinoma | 2006 | [161,162,163,164] |
| Gastrointestinal stromal tumor | ||||
 | Ropinirole | Parkinson’s disease | 1997 | [165,166] |
| Restless leg syndrome | ||||
 | Ziprasidone | Schizophrenia | 2001 | [167] |
| Bipolar disorder |
| Identifier | Phase | Drug(s) | Drug Class | Size | Status |
|---|---|---|---|---|---|
| NCT00746941 | II | Mefloquine | Small Molecule | 37 | Terminated |
| NCT00001048 | II | Cytarabine | Small Molecule | 90 | Terminated |
| NCT00002395 | II | Topotecan | Small Molecule | 54 | Completed |
| NCT04091932 | II | Pembrolizumab | Antibody | 10 | Unknown |
| NCT04781309 | I | NT-I7 | Recombinant interleukin | 12 | Recruiting |
| NCT00002270 | N/A | α interferon, zidovudine | Combination | Unknown | Unknown |
| NCT00000945 | N/A | Cidofovir | Small Molecule | 24 | Completed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaiserman, J.; O’Hara, B.A.; Haley, S.A.; Atwood, W.J. An Elusive Target: Inhibitors of JC Polyomavirus Infection and Their Development as Therapeutics for the Treatment of Progressive Multifocal Leukoencephalopathy. Int. J. Mol. Sci. 2023, 24, 8580. https://doi.org/10.3390/ijms24108580
Kaiserman J, O’Hara BA, Haley SA, Atwood WJ. An Elusive Target: Inhibitors of JC Polyomavirus Infection and Their Development as Therapeutics for the Treatment of Progressive Multifocal Leukoencephalopathy. International Journal of Molecular Sciences. 2023; 24(10):8580. https://doi.org/10.3390/ijms24108580
Chicago/Turabian StyleKaiserman, Jacob, Bethany A. O’Hara, Sheila A. Haley, and Walter J. Atwood. 2023. "An Elusive Target: Inhibitors of JC Polyomavirus Infection and Their Development as Therapeutics for the Treatment of Progressive Multifocal Leukoencephalopathy" International Journal of Molecular Sciences 24, no. 10: 8580. https://doi.org/10.3390/ijms24108580