
Isolation and NMR Scaling Factors for the Structure Determination of Lobatolide H, a Flexible Sesquiterpene from Neurolaena lobata †
 , , , , , and
, , , , , and
Abstract
:1. Introduction
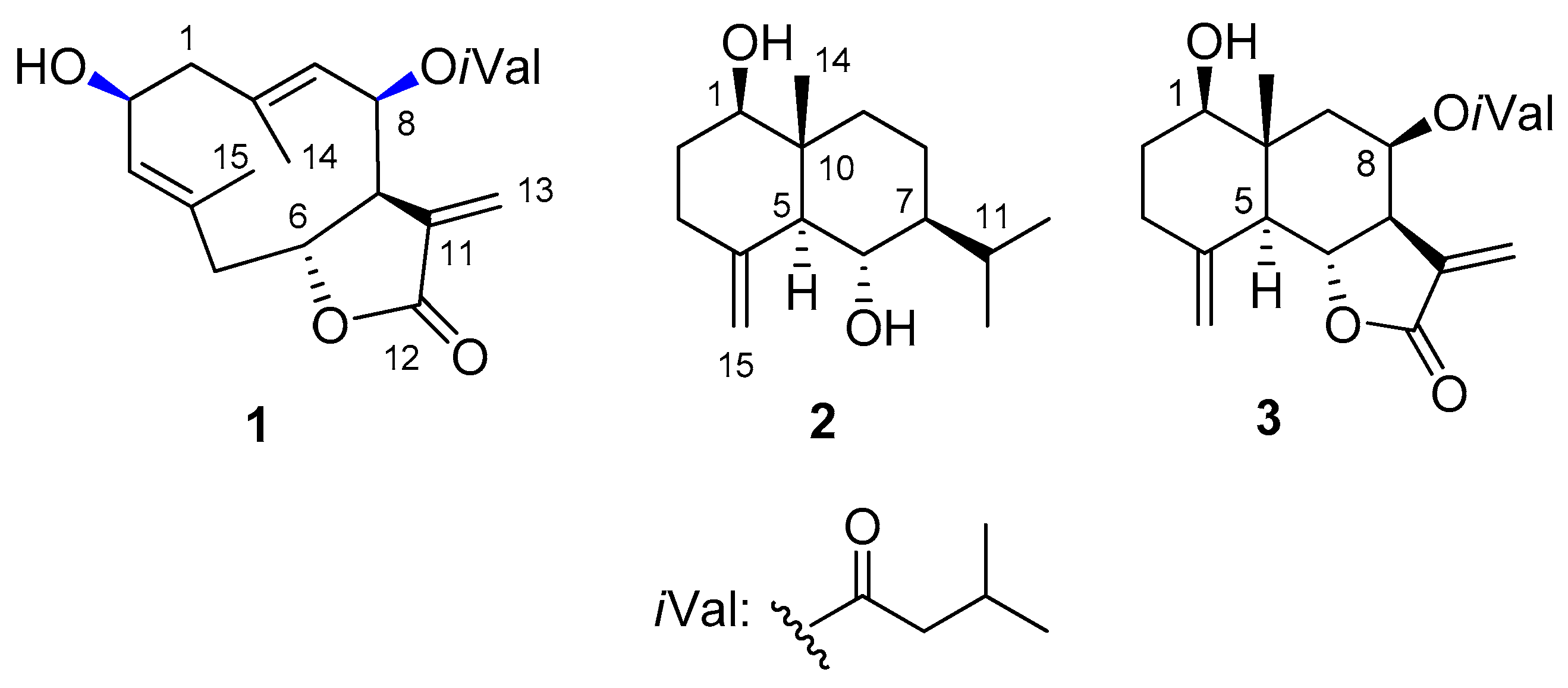
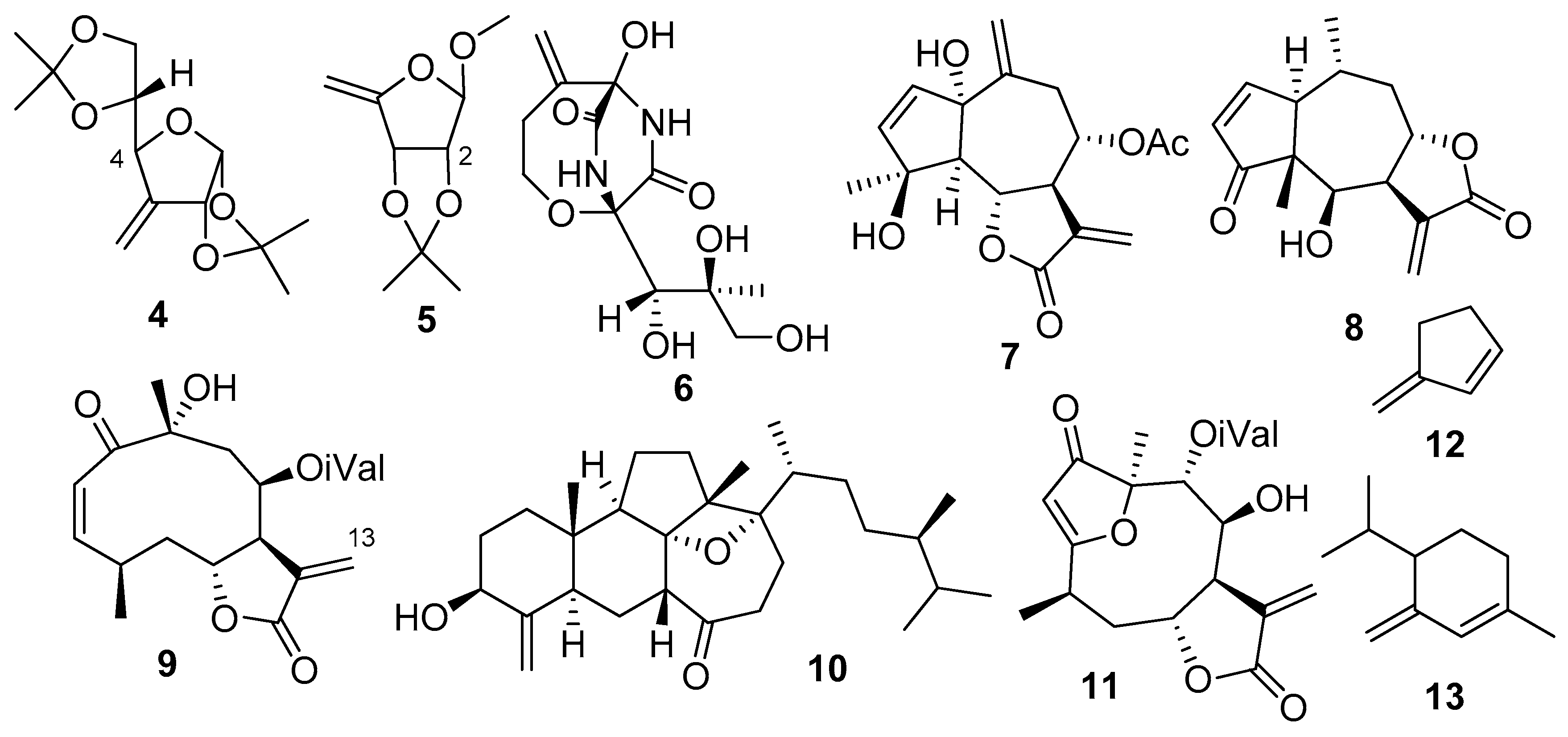
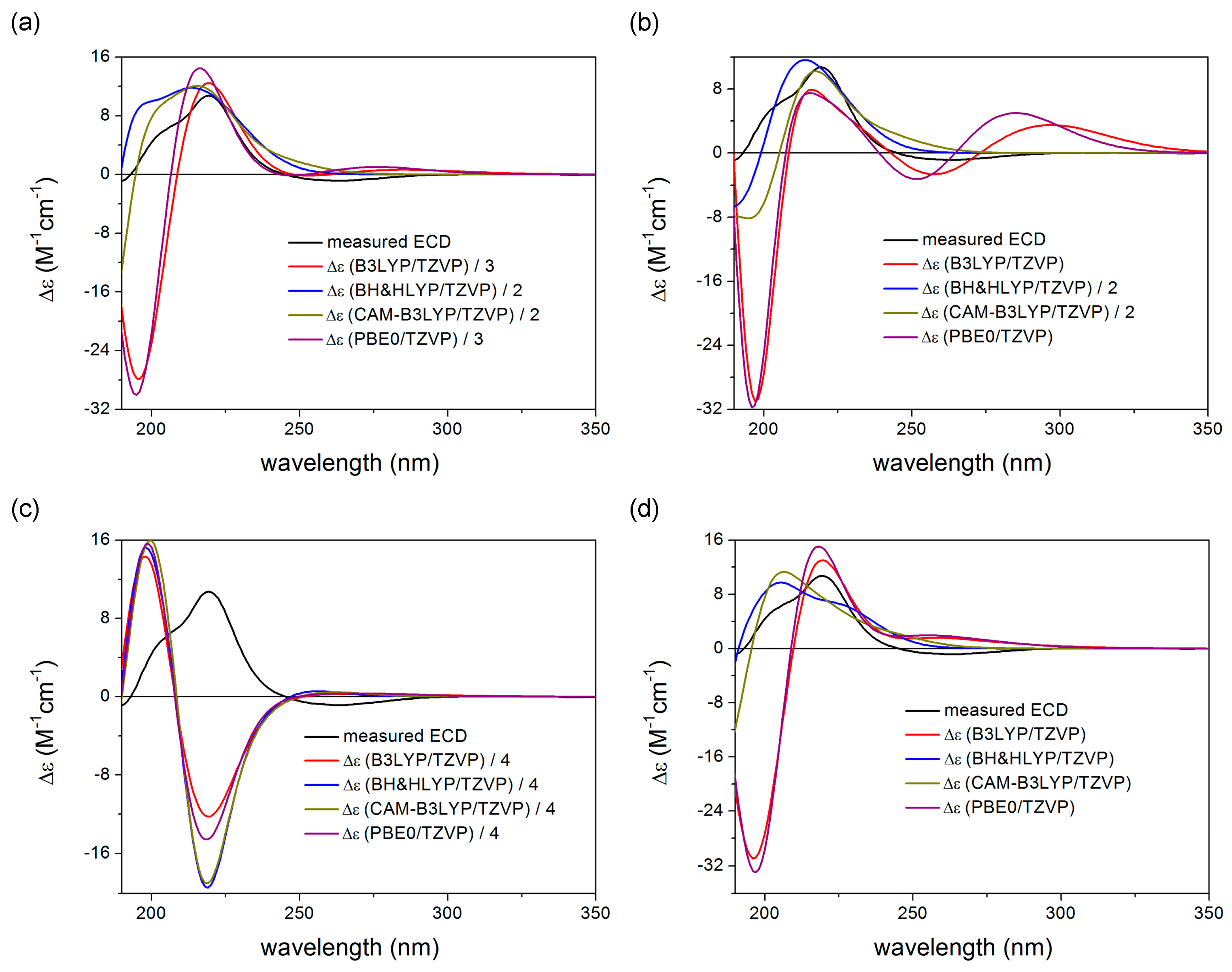
2. Results and Discussion
3. Materials and Methods
3.1. General Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Physical Characteristics of the New Compound
