
Potassium Channels, Glucose Metabolism and Glycosylation in Cancer Cells
 , , , ,
, , , ,
Abstract
: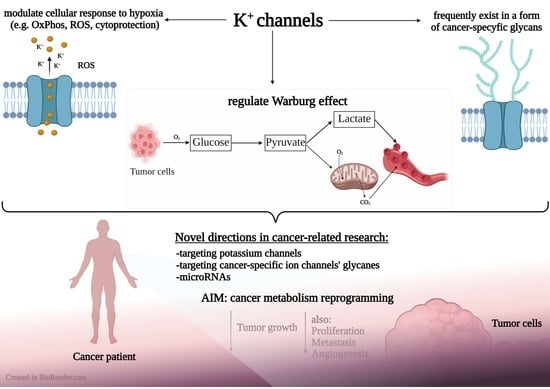
1. Introduction
2. The Role of Potassium Channels in the Metabolism of Cancer Cells
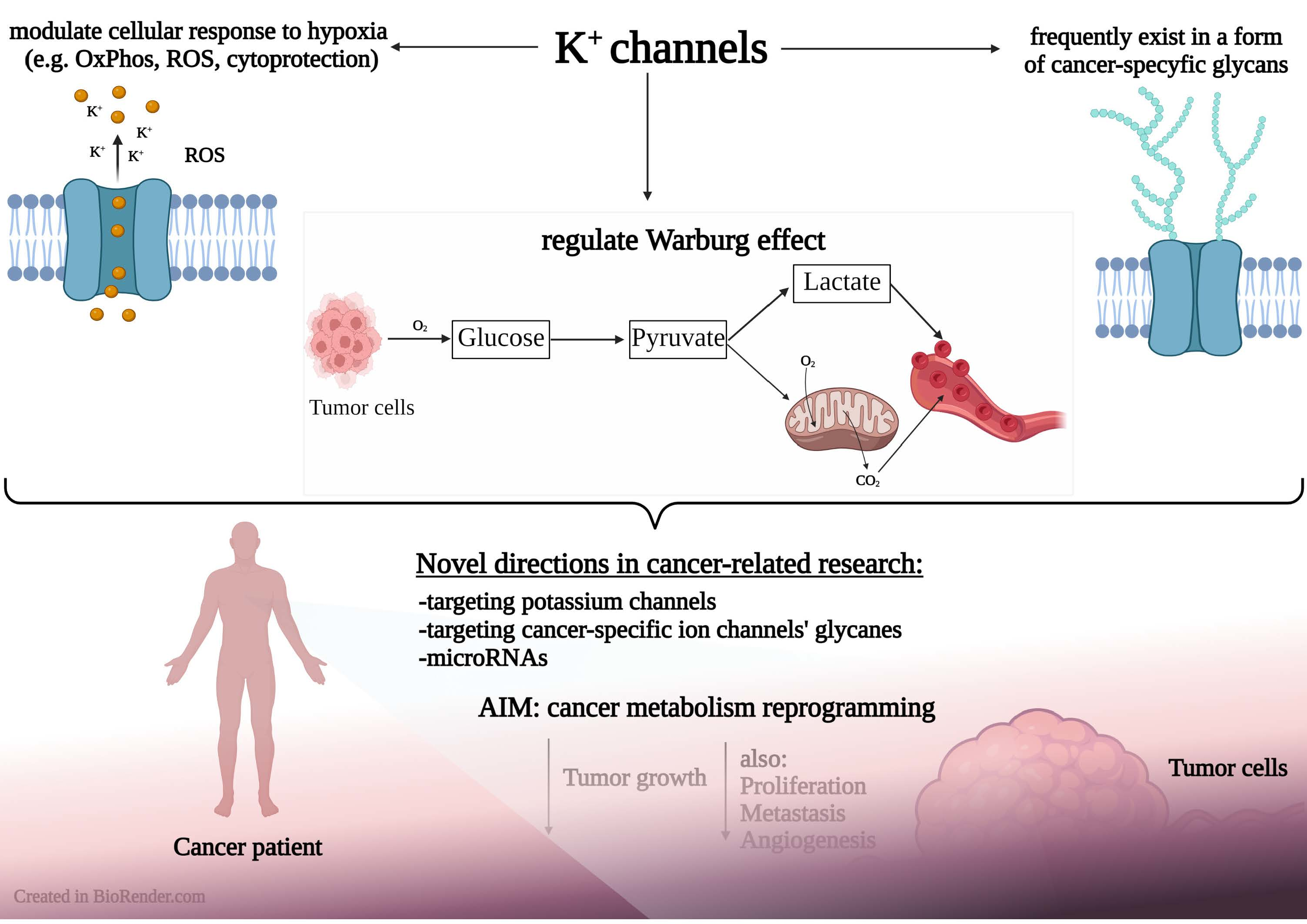
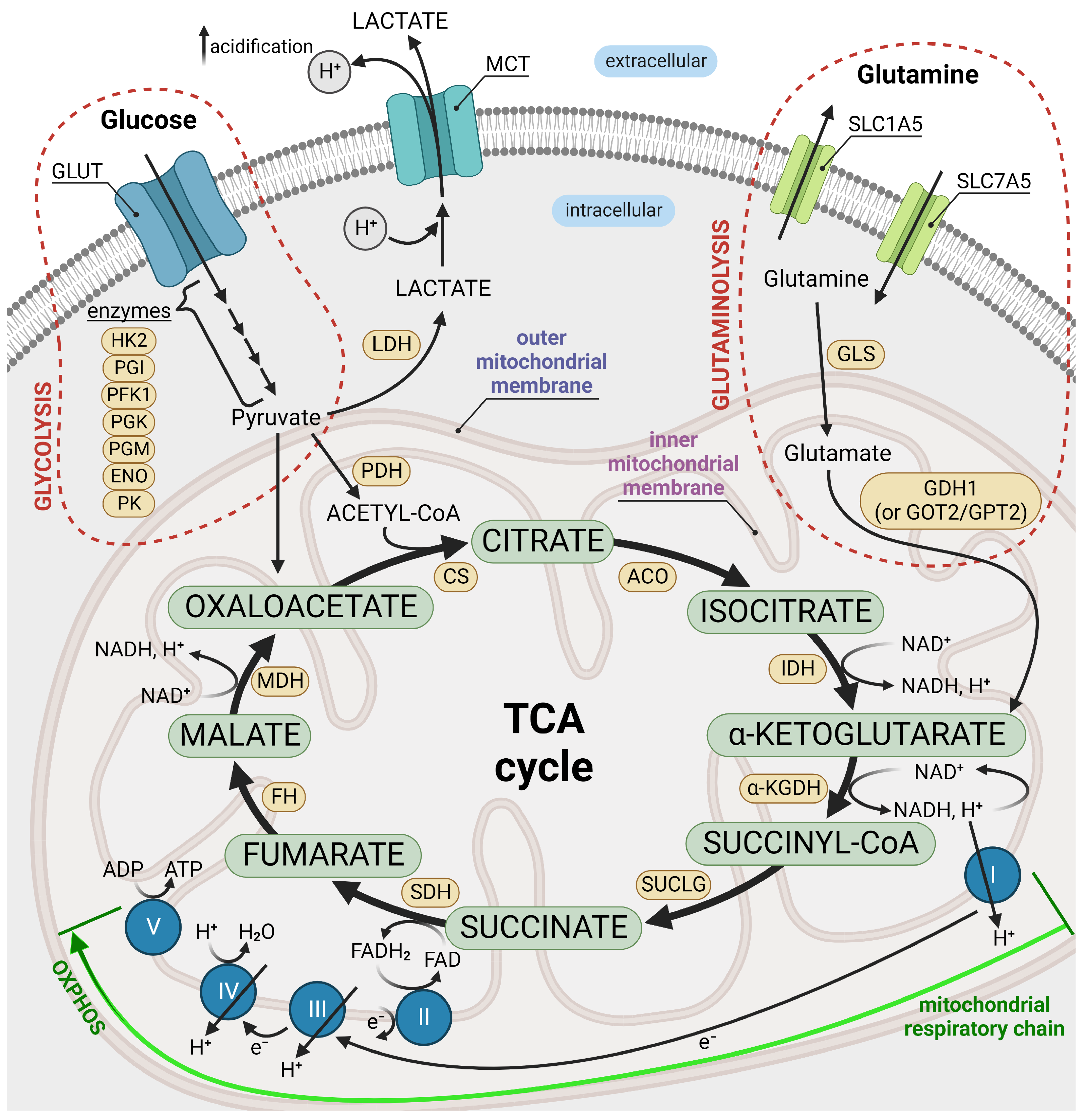
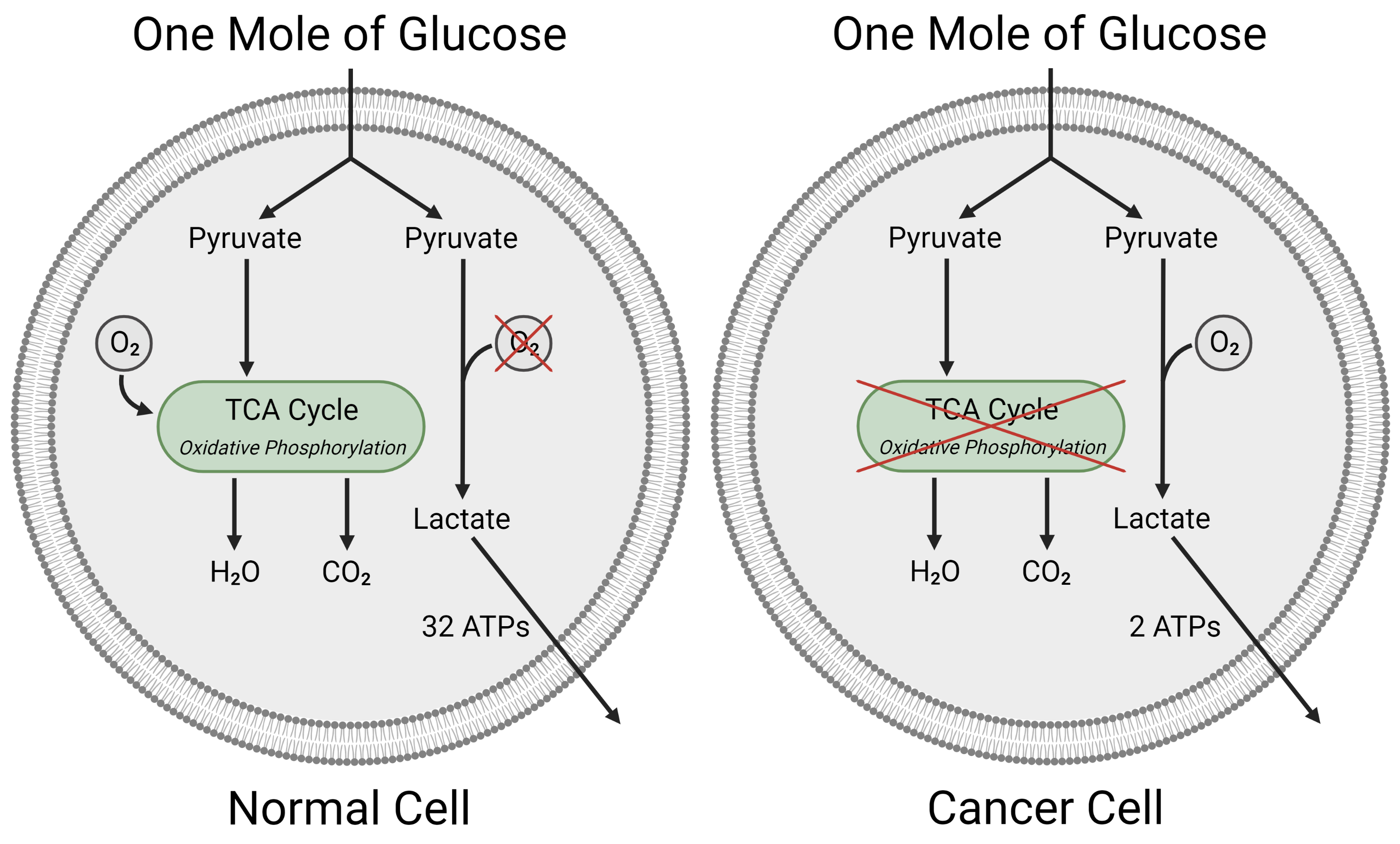
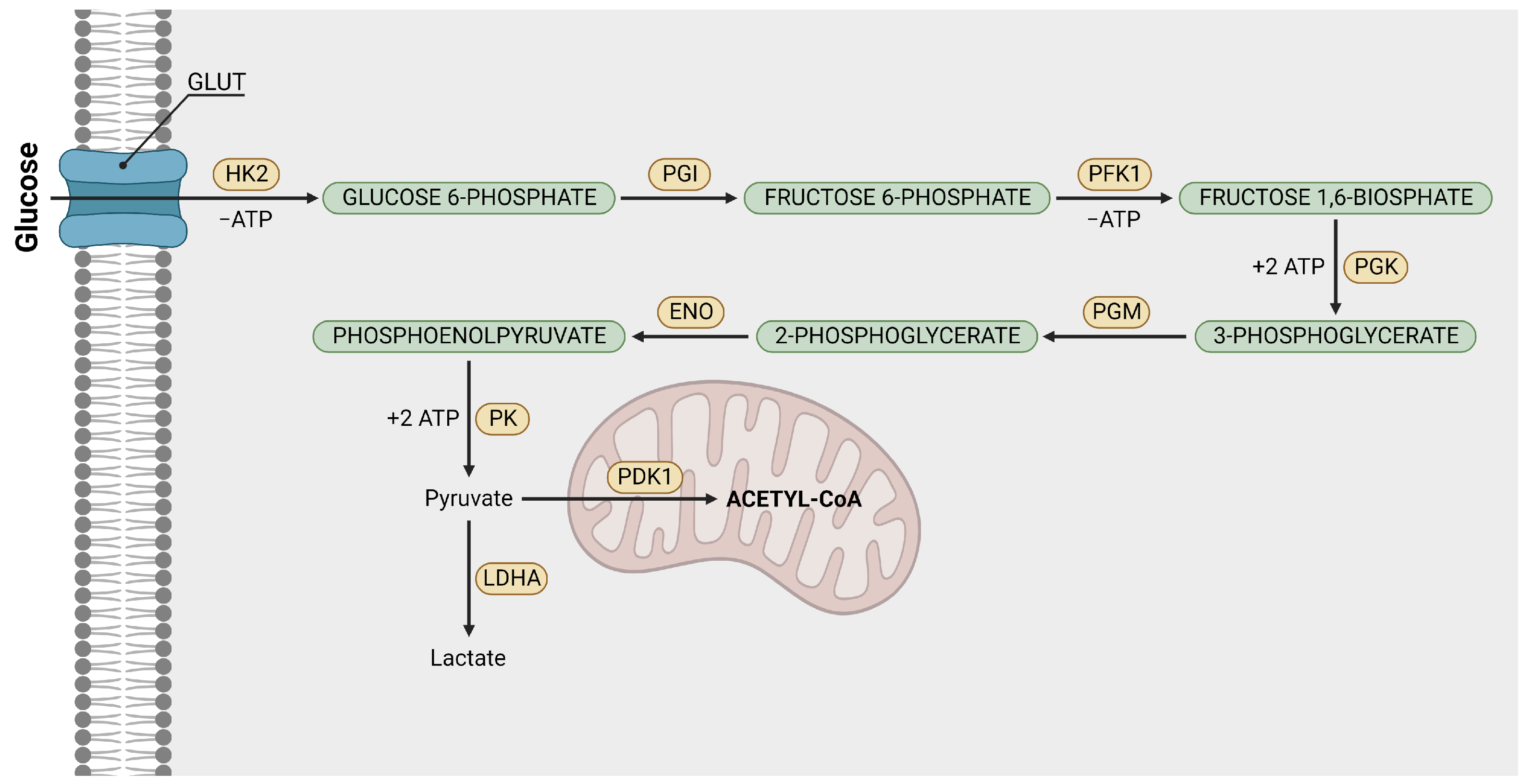
2.1. Glycolysis
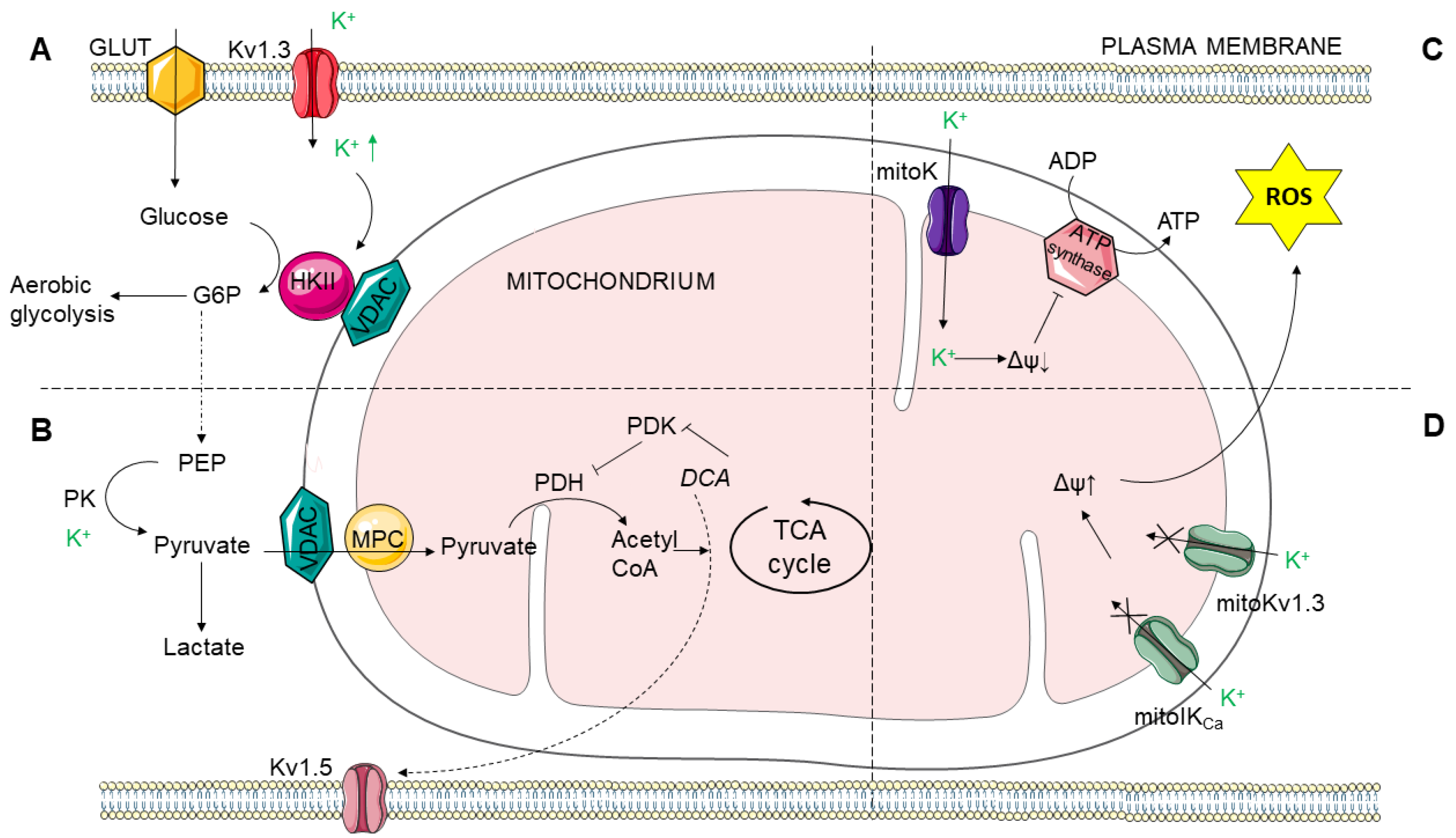
2.2. Mitochondrial Link to Cancer Cell Metabolism of Glucose and Potassium Channels
2.2.1. Mitochondrial Function
2.2.2. Glutaminolysis and the TCA Cycle
2.2.3. Hexokinase
2.2.4. Pyruvate Kinase
2.2.5. Pyruvate Dehydrogenase
2.2.6. Mitochondrial Potassium Channels
2.3. Effects of Hypoxia on the Potassium Channels Activity
3. Glycosylation-Dependent Alterations in Properties of Potassium Channels in Cancer Cells
3.1. Glycosylation in Cancer Cells
3.2. Functional Role of Glycans in Potassium Channels
3.3. miRNAs as a Common Element in the Regulation of Potassium Channels and Sialotransferases
