
Genomic Landscape of Normal and Breast Cancer Tissues in a Hungarian Pilot Cohort
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
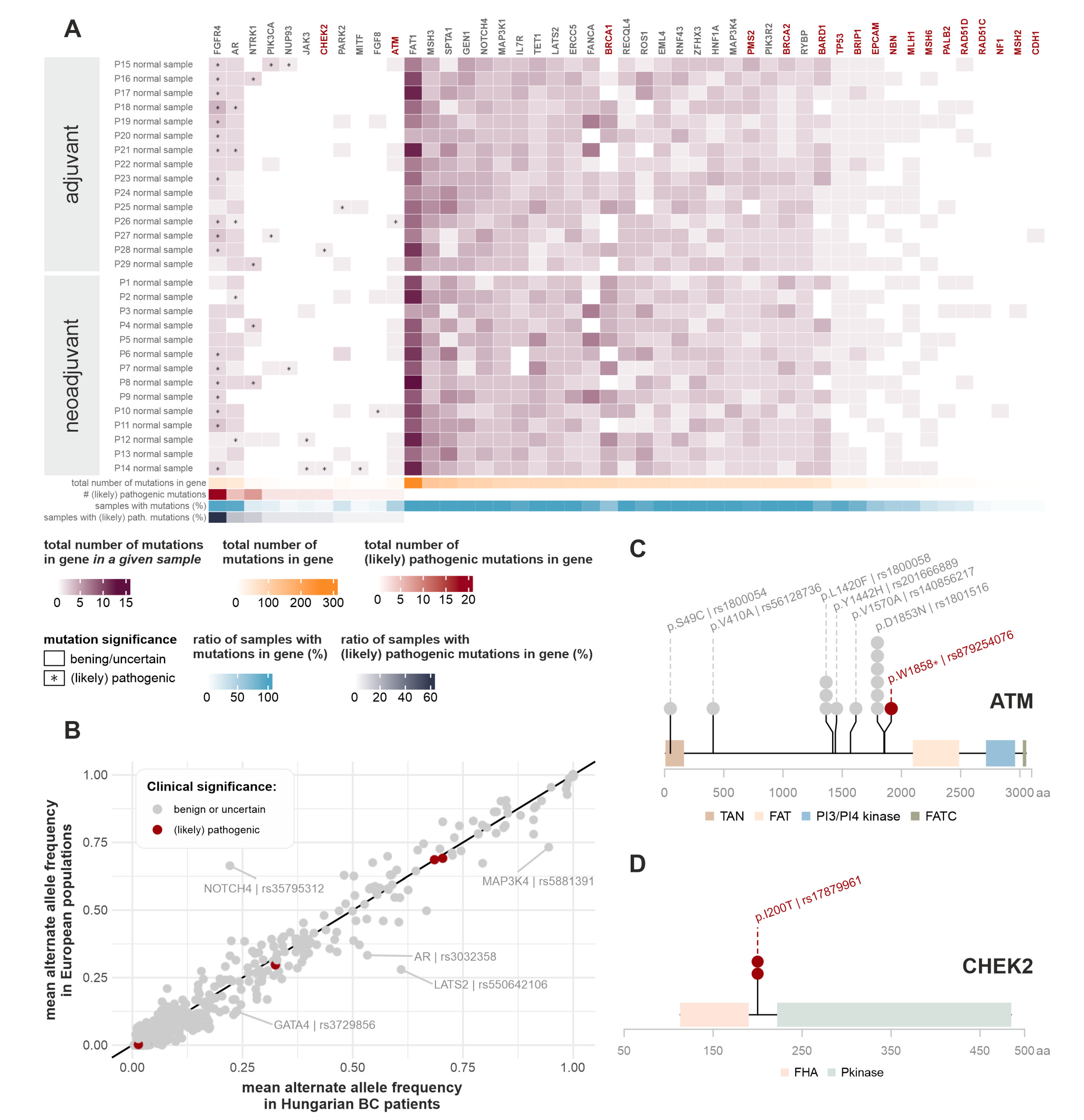
2.2. Germline Short Variants
2.2.1. Most Frequently Mutated Genes in the Germline Genomes
2.2.2. Comparison of Germline Mutation Incidence Rate with Other Patient Cohorts
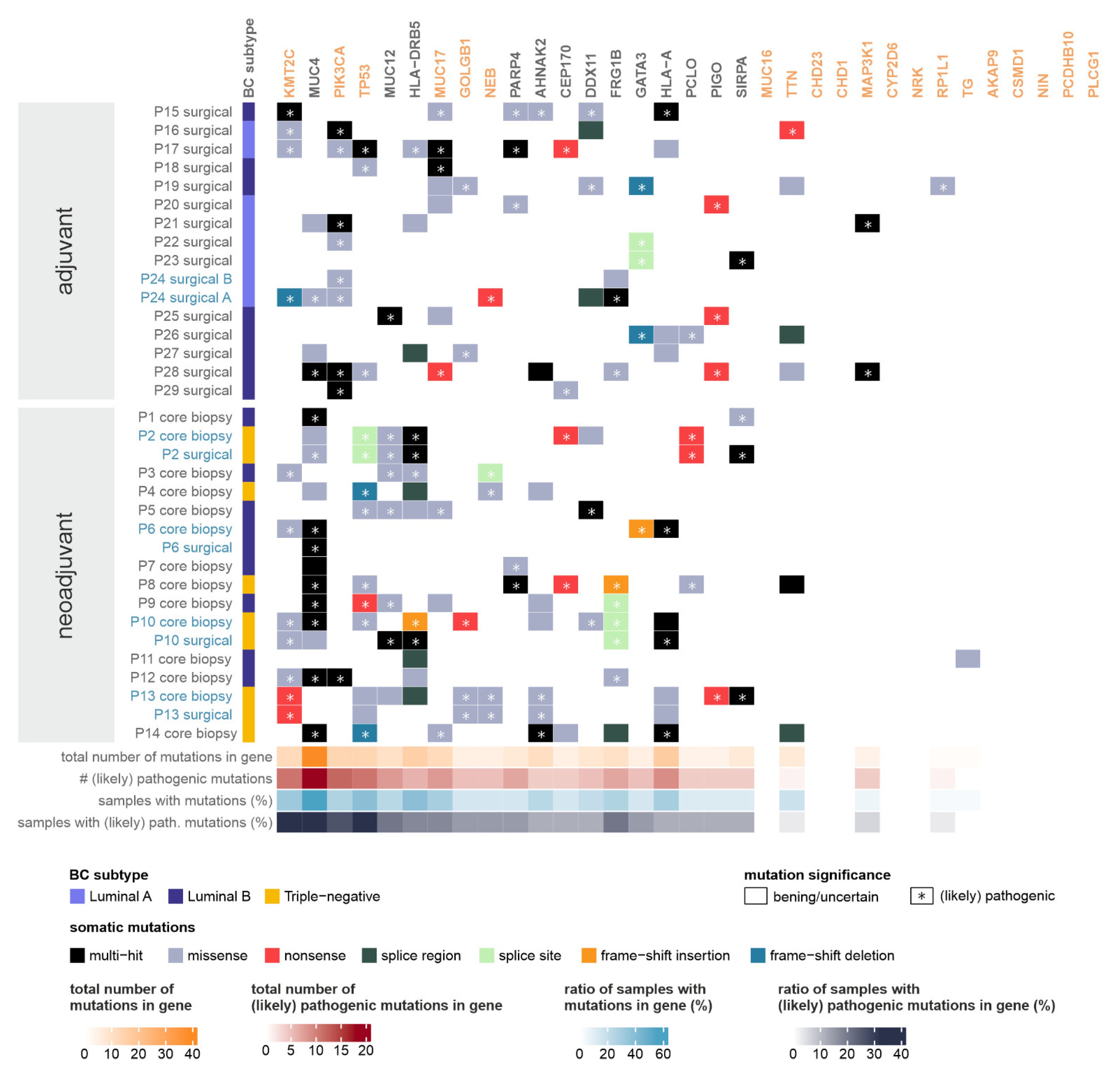
2.3. Somatic Mutations
2.3.1. Somatic Tumor Mutational Burden (TMB) and Distribution of Mutational Subtypes
2.3.2. Genes Most Affected by Somatic Short Variants
2.4. Somatic Copy Number Variations
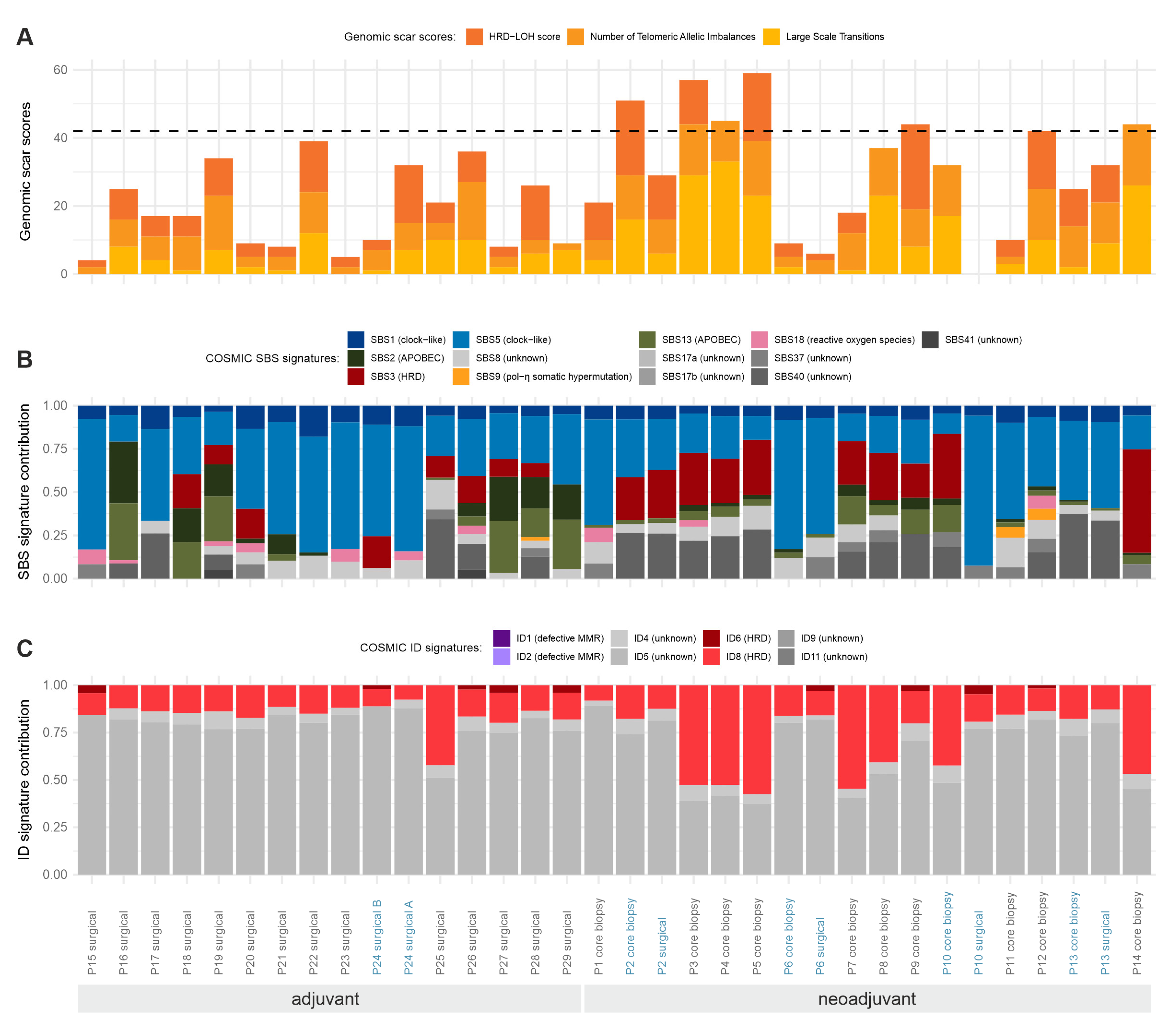
2.5. Statistical Patterns of Somatic Mutations
2.6. Somatic Fusion Events and Splice Variants
2.7. Validation of Identified Short Variants
3. Discussion
3.1. Germline Mutations
3.2. Somatic Mutations
3.3. Tumor Mutation Burden
3.4. Somatic Mutational Signatures
3.5. Somatic Copy Number Variations
4. Materials and Methods
4.1. Patients and DNA Extraction
4.2. Whole-Genome Sequencing
4.3. Bioinformatic Analysis—Short Variant and CNV Detection
4.4. Validation and RNA Variants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ER | Estrogen Receptor |
| ICGC | International Cancer Genetics Consortium |
| ID | Indel |
| GATK | Genome Analysis Toolkit |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HRD | Homologous Recombination Deficiency |
| IDC-NST | Invasive Ductal Carcinoma of No Special Type |
| ILC | Invasive Lobular Carcinoma |
| PR | Progesterone Receptor |
| SNP | Single-Nucleotide Polymorphism |
| TCGA | The Cancer Genome Atlas |
| TSO | TruSight Oncology |
| TMB | Tumor Mutation Burden |
| VUS | Variant of Uncertain Significance |
| WGS | Whole-Genome Sequencing |
| WES | Whole-Exome Sequencing |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef]
- Dafni, U.; Tsourti, Z.; Alatsathianos, I. Breast Cancer Statistics in the European Union: Incidence and Survival across European Countries. Breast Care 2019, 14, 344–353. [Google Scholar] [CrossRef]