3.5. Computational Section
3.6. Antiproliferative MTT Assay
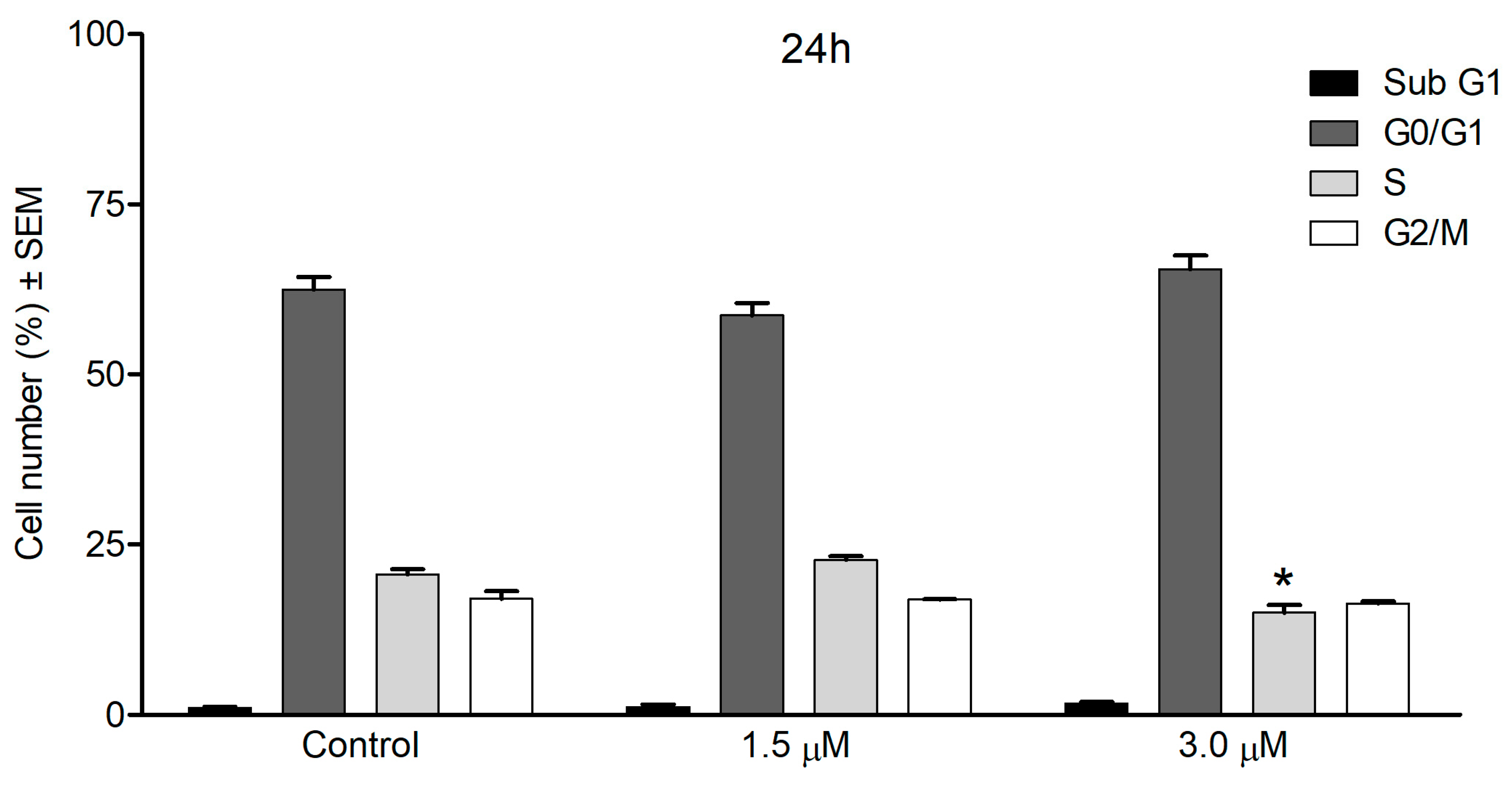
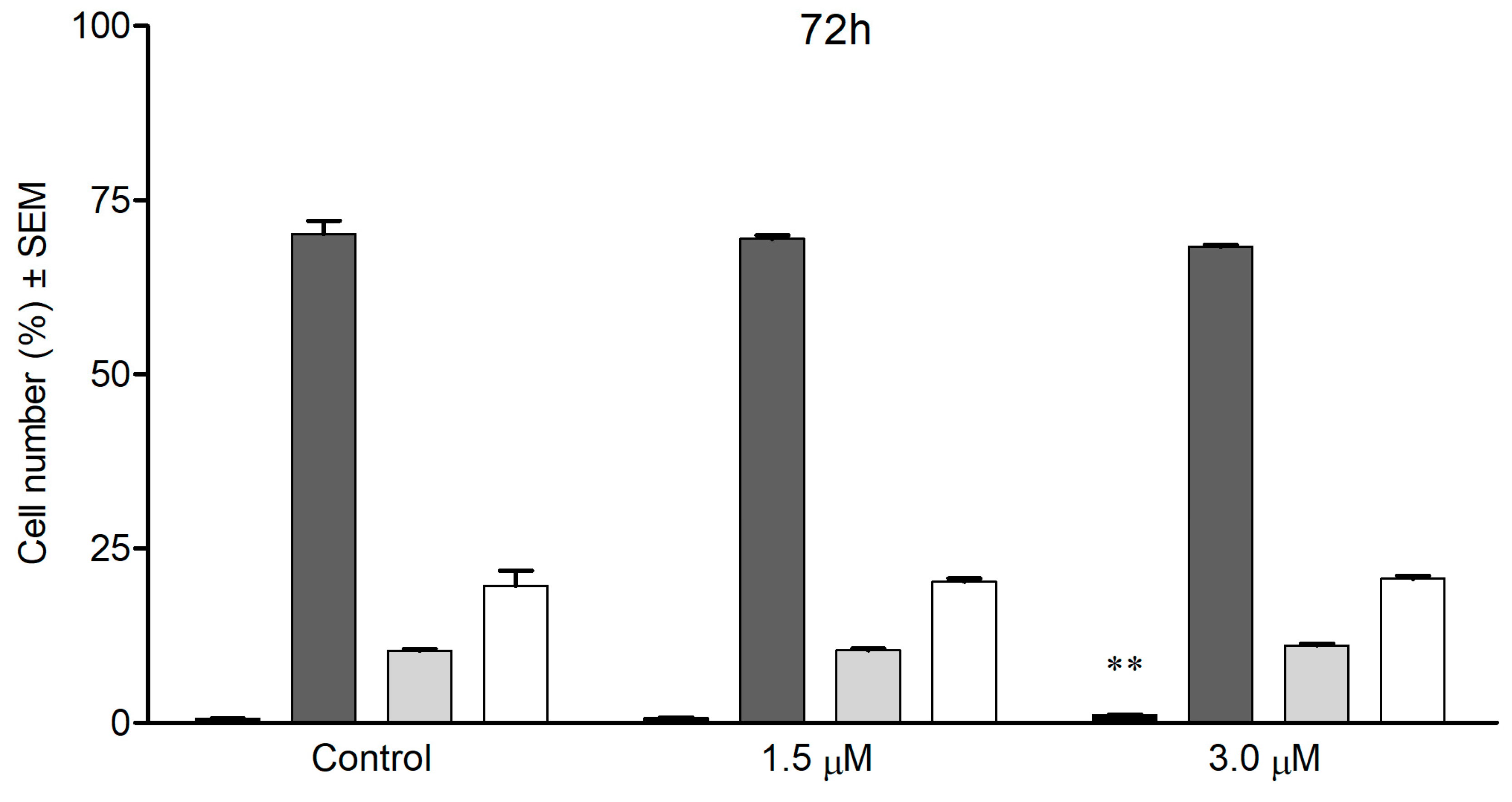
3.7. Cell Cycle Analysis by Flow Cytometry
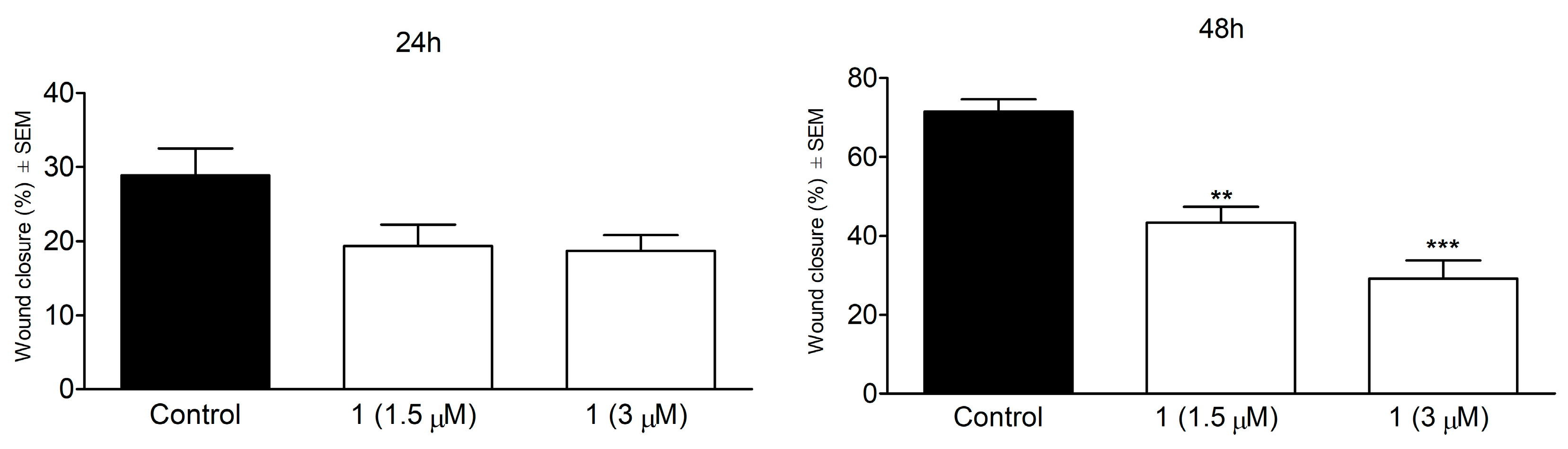
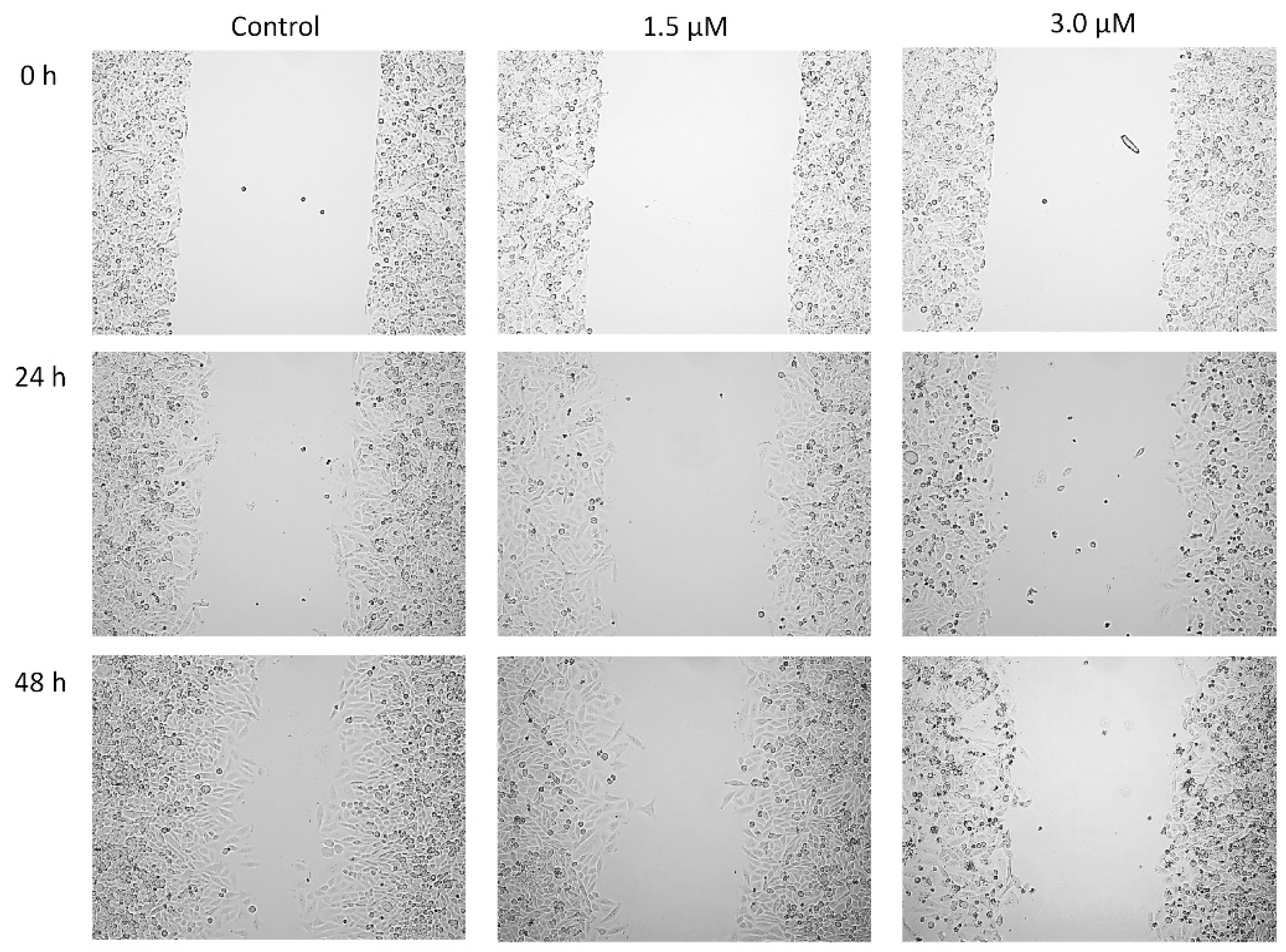
3.8. Wound Healing Assay
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Babaei, G.; Aliarab, A.; Abroon, S.; Rasmi, Y.; Aziz, S.G.G. Application of sesquiterpene lactone: A new promising way for cancer therapy based on anticancer activity. Biomed. Pharmacother. 2018, 106, 239–246. [Google Scholar] [CrossRef]
- Paço, A.; Brás, T.; Santos, J.O.; Sampaio, P.; Gomes, A.C.; Duarte, M.F. Anti-Inflammatory and Immunoregulatory Action of Sesquiterpene Lactones. Molecules 2022, 27, 1142. [Google Scholar] [CrossRef] [PubMed]
- Shulha, O.; Zidorn, C. Sesquiterpene lactones and their precursors as chemosystematic markers in the tribe Cichorieae of the Asteraceae revisited: An update (2008–2017). Phytochemistry 2019, 163, 149–177. [Google Scholar] [CrossRef]
- Zorrilla, J.G.; Cala, A.; Rial, C.; Mejias, F.J.R.; Molinillo, J.M.G.; Varela, R.M.; Macias, F.A. Synthesis of Active Strigolactone Analogues Based on Eudesmane- and Guaiane-Type Sesquiterpene Lactones. J. Agric. Food Chem. 2020, 68, 9636–9645. [Google Scholar] [CrossRef] [PubMed]
- Laurella, L.C.; Mirakian, N.T.; Garcia, M.N.; Grasso, D.H.; Sülsen, V.P.; Papademetrio, D.L. Sesquiterpene Lactones as Promising Candidates for Cancer Therapy: Focus on Pancreatic Cancer. Molecules 2022, 27, 3492. [Google Scholar] [CrossRef] [PubMed]
- Moujir, L.; Callies, O.; Sousa, P.M.C.; Sharopov, F.; Seca, A.M.L. Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases. Appl. Sci. 2020, 10, 3001. [Google Scholar] [CrossRef]
- Liu, J.; Yang, Z.; Kong, Y.; He, Y.; Xu, Y.; Cao, X. Antitumor activity of alantolactone in lung cancer cell lines NCI-H1299 and Anip973. J. Food Biochem. 2019, 43, 12972. [Google Scholar] [CrossRef]
- Zhu, N.L.; Tang, C.; Xu, C.; Ke, C.Q.; Lin, G.; Jenis, J.; Yao, S.; Liu, H.; Ye, Y. Cytotoxic germacrane-type sesquiterpene lactones from the whole plant of Carpesium lipskyi. J. Nat. Prod. 2019, 82, 919–927. [Google Scholar] [CrossRef]
- Wang, J.; Su, S.; Zhang, S.; Zhai, S.; Sheng, R.; Wu, W.; Guo, R. Structure-activity relationship and synthetic methodologies of α-santonin derivatives with diverse bioactivities: A mini-review. Eur. J. Med. Chem. 2019, 175, 215–233. [Google Scholar]
- Matos, M.S.; Anastácio, J.D.; dos Santos, C.N. Sesquiterpene Lactones: Promising Natural Compounds to Fight Inflammation. Pharmaceutics 2021, 13, 991. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wen, X.; Ke, C.Q.; Zhang, T.; Lin, L.; