

Identification of 7-Ketocholesterol-Modulated Pathways and Sterculic Acid Protective Effect in Retinal Pigmented Epithelium Cells by Using Genome-Wide Transcriptomic Analysis
 , , and
, , and
Abstract
:1. Introduction
2. Results
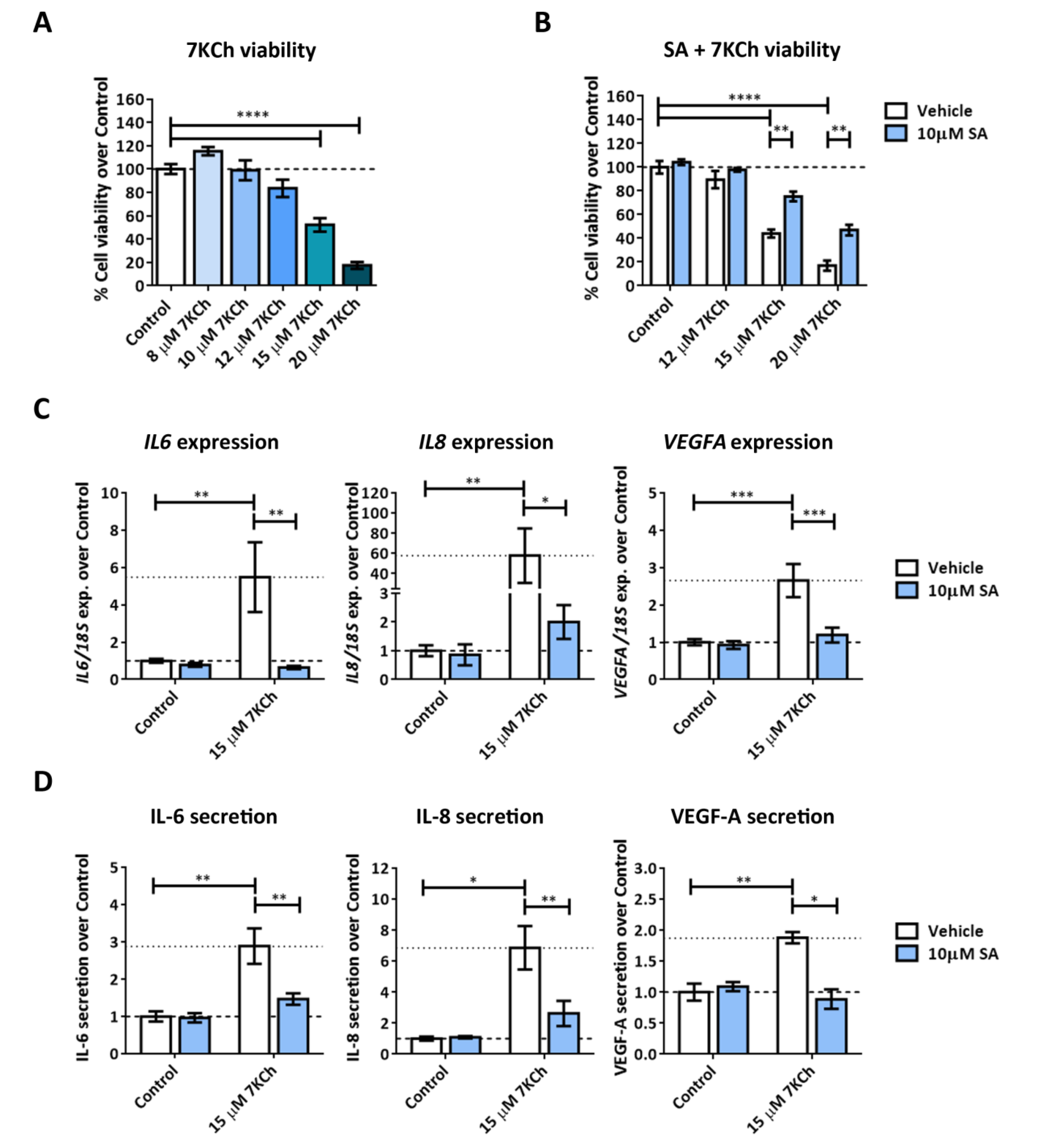
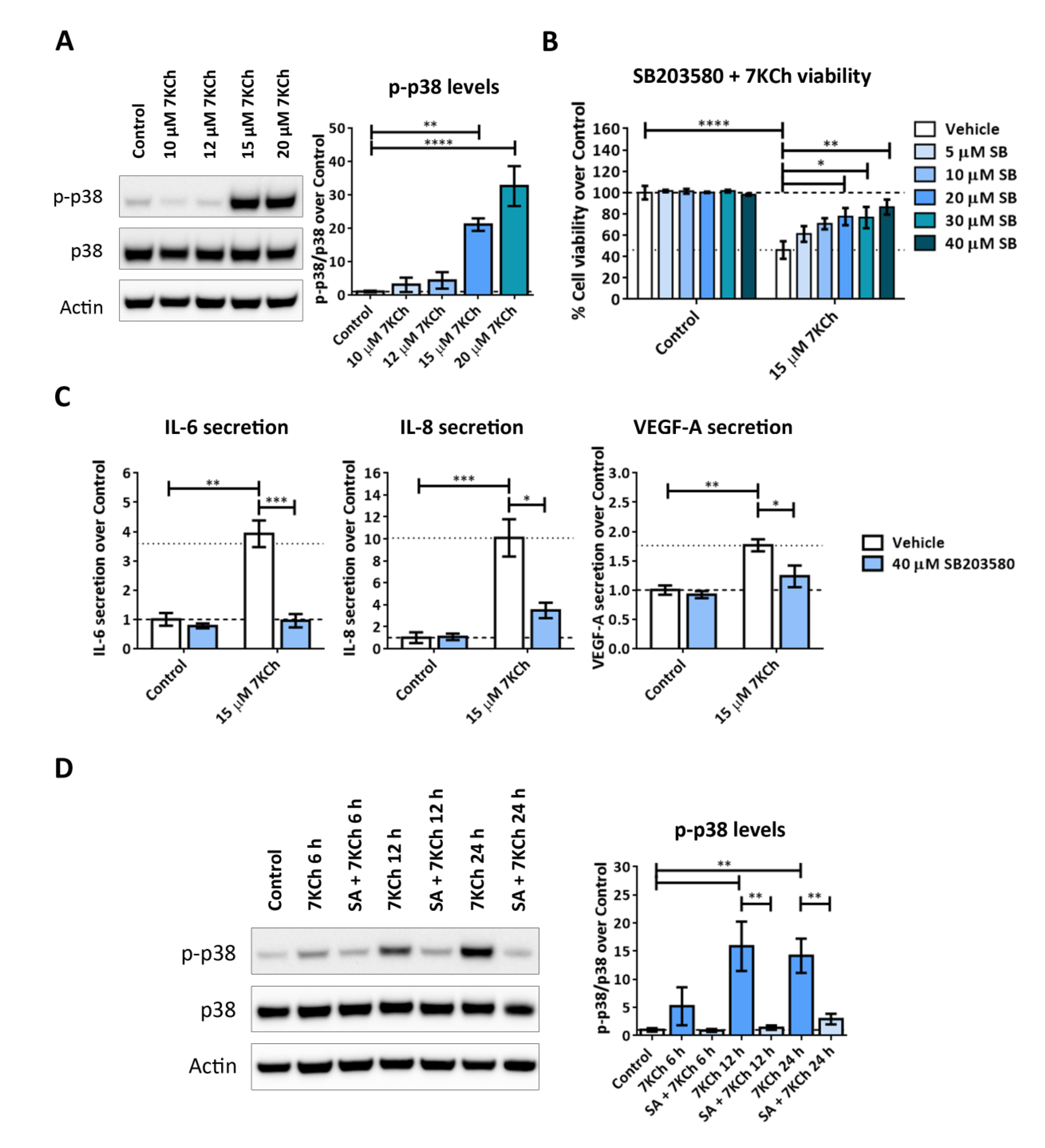
2.1. 7KCh Induces Cell Death and Inflammatory Responses in mRPE Cells, Which Are Attenuated by SA Administration
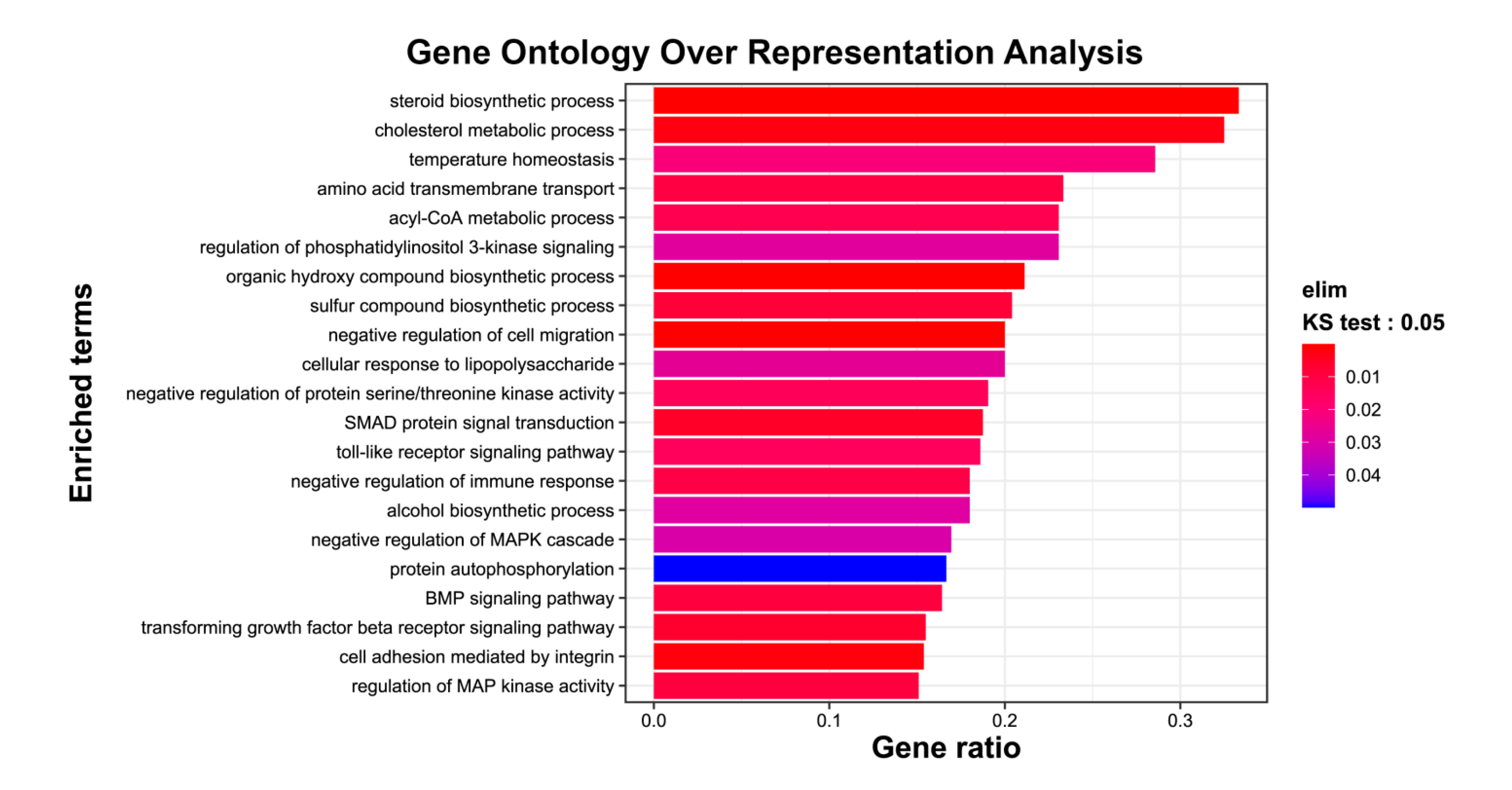
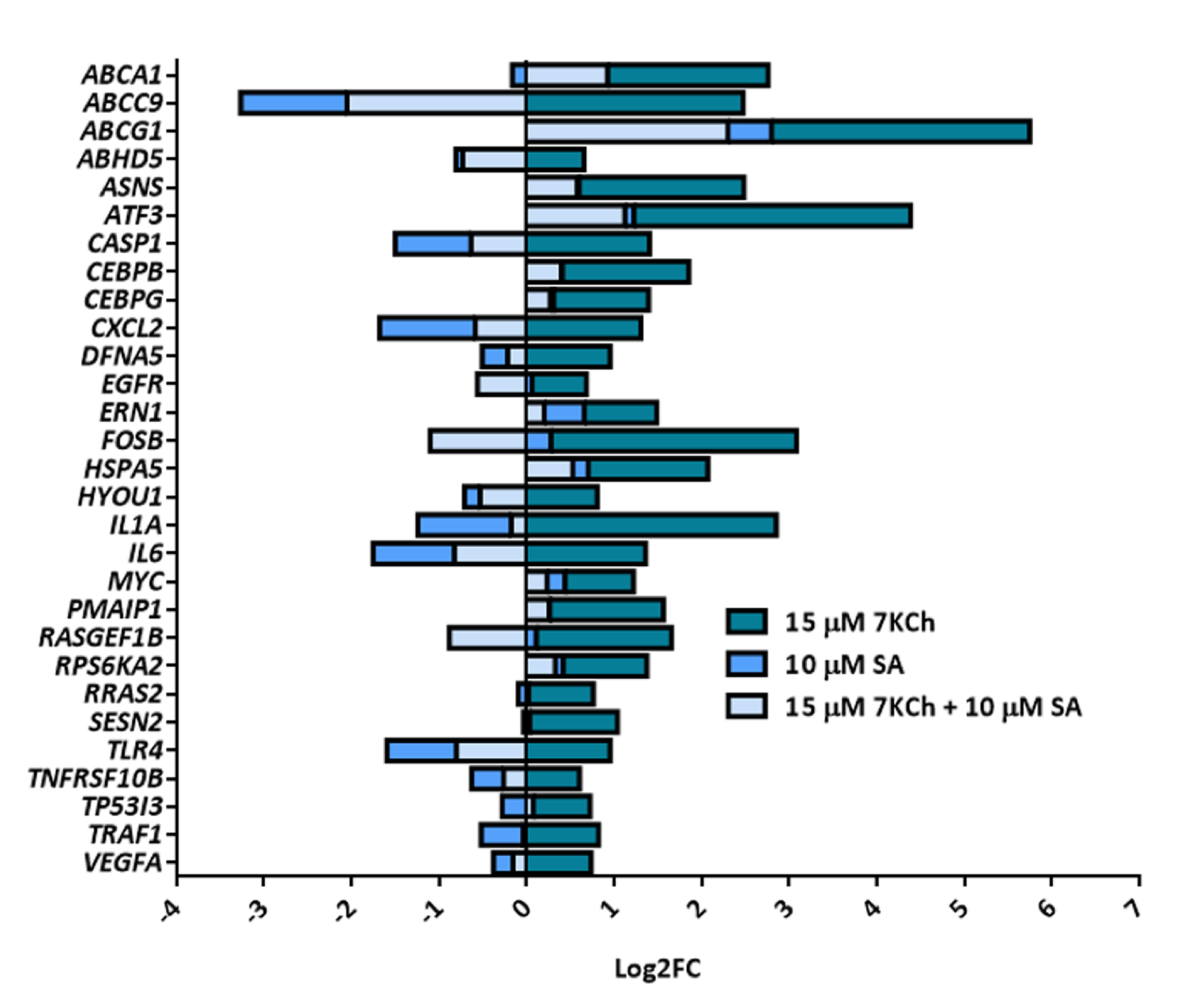
2.2. Exposure of mRPE Cells to 7KCh Causes Modulation of Genes Associated with Lipid Metabolism, ER (Endoplasmic Reticulum) Stress, Inflammation and Cell Death
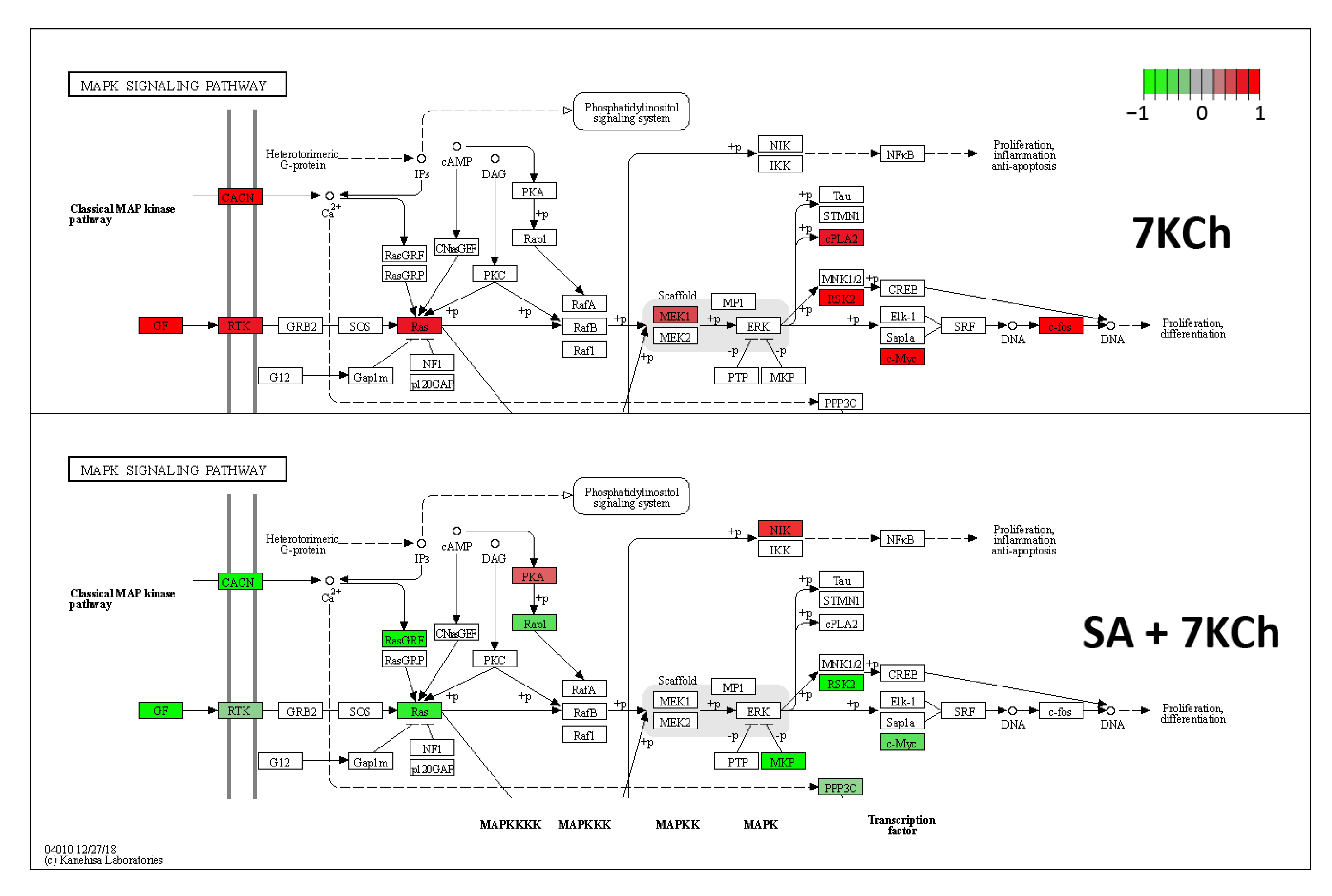
2.3. SA Exerts a Reverse Effect on the Modulation of Part of the Genes Altered by 7KCh
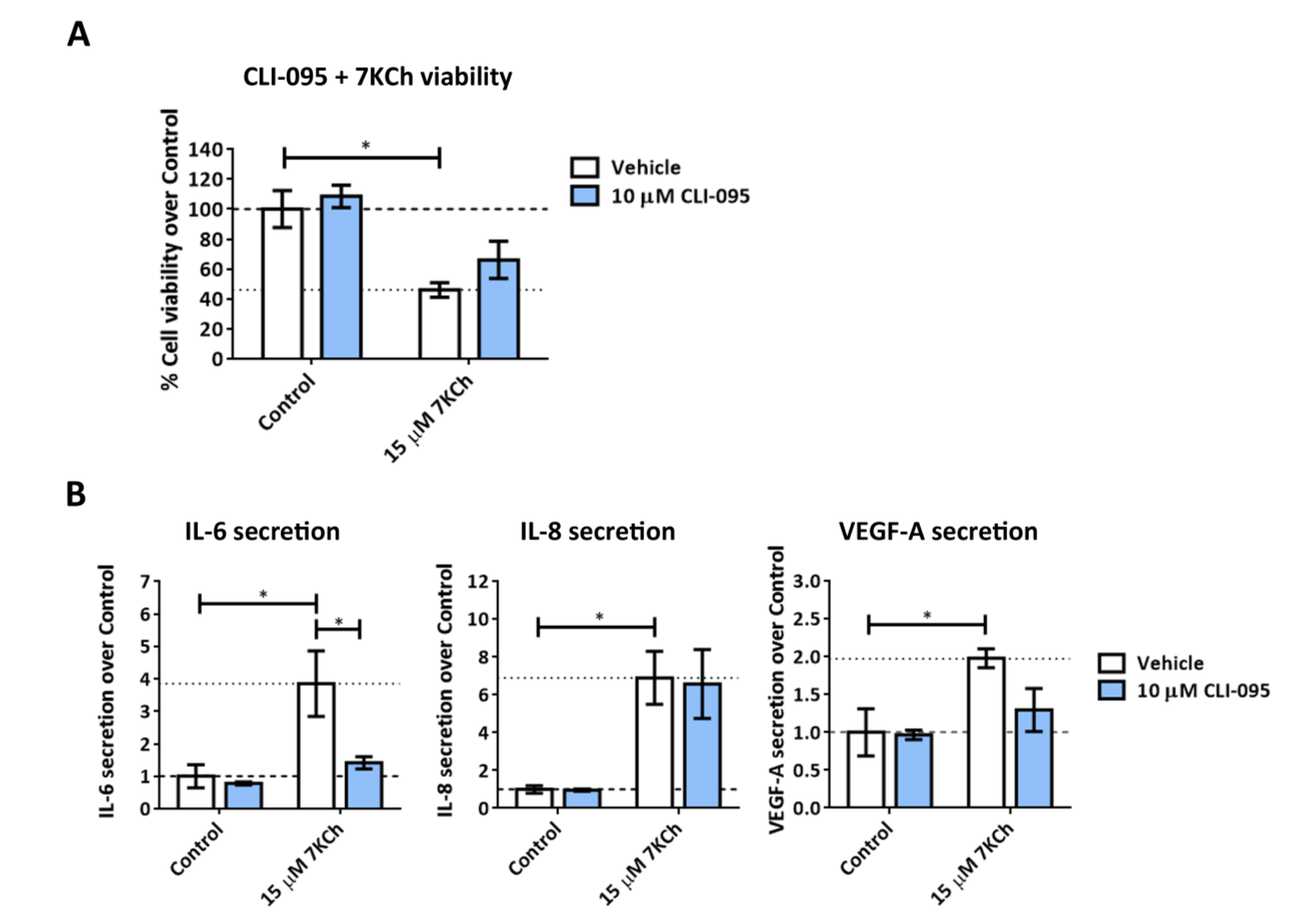
2.4. TLR4 Does Not Have an Important Role in Mediating 7KCh-Response in mRPE Cells
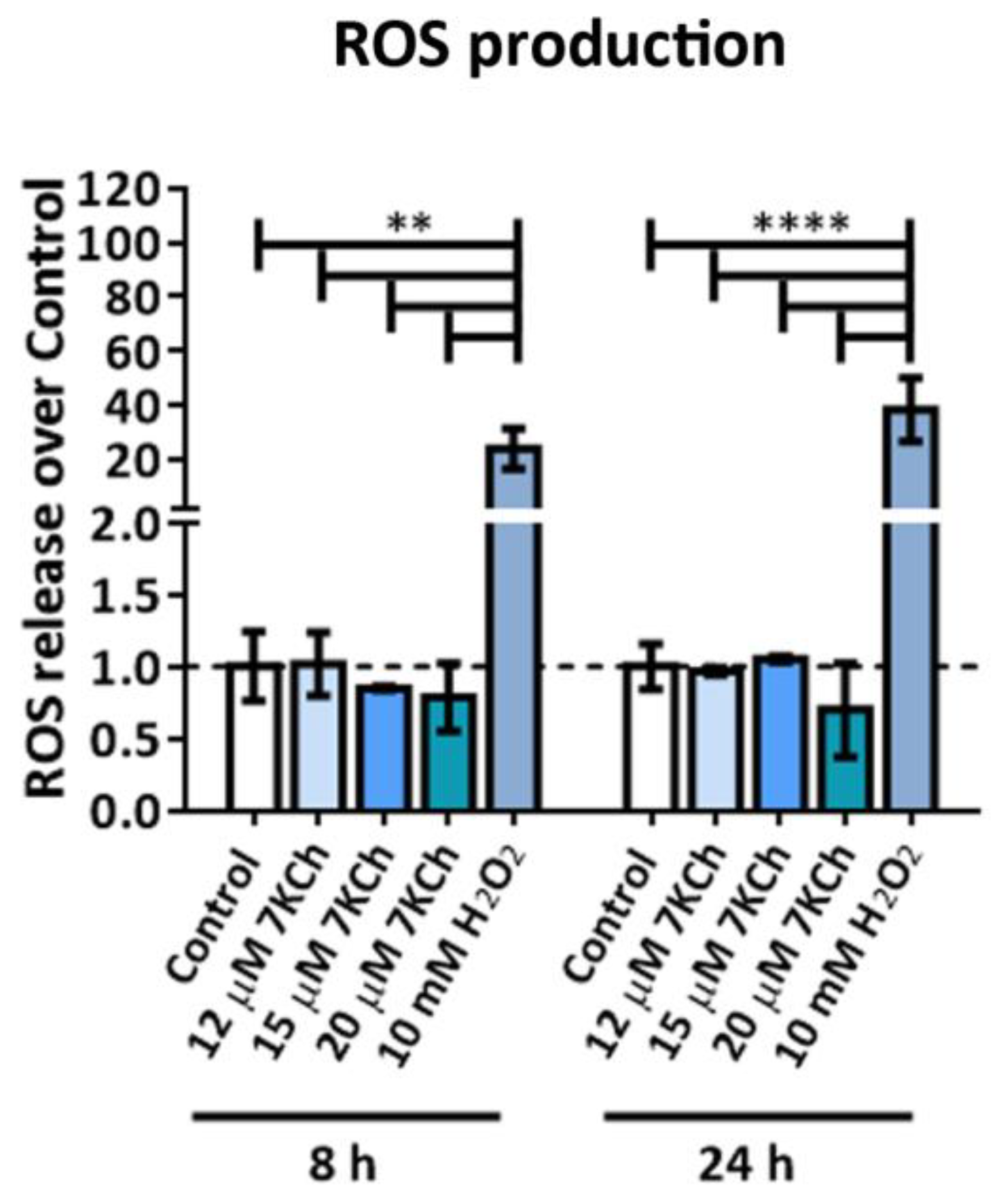
2.5. 7KCh Does Not Induce ROS Release in mRPE Cells
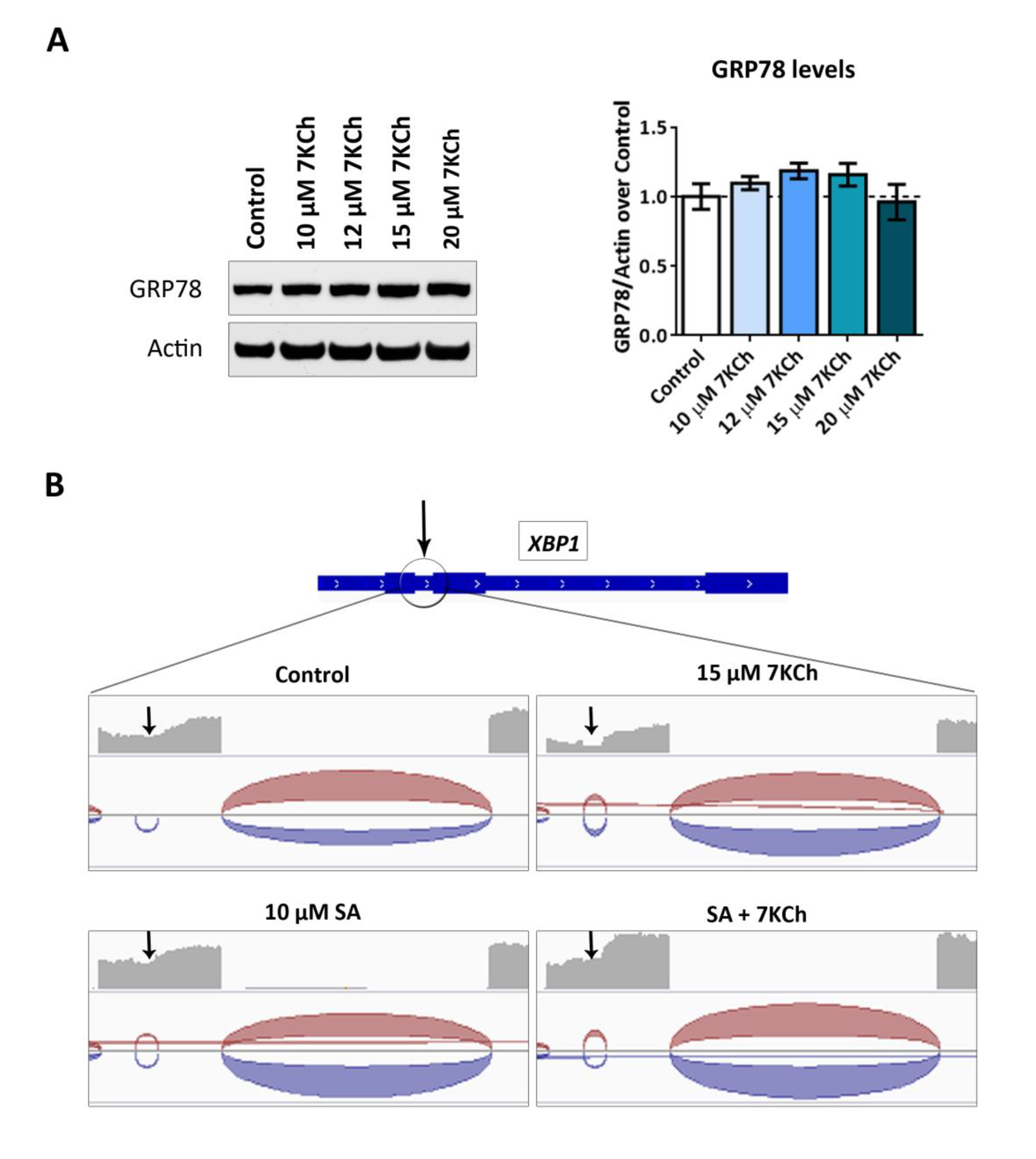
2.6. ER Stress Induced by 7KCh in mRPE Cells Is Mediated by the Unfolded Protein Response
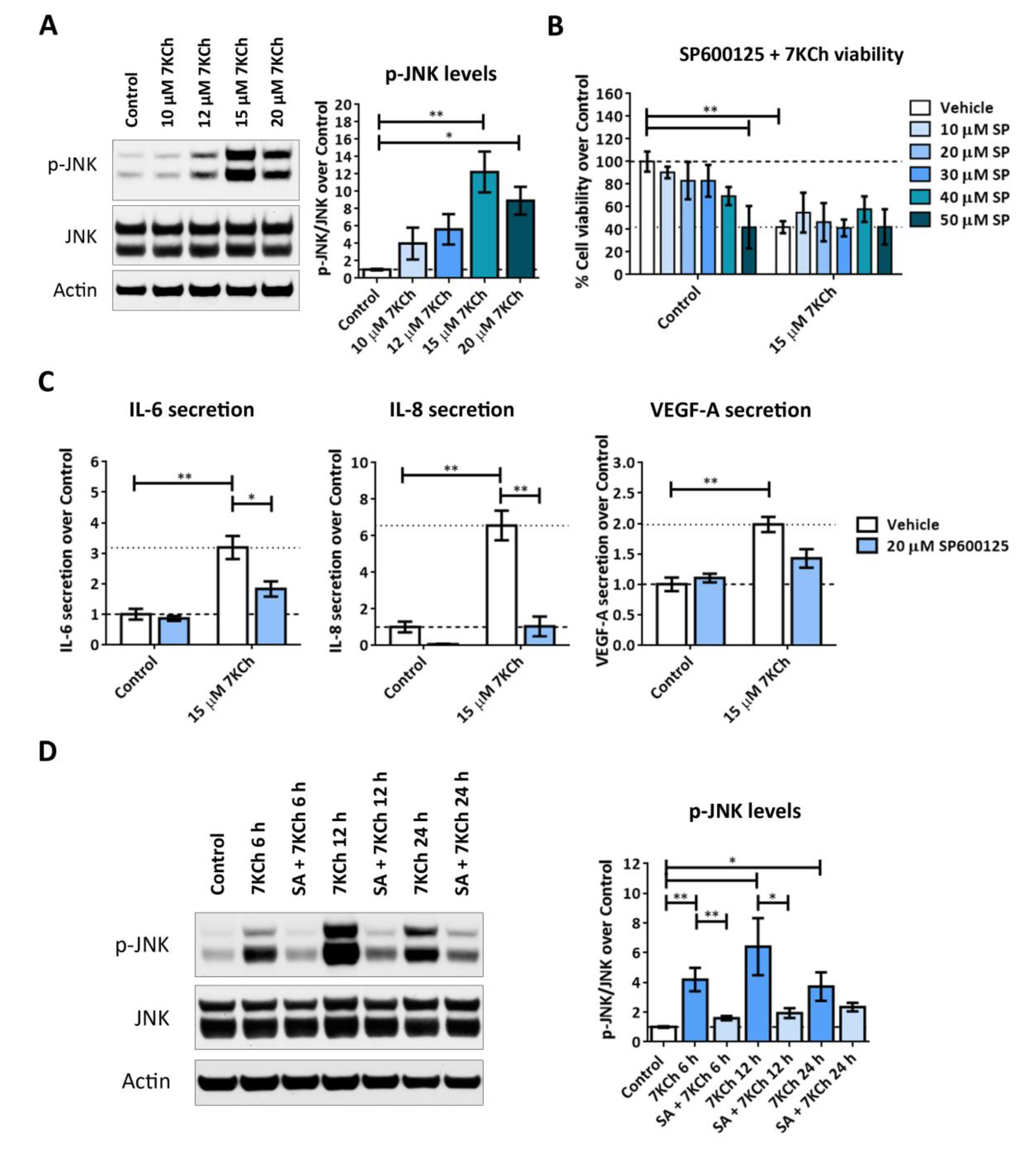
2.7. Both JNK and p38 Pathways Are Involved in the Inflammatory Response to 7KCh in mRPE Cells, but Only p38 Participates in the Cell Death Response
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Cell Treatments
4.3. Cell Viability Assays
4.4. ROS Detection Assay
4.5. RNA Purification
4.6. Quantitative Real-Time PCR
4.7. Next-Generation Sequencing
4.8. Western Blotting
4.9. ELISAs
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 7KCh | 7-Ketocholesterol |
| AMD | age-related macular degeneration |
| CNV | choroidal neovascularization |
| DEG | differentially expressed genes |
| ER | endoplasmic reticulum |
| FC | fold change |
| FDR | false discovery rate |
| GO | gene ontology |
| IGV | Integrative Genome Viewer |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LXR | liver X receptor |
| mRPE | monkey retinal pigment epithelium |
| qRT-PCR | real-time polymerase chain reaction |
| RPE | retinal pigment epithelium |
| SA | sterculic acid |
| SCD1 | stearoyl coenzyme-A desaturase 1 |
| SEM | standard error of the means |
| TBS | tris-buffered saline |
| UPR | unfolded protein response |
References
- Deng, Y.; Qiao, L.; Du, M.; Qu, C.; Wan, L.; Li, J.; Huang, L. Age-related macular degeneration: Epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes Dis. 2022, 9, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Mehrzadi, S.; Hemati, K.; Reiter, R.J.; Hosseinzadeh, A. Mitochondrial dysfunction in age-related macular degeneration: Melatonin as a potential treatment. Expert Opin. Ther. Targets 2020, 24, 359–378. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A. Introduction to the issue regarding research regarding age related macular degeneration. Mol. Asp. Med. 2012, 33, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Cao, S.; Rajapakse, S.; Matsubara, J.A. Understanding AMD by analogy: Systematic review of lipid-related common pathogenic mechanisms in AMD, AD, AS and GN. Lipids Health Dis. 2018, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A. The Diagnosis and Treatment of Age-Related Macular Degeneration. Dtsch. Arztebl. Int. 2020, 117, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.J.; Mirza, R.G.; Gill, M.K. Age-Related Macular Degeneration. Med. Clin. North Am. 2021, 105, 473–491. [Google Scholar] [CrossRef]
- Curcio, C.A. Antecedents of Soft Drusen, the Specific Deposits of Age-Related Macular Degeneration, in the Biology of Human Macula. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD182–AMD194. [Google Scholar] [CrossRef]
- Curcio, C.A. Soft Drusen in Age-Related Macular Degeneration: Biology and Targeting Via the Oil Spill Strategies. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD160–AMD181. [Google Scholar] [CrossRef]
- Spaide, R.F.; Ooto, S.; Curcio, C.A. Subretinal drusenoid deposits AKA pseudodrusen. Surv. Ophthalmol. 2018, 63, 782–815. [Google Scholar] [CrossRef]
- Curcio, C.A.; Johnson, M.; Rudolf, M.; Huang, J.D. The oil spill in ageing Bruch membrane. Br. J. Ophthalmol. 2011, 95, 1638–1645. [Google Scholar] [CrossRef]
- Abokyi, S.; To, C.H.; Lam, T.T.; Tse, D.Y. Central Role of Oxidative Stress in Age-Related Macular Degeneration: Evidence from a Review of the Molecular Mechanisms and Animal Models. Oxidative Med. Cell. Longev. 2020, 2020, 7901270. [Google Scholar] [CrossRef] [PubMed]
- Pfau, M.; von der Emde, L.; de Sisternes, L.; Hallak, J.A.; Leng, T.; Schmitz-Valckenberg, S.; Holz, F.G.; Fleckenstein, M.; Rubin, D.L. Progression of Photoreceptor Degeneration in Geographic Atrophy Secondary to Age-related Macular Degeneration. JAMA Ophthalmol. 2020, 138, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 4271. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, J.; Usategui-Martin, R.; Sanabria, M.R.; Fernandez-Perez, E.; Telleria, J.J.; Coco-Martin, R.M. Pathophysiology of Age-Related Macular Degeneration. Implications for Treatment. Ophthalmic Res. 2022, 65, 615–636. [Google Scholar] [CrossRef] [PubMed]
- Moreira, E.F.; Larrayoz, I.M.; Lee, J.W.; Rodriguez, I.R. 7-Ketocholesterol is present in lipid deposits in the primate retina: Potential implication in the induction of VEGF and CNV formation. Investig. Ophthalmol. Vis. Sci. 2009, 50, 523–532. [Google Scholar] [CrossRef]
- Bobadilla, M.; Pariente, A.; Oca, A.I.; Pelaez, R.; Perez-Sala, A.; Larrayoz, I.M. Biomarkers as Predictive Factors of Anti-VEGF Response. Biomedicines 2022, 10, 1003. [Google Scholar] [CrossRef]
- Oca, A.I.; Perez-Sala, A.; Pariente, A.; Ochoa, R.; Velilla, S.; Pelaez, R.; Larrayoz, I.M. Predictive Biomarkers of Age-Related Macular Degeneration Response to Anti-VEGF Treatment. J. Pers. Med. 2021, 11, 1329. [Google Scholar] [CrossRef]
- Mettu, P.S.; Allingham, M.J.; Cousins, S.W. Incomplete response to Anti-VEGF therapy in neovascular AMD: Exploring disease mechanisms and therapeutic opportunities. Prog. Retin. Eye Res. 2021, 82, 100906. [Google Scholar] [CrossRef]
- Huang, J.D.; Amaral, J.; Lee, J.W.; Larrayoz, I.M.; Rodriguez, I.R. Sterculic acid antagonizes 7-ketocholesterol-mediated inflammation and inhibits choroidal neovascularization. Biochim. Et Biophys. Acta 2012, 1821, 637–646. [Google Scholar] [CrossRef]
- Huang, J.D.; Amaral, J.; Lee, J.W.; Rodriguez, I.R. 7-Ketocholesterol-induced inflammation signals mostly through the TLR4 receptor both in vitro and in vivo. PLoS ONE 2014, 9, e100985. [Google Scholar] [CrossRef]
- Leoni, V.; Nury, T.; Vejux, A.; Zarrouk, A.; Caccia, C.; Debbabi, M.; Fromont, A.; Sghaier, R.; Moreau, T.; Lizard, G. Mitochondrial dysfunctions in 7-ketocholesterol-treated 158N oligodendrocytes without or with alpha-tocopherol: Impacts on the cellular profil of tricarboxylic cycle-associated organic acids, long chain saturated and unsaturated fatty acids, oxysterols, cholesterol and cholesterol precursors. J. Steroid Biochem. Mol. Biol. 2017, 169, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Sul, O.J.; Li, G.; Kim, J.E.; Kim, E.S.; Choi, H.S. 7-ketocholesterol enhances autophagy via the ROS-TFEB signaling pathway in osteoclasts. J. Nutr. Biochem. 2021, 96, 108783. [Google Scholar] [CrossRef] [PubMed]
- Tani, M.; Kamata, Y.; Deushi, M.; Osaka, M.; Yoshida, M. 7-Ketocholesterol enhances leukocyte adhesion to endothelial cells via p38MAPK pathway. PLoS ONE 2018, 13, e0200499. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Xie, L.; Gu, Q.; Qiu, Q.; Wu, X.; Yin, L. 7-Ketocholesterol disturbs RPE cells phagocytosis of the outer segment of photoreceptor and induces inflammation through ERK signaling pathway. Exp. Eye Res. 2019, 189, 107849. [Google Scholar] [CrossRef]
- Nury, T.; Sghaier, R.; Zarrouk, A.; Menetrier, F.; Uzun, T.; Leoni, V.; Caccia, C.; Meddeb, W.; Namsi, A.; Sassi, K.; et al. Induction of peroxisomal changes in oligodendrocytes treated with 7-ketocholesterol: Attenuation by alpha-tocopherol. Biochimie 2018, 153, 181–202. [Google Scholar] [CrossRef]
- Wang, H.; Ramshekar, A.; Kunz, E.; Hartnett, M.E. 7-ketocholesterol induces endothelial-mesenchymal transition and promotes fibrosis: Implications in neovascular age-related macular degeneration and treatment. Angiogenesis 2021, 24, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Vejux, A.; Abed-Vieillard, D.; Hajji, K.; Zarrouk, A.; Mackrill, J.J.; Ghosh, S.; Nury, T.; Yammine, A.; Zaibi, M.; Mihoubi, W.; et al. 7-Ketocholesterol and 7beta-hydroxycholesterol: In vitro and animal models used to characterize their activities and to identify molecules preventing their toxicity. Biochem. Pharmacol. 2020, 173, 113648. [Google Scholar] [CrossRef]
- Brahmi, F.; Vejux, A.; Sghaier, R.; Zarrouk, A.; Nury, T.; Meddeb, W.; Rezig, L.; Namsi, A.; Sassi, K.; Yammine, A.; et al. Prevention of 7-ketocholesterol-induced side effects by natural compounds. Crit. Rev. Food Sci. Nutr. 2019, 59, 3179–3198. [Google Scholar] [CrossRef]
- Poli, G.; Biasi, F.; Leonarduzzi, G. Oxysterols in the pathogenesis of major chronic diseases. Redox Biol. 2013, 1, 125–130. [Google Scholar] [CrossRef]
- Indaram, M.; Ma, W.; Zhao, L.; Fariss, R.N.; Rodriguez, I.R.; Wong, W.T. 7-Ketocholesterol increases retinal microglial migration, activation, and angiogenicity: A potential pathogenic mechanism underlying age-related macular degeneration. Sci. Rep. 2015, 5, 9144. [Google Scholar] [CrossRef]
- Shi, G.; Chen, S.; Wandu, W.S.; Ogbeifun, O.; Nugent, L.F.; Maminishkis, A.; Hinshaw, S.J.; Rodriguez, I.R.; Gery, I. Inflammasomes Induced by 7-Ketocholesterol and Other Stimuli in RPE and in Bone Marrow-Derived Cells Differ Markedly in Their Production of IL-1beta and IL-18. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.; Lee, J.W.; Chou, J.; Campos, M.M.; Rodriguez, I.R. 7-Ketocholesterol induces inflammation and angiogenesis In Vivo: A novel rat model. PLoS ONE 2013, 8, e56099. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, I.R.; Clark, M.E.; Lee, J.W.; Curcio, C.A. 7-ketocholesterol accumulates in ocular tissues as a consequence of aging and is present in high levels in drusen. Exp. Eye Res. 2014, 128, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Olivier, E.; Dutot, M.; Regazzetti, A.; Leguillier, T.; Dargere, D.; Auzeil, N.; Laprevote, O.; Rat, P. P2X7-pannexin-1 and amyloid beta-induced oxysterol input in human retinal cell: Role in age-related macular degeneration? Biochimie 2016, 127, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Pariente, A.; Pelaez, R.; Perez-Sala, A.; Larrayoz, I.M. Inflammatory and cell death mechanisms induced by 7-ketocholesterol in the retina. Implications for age-related macular degeneration. Exp. Eye Res. 2019, 187, 107746. [Google Scholar] [CrossRef]
- Herrera-Meza, M.S.; Mendoza-Lopez, M.R.; Garcia-Barradas, O.; Sanchez-Otero, M.G.; Silva-Hernandez, E.R.; Angulo, J.O.; Oliart-Ros, R.M. Dietary anhydrous milk fat naturally enriched with conjugated linoleic acid and vaccenic acid modify cardiovascular risk biomarkers in spontaneously hypertensive rats. Int. J. Food Sci. Nutr. 2013, 64, 575–586. [Google Scholar] [CrossRef]
- Pelaez, R.; Pariente, A.; Perez-Sala, A.; Larrayoz, I.M. Sterculic Acid: The Mechanisms of Action beyond Stearoyl-CoA Desaturase Inhibition and Therapeutic Opportunities in Human Diseases. Cells 2020, 9, 140. [Google Scholar] [CrossRef]
- Ortinau, L.C.; Nickelson, K.J.; Stromsdorfer, K.L.; Naik, C.Y.; Pickering, R.T.; Haynes, R.A.; Fritsche, K.L.; Perfield, J.W., 2nd. Sterculic oil, a natural inhibitor of SCD1, improves the metabolic state of obese OLETF rats. Obesity 2013, 21, 344–352. [Google Scholar] [CrossRef]
- Pariente, A.; Perez-Sala, A.; Ochoa, R.; Pelaez, R.; Larrayoz, I.M. Genome-Wide Transcriptomic Analysis Identifies Pathways Regulated by Sterculic Acid in Retinal Pigmented Epithelium Cells. Cells 2020, 9, 1187. [Google Scholar] [CrossRef]
- Glickman, M.E.; Rao, S.R.; Schultz, M.R. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J. Clin. Epidemiol. 2014, 67, 850–857. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Kang, T.I.; So, J.S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines 2021, 9, 791. [Google Scholar] [CrossRef]
- Larrayoz, I.M.; Huang, J.D.; Lee, J.W.; Pascual, I.; Rodriguez, I.R. 7-ketocholesterol-induced inflammation: Involvement of multiple kinase signaling pathways via NFkappaB but independently of reactive oxygen species formation. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4942–4955. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Dean, R.T.; Jessup, W. Free and esterified oxysterol: Formation during copper-oxidation of low density lipoprotein and uptake by macrophages. J. Lipid Res. 1996, 37, 320–335. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Wang, Y. Cholesterol metabolism: From lipidomics to immunology. J. Lipid Res. 2022, 63, 100165. [Google Scholar] [CrossRef]
- Yanagisawa, R.; He, C.; Asai, A.; Hellwig, M.; Henle, T.; Toda, M. The Impacts of Cholesterol, Oxysterols, and Cholesterol Lowering Dietary Compounds on the Immune System. Int. J. Mol. Sci. 2022, 23, 12236. [Google Scholar] [CrossRef]
- Xie, L.; Gu, Q.; Wu, X.; Yin, L. Activation of LXRs Reduces Oxysterol Lipotoxicity in RPE Cells by Promoting Mitochondrial Function. Nutrients 2022, 14, 2473. [Google Scholar] [CrossRef]
- Iborra, R.T.; Machado-Lima, A.; Castilho, G.; Nunes, V.S.; Abdalla, D.S.; Nakandakare, E.R.; Passarelli, M. Advanced glycation in macrophages induces intracellular accumulation of 7-ketocholesterol and total sterols by decreasing the expression of ABCA-1 and ABCG-1. Lipids Health Dis. 2011, 10, 172. [Google Scholar] [CrossRef]
- Okabe, A.; Urano, Y.; Itoh, S.; Suda, N.; Kotani, R.; Nishimura, Y.; Saito, Y.; Noguchi, N. Adaptive responses induced by 24S-hydroxycholesterol through liver X receptor pathway reduce 7-ketocholesterol-caused neuronal cell death. Redox Biol. 2013, 2, 28–35. [Google Scholar] [CrossRef]
- Levy, D.; de Melo, T.C.; Oliveira, B.A.; Paz, J.L.; de Freitas, F.A.; Reichert, C.O.; Rodrigues, A.; Bydlowski, S.P. 7-Ketocholesterol and cholestane-triol increase expression of SMO and LXRalpha signaling pathways in a human breast cancer cell line. Biochem. Biophys. Rep. 2019, 19, 100604. [Google Scholar] [CrossRef]
- William, S.; Duncan, T.; Redmond, T.M. Pretreatment of human retinal pigment epithelial cells with sterculic acid forestalls fenretinide-induced apoptosis. Sci. Rep. 2022, 12, 22442. [Google Scholar] [CrossRef] [PubMed]
- Aye, I.L.; Waddell, B.J.; Mark, P.J.; Keelan, J.A. Oxysterols exert proinflammatory effects in placental trophoblasts via TLR4-dependent, cholesterol-sensitive activation of NF-kappaB. Mol. Hum. Reprod. 2012, 18, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Cano, M.; Wang, L.; Wan, J.; Barnett, B.P.; Ebrahimi, K.; Qian, J.; Handa, J.T. Oxidative stress induces mitochondrial dysfunction and a protective unfolded protein response in RPE cells. Free. Radic. Biol. Med. 2014, 69, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pedruzzi, E.; Guichard, C.; Ollivier, V.; Driss, F.; Fay, M.; Prunet, C.; Marie, J.C.; Pouzet, C.; Samadi, M.; Elbim, C.; et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol. Cell. Biol. 2004, 24, 10703–10717. [Google Scholar] [CrossRef]
- Yammine, A.; Nury, T.; Vejux, A.; Latruffe, N.; Vervandier-Fasseur, D.; Samadi, M.; Greige-Gerges, H.; Auezova, L.; Lizard, G. Prevention of 7-Ketocholesterol-Induced Overproduction of Reactive Oxygen Species, Mitochondrial Dysfunction and Cell Death with Major Nutrients (Polyphenols, omega3 and omega9 Unsaturated Fatty Acids) of the Mediterranean Diet on N2a Neuronal Cells. Molecules 2020, 25, 2296. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell's response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Guina, T.; Deiana, M.; Calfapietra, S.; Cabboi, B.; Maina, M.; Tuberoso, C.I.; Leonarduzzi, G.; Gamba, P.; Gargiulo, S.; Testa, G.; et al. The role of p38 MAPK in the induction of intestinal inflammation by dietary oxysterols: Modulation by wine phenolics. Food Funct. 2015, 6, 1218–1228. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef]
- Yue, J.; Lopez, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- Sung, S.C.; Kim, K.; Lee, K.A.; Choi, K.H.; Kim, S.M.; Son, Y.H.; Moon, Y.S.; Eo, S.K.; Rhim, B.Y. 7-Ketocholesterol upregulates interleukin-6 via mechanisms that are distinct from those of tumor necrosis factor-alpha, in vascular smooth muscle cells. J. Vasc. Res. 2009, 46, 36–44. [Google Scholar] [CrossRef]
- Suzuki, K.; Sakiyama, Y.; Usui, M.; Obama, T.; Kato, R.; Itabe, H.; Yamamoto, M. Oxidized low-density lipoprotein increases interleukin-8 production in human gingival epithelial cell line Ca9-22. J. Periodontal Res. 2010, 45, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Hyttinen, J.M.; Toropainen, E.; Kaarniranta, K. Endoplasmic reticulum stress in age-related macular degeneration: Trigger for neovascularization. Mol. Med. 2010, 16, 535–542. [Google Scholar] [CrossRef] [PubMed]
- SanGiovanni, J.P.; Lee, P.H. AMD-associated genes encoding stress-activated MAPK pathway constituents are identified by interval-based enrichment analysis. PLoS ONE 2013, 8, e71239. [Google Scholar] [CrossRef] [PubMed]
- Kheitan, S.; Minuchehr, Z.; Soheili, Z.S. Exploring the cross talk between ER stress and inflammation in age-related macular degeneration. PLoS ONE 2017, 12, e0181667. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- Becerra, S.P.; Fariss, R.N.; Wu, Y.Q.; Montuenga, L.M.; Wong, P.; Pfeffer, B.A. Pigment epithelium-derived factor in the monkey retinal pigment epithelium and interphotoreceptor matrix: Apical secretion and distribution. Exp. Eye Res. 2004, 78, 223–234. [Google Scholar] [CrossRef]
- Larrayoz, I.M.; de Luis, A.; Rua, O.; Velilla, S.; Cabello, J.; Martinez, A. Molecular effects of doxycycline treatment on pterygium as revealed by massive transcriptome sequencing. PLoS ONE 2012, 7, e39359. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Gueguen, L.; Coppee, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2013, 42, D199–D205. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Gene ID | Log2FC | FDR | Gene Description |
|---|---|---|---|---|
| Lipid metabolism | ACACA | −0.69 | 4.43 × 10−5 | acetyl-CoA carboxylase 1 |
| ABHD5 | 0.66 | 2.64 × 10−2 | abhydrolase domain containing 5 | |
| ACLY | −0.76 | 7.19 × 10−8 | ATP-citrate synthase | |
| CAV1 | −0.53 | 4.64 × 10−2 | Caveolin-1 | |
| ELOVL4 | 0.99 | 2.57 × 10−2 | Elongation very long chain fatty acids protein 4 | |
| FADS1 | −0.82 | 1.86 × 10−5 | fatty acid desaturase 1 | |
| FADS2 | −1.02 | 1.05 × 10−7 | fatty acid desaturase 2 | |
| FASN | −1.17 | 2.21 × 10−8 | Fatty acid synthase | |
| SCD | −0.93 | 2.26 × 10−7 | Stearoyl-CoA desaturase | |
| Sterol biosynthesis and transport | ABCA1 | 1.83 | 1.91 × 10−14 | ATP binding cassette subfamily A member 1 |
| ABCG1 | 2.95 | 9.60 × 10−21 | ATP binding cassette subfamily G member 1 | |
| ABCC9 | 2.48 | 6.32 × 10−5 | ATP binding cassette subfamily C member 9 | |
| FDFT1 | −1.45 | 1.38 × 10−26 | squalene synthase | |
| HMGCR | −1.65 | 3.23 × 10−17 | 3-hydroxy-3-methylglutaryl coenzyme A reductase | |
| HMGCS1 | −3.1 | 2.56 × 10−51 | hydroxymethylglutaryl-CoA synthase | |
| INSIG1 | −2.09 | 9.80 × 10−28 | insulin-induced gene protein | |
| LDLR | −0.8 | 4.65 × 10−3 | low-density lipoprotein receptor | |
| LSS | −2.08 | 6.49 × 10−44 | terpene cyclase/mutase family member | |
| MSMO1 | −2.2 | 2.28 × 10−42 | methylsterol monooxygenase 1 | |
| MVD | −2.4 | 4.33 × 10−37 | diphosphomevalonate decarboxylase | |
| MVK | −1.13 | 4.66 × 10−8 | mevalonate kinase | |
| SQLE | −2.11 | 1.36 × 10−44 | squalene monooxygenase | |
| ER stress | ASNS | 1.88 | 3.62 × 10−7 | glutamine-dependent asparagine synthetase |
| ATF3 | 3.16 | 1.58 × 10−8 | activating transcription factor 3 | |
| CEBPB | 1.44 | 3.54 × 10−4 | CCAAT/enhancer-binding protein beta | |
| CEBPG | 1.09 | 1.71 × 10−4 | CCAAT/enhancer-binding protein gamma | |
| EIF2S2 | 0.46 | 3.81 × 10−2 | eukaryotic translation initiation factor 2 subunit beta | |
| ERN1 | 0.83 | 1.33 × 10−3 | non-specific serine/threonine protein kinase | |
| GFPT1 | 0.91 | 2.52 × 10−3 | glutamine--fructose-6-phosphate transaminase 1 | |
| HSPA5 | 1.36 | 1.94 × 10−5 | heat Shock Protein Family A (Hsp70) Member 5 | |
| HYOU1 | 0.81 | 1.40 × 10−3 | hypoxia up-regulated 1 | |
| PREB | 0.63 | 8.42 × 10−3 | prolactin regulatory element binding | |
| SRPRB | 0.82 | 4.86 × 10−4 | SRP receptor subunit beta | |
| SSR1 | 0.47 | 2.21 × 10−2 | signal sequence receptor subunit 1 | |
| NFκB and MAPK inflammatory signaling | CXCL2 | 1.3 | 1.99 × 10−2 | C-X-C motif chemokine 2 |
| EGFR | 0.62 | 2.55 × 10−2 | receptor protein-tyrosine kinase | |
| FOSB | 2.81 | 2.50 × 10−2 | FosB proto-oncogene | |
| IL1A | 2.85 | 4.87 × 10−5 | interleukin-1 alpha | |
| IL6 | 1.36 | 4.98 × 10−2 | interleukin-6 | |
| MAP2K1 | 0.46 | 1.83 × 10−2 | mitogen-activated protein kinase kinase 1 | |
| MAP3K8 | 0.89 | 4.25 × 10−2 | mitogen-activated protein kinase kinase kinase 8 | |
| MYC | 0.78 | 4.90 × 10−5 | mYC proto-oncogene | |
| NFKBIA | 0.55 | 1.83 × 10−2 | NF-kappa-B inhibitor alpha | |
| NFKBIB | 1.03 | 7.77 × 10−5 | NF-kappa-B inhibitor beta | |
| NFKBIERPS6KA1 | 0.540.95 | 4.83 × 10−22.22 × 10−3 | NF-kappa-B inhibitor epsilonribosomal Protein S6 Kinase A1 | |
| RRAS2 | 0.74 | 1.14 × 10−3 | Ras related 2 | |
| TLR4 | 0.96 | 2.31 × 10−3 | toll-like receptor 4 | |
| TRAF1 | 0.82 | 2.35 × 10−2 | TNF Receptor Associated Factor 1 | |
| VEGFA | 0.74 | 3.42 × 10−2 | vascular endothelial growth factor A | |
| Cell death signaling | APAF1 | −0.45 | 3.16 × 10−2 | apoptotic peptidase activating factor 1 |
| CASP1 | 1.40 | 3.21 × 10−7 | caspase-1 | |
| DFNA5 | 0.95 | 9.87 × 10−4 | non-syndromic hearing impairment protein 5 isoform A | |
| GADD45A | 0.70 | 1.99 × 10−5 | growth arrest and DNA damage-inducible alpha | |
| PMAIP1 | 1.30 | 9.07 × 10−6 | phorbol-12-Myristate-13-Acetate-Induced Protein 1 | |
| TNFRSF10B | 0.60 | 2.36 × 10−2 | TNF receptor superfamily member 10b | |
| TP53I3 | 0.65 | 2.57 × 10−2 | tumor protein p53 inducible protein 3 |
| Gene | Oligonucleotide Sequence |
|---|---|
| 18S—Forward | 5′-ATGCTCTTAGCTGAGTGTCCCG-3′ |
| 18S—Reverse | 5′-ATTCCTAGCTGCGGTATCCAGG-3′ |
| IL8—Forward | 5′-ACCAAGGAAATCGGCCCCTA-3′ |
| IL8—Reverse | 5′-CCATACCTCTAGGCTGGCTATC-3′ |
| IL6—Forward | 5′-CTGGTCTTTTGGAGTTTGAGGT-3′ |
| IL6—Reverse | 5′-GCTGGCATTTGTGGTTGGT-3′ |
| VEGFA—Forward | 5′-TTCACCCTCGTCCTCTTCCT-3′ |
| VEGFA—Reverse | 5′-ATCCTGCCCTGTCTCTCTGT-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pariente, A.; Pérez-Sala, Á.; Ochoa, R.; Bobadilla, M.; Villanueva-Martínez, Á.; Peláez, R.; Larráyoz, I.M. Identification of 7-Ketocholesterol-Modulated Pathways and Sterculic Acid Protective Effect in Retinal Pigmented Epithelium Cells by Using Genome-Wide Transcriptomic Analysis. Int. J. Mol. Sci. 2023, 24, 7459. https://doi.org/10.3390/ijms24087459
Pariente A, Pérez-Sala Á, Ochoa R, Bobadilla M, Villanueva-Martínez Á, Peláez R, Larráyoz IM. Identification of 7-Ketocholesterol-Modulated Pathways and Sterculic Acid Protective Effect in Retinal Pigmented Epithelium Cells by Using Genome-Wide Transcriptomic Analysis. International Journal of Molecular Sciences. 2023; 24(8):7459. https://doi.org/10.3390/ijms24087459
Chicago/Turabian StylePariente, Ana, Álvaro Pérez-Sala, Rodrigo Ochoa, Miriam Bobadilla, Ángela Villanueva-Martínez, Rafael Peláez, and Ignacio M. Larráyoz. 2023. "Identification of 7-Ketocholesterol-Modulated Pathways and Sterculic Acid Protective Effect in Retinal Pigmented Epithelium Cells by Using Genome-Wide Transcriptomic Analysis" International Journal of Molecular Sciences 24, no. 8: 7459. https://doi.org/10.3390/ijms24087459