- Dyba, T.; Randi, G.; Bray, F.; Martos, C.; Giusti, F.; Nicholson, N.; Gavin, A.; Flego, M.; Neamtiu, L.; Dimitrova, N.; et al. The European cancer burden in 2020: Incidence and mortality estimates for 40 countries and 25 major cancers. Eur. J. Cancer 2021, 157, 308–347. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabe, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Wijewardhane, N.; Dressler, L.; Ciccarelli, F.D. Normal Somatic Mutations in Cancer Transformation. Cancer Cell 2021, 39, 125–129. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gusev, A.; Heng, Y.J.; Alexandrov, L.B.; Kraft, P. Somatic mutational profiles and germline polygenic risk scores in human cancer. Genome Med. 2022, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.; Johnstone, K.J.; McCart Reed, A.E.; Simpson, P.T.; Lakhani, S.R. Hereditary breast cancer: Syndromes, tumour pathology and molecular testing. Histopathology 2023, 82, 70–82. [Google Scholar] [CrossRef]
- Nones, K.; Johnson, J.; Newell, F.; Patch, A.M.; Thorne, H.; Kazakoff, S.H.; de Luca, X.M.; Parsons, M.T.; Ferguson, K.; Reid, L.E.; et al. Whole-genome sequencing reveals clinically relevant insights into the aetiology of familial breast cancers. Ann. Oncol. 2019, 30, 1071–1079. [Google Scholar] [CrossRef]
- Loveday, C.; Garrett, A.; Law, P.; Hanks, S.; Poyastro-Pearson, E.; Adlard, J.W.; Barwell, J.; Berg, J.; Brady, A.F.; Brewer, C.; et al. Analysis of rare disruptive germline mutations in 2135 enriched BRCA-negative breast cancers excludes additional high-impact susceptibility genes. Ann. Oncol. 2022, 33, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.W.; Zabidi, M.M.A.; Ng, P.S.; Meng, M.Y.; Hasan, S.N.; Sandey, B.; Sammut, S.J.; Yip, C.H.; Rajadurai, P.; Rueda, O.M.; et al. The molecular landscape of Asian breast cancers reveals clinically relevant population-specific differences. Nat. Commun. 2020, 11, 6433. [Google Scholar] [CrossRef]
- Yap, Y.S.; Lu, Y.S.; Tamura, K.; Lee, J.E.; Ko, E.Y.; Park, Y.H.; Cao, A.Y.; Lin, C.H.; Toi, M.; Wu, J.; et al. Insights Into Breast Cancer in the East vs the West: A Review. JAMA Oncol. 2019, 5, 1489–1496. [Google Scholar] [CrossRef]
- Mathioudaki, A.; Ljungstrom, V.; Melin, M.; Arendt, M.L.; Nordin, J.; Karlsson, A.; Muren, E.; Saksena, P.; Meadows, J.R.S.; Marinescu, V.D.; et al. Targeted sequencing reveals the somatic mutation landscape in a Swedish breast cancer cohort. Sci. Rep. 2020, 10, 19304. [Google Scholar] [CrossRef]
- Helgadottir, H.T.; Thutkawkorapin, J.; Lagerstedt-Robinson, K.; Lindblom, A. Sequencing for germline mutations in Swedish breast cancer families reveals novel breast cancer risk genes. Sci. Rep. 2021, 11, 14737. [Google Scholar] [CrossRef] [PubMed]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thurlimann, B.; Senn, H.J. Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef] [PubMed]
- Pinder, S.E.; Provenzano, E.; Earl, H.; Ellis, I.O. Laboratory handling and histology reporting of breast specimens from patients who have received neoadjuvant chemotherapy. Histopathology 2007, 50, 409–417. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Sammut, S.J.; Ross, E.M.; Bashford-Rogers, R.; Greenstein, E.; Markus, H.; Morganella, S.; Teng, Y.; Maruvka, Y.; Pereira, B.; et al. The Genomic and Immune Landscapes of Lethal Metastatic Breast Cancer. Cell Rep. 2019, 27, 2690–2708.e10. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. New WHO Classification for Breast Tumours, 5th ed.; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Belsare, S.; Levy-Sakin, M.; Mostovoy, Y.; Durinck, S.; Chaudhuri, S.; Xiao, M.; Peterson, A.S.; Kwok, P.Y.; Seshagiri, S.; Wall, J.D. Evaluating the quality of the 1000 genomes project data. BMC Genom. 2019, 20, 620. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Thulin, A.; Andersson, C.; Werner Ronnerman, E.; De Lara, S.; Chamalidou, C.; Schoenfeld, A.; Kovacs, A.; Fagman, H.; Enlund, F.; Linderholm, B.K. Discordance of PIK3CA and TP53 mutations between breast cancer brain metastases and matched primary tumors. Sci. Rep. 2021, 11, 23548. [Google Scholar] [CrossRef]