4. Discussion
4.1. Pharmacological Modulation of Potassium Channels in Cancer
4.2. Is It Possible to Reprogram the Cancer Metabolism via Modulation of Potassium Channels?
4.3. The Therapeutic Potential of Controlling Potassium Channels Glycosylation
4.4. Challenges and Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Acetyl-CoA | acetyl coenzyme A |
| ADP | adenosine diphosphate |
| ATP | adenosine triphosphate |
| BK | big conductance Ca-activated potassium channel |
| CLIC1 | chloride intracellular channel 1 |
| DCA | dichloroacetate |
| Eno | enolase |
| ETC | electron transport chain |
| FADH | reduced form of flavin adenine dinucleotide |
| G6P | glucose-6-phosphate |
| GDH | glutamate dehydrogenase |
| GLS | glutaminase |
| GLT-1 | glutamate transporter-1 |
| GSH | glutathione |
| hERG | human rapid delayed rectifier potassium channel |
| HIF-1 | hypoxia-inducible-factor-1 |
| HK1 (HKI) | hexokinase-1 |
| HK2 (HKII) | hexokinase-2 |
| HO-1 | heme oxygenase-1 |
| Hsp90 | Heat Shock Protein 90 |
| IKCa | intermediate calcium activated potassium channel |
| two-pore domain potassium channel | |
| inward rectifier potassium channel | |
| K | voltage-gated potassium channel |
| LDHA | lactate dehydrogenase A |
| LCSC | liver cancer stem cells |
| MCT4 | monocarboxylate transporter-4 |
| mitoK | mitochondrial ATP-sensitive potassium channel |
| mitoBK | mitochondrial big potassium channel |
| mitoBK | Ca-activated mitochondrial big potassium channel |
| mitoK | ATP-sensitive mitochondrial potassium channel |
| mitoSK | Ca-activated mitochondrial small-conductance potassium channel |
| mitoSLO2 | mitochondrial sodium-activated potassium channel |
| mitoTASK-3 | mitochondrial TWIK-related acid-sensitive potassium channel |
| MPC | mitochondrial pyruvate carrier |
| MPTP | mitochondrial permeability transition pore |
| NADH | reduced form of Nicotinamide adenine dinucleotide |
| NOX4 | NADPH oxidase 4 |
| O-GLCNAc | O-linked-N-Acetyl-D-glucosamine |
| O-GlcNAcylation | O-linked-N-acetylglucosaminylation |
| OxPhos | oxidative phosphorylation |
| PAH | pulmonary artery hypertension |
| PDAC | ductal pancreas adenocarcinoma |
| PDH | pyruvate dehydrogenase |
| PDK1 | pyruvate dehydrogenase kinase-1 |
| PDK4 | pyruvate dehydrogenase kinase-4 |
| PEP | phosphoenolopyruvate |
| PFK1 | 6-phosphofructokinase-1 |
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 |
| PGI | phosphoglucose isomerase |
| PGK | phoglycerate kinase |
| PGM | phosphoglycerate mutase |
| PK | pyruvate kinase |
| PKM2 | pyruvate kinase isoenzyme type M2 |
| PRKAA1 | AMP-activated, alpha 1 catalytic subunit |
| ROS | reactive oxygen species |
| TCA | tricarboxylic acid (cycle) |
| SK | small conductance calcium-activated potassium channel |
| Sp1 | transcription factor specificity protein 1 |
| TME | tumor microenvironment |
| UDP-GlcNAc | UDP-N-acetylglucosamine |
| VDAC | voltage-dependent anion channel |
| VEGF | vascular endothelial growth factor |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels in cancer: Are cancer hallmarks oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Biasutto, L.; Managò, A.; Gulbins, E.; Zoratti, M.; Szabò, I. Intracellular ion channels and cancer. Front. Physiol. 2013, 4, 227. [Google Scholar] [CrossRef] [PubMed]
- Kischel, P.; Girault, A.; Rodat-Despoix, L.; Chamlali, M.; Radoslavova, S.; Abou Daya, H.; Lefebvre, T.; Foulon, A.; Rybarczyk, P.; Hague, F.; et al. Ion channels: New actors playing in chemotherapeutic resistance. Cancers 2019, 11, 376. [Google Scholar] [CrossRef] [PubMed]
- Capatina, A.L.; Lagos, D.; Brackenbury, W.J. Targeting ion channels for cancer treatment: Current progress and future challenges. In Targets of Cancer Diagnosis and Treatment; Springer: Berlin/Heidelberg, Germany, 2020; pp. 1–43. [Google Scholar]
- Li, M.; Tian, P.; Zhao, Q.; Ma, X.; Zhang, Y. Potassium channels: Novel targets for tumor diagnosis and chemoresistance. Front. Oncol. 2022, 12, 1074469. [Google Scholar] [CrossRef]
- Huang, X.; Jan, L.Y. Targeting potassium channels in cancer. J. Cell Biol. 2014, 206, 151–162. [Google Scholar] [CrossRef]
- Prosdocimi, E.; Checchetto, V.; Leanza, L. Targeting the mitochondrial potassium channel KV1.3 to kill cancer cells: Drugs, strategies, and new perspectives. SLAS DISCOVERY: Adv. Life Sci. R&D 2019, 24, 882–892. [Google Scholar]
- Szabo, I.; Zoratti, M.; Biasutto, L. Targeting mitochondrial ion channels for cancer therapy. Redox Biol. 2021, 42, 101846. [Google Scholar] [CrossRef]
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Managò, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct pharmacological targeting of a mitochondrial ion channel selectively kills tumor cells in vivo. Cancer Cell 2017, 31, 516–531. [Google Scholar] [CrossRef]
- Bugan, I.; Kucuk, S.; Karagoz, Z.; Fraser, S.P.; Kaya, H.; Dodson, A.; Foster, C.S.; Altun, S.; Djamgoz, M.B. Anti-metastatic effect of ranolazine in an in vivo rat model of prostate cancer, and expression of voltage-gated sodium channel protein in human prostate. Prostate Cancer Prostatic Dis. 2019, 22, 569–579. [Google Scholar] [CrossRef]
- Driffort, V.; Gillet, L.; Bon, E.; Marionneau-Lambot, S.; Oullier, T.; Joulin, V.; Collin, C.; Pagès, J.C.; Jourdan, M.L.; Chevalier, S.; et al. Ranolazine inhibits Na v 1.5-mediated breast cancer cell invasiveness and lung colonization. Mol. Cancer 2014, 13, 264. [Google Scholar] [CrossRef]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Arismendi-Morillo, G.; Mukherjee, P.; Chinopoulos, C. On the origin of ATP synthesis in cancer. Iscience 2020, 23, 101761. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Pollard, P.; Wortham, N.; Tomlinson, I. The TCA cycle and tumorigenesis: The examples of fumarate hydratase and succinate dehydrogenase. Ann. Med. 2003, 35, 634–635. [Google Scholar] [CrossRef]
- Gottlieb, E.; Tomlinson, I.P. Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 2005, 5, 857–866. [Google Scholar] [CrossRef]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Thomas, D.; Rathinavel, A.K.; Radhakrishnan, P. Altered glycosylation in cancer: A promising target for biomarkers and therapeutics. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1875, 188464. [Google Scholar] [CrossRef] [PubMed]
- Berois, N.; Pittini, A.; Osinaga, E. Targeting tumor glycans for cancer therapy: Successes, limitations, and perspectives. Cancers 2022, 14, 645. [Google Scholar] [CrossRef] [PubMed]
- Abdel Rahman, A.M.; Ryczko, M.; Pawling, J.; Dennis, J.W. Probing the hexosamine biosynthetic pathway in human tumor cells by multitargeted tandem mass spectrometry. ACS Chem. Biol. 2013, 8, 2053–2062. [Google Scholar] [CrossRef]
- Nagel, A.K.; Ball, L.E. Intracellular protein O-GlcNAc modification integrates nutrient status with transcriptional and metabolic regulation. Adv. Cancer Res. 2015, 126, 137–166. [Google Scholar] [PubMed]
- Lee, J.B.; Pyo, K.H.; Kim, H.R. Role and function of O-GlcNAcylation in cancer. Cancers 2021, 13, 5365. [Google Scholar] [CrossRef]
- Bischof, H.; Burgstaller, S.; Springer, A.; Matt, L.; Rauter, T.; Bachkönig, O.A.; Schmidt, T.; Groschner, K.; Schindl, R.; Madl, T.; et al. Potassium ions promote hexokinase-II dependent glycolysis. Iscience 2021, 24, 102346. [Google Scholar] [CrossRef]
- Purushothaman, A.; Mohajeri, M.; Lele, T.P. The role of glycans in the mechanobiology of cancer. J. Biol. Chem. 2023, 299, 102935. [Google Scholar] [CrossRef]
- Dart, C. Symposium Review: Lipid microdomains and the regulation of ion channel function. J. Physiol. 2010, 588, 3169–3178. [Google Scholar] [CrossRef]
- Corradi, V.; Sejdiu, B.I.; Mesa-Galloso, H.; Abdizadeh, H.; Noskov, S.Y.; Marrink, S.J.; Tieleman, D.P. Emerging diversity in lipid–protein interactions. Chem. Rev. 2019, 119, 5775–5848. [Google Scholar] [CrossRef]
- Hall, M.K.; Cartwright, T.A.; Fleming, C.M.; Schwalbe, R.A. Importance of glycosylation on function of a potassium channel in neuroblastoma cells. PLoS ONE 2011, 6, e19317. [Google Scholar] [CrossRef]
- Sprovieri, P.; Martino, G. The role of the carbohydrates in plasmatic membrane. Physiol. Res. 2018, 67, 1–11. [Google Scholar] [CrossRef]
- Mant, A.; Williams, S.; Roncoroni, L.; Lowry, E.; Johnson, D.; O’Kelly, I. N-glycosylation-dependent control of functional expression of background potassium channels K2P3. 1 and K2P9. 1. J. Biol. Chem. 2013, 288, 3251–3264. [Google Scholar] [CrossRef]
- Zhu, J.; Yan, J.; Thornhill, W.B. N-glycosylation promotes the cell surface expression of Kv1. 3 potassium channels. FEBS J. 2012, 279, 2632–2644. [Google Scholar] [CrossRef]
- Baycin-Hizal, D.; Gottschalk, A.; Jacobson, E.; Mai, S.; Wolozny, D.; Zhang, H.; Krag, S.S.; Betenbaugh, M.J. Physiologic and pathophysiologic consequences of altered sialylation and glycosylation on ion channel function. Biochem. Biophys. Res. Commun. 2014, 453, 243–253. [Google Scholar] [CrossRef]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef]
- Gyamfi, J.; Kim, J.; Choi, J. Cancer as a metabolic disorder. Int. J. Mol. Sci. 2022, 23, 1155. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Semenza, G.L.; Artemov, D.; Bedi, A.; Bhujwalla, Z.; Chiles, K.; Feldser, D.; Laughner, E.; Ravi, R.; Simons, J.; Taghavi, P.; et al. ‘The metabolism of tumours’: 70 years later. In The Tumour Microenvironment: Causes and Consequences of Hypoxia and Acidity: Novartis Foundation Symposium 240; Wiley Online Library: Hoboken, NJ, USA, 2001; Volume 240, pp. 251–264. [Google Scholar]
- Li, X.b.; Gu, J.d.; Zhou, Q.h. Review of aerobic glycolysis and its key enzymes—New targets for lung cancer therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Gillies, R.J.; Robey, I.; Gatenby, R.A. Causes and consequences of increased glucose metabolism of cancers. J. Nucl. Med. 2008, 49, 24S–42S. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7, 7. [Google Scholar] [CrossRef]
- Ratcliffe, P.J. Fumarate hydratase deficiency and cancer: Activation of hypoxia signaling? Cancer Cell 2007, 11, 303–305. [Google Scholar] [CrossRef]
- Rasheed, M.R.H.A.; Tarjan, G. Succinate dehydrogenase complex: An updated review. Arch. Pathol. Lab. Med. 2018, 142, 1564–1570. [Google Scholar] [CrossRef]
- Smith, D.G.; Sturmey, R.G. Parallels between embryo and cancer cell metabolism. Biochem. Soc. Trans. 2013, 41, 664–669. [Google Scholar] [CrossRef]
- King, A.; Selak, M.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.; Xu, R.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef]
- Pan, J.G.; Mak, T.W. Metabolic targeting as an anticancer strategy: Dawn of a new era? Sci. STKE 2007, 2007, pe14. [Google Scholar] [CrossRef]
- Altenberg, B.; Greulich, K. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef]
- Bygrave, F.L. The ionic environment and metabolic control. Nature 1967, 214, 667–671. [Google Scholar] [CrossRef]
- Gohara, D.W.; Di Cera, E. Molecular mechanisms of enzyme activation by monovalent cations. J. Biol. Chem. 2016, 291, 20840–20848. [Google Scholar] [CrossRef]
- Burgstaller, S.; Bischof, H.; Matt, L.; Lukowski, R. Assessing K+ ions and K+ channel functions in cancer cell metabolism using fluorescent biosensors. Free. Radic. Biol. Med. 2022, 181, 43–51. [Google Scholar] [CrossRef]
- Bischof, H.; Burgstaller, S.; Graier, W.F.; Lukowski, R.; Malli, R. Unveiling the K+-sensitivity of cell metabolism using genetically encoded, FRET-based K+, glucose, and ATP biosensors. STAR Protoc. 2021, 2, 100843. [Google Scholar] [CrossRef]
- Jackson, J.G.; O’Donnell, J.C.; Krizman, E.; Robinson, M.B. Displacing hexokinase from mitochondrial voltage-dependent anion channel impairs GLT-1-mediated glutamate uptake but does not disrupt interactions between GLT-1 and mitochondrial proteins. J. Neurosci. Res. 2015, 93, 999–1008. [Google Scholar] [CrossRef]
- Lin, G.; Lin, L.; Lin, H.; Chen, W.; Chen, L.; Chen, X.; Chen, S.; Lin, Q.; Xu, Y.; Zeng, Y. KCNK3 inhibits proliferation and glucose metabolism of lung adenocarcinoma via activation of AMPK-TXNIP pathway. Cell Death Discov. 2022, 8, 360. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Parry, S.; Xiao, Y.; Zhou, S.; Liu, Q. Molecular targets of the Warburg effect and inflammatory cytokines in the pathogenesis of pulmonary artery hypertension. Clin. Chim. Acta 2017, 466, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Bersin, R.M.; Stacpoole, P.W. Dichloroacetate as metabolic therapy for myocardial ischemia and failure. Am. Heart J. 1997, 134, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar] [CrossRef]
- Krabbendam, I.E.; Honrath, B.; Dilberger, B.; Iannetti, E.F.; Branicky, R.S.; Meyer, T.; Evers, B.; Dekker, F.J.; Koopman, W.J.; Beyrath, J.; et al. SK channel-mediated metabolic escape to glycolysis inhibits ferroptosis and supports stress resistance in C. elegans. Cell Death Dis. 2020, 11, 263. [Google Scholar] [CrossRef]
- Sotelo-Hitschfeld, T.; Niemeyer, M.I.; Machler, P.; Ruminot, I.; Lerchundi, R.; Wyss, M.T.; Stobart, J.; Fernández-Moncada, I.; Valdebenito, R.; Garrido-Gerter, P.; et al. Channel-mediated lactate release by K+-stimulated astrocytes. J. Neurosci. 2015, 35, 4168–4178. [Google Scholar] [CrossRef]
- Honasoge, A.; Shelton, K.A.; Sontheimer, H. Autocrine regulation of glioma cell proliferation via pHe-sensitive K+ channels. Am. J. Physiol. Cell Physiol. 2014, 306, C493–C505. [Google Scholar] [CrossRef]
- Fan, J.; Tian, R.; Yang, X.; Wang, H.; Shi, Y.; Fan, X.; Zhang, J.; Chen, Y.; Zhang, K.; Chen, Z.; et al. KCNN4 promotes the stemness potentials of liver cancer stem cells by enhancing glucose metabolism. Int. J. Mol. Sci. 2022, 23, 6958. [Google Scholar] [CrossRef]
- Lastraioli, E.; Bencini, L.; Bianchini, E.; Romoli, M.R.; Crociani, O.; Giommoni, E.; Messerini, L.; Gasperoni, S.; Moretti, R.; Di Costanzo, F.; et al. hERG1 channels and Glut-1 as independent prognostic indicators of worse outcome in stage I and II colorectal cancer: A pilot study. Transl. Oncol. 2012, 5, 105–112. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Mirzaei, H.; Hamblin, M.R. Regulation of glycolysis by non-coding RNAs in cancer: Switching on the Warburg effect. Mol. Ther.-Oncolytics 2020, 19, 218–239. [Google Scholar] [CrossRef]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of microRNA targeting efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.D.; Li, J.; Huang, K.Y.; Shrestha, S.; Hong, H.C.; Tang, Y.; Chen, Y.G.; Jin, C.N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA–target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.D.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. mmiRTarBase update 2022: An informative resource for experimentally validated miRNA–target interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]
- Snezhkina, A.; Krasnov, G.; Zhikrivetskaya, S.; Karpova, I.Y.; Fedorova, M.; Nyushko, K.; Belyakov, M.; Gnuchev, N.; Sidorov, D.; Alekseev, B.Y.; et al. Overexpression of microRNAs miR-9,-98, and-199 correlates with the downregulation of HK2 expression in colorectal cancer. Mol. Biol. 2018, 52, 190–199. [Google Scholar] [CrossRef]
- Song, W.; Chen, Y.; Zhu, G.; Xie, H.; Yang, Z.; Li, L. Exosome-mediated miR-9-5p promotes proliferation and migration of renal cancer cells both in vitro and in vivo by targeting SOCS4. Biochem. Biophys. Res. Commun. 2020, 529, 1216–1224. [Google Scholar] [CrossRef]
- Riancho, J.; Vázquez-Higuera, J.L.; Pozueta, A.; Lage, C.; Kazimierczak, M.; Bravo, M.; Calero, M.; Gonalezález, A.; Rodríguez, E.; Lleó, A.; et al. MicroRNA profile in patients with Alzheimer’s disease: Analysis of miR-9-5p and miR-598 in raw and exosome enriched cerebrospinal fluid samples. J. Alzheimer’S Dis. 2017, 57, 483–491. [Google Scholar] [CrossRef]
- Lu, J.; Wang, L.; Chen, W.; Wang, Y.; Zhen, S.; Chen, H.; Cheng, J.; Zhou, Y.; Li, X.; Zhao, L. miR-603 targeted hexokinase-2 to inhibit the malignancy of ovarian cancer cells. Arch. Biochem. Biophys. 2019, 661, 1–9. [Google Scholar] [CrossRef]
- Ling, Z.; Liu, D.; Zhang, G.; Liang, Q.; Xiang, P.; Xu, Y.; Han, C.; Tao, T. miR-361-5p modulates metabolism and autophagy via the Sp1-mediated regulation of PKM2 in prostate cancer. Oncol. Rep. 2017, 38, 1621–1628. [Google Scholar] [CrossRef]
- Tao, Y.; Zhou, J.; Wang, Z.; Tao, H.; Bai, J.; Ge, G.; Li, W.; Zhang, W.; Hao, Y.; Yang, X.; et al. Human bone mesenchymal stem cells-derived exosomal miRNA-361-5p alleviates osteoarthritis by downregulating DDX20 and inactivating the NF-κB signaling pathway. Bioorg. Chem. 2021, 113, 104978. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Mao, L.f.; Zhang, Z.p.; Lv, W.w.; Feng, X.p.; Liao, H.j.; Dong, C.; Kaluba, B.; Tang, X.f.; Chang, S. Down-regulated miR-125a-5p promotes the reprogramming of glucose metabolism and cell malignancy by increasing levels of CD147 in thyroid cancer. Thyroid 2018, 28, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Lv, A.; Tu, Z.; Huang, Y.; Lu, W.; Xie, B. Circulating exosomal miR-125a-5p as a novel biomarker for cervical cancer. Oncol. Lett. 2021, 21, 54. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Pei, Y.; Zhang, C.; Qi, Y.; Xia, J.; Hao, C.; Yao, W. Exosomal miR-125a-5p regulates T lymphocyte subsets to promote silica-induced pulmonary fibrosis by targeting TRAF6. Ecotoxicol. Environ. Saf. 2023, 249, 114401. [Google Scholar] [CrossRef]
- Xueya, Z.; Yamei, L.; Sha, C.; Dan, C.; Hong, S.; Xingyu, Y.; Weiwei, C. Exosomal encapsulation of miR-125a-5p inhibited trophoblast cell migration and proliferation by regulating the expression of VEGFA in preeclampsia. Biochem. Biophys. Res. Commun. 2020, 525, 646–653. [Google Scholar] [CrossRef]
- Wang, D.; Hao, C.; Zhang, L.; Zhang, J.; Liu, S.; Li, Y.; Qu, Y.; Zhao, Y.; Huang, R.; Wei, J.; et al. Exosomal miR-125a-5p derived from silica-exposed macrophages induces fibroblast transdifferentiation. Ecotoxicol. Environ. Saf. 2020, 192, 110253. [Google Scholar] [CrossRef]
- Hui, L.; Zhang, J.; Guo, X. MiR-125b-5p suppressed the glycolysis of laryngeal squamous cell carcinoma by down-regulating hexokinase-2. Biomed. Pharmacother. 2018, 103, 1194–1201. [Google Scholar] [CrossRef]
- Cao, J.Y.; Wang, B.; Tang, T.T.; Wen, Y.; Li, Z.L.; Feng, S.T.; Wu, M.; Liu, D.; Yin, D.; Ma, K.L.; et al. Exosomal miR-125b-5p deriving from mesenchymal stem cells promotes tubular repair by suppression of p53 in ischemic acute kidney injury. Theranostics 2021, 11, 5248. [Google Scholar] [CrossRef]
- Shen, K.; Wang, X.; Wang, Y.; Jia, Y.; Zhang, Y.; Wang, K.; Luo, L.; Cai, W.; Li, J.; Li, S.; et al. miR-125b-5p in adipose derived stem cells exosome alleviates pulmonary microvascular endothelial cells ferroptosis via Keap1/Nrf2/GPX4 in sepsis lung injury. Redox Biol. 2023, 62, 102655. [Google Scholar] [CrossRef]
- Wu, Q.Q.; Zheng, B.; Weng, G.B.; Yang, H.M.; Ren, Y.; Weng, X.J.; Zhang, S.W.; Zhu, W.Z. Downregulated NOX4 underlies a novel inhibitory role of microRNA-137 in prostate cancer. J. Cell. Biochem. 2019, 120, 10215–10227. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, J.; Chen, L.; Jin, Y.; Zhang, G.; Lin, Z.; Du, S.; Fu, Z.; Chen, T.; Qin, Y.; et al. Serum secreted miR-137-containing exosomes affects oxidative stress of neurons by regulating OXR1 in Parkinson’s disease. Brain Res. 2019, 1722, 146331. [Google Scholar] [CrossRef]
- Zhang, D.; Cai, G.; Liu, K.; Zhuang, Z.; Jia, K.; Pei, S.; Wang, X.; Wang, H.; Xu, S.; Cui, C.; et al. Microglia exosomal miRNA-137 attenuates ischemic brain injury through targeting Notch1. Aging 2021, 13, 4079. [Google Scholar] [CrossRef]
- Lv, N.; Hao, S.; Luo, C.; Abukiwan, A.; Hao, Y.; Gai, F.; Huang, W.; Huang, L.; Xiao, X.; Eichmuller, S.B.; et al. miR-137 inhibits melanoma cell proliferation through downregulation of GLO1. Sci. China Life Sci. 2018, 61, 541–549. [Google Scholar] [CrossRef]
- Ferguson, S.W.; Wang, J.; Lee, C.J.; Liu, M.; Neelamegham, S.; Canty, J.M.; Nguyen, J. The microRNA regulatory landscape of MSC-derived exosomes: A systems view. Sci. Rep. 2018, 8, 1419. [Google Scholar] [CrossRef]
- Zhou, W.; Xu, M.; Wang, Z.; Yang, M. Engineered exosomes loaded with miR-449a selectively inhibit the growth of homologous non-small cell lung cancer. Cancer Cell Int. 2021, 21, 485. [Google Scholar] [CrossRef]
- Guo, C.J.; Cao, X.L.; Zhang, Y.F.; Yue, K.Y.; Han, J.; Yan, H.; Han, H.; Zheng, M.H. Exosome-mediated inhibition of microRNA-449a promotes the amplification of mouse retinal progenitor cells and enhances their transplantation in retinal degeneration mouse models. Mol. Ther.-Nucleic Acids 2023, 31, 763–778. [Google Scholar] [CrossRef]
- Hua, S.; Lei, L.; Deng, L.; Weng, X.; Liu, C.; Qi, X.; Wang, S.; Zhang, D.; Zou, X.; Cao, C.; et al. miR-139-5p inhibits aerobic glycolysis, cell proliferation, migration, and invasion in hepatocellular carcinoma via a reciprocal regulatory interaction with ETS1. Oncogene 2018, 37, 1624–1636. [Google Scholar] [CrossRef]
- Liu, H.; Wang, F.; Zhang, Y.; Xing, Y.; Wang, Q. Exosomal microRNA-139-5p from mesenchymal stem cells accelerates trophoblast cell invasion and migration by motivation of the ERK/MMP-2 pathway via downregulation of protein tyrosine phosphatase. J. Obstet. Gynaecol. Res. 2020, 46, 2561–2572. [Google Scholar] [CrossRef]
- Sun, H.; Dai, J.; Chen, M.; Chen, Q.; Xie, Q.; Zhang, W.; Li, G.; Yan, M. miR-139-5p Was Identified as Biomarker of Different Molecular Subtypes of Breast Carcinoma. Front. Oncol. 2022, 12, 857714. [Google Scholar] [CrossRef]
- Sun, K.; Hu, P.; Xu, F. LINC00152/miR-139-5p regulates gastric cancer cell aerobic glycolysis by targeting PRKAA1. Biomed. Pharmacother. 2018, 97, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Shen, Y.; He, Y.; Pan, X.; Xu, J.; Jiang, Y.; Zhang, Q.; Wang, S.; Kong, F.; Zhao, S.; et al. The miR-15b-5p/PDK4 axis regulates osteosarcoma proliferation through modulation of the Warburg effect. Biochem. Biophys. Res. Commun. 2018, 503, 2749–2757. [Google Scholar] [CrossRef] [PubMed]
- Yadava, S.M.; Feng, A.; Parobchak, N.; Wang, B.; Rosen, T. miR-15b-5p promotes expression of proinflammatory cytokines in human placenta by inhibiting Apelin signaling pathway. Placenta 2021, 104, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Khoshbakht, T.; Hussen, B.M.; Jamal, H.H.; Taheri, M.; Hajiesmaeili, M. A comprehensive review on function of miR-15b-5p in malignant and non-malignant disorders. Front. Oncol. 2022, 12, 1874. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, T.; Chen, J.; Li, R.; Chen, H.; Luo, S.; Chen, D.; Cai, C.; Li, W. The role of Exosomal miRNAs in cancer. J. Transl. Med. 2022, 20, 6. [Google Scholar] [CrossRef]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA mutations in human cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef]
- Gaude, E.; Frezza, C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014, 2, 10. [Google Scholar] [CrossRef]
- Grasso, D.; Zampieri, L.X.; Capelôa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Zhang, G.F.; Jensen, M.V.; Gray, S.M.; El, K.; Wang, Y.; Lu, D.; Becker, T.C.; Campbell, J.E.; Newgard, C.B. Reductive TCA cycle metabolism fuels glutamine-and glucose-stimulated insulin secretion. Cell Metab. 2021, 33, 804–817. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef]
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Rakha, E.A.; Green, A.R.; Craze, M.L. The role of glutaminase in cancer. Histopathology 2020, 76, 498–508. [Google Scholar] [CrossRef]
- Eniafe, J.; Jiang, S. The functional roles of TCA cycle metabolites in cancer. Oncogene 2021, 40, 3351–3363. [Google Scholar] [CrossRef]
- Jin, L.; Alesi, G.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef]
- Fernandez-Checa, J.C.; Kaplowitz, N.; Garcia-Ruiz, C.; Colell, A.; Miranda, M.; MARi, M.; Ardite, E.; Morales, A. GSH transport in mitochondria: Defense against TNF-induced oxidative stress and alcohol-induced defect. Am. J. Physiol.-Gastrointest. Liver Physiol. 1997, 273, G7–G17. [Google Scholar] [CrossRef]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free. Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Zaid, H.; Israelson, A.; Nahon, E.; Shoshan-Barmatz, V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: Mapping the site of binding. J. Biol. Chem. 2008, 283, 13482–13490. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.; Ko, Y.; Pedersen, P. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, E.; Pedersen, P.L. High aerobic glycolysis of rat hepatoma cells in culture: Role of mitochondrial hexokinase. Proc. Natl. Acad. Sci. USA 1977, 74, 3735–3739. [Google Scholar] [CrossRef] [PubMed]
- Zahra, K.; Dey, T.; Mishra, S.P.; Pandey, U. Pyruvate kinase M2 and cancer: The role of PKM2 in promoting tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, A.; Bednarczyk, P.; Jedraszko, J.; Kampa, R.P.; Koprowski, P.; Krajewska, M.; Kucman, S.; Kulawiak, B.; Laskowski, M.; Rotko, D.; et al. Mitochondrial potassium channels—An overview. Postep. Biochem. 2018, 64, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Pan, M.; Ma, J.; Song, X.; Cao, W.; Zhang, P. Recent progress in the use of mitochondrial membrane permeability transition pore in mitochondrial dysfunction-related disease therapies. Mol. Cell. Biochem. 2021, 476, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the permeability transition pore: Molecular mechanisms in human pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef]
- Szabó, I.; Bock, J.; Grassmé, H.; Soddemann, M.; Wilker, B.; Lang, F.; Zoratti, M.; Gulbins, E. Mitochondrial potassium channel Kv1. 3 mediates Bax-induced apoptosis in lymphocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 14861–14866. [Google Scholar] [CrossRef]
- Peruzzo, R.; Mattarei, A.; Romio, M.; Paradisi, C.; Zoratti, M.; Szabò, I.; Leanza, L. Regulation of proliferation by a mitochondrial potassium channel in pancreatic ductal adenocarcinoma cells. Front. Oncol. 2017, 7, 239. [Google Scholar] [CrossRef]
- Leanza, L.; Henry, B.; Sassi, N.; Zoratti, M.; Chandy, K.G.; Gulbins, E.; Szabò, I. Inhibitors of mitochondrial Kv1. 3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol. Med. 2012, 4, 577–593. [Google Scholar] [CrossRef]
- Turner, K.L.; Honasoge, A.; Robert, S.M.; McFerrin, M.M.; Sontheimer, H. A proinvasive role for the Ca2+-activated K+ channel KCa3. 1 in malignant glioma. Glia 2014, 62, 971–981. [Google Scholar] [CrossRef]
- Bulk, E.; Ay, A.S.; Hammadi, M.; Ouadid-Ahidouch, H.; Schelhaas, S.; Hascher, A.; Rohde, C.; Thoennissen, N.H.; Wiewrodt, R.; Schmidt, E.; et al. Epigenetic dysregulation of KCa3. 1 channels induces poor prognosis in lung cancer. Int. J. Cancer 2015, 137, 1306–1317. [Google Scholar] [CrossRef]
- De Marchi, U.; Sassi, N.; Fioretti, B.; Catacuzzeno, L.; Cereghetti, G.M.; Szabò, I.; Zoratti, M. Intermediate conductance Ca2+-activated potassium channel (KCa3. 1) in the inner mitochondrial membrane of human colon cancer cells. Cell Calcium 2009, 45, 509–516. [Google Scholar] [CrossRef]
- Bulk, E.; Todesca, L.M.; Bachmann, M.; Szabo, I.; Rieke, M.; Schwab, A. Functional expression of mitochondrial KCa3. 1 channels in non-small cell lung cancer cells. Pflugers Arch.-Eur. J. Physiol. 2022, 474, 1147–1157. [Google Scholar] [CrossRef]
- Bauer, D.; Werth, F.; Nguyen, H.A.; Kiecker, F.; Eberle, J. Critical role of reactive oxygen species (ROS) for synergistic enhancement of apoptosis by vemurafenib and the potassium channel inhibitor TRAM-34 in melanoma cells. Cell Death Dis. 2017, 8, e2594. [Google Scholar] [CrossRef]
- Kovalenko, I.; Glasauer, A.; Schockel, L.; Sauter, D.R.; Ehrmann, A.; Sohler, F.; Hagebarth, A.; Novak, I.; Christian, S. Identification of KCa3. 1 channel as a novel regulator of oxidative phosphorylation in a subset of pancreatic carcinoma cell lines. PLoS ONE 2016, 11, e0160658. [Google Scholar] [CrossRef]
- Malinska, D.; Mirandola, S.R.; Kunz, W.S. Mitochondrial potassium channels and reactive oxygen species. FEBS Lett. 2010, 584, 2043–2048. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Laskowski, M.; Augustynek, B.; Kulawiak, B.; Koprowski, P.; Bednarczyk, P.; Jarmuszkiewicz, W.; Szewczyk, A. What do we not know about mitochondrial potassium channels? Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 1247–1257. [Google Scholar] [CrossRef]
- Garthwaite, G.; Williams, G.D.; Garthwaite, J. Glutamate toxicity: An experimental and theoretical analysis. Eur. J. Neurosci. 1992, 4, 353–360. [Google Scholar] [CrossRef]
- Kulawiak, B.; Szewczyk, A. Glutamate-induced cell death in HT22 mouse hippocampal cells is attenuated by paxilline, a BK channel inhibitor. Mitochondrion 2012, 12, 169–172. [Google Scholar] [CrossRef]
- Iorio, J.; Petroni, G.; Duranti, C.; Lastraioli, E. Potassium and sodium channels and the Warburg effect: Biophysical regulation of cancer metabolism. Bioelectricity 2019, 1, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 2007, 80, 51. [Google Scholar] [PubMed]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Honore, E. Molecular physiology of oxygen-sensitive potassium channels. Eur. Respir. J. 2001, 18, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Park, S.M.; Park, J.S.; Byun, J.H.; Jin, H.J.; Seo, S.H.; Ryu, P.D.; Lee, S.Y. Kv3.1 and Kv3.4, are involved in cancer cell migration and invasion. Int. J. Mol. Sci. 2018, 19, 1061. [Google Scholar] [CrossRef]