- Fumagalli, C.; Ranghiero, A.; Gandini, S.; Corso, F.; Taormina, S.; De Camilli, E.; Rappa, A.; Vacirca, D.; Viale, G.; Guerini-Rocco, E.; et al. Inter-tumor genomic heterogeneity of breast cancers: Comprehensive genomic profile of primary early breast cancers and relapses. Breast Cancer Res. 2020, 22, 107. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Chen, Y.; Zhang, D.; Wei, Y.; Li, Z.; Li, Q.; Xu, B. Homologous Recombination Deficiency (HRD) and BRCA 1/2 Gene Mutation for Predicting the Effect of Platinum-Based Neoadjuvant Chemotherapy of Early-Stage Triple-Negative Breast Cancer (TNBC): A Systematic Review and Meta-Analysis. J. Pers. Med. 2022, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- How, J.A.; Jazaeri, A.A.; Fellman, B.; Daniels, M.S.; Penn, S.; Solimeno, C.; Yuan, Y.; Schmeler, K.; Lanchbury, J.S.; Timms, K.; et al. Modification of Homologous Recombination Deficiency Score Threshold and Association with Long-Term Survival in Epithelial Ovarian Cancer. Cancers 2021, 13, 946. [Google Scholar] [CrossRef]
- Kalimutho, M.; Nones, K.; Srihari, S.; Duijf, P.H.G.; Waddell, N.; Khanna, K.K. Patterns of Genomic Instability in Breast Cancer. Trends Pharmacol. Sci. 2019, 40, 198–211. [Google Scholar] [CrossRef]
- Inagaki-Kawata, Y.; Yoshida, K.; Kawaguchi-Sakita, N.; Kawashima, M.; Nishimura, T.; Senda, N.; Shiozawa, Y.; Takeuchi, Y.; Inoue, Y.; Sato-Otsubo, A.; et al. Genetic and clinical landscape of breast cancers with germline BRCA1/2 variants. Commun. Biol. 2020, 3, 578. [Google Scholar] [CrossRef]
- Van Marcke, C.; Helaers, R.; De Leener, A.; Merhi, A.; Schoonjans, C.A.; Ambroise, J.; Galant, C.; Delree, P.; Rothe, F.; Bar, I.; et al. Tumor sequencing is useful to refine the analysis of germline variants in unexplained high-risk breast cancer families. Breast Cancer Res. 2020, 22, 36. [Google Scholar] [CrossRef]
- Shahi, R.B.; De Brakeleer, S.; Caljon, B.; Pauwels, I.; Bonduelle, M.; Joris, S.; Fontaine, C.; Vanhoeij, M.; Van Dooren, S.; Teugels, E.; et al. Identification of candidate cancer predisposing variants by performing whole-exome sequencing on index patients from BRCA1 and BRCA2-negative breast cancer families. BMC Cancer 2019, 19, 313. [Google Scholar] [CrossRef]
- Hempel, D.; Ebner, F.; Garg, A.; Trepotec, Z.; Both, A.; Stein, W.; Gaumann, A.; Guttler, L.; Janni, W.; DeGregorio, A.; et al. Real world data analysis of next generation sequencing and protein expression in metastatic breast cancer patients. Sci. Rep. 2020, 10, 10459. [Google Scholar] [CrossRef]
- Powles, R.L.; Wali, V.B.; Li, X.; Barlow, W.E.; Nahleh, Z.; Thompson, A.M.; Godwin, A.K.; Hatzis, C.; Pusztai, L. Analysis of Pre- and Posttreatment Tissues from the SWOG S0800 Trial Reveals an Effect of Neoadjuvant Chemotherapy on the Breast Cancer Genome. Clin. Cancer Res. 2020, 26, 1977–1984. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Berry, D.A.; Cirrincione, C.T.; Barry, W.T.; Pitcher, B.N.; Harris, L.N.; Ollila, D.W.; Krop, I.E.; Henry, N.L.; Weckstein, D.J.; et al. Molecular Heterogeneity and Response to Neoadjuvant Human Epidermal Growth Factor Receptor 2 Targeting in CALGB 40601, a Randomized Phase III Trial of Paclitaxel Plus Trastuzumab With or Without Lapatinib. J. Clin. Oncol. 2016, 34, 542–549. [Google Scholar] [CrossRef]
- Dorling, L.; Carvalho, S.; Allen, J.; Gonzalez-Neira, A.; Luccarini, C.; Wahlstrom, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; Wang, Q.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef]
- Lee, K.; Seifert, B.A.; Shimelis, H.; Ghosh, R.; Crowley, S.B.; Carter, N.J.; Doonanco, K.; Foreman, A.K.; Ritter, D.I.; Jimenez, S.; et al. Clinical validity assessment of genes frequently tested on hereditary breast and ovarian cancer susceptibility sequencing panels. Genet. Med. 2019, 21, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Greville-Heygate, S.L.; Maishman, T.; Tapper, W.J.; Cutress, R.I.; Copson, E.; Dunning, A.M.; Haywood, L.; Jones, L.J.; Eccles, D.M. Pathogenic Variants in CHEK2 Are Associated With an Adverse Prognosis in Symptomatic Early-Onset Breast Cancer. JCO Precis. Oncol. 2020, 4, 472–485. [Google Scholar] [CrossRef]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kim, S.; Hundal, J.; Herndon, J.M.; Li, S.; Petti, A.A.; Soysal, S.D.; Li, L.; McLellan, M.D.; Hoog, J.; et al. Breast Cancer Neoantigens Can Induce CD8(+) T-Cell Responses and Antitumor Immunity. Cancer Immunol. Res. 2017, 5, 516–523. [Google Scholar] [CrossRef]
- Denkert, C.; Untch, M.; Benz, S.; Schneeweiss, A.; Weber, K.E.; Schmatloch, S.; Jackisch, C.; Sinn, H.P.; Golovato, J.; Karn, T.; et al. Reconstructing tumor history in breast cancer: Signatures of mutational processes and response to neoadjuvant chemotherapy (small star, filled). Ann. Oncol. 2021, 32, 500–511. [Google Scholar] [CrossRef]
- Kostecka, A.; Nowikiewicz, T.; Olszewski, P.; Koczkowska, M.; Horbacz, M.; Heinzl, M.; Andreou, M.; Salazar, R.; Mair, T.; Madanecki, P.; et al. High prevalence of somatic PIK3CA and TP53 pathogenic variants in the normal mammary gland tissue of sporadic breast cancer patients revealed by duplex sequencing. NPJ Breast Cancer 2022, 8, 76. [Google Scholar] [CrossRef]
- Chang, C.S.; Kitamura, E.; Johnson, J.; Bollag, R.; Hawthorn, L. Genomic analysis of racial differences in triple negative breast cancer. Genomics 2019, 111, 1529–1542. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Addeo, A. The FDA approval of pembrolizumab for patients with TMB >10 mut/Mb: Was it a wise decision? No. Ann. Oncol. 2020, 31, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Pilie, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Lim, B.; et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef]
- Narang, P.; Chen, M.; Sharma, A.A.; Anderson, K.S.; Wilson, M.A. The neoepitope landscape of breast cancer: Implications for immunotherapy. BMC Cancer 2019, 19, 200. [Google Scholar] [CrossRef] [PubMed]
- Barroso-Sousa, R.; Jain, E.; Cohen, O.; Kim, D.; Buendia-Buendia, J.; Winer, E.; Lin, N.; Tolaney, S.M.; Wagle, N. Prevalence and mutational determinants of high tumor mutation burden in breast cancer. Ann. Oncol. 2020, 31, 387–394. [Google Scholar] [CrossRef]
- Dennis, J.; Tyrer, J.P.; Walker, L.C.; Michailidou, K.; Dorling, L.; Bolla, M.K.; Wang, Q.; Ahearn, T.U.; Andrulis, I.L.; Anton-Culver, H.; et al. Rare germline copy number variants (CNVs) and breast cancer risk. Commun. Biol. 2022, 5, 65. [Google Scholar] [CrossRef]
- Morisaki, T.; Kubo, M.; Umebayashi, M.; Yew, P.Y.; Yoshimura, S.; Park, J.H.; Kiyotani, K.; Kai, M.; Yamada, M.; Oda, Y.; et al. Neoantigens elicit T cell responses in breast cancer. Sci. Rep. 2021, 11, 13590. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Davies, H.; Morganella, S.; Purdie, C.A.; Jang, S.J.; Borgen, E.; Russnes, H.; Glodzik, D.; Zou, X.; Viari, A.; Richardson, A.L.; et al. Whole-Genome Sequencing Reveals Breast Cancers with Mismatch Repair Deficiency. Cancer Res. 2017, 77, 4755–4762. [Google Scholar] [CrossRef]
- Li, J.; Lu, H.; Ng, P.K.; Pantazi, A.; Ip, C.K.M.; Jeong, K.J.; Amador, B.; Tran, R.; Tsang, Y.H.; Yang, L.; et al. A functional genomic approach to actionable gene fusions for precision oncology. Sci. Adv. 2022, 8, eabm2382. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999, 9, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Clarke, L.; Fairley, S.; Zheng-Bradley, X.; Streeter, I.; Perry, E.; Lowy, E.; Tasse, A.M.; Flicek, P. The international Genome sample resource (IGSR): A worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2017, 45, D854–D859. [Google Scholar] [CrossRef]
- Favero, F.; Joshi, T.; Marquard, A.M.; Birkbak, N.J.; Krzystanek, M.; Li, Q.; Szallasi, Z.; Eklund, A.C. Sequenza: Allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 2015, 26, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef]
- Popova, T.; Manie, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [PubMed]
- Sztupinszki, Z.; Diossy, M.; Krzystanek, M.; Reiniger, L.; Csabai, I.; Favero, F.; Birkbak, N.J.; Eklund, A.C.; Syed, A.; Szallasi, Z. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer 2018, 4, 16. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variable | Adjuvant Cohort (n = 15) | Neoadjuvant Cohort (n = 14) | |
|---|---|---|---|
| Age (years) | |||
| Mean ± SD | 58.93 ± 13.23 | 54.71 ± 9.93 | |
| Range | 32–80 | 37–67 | |
| Tumor tissue histological subtypes | 16 tumor samples | 18 tumor samples | |
| core biopsy | surgical | ||
| Invasive lobular carcinoma (ILC) | 4 (25.0%) | 0 (0%) | 0 (0%) |
| Invasive breast carcinoma of no special type (NST) | 11 (68.8%) | 14 (77.8%) | 3 (16.7%) |
| Mixed ductal and lobular carcinoma | 1 (6.2) | ||
| Metaplastic carcinoma | 0 (0%) | 0 (0%) | 1 (5.5) |
| Pathological tumor size (pT) | pT | ypT | |