- Downie, B.R.; Sánchez, A.; Knötgen, H.; Contreras-Jurado, C.; Gymnopoulos, M.; Weber, C.; Stühmer, W.; Pardo, L.A. Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors. J. Biol. Chem. 2008, 283, 36234–36240. [Google Scholar] [CrossRef]
- Crociani, O.; Zanieri, F.; Pillozzi, S.; Lastraioli, E.; Stefanini, M.; Fiore, A.; Fortunato, A.; D’Amico, M.; Masselli, M.; De Lorenzo, E.; et al. hERG1 channels modulate integrin signaling to trigger angiogenesis and tumor progression in colorectal cancer. Sci. Rep. 2013, 3, 3308. [Google Scholar] [CrossRef]
- Lai, Q.; Wang, T.; Guo, Q.; Zhang, Y.; Wang, Y.; Yuan, L.; Ling, R.; He, Y.; Wang, W. Positive correlation between the expression of hEag1 and HIF-1α in breast cancers: An observational study. BMJ Open 2014, 4, e005049. [Google Scholar] [CrossRef]
- Girault, A.; Ahidouch, A.; Ouadid-Ahidouch, H. Roles for Ca2+ and K+ channels in cancer cells exposed to the hypoxic tumour microenvironment. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2020, 1867, 118644. [Google Scholar] [CrossRef]
- Leithner, K.; Hirschmugl, B.; Li, Y.; Tang, B.; Papp, R.; Nagaraj, C.; Stacher, E.; Stiegler, P.; Lindenmann, J.; Olschewski, A.; et al. TASK-1 regulates apoptosis and proliferation in a subset of non-small cell lung cancers. PLoS ONE 2016, 11, e0157453. [Google Scholar] [CrossRef]
- Mu, D.; Chen, L.; Zhang, X.; See, L.H.; Koch, C.M.; Yen, C.; Tong, J.J.; Spiegel, L.; Nguyen, K.C.; Servoss, A.; et al. Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell 2003, 3, 297–302. [Google Scholar] [CrossRef]
- Vaddi, D.R.; Piao, L.; Khan, S.A.; Wang, N.; Prabhakar, N.R.; Nanduri, J. Hypoxia induced hERG trafficking defect linked to cell cycle arrest in SH-SY5Y cells. PLoS ONE 2019, 14, e0215905. [Google Scholar] [CrossRef]
- Al-Owais, M.M.; Scragg, J.L.; Dallas, M.L.; Boycott, H.E.; Warburton, P.; Chakrabarty, A.; Boyle, J.P.; Peers, C. Carbon monoxide mediates the anti-apoptotic effects of heme oxygenase-1 in medulloblastoma DAOY cells via K+ channel inhibition. J. Biol. Chem. 2012, 287, 24754–24764. [Google Scholar] [CrossRef]
- Ryland, K.E.; Svoboda, L.K.; Vesely, E.D.; McIntyre, J.C.; Zhang, L.; Martens, J.R.; Lawlor, E.R. Polycomb-dependent repression of the potassium channel-encoding gene KCNA5 promotes cancer cell survival under conditions of stress. Oncogene 2015, 34, 4591–4600. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, Y.; Xu, Y.; Pan, Y.; Shao, C. Hydrogen sulfide contributes to hypoxia-induced radioresistance on hepatoma cells. J. Radiat. Res. 2011, 52, 622–628. [Google Scholar] [CrossRef]
- Dutta, D.; Mandal, C.; Mandal, C. Unusual glycosylation of proteins: Beyond the universal sequon and other amino acids. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2017, 1861, 3096–3108. [Google Scholar] [CrossRef]
- Patel, C.; Saad, H.; Shenkman, M.; Lederkremer, G.Z. Oxidoreductases in glycoprotein glycosylation, folding, and ERAD. Cells 2020, 9, 2138. [Google Scholar] [CrossRef]
- Cohen, M.; Varki, A. Modulation of glycan recognition by clustered saccharide patches. Int. Rev. Cell Mol. Biol. 2014, 308, 75–125. [Google Scholar]
- Barchi Jr, J.J.; Strain, C.N. The effect of a methyl group on structure and function: Serine vs. threonine glycosylation and phosphorylation. Front. Mol. Biosci. 2023, 10, 1117850. [Google Scholar] [CrossRef]
- Mereiter, S.; Balmaña, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the era of cancer-targeted therapy: Where are we heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Doud, E.H.; Yeh, E.S. Mass Spectrometry-Based Glycoproteomic Workflows for Cancer Biomarker Discovery. Technol. Cancer Res. Treat. 2023, 22, 15330338221148811. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M. Cancer glycan epitopes: Biosynthesis, structure and function. Glycobiology 2018, 28, 670–696. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Jia, W.; Yang, J.; Zhan, X. Cancer glycomics offers potential biomarkers and therapeutic targets in the framework of 3P medicine. Biomol. Modif. -Endocr.-Relat. Cancers 2023, 16648714, 80. [Google Scholar] [CrossRef] [PubMed]
- Peric, L.; Vukadin, S.; Petrovic, A.; Kuna, L.; Puseljic, N.; Sikora, R.; Rozac, K.; Vcev, A.; Smolic, M. Glycosylation Alterations in Cancer Cells, Prognostic Value of Glycan Biomarkers and Their Potential as Novel Therapeutic Targets in Breast Cancer. Biomedicines 2022, 10, 3265. [Google Scholar] [CrossRef]
- Capera, J.; Serrano-Novillo, C.; Navarro-Pérez, M.; Cassinelli, S.; Felipe, A. The potassium channel odyssey: Mechanisms of traffic and membrane arrangement. Int. J. Mol. Sci. 2019, 20, 734. [Google Scholar] [CrossRef]
- Schwetz, T.A.; Norring, S.A.; Ednie, A.R.; Bennett, E.S. Sialic acids attached to O-glycans modulate voltage-gated potassium channel gating. J. Biol. Chem. 2011, 286, 4123–4132. [Google Scholar] [CrossRef]
- Lazniewska, J.; Weiss, N. The “Sweet” Side of Ion Channels. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2014; Volume 167, pp. 67–114. [Google Scholar]
- Noma, K.; Kimura, K.; Minatohara, K.; Nakashima, H.; Nagao, Y.; Mizoguchi, A.; Fujiyoshi, Y. Triple N-glycosylation in the long S5-P loop regulates the activation and trafficking of the Kv12. 2 potassium channel. J. Biol. Chem. 2009, 284, 33139–33150. [Google Scholar] [CrossRef]
- Wiedmann, F.; Schlund, D.; Voigt, N.; Ratte, A.; Kraft, M.; Katus, H.A.; Schmidt, C. N-glycosylation–dependent regulation of hK2P17. 1 currents. Mol. Biol. Cell 2019, 30, 1425–1436. [Google Scholar] [CrossRef]
- Carrington, S.J.; Hernandez, C.C.; Swale, D.R.; Aluko, O.A.; Denton, J.S.; Cone, R.D. G protein–coupled receptors differentially regulate glycosylation and activity of the inwardly rectifying potassium channel Kir7. 1. J. Biol. Chem. 2018, 293, 17739–17753. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, A.; Holmgren, M. Deglycosylation of Shaker KV channels affects voltage sensing and the open–closed transition. J. Gen. Physiol. 2018, 150, 1025–1034. [Google Scholar] [CrossRef]
- Brooks, N.L.; Corey, M.J.; Schwalbe, R.A. Characterization of N-glycosylation consensus sequences in the Kv3. 1 channel. FEBS J. 2006, 273, 3287–3300. [Google Scholar] [CrossRef]
- Schwetz, T.A.; Norring, S.A.; Bennett, E.S. N-glycans modulate Kv1. 5 gating but have no effect on Kv1. 4 gating. Biochim. Biophys. Acta (BBA)-Biomembr. 2010, 1798, 367–375. [Google Scholar] [CrossRef]
- Hall, M.K.; Weidner, D.A.; Dayal, S.; Pak, E.; Murashov, A.K.; Schwalbe, R.A. Membrane Distribution and Activity of a Neuronal Voltage-Gated K+ Channel is Modified by Replacement of Complex Type N-Glycans with Hybrid Type. J. Glycobiol. 2017, 6, 128. [Google Scholar]
- Wiedmann, F.; Schlund, D.; Faustino, F.; Kraft, M.; Ratte, A.; Thomas, D.; Katus, H.A.; Schmidt, C. N-glycosylation of TREK-1/hK2P2. 1 two-pore-domain potassium (K2P) channels. Int. J. Mol. Sci. 2019, 20, 5193. [Google Scholar] [CrossRef]
- Pabon, A.; Chan, K.W.; Sui, J.L.; Wu, X.; Logothetis, D.E.; Thornhill, W.B. Glycosylation of GIRK1 at Asn119 and ROMK1 at Asn117 has different consequences in potassium channel function. J. Biol. Chem. 2000, 275, 30677–30682. [Google Scholar] [CrossRef]
- Patil, N.; Allgayer, H.; Leupold, J.H. MicroRNAs in the tumor microenvironment. Tumor Microenviron. Mol. Play. Part B 2020, 1277, 1–31. [Google Scholar]
- Tang, Y.; Zong, S.; Zeng, H.; Ruan, X.; Yao, L.; Han, S.; Hou, F. MicroRNAs and angiogenesis: A new era for the management of colorectal cancer. Cancer Cell Int. 2021, 21, 221. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Q.; Wang, X.; Yao, X.; Zhang, B.; Wu, J.; Sun, C. Exosomal ncRNAs facilitate interactive ‘dialogue’between tumor cells and tumor-associated macrophages. Cancer Lett. 2022, 552, 215975. [Google Scholar] [CrossRef]
- Hassinen, A.; Khoder-Agha, F.; Khosrowabadi, E.; Mennerich, D.; Harrus, D.; Noel, M.; Dimova, E.Y.; Glumoff, T.; Harduin-Lepers, A.; Kietzmann, T.; et al. A Golgi-associated redox switch regulates catalytic activation and cooperative functioning of ST6Gal-I with B4GalT-I. Redox Biol. 2019, 24, 101182. [Google Scholar] [CrossRef]
- Jones, R.B.; Dorsett, K.A.; Hjelmeland, A.B.; Bellis, S.L. The ST6Gal-I sialyltransferase protects tumor cells against hypoxia by enhancing HIF-1α signaling. J. Biol. Chem. 2018, 293, 5659–5667. [Google Scholar] [CrossRef] [PubMed]
- Rencelj, A.; Gvozdenovic, N.; Cemazar, M. MitomiRs: Their roles in mitochondria and importance in cancer cell metabolism. Radiol. Oncol. 2021, 55, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Yan, H.; Zhang, Y.; Hao, L.; Zhu, X.; Mei, Q.; Sun, S. c-Myc-mediated repression of miR-15-16 in hypoxia is induced by increased HIF-2α and promotes tumor angiogenesis and metastasis by upregulating FGF2. Oncogene 2015, 34, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Guo, W.; Liu, W. Exosomes derived from bone marrow mesenchymal stem cells inhibit human aortic vascular smooth muscle cells calcification via the miR-15a/15b/16/NFATc3/OCN axis. Biochem. Biophys. Res. Commun. 2022, 635, 65–76. [Google Scholar] [CrossRef]
- Li, C.; Li, Y.; Lu, Y.; Niu, Z.; Zhao, H.; Peng, Y.; Li, M. miR-26 family and its target genes in tumorigenesis and development. Crit. Rev. Oncol. 2021, 157, 103124. [Google Scholar] [CrossRef]
- Chettimada, S.; Lorenz, D.R.; Misra, V.; Wolinsky, S.M.; Gabuzda, D. Small RNA sequencing of extracellular vesicles identifies circulating miRNAs related to inflammation and oxidative stress in HIV patients. BMC Immunol. 2020, 21, 57. [Google Scholar] [CrossRef]
- Xu, Y.; Ge, Y.; Chen, X.; Zhang, Y.; Chen, H.; Liu, D.; Lu, Y.; Liu, Y.; Tu, W. Hypoxic Cell-Derived Extracellular Vesicles Aggravate Rectal Injury Following Radiotherapy via MiR-122-5p. Front. Cell Dev. Biol. 2022, 10, 908. [Google Scholar] [CrossRef]
- Li, L.; Wang, Q.; Yuan, Z.; Chen, A.; Liu, Z.; Wang, Z.; Li, H. LncRNA-MALAT1 promotes CPC proliferation and migration in hypoxia by up-regulation of JMJD6 via sponging miR-125. Biochem. Biophys. Res. Commun. 2018, 499, 711–718. [Google Scholar] [CrossRef]
- Kot, A.; Kaczmarek, R. Exosomal miRNA Profiling in Vitreous Humor in Proliferative Diabetic Retinopathy. Cells 2023, 12, 123. [Google Scholar] [CrossRef]
- Parikh, M.; Pierce, G.N. A brief review on the biology and effects of cellular and circulating micrornas on cardiac remodeling after infarction. Int. J. Mol. Sci. 2021, 22, 4995. [Google Scholar] [CrossRef]
- Behera, J.; Govindan, S.; Ramasamy, M. Nitric oxide promotes cell-matrix adhesion of endothelial progenitor cells under hypoxia condition via ITGA5 CpG promoter demethylation. Biochem. Biophys. Res. Commun. 2023, 644, 162–170. [Google Scholar] [CrossRef]
- Zhao, Z.; Wu, C.; He, X.; Zhao, E.; Hu, S.; Han, Y.; Wang, T.; Chen, Y.; Liu, T.; Huang, S. miR-152-3p aggravates vascular endothelial cell dysfunction by targeting DEAD-box helicase 6 (DDX6) under hypoxia. Bioengineered 2021, 12, 4899–4910. [Google Scholar] [CrossRef]
- Li, Y.; Wu, A.; Dai, W.; Liu, R.; Jiang, B.; Zhou, R. Cerebrospinal fluid exosomal miR-152-3p predicts the occurrence of subarachnoid haemorrhage and regulates vascular smooth muscle cell dysfunction. Folia Neuropathol. 2022, 60, 185–194. [Google Scholar] [CrossRef]
- Blissenbach, B.; Nakas, C.T.; Kronke, M.; Geiser, T.; Merz, T.M.; Pichler Hefti, J. Hypoxia-induced changes in plasma micro-RNAs correlate with pulmonary artery pressure at high altitude. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 314, L157–L164. [Google Scholar] [CrossRef]
- Sotillo, J.; Robinson, M.W.; Kimber, M.J.; Cucher, M.; Ancarola, M.E.; Nejsum, P.; Marcilla, A.; Eichenberger, R.M.; Tritten, L. The protein and microRNA cargo of extracellular vesicles from parasitic helminths–current status and research priorities. Int. J. Parasitol. 2020, 50, 635–645. [Google Scholar] [CrossRef]
- Lin, J.; Maimaitiyiming, A.; Chen, S.; Xiao, M.; Xian, Z. Hypoxia-induced miR-27 and miR-195 regulate ATP consumption, viability, and metabolism of rat cardiomyocytes by targeting PPARγ and FASN expression. Aging 2021, 13, 10158. [Google Scholar] [CrossRef]
- Cheng, C.; Guo, F.; Yang, H.; Ma, J.; Li, H.; Yin, L.; Li, M.; Liu, S. Identification and analysis of the predictive urinary exosomal miR-195-5p in lupus nephritis based on renal miRNA-mRNA co-expression network. Lupus 2022, 31, 1786–1799. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, D.; Hui, B.; Shu, L.; Tang, X.; Wang, C.; Xie, J.; Yin, Y.; Sagnelli, M.; Yang, N.; et al. A novel regulatory mechanism network mediated by lncRNA TUG1 that induces the impairment of spiral artery remodeling in preeclampsia. Mol. Ther. 2022, 30, 1692–1705. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Y.; Zhou, M.; Zhang, Y.; Wang, P.; Li, X.; Yang, J.; Wang, H.; Ding, Z. HOXA9 inhibits HIF-1α-mediated glycolysis through interacting with CRIP2 to repress cutaneous squamous cell carcinoma development. Nat. Commun. 2018, 9, 1480. [Google Scholar] [CrossRef]
- Coon, J.; Kingsley, K.; Howard, K.M. miR-365 (microRNA): Potential biomarker in oral squamous cell carcinoma exosomes and extracellular vesicles. Int. J. Mol. Sci. 2020, 21, 5317. [Google Scholar] [CrossRef]
- Cui, C.; Li, Y.; Liu, Y. Down-regulation of miR-377 suppresses high glucose and hypoxia-induced angiogenesis and inflammation in human retinal endothelial cells by direct up-regulation of target gene SIRT1. Human Cell 2019, 32, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Song, X.; Yu, M.; Niu, L.; Zhao, Y.; Tang, Y.; Zheng, B.; Song, X.; Xie, L. Serum exosomal miR-377-3p and miR-381-3p as diagnostic biomarkers in colorectal cancer. Future Oncol. 2022, 18, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.H.; Huang, P.H.; Hsu, Y.J.; Chen, Y.W.; Wang, J.C.; Chen, Y.H.; Lin, S.J. Roles of the Hypoximir microRNA-424/322 on Acute Hypoxia and Hypoxia-Induced Pulmonary Vascular Leakage. EBioMedicine 2018. [Google Scholar] [CrossRef]
- Wang, Z.; Jiao, P.; Zhong, Y.; Ji, H.; Zhang, Y.; Song, H.; Du, H.; Ding, X.; Wu, H. The Endoplasmic Reticulum-Stressed Head and Neck Squamous Cell Carcinoma Cells Induced Exosomal miR-424-5p Inhibits Angiogenesis and Migration of Humanumbilical Vein Endothelial Cells Through LAMC1-Mediated Wnt/β-Catenin Signaling Pathway. Cell Transplant. 2022, 31, 09636897221083549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ye, W.; Li, Y. The effect of MiR-497 on the expression of target genes BCL-2 and LC3B on cardiomyocytes injured by hypoxia/reoxygenation. Cell. Mol. Biol. 2022, 68, 170–176. [Google Scholar] [CrossRef]
- Abdelrahman, A.; Negroni, C.; Sahm, F.; Adams, C.L.; Urbanic-Purkart, T.; Khalil, M.; Vergura, R.; Morelli, C.; Hanemann, C.O. miR-497 and 219 in blood aid meningioma classification. J. Neuro-Oncol. 2022, 160, 137–147. [Google Scholar] [CrossRef]
- Lin, X.; Shan, S.K.; Xu, F.; Zhong, J.Y.; Wu, F.; Duan, J.Y.; Guo, B.; Li, F.X.Z.; Wang, Y.; Zheng, M.H.; et al. The crosstalk between endothelial cells and vascular smooth muscle cells aggravates high phosphorus-induced arterial calcification. Cell Death Dis. 2022, 13, 650. [Google Scholar] [CrossRef]
- Luo, X.; Luo, S.Z.; Xu, Z.X.; Zhou, C.; Li, Z.H.; Zhou, X.Y.; Xu, M.Y. Lipotoxic hepatocyte-derived exosomal miR-1297 promotes hepatic stellate cell activation through the PTEN signaling pathway in metabolic-associated fatty liver disease. World J. Gastroenterol. 2021, 27, 1419. [Google Scholar] [CrossRef]
- Cao, W.; Zeng, Z.; He, Z.; Lei, S. Hypoxic pancreatic stellate cell-derived exosomal mirnas promote proliferation and invasion of pancreatic cancer through the PTEN/AKT pathway. Aging 2021, 13, 7120. [Google Scholar] [CrossRef]
- Zhang, G.; Ding, L.; Sun, G.; Liu, Z.; Ou, W.; Wang, B.; Sun, Y. LncRNA AZIN1-AS1 ameliorates myocardial ischemia–reperfusion injury by targeting miR-6838-5p/WNT3A axis to activate Wnt-β/catenin signaling pathway. In Vitr. Cell. Dev. Biol.-Anim. 2022, 58, 54–68. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Xiang, S.; Zheng, Z.; Bian, Y.; Feng, B.; Weng, X. Chondrocytes-derived exosomal miR-8485 regulated the Wnt/β-catenin pathways to promote chondrogenic differentiation of BMSCs. Biochem. Biophys. Res. Commun. 2020, 523, 506–513. [Google Scholar] [CrossRef]
- Bachmann, M.; Rossa, A.; Varanita, T.; Fioretti, B.; Biasutto, L.; Milenkovic, S.; Checchetto, V.; Peruzzo, R.; Ahmad, S.A.; Patel, S.H.; et al. Pharmacological targeting of the mitochondrial calcium-dependent potassium channel KCa3. 1 triggers cell death and reduces tumor growth and metastasis in vivo. Cell Death Dis. 2022, 13, 1055. [Google Scholar] [CrossRef]
- Bachmann, M.; Rossa, A.; Antoniazzi, G.; Biasutto, L.; Carrer, A.; Campagnaro, M.; Leanza, L.; Gonczi, M.; Csernoch, L.; Paradisi, C.; et al. Synthesis and cellular effects of a mitochondria-targeted inhibitor of the two-pore potassium channel TASK-3. Pharmacol. Res. 2021, 164, 105326. [Google Scholar] [CrossRef]
- Severin, F.; Urbani, A.; Varanita, T.; Bachmann, M.; Azzolini, M.; Martini, V.; Pizzi, M.; Tos, A.P.D.; Frezzato, F.; Mattarei, A.; et al. Pharmacological modulation of Kv1. 3 potassium channel selectively triggers pathological B lymphocyte apoptosis in vivo in a genetic CLL model. J. Exp. Clin. Cancer Res. 2022, 41, 64. [Google Scholar] [CrossRef]
- Li, W.; Wilson, G.C.; Bachmann, M.; Wang, J.; Mattarei, A.; Paradisi, C.; Edwards, M.J.; Szabo, I.; Gulbins, E.; Ahmad, S.A.; et al. Inhibition of a mitochondrial potassium channel in combination with gemcitabine and abraxane drastically reduces pancreatic ductal adenocarcinoma in an immunocompetent orthotopic murine model. Cancers 2022, 14, 2618. [Google Scholar] [CrossRef]
- Payne, S.L.; Ram, P.; Srinivasan, D.H.; Le, T.T.; Levin, M.; Oudin, M.J. Potassium channel-driven bioelectric signalling regulates metastasis in triple-negative breast cancer. EBioMedicine 2022, 75, 103767. [Google Scholar] [CrossRef]
- Lastraioli, E. Focus on triple-negative breast cancer: Potassium channel expression and clinical correlates. Front. Pharmacol. 2020, 11, 725. [Google Scholar] [CrossRef]
- Hou, X.; Tang, L.; Li, X.; Xiong, F.; Mo, Y.; Jiang, X.; Deng, X.; Peng, M.; Wu, P.; Zhao, M.; et al. Potassium channel protein KCNK6 promotes breast cancer cell proliferation, invasion, and migration. Front. Cell Dev. Biol. 2021, 9, 616784. [Google Scholar] [CrossRef]
- Francisco, M.A.; Wanggou, S.; Fan, J.J.; Dong, W.; Chen, X.; Momin, A.; Abeysundara, N.; Min, H.K.; Chan, J.; McAdam, R.; et al. Chloride intracellular channel 1 cooperates with potassium channel EAG2 to promote medulloblastoma growth. J. Exp. Med. 2020, 217, e20190971. [Google Scholar] [CrossRef]
- Bergers, G.; Fendt, S.M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef] [PubMed]
- Ghanavat, M.; Shahrouzian, M.; Zayeri, Z.D.; Banihashemi, S.; Kazemi, S.M.; Saki, N. Digging deeper through glucose metabolism and its regulators in cancer and metastasis. Life Sci. 2021, 264, 118603. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Baek, K.H. Regulation of cancer metabolism by deubiquitinating enzymes: The Warburg effect. Int. J. Mol. Sci. 2021, 22, 6173. [Google Scholar] [CrossRef] [PubMed]
- Urbańska, K.; Orzechowski, A. Unappreciated role of LDHA and LDHB to control apoptosis and autophagy in tumor cells. Int. J. Mol. Sci. 2019, 20, 2085. [Google Scholar] [CrossRef]
- Sharma, D.; Singh, M.; Rani, R. Role of LDH in tumor glycolysis: Regulation of LDHA by small molecules for cancer therapeutics. Semin. Cancer Biol. 2022, 87, 184–995. [Google Scholar] [CrossRef]
- Hui, A.S.; Bauer, A.L.; Striet, J.B.; Schnell, P.O.; Czyzyk-Krzeska, M.F. Calcium signaling stimulates translation of HIF-α during hypoxia. FASEB J. 2006, 20, 466–475. [Google Scholar] [CrossRef]
- Ednie, A.R.; Bennett, E.S. Reduced sialylation impacts ventricular repolarization by modulating specific K+ channel isoforms distinctly. J. Biol. Chem. 2015, 290, 2769–2783. [Google Scholar] [CrossRef]
- Nag, S.; Mandal, A.; Joshi, A.; Jain, N.; Srivastava, R.S.; Singh, S.; Khattri, A. Sialyltransferases and Neuraminidases: Potential Targets for Cancer Treatment. Diseases 2022, 10, 114. [Google Scholar] [CrossRef]
- Munkley, J. Aberrant Sialylation in Cancer: Therapeutic Opportunities. Cancers 2022, 14, 4248. [Google Scholar] [CrossRef]
- Stockmann, H.; Marin, V.L.; Nimmer, P.; Balut, C.M.; Davidson, D.J.; Richardson, P.L.; Vasudevan, A. Glycan-Mediated, Ligand-Controlled Click Chemistry for Drug-Target Identification. ChemBioChem 2016, 17, 150–154. [Google Scholar] [CrossRef]
- Thu, C.T.; Mahal, L.K. Sweet control: MicroRNA regulation of the glycome. Biochemistry 2019, 59, 3098–3110. [Google Scholar] [CrossRef]
- Falcone, G.; Felsani, A.; D’Agnano, I. Signaling by exosomal microRNAs in cancer. J. Exp. Clin. Cancer Res. 2015, 34, 32. [Google Scholar] [CrossRef]
- Garcia-Martin, R.; Wang, G.; Brandão, B.B.; Zanotto, T.M.; Shah, S.; Kumar Patel, S.; Schilling, B.; Kahn, C.R. MicroRNA sequence codes for small extracellular vesicle release and cellular retention. Nature 2022, 601, 446–451. [Google Scholar] [CrossRef]
- Barile, L.; Vassalli, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol. Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef]
- Toplak, Ž.; Hendrickx, L.A.; Abdelaziz, R.; Shi, X.; Peigneur, S.; Tomašič, T.; Tytgat, J.; Peterlin-Mašič, L.; Pardo, L.A. Overcoming challenges of HERG potassium channel liability through rational design: Eag1 inhibitors for cancer treatment. Med. Res. Rev. 2022, 42, 183–226. [Google Scholar] [CrossRef]
- Preußat, K.; Beetz, C.; Schrey, M.; Kraft, R.; Wolfl, S.; Kalff, R.; Patt, S. Expression of voltage-gated potassium channels Kv1. 3 and Kv1. 5 in human gliomas. Neurosci. Lett. 2003, 346, 33–36. [Google Scholar] [CrossRef]
- Bielanska, J.; Hernandez-Losa, J.; Perez-Verdaguer, M.; Moline, T.; Somoza, R.; Cajal, S.; Condom, E.; Ferreres, J.; Felipe, A. Voltage-dependent potassium channels Kv1. 3 and Kv1. 5 in human cancer. Curr. Cancer Drug Targets 2009, 9, 904–914. [Google Scholar] [CrossRef]
- Than, B.L.; Goos, J.; Sarver, A.L.; O’Sullivan, M.G.; Rod, A.; Starr, T.K.; Fijneman, R.J.; Meijer, G.A.; Zhao, L.; Zhang, Y.; et al. The role of KCNQ1 in mouse and human gastrointestinal cancers. Oncogene 2014, 33, 3861–3868. [Google Scholar] [CrossRef]
- Rosa, P.; Sforna, L.; Carlomagno, S.; Mangino, G.; Miscusi, M.; Pessia, M.; Franciolini, F.; Calogero, A.; Catacuzzeno, L. Overexpression of large-conductance calcium-activated potassium channels in human glioblastoma stem-like cells and their role in cell migration. J. Cell. Physiol. 2017, 232, 2478–2488. [Google Scholar] [CrossRef]
- Wrzosek, A.; Augustynek, B.; Żochowska, M.; Szewczyk, A. Mitochondrial potassium channels as druggable targets. Biomolecules 2020, 10, 1200. [Google Scholar] [CrossRef]
- Augustynek, B.; Koprowski, P.; Rotko, D.; Kunz, W.S.; Szewczyk, A.; Kulawiak, B. Mitochondrial BK channel openers CGS7181 and CGS7184 exhibit cytotoxic properties. Int. J. Mol. Sci. 2018, 19, 353. [Google Scholar] [CrossRef] [PubMed]
- Moreels, L.; Bhat, C.; Voráčová, M.; Peigneur, S.; Goovaerts, H.; Maki-Lohiluoma, E.; Zahed, F.; Pardo, L.A.; Yli-Kauhaluoma, J.; Kiuru, P.; et al. Synthesis of novel purpurealidin analogs and evaluation of their effect on the cancer-relevant potassium channel KV10. 1. PLoS ONE 2017, 12, e0188811. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Wang, P.; Zhu, X.; Morishima, M.; Liu, Y.; Zheng, M.; Liu, G.; Osanai, H.; Yoshimura, K.; Kume, S.; et al. Electrophysiological evaluation of an anticancer drug gemcitabine on cardiotoxicity revealing down-regulation and modification of the activation gating properties in the human rapid delayed rectifier potassium channel. PLoS ONE 2023, 18, e0280656. [Google Scholar] [CrossRef] [PubMed]
- Poupon, L.; Lamoine, S.; Pereira, V.; Barriere, D.A.; Lolignier, S.; Giraudet, F.; Aissouni, Y.; Meleine, M.; Prival, L.; Richard, D.; et al. Targeting the TREK-1 potassium channel via riluzole to eliminate the neuropathic and depressive-like effects of oxaliplatin. Neuropharmacology 2018, 140, 43–61. [Google Scholar] [CrossRef]
- Kale, V.P.; Amin, S.G.; Pandey, M.K. Targeting ion channels for cancer therapy by repurposing the approved drugs. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 2747–2755. [Google Scholar] [CrossRef]
- Patil, V.M.; Gaurav, A.; Garg, P.; Masand, N. Non-cancer to anti-cancer: Investigation of human ether-a-go-go-related gene potassium channel inhibitors as potential therapeutics. J. Egypt. Natl. Cancer Inst. 2021, 33, 33. [Google Scholar] [CrossRef]
- Loza-Huerta, A.; Milo, E.; Picones, A.; Hernández-Cruz, A.; Luis, E. Thallium-sensitive fluorescent assay reveals loperamide as a new inhibitor of the potassium channel Kv10.1. Pharmacol. Rep. 2021, 73, 1744–1753. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potassium Channels | microRNA | Target | Cancer (Model) | Expression | References | References [miRNA–Exosomes] |
|---|---|---|---|---|---|---|
| Kir2.1, K4.1 | miR-9-5p | HK2 | Colorectal cancer (human) | ↑ | [79] | [80,81] |
| Kir2.2 | miR-603 | HK2 | Ovarian cancer (in vitro, in vivo, human) | ↑ | [82] | – |
| Ovarian cancer (human) | ↓ | |||||
| Kir3.1 | miR-361-5p | Sp1/PKM2 | Bladder cancer (in vitro) | ↓ | [83] | [84] |
| Kv12.3, Kv9.3 | miR-125a-5p | CD147 | Thyroid cancer (in vitro) | ↓ | [85] | [86,87,88,89] |
| Kv1.1, Kv12.3, Kv9.3 | miR-125b-5p | HK2 | Laryngeal squamous (in vitro, human) | ↓ | [90] | [91,92] |
| Kv1.2 | miR-137 | NOX4 | Prostate cancer (in vitro) | ↑ | [93] | [94,95] |
| GLO1 | Melanoma (in vitro) | ↓ | [96] | |||
| Kv7.3 | miR-449a | LDHA | Lung cancer (in vitro) | ↓ | [97] | [98,99] |
| Kv7.5 | miR-139-5p | HK1, PFKFB3 | Liver cancer (in vitro, in vivo) | ↓ | [100] | [101,102] |
| PRKAA1 | Gastric cancer (in vitro) | ↓ | [103] | |||
| KCa3.1 | miR-15b-5p | PDK4 | Osteosarcoma (in vitro) | ↓ | [104] | [105,106] |
| microRNA | Sialyltransferase | Potassium Channel | References [miRNA–Hypoxia] | References [miRNA–Exosomes] |
|---|---|---|---|---|
| miR-15 | ST8SIA3 | K3.1 | Xue et al. (2015) [186] | Luo et al. (2022) [187] |
| miR-16 | ST8SIA3 | K3.1 | Xue et al. (2015) [186] | Luo et al. (2022) [187] |
| miR-26 | ST6GAL2 | Kv11.3 | Li et al. (2021) [188] | Chettimada et al. (2020) [189] |
| Kv7.4 | ||||
| K1.1 | ||||
| miR-122 | ST6GALNAC4 | Kir7.1 | Xu et al. (2022) [190] | Xu et al. (2022) [190] |
| miR-125 | ST6GAL1 | Kv1.1 | Li et al. (2018) [191] | Kot et al. (2023) [192] |
| Kv9.3 | ||||
| Kv12.3 | ||||
| ST6GALNAC6 | Kv1.1 | |||
| Kv12.3 | ||||
| Kv9.3 | ||||
| miR-135 | ST6GAL2 | Kv4.1 | — | Parikh et al. (2021) [193] |
| Kir3.2 | ||||
| ST8SIA3 | Kv4.1 | |||
| Kir3.1 | ||||
| miR-148 | ST8SIA3 | Kv4.3 | Behara et al. (2023) [194] | Chettimada et al (2020) [189] |
| Kir3.1 | ||||
| miR-152 | ST8SIA3 | Kv4.3 | Zhao et al. (2021) [195] | Li et al. (2022) [196] |
| miR-190 | ST6GAL2 | Kv7.5 | Blissenbach et al. (2018) [197] | Sotillo et al. (2020) [198] |
| miR-195 | ST8SIA3 | K3.1 | Lin et al. (2021) [199] | Cheng et al. (2022) [200] |
| miR-218 | ST8SIA5 | Kv4.2 | Xu et al. (2022) [201] | Cheng et al. (2022) [200] |
| K15.1 | ||||
| miR-365 | ST6GAL2 | Kir3.1 | Zhou et al. (2018) [202] | Coon et al. (2020) [203] |
| Kv11.1 | ||||
| Kv7.1 | ||||
| miR-377 | ST6GALNAC5 | Kv1.4 | Cui et al. (2019) [204] | Wang et al. (2022) [205] |
| miR-424 | ST8SIA3 | K3.1 | Tsai et al. (2018) [206] | Wang et al. (2022) [207] |
| miR-497 | ST8SIA3 | K3.1 | Ye et al. (2022) [208] | Abdelrahma et al. (2022) [209] |
| miR-670 | ST8SIA3 | Kv9.1 | – | Lin et al. (2022) [210] |
| miR-1297 | ST6GAL2 | Kv11.3 | – | Luo et al. (2021) [211] |
| Kv7.4 | ||||
| K1.1 | ||||
| miR-4319 | ST6GAL1 | Kv9.3 | – | – |
| Kv12.3 | ||||
| ST6GALNAC6 | Kv12.3 | – | – | |
| Kv9.3 | ||||
| miR-4465 | ST6GAL2 | Kv7.4 | Cao et al. (2021) [212] | Cao et al. (2021) [212] |
| K1.1 | ||||
| miR-6838 | ST8SIA3 | K3.1 | Zhang et al. (2022) [213] | – |
| miR-8485 | ST8SIA4 | Kir2.2 | – | Li et al. (2020) [214] |
| Kir3.1 | ||||
| Kir4.1 | ||||
| Kv1.4 | ||||
| Kv2.1 | ||||
| Kv3.4 | ||||
| K5.1 | ||||
| K10.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wawrzkiewicz-Jałowiecka, A.; Lalik, A.; Lukasiak, A.; Richter-Laskowska, M.; Trybek, P.; Ejfler, M.; Opałka, M.; Wardejn, S.; Delfino, D.V. Potassium Channels, Glucose Metabolism and Glycosylation in Cancer Cells. Int. J. Mol. Sci. 2023, 24, 7942. https://doi.org/10.3390/ijms24097942
Wawrzkiewicz-Jałowiecka A, Lalik A, Lukasiak A, Richter-Laskowska M, Trybek P, Ejfler M, Opałka M, Wardejn S, Delfino DV. Potassium Channels, Glucose Metabolism and Glycosylation in Cancer Cells. International Journal of Molecular Sciences. 2023; 24(9):7942. https://doi.org/10.3390/ijms24097942
Chicago/Turabian StyleWawrzkiewicz-Jałowiecka, Agata, Anna Lalik, Agnieszka Lukasiak, Monika Richter-Laskowska, Paulina Trybek, Maciej Ejfler, Maciej Opałka, Sonia Wardejn, and Domenico V. Delfino. 2023. "Potassium Channels, Glucose Metabolism and Glycosylation in Cancer Cells" International Journal of Molecular Sciences 24, no. 9: 7942. https://doi.org/10.3390/ijms24097942