| T0 | 0 (0%) | 4 (28.5%) | |
| T1 | 4 (27%) | 5 (36%) | |
| T2 | 10 (67%) | 4 (28.5%) | |
| T3 | 1 (6%) | 1 (7%) | |
| Pathological lymph node status (pN) | pN | ypN | |
| pN0 | 9 (60%) | 7 (50%) | |
| pN1 | 3 (20%) | 5 (36%) | |
| pN2 | 2 (13%) | 1 (7%) | |
| pN3 | 1 (7%) | 1 (7%) | |
| No | 14 (93%) | 11 (78%) | |
| Yes | 1 (7%) | 2 (14%) | |
| NA | 0 (0%) | 1 (7%) | |
| ER status | |||
| Positive | 15 (100%) | 8 (57%) | |
| Negative | 0 (0%) | 6 (43%) | |
| PR status | |||
| Positive | 11 (73%) | 6 (43%) | |
| Negative | 4 (27%) | 8 (57%) | |
| HER2 | |||
| Positive | 1 (7%) | 0 (0%) | |
| Negative | 14 (93%) | 14 (100%) | |
| IHC molecular subtype 1 | |||
| Luminal A | 7 (47%) | 0 (0%) | |
| Luminal B | 8 (53%) | 8 (57%) | |
| Triple negative | 0 (0%) | 6 (43%) | |
| Tumor response to neoadjuvant therapy 2 | |||
| TR1a | 4 (28.5%) | ||
| TR1b | 0 | ||
| TR2a | 0 | ||
| TR2b | 5 (36%) | ||
| TR2c | 4 (28.5%) | ||
| TR3 | 0 | ||
| NA | 1 (7%) | ||
| Lymph node response to neoadjuvant therapy 2 | |||
| NR1 | 0 | ||
| NR2 | 1 (7%) | ||
| NR3 | 4 (28.5%) | ||
| NR4 | 0 | ||
| NA | 9 (64.5%) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pipek, O.; Alpár, D.; Rusz, O.; Bödör, C.; Udvarnoki, Z.; Medgyes-Horváth, A.; Csabai, I.; Szállási, Z.; Madaras, L.; Kahán, Z.; et al. Genomic Landscape of Normal and Breast Cancer Tissues in a Hungarian Pilot Cohort. Int. J. Mol. Sci. 2023, 24, 8553. https://doi.org/10.3390/ijms24108553
Pipek O, Alpár D, Rusz O, Bödör C, Udvarnoki Z, Medgyes-Horváth A, Csabai I, Szállási Z, Madaras L, Kahán Z, et al. Genomic Landscape of Normal and Breast Cancer Tissues in a Hungarian Pilot Cohort. International Journal of Molecular Sciences. 2023; 24(10):8553. https://doi.org/10.3390/ijms24108553
Chicago/Turabian StylePipek, Orsolya, Donát Alpár, Orsolya Rusz, Csaba Bödör, Zoltán Udvarnoki, Anna Medgyes-Horváth, István Csabai, Zoltán Szállási, Lilla Madaras, Zsuzsanna Kahán, and et al. 2023. "Genomic Landscape of Normal and Breast Cancer Tissues in a Hungarian Pilot Cohort" International Journal of Molecular Sciences 24, no. 10: 8553. https://doi.org/10.3390/ijms24108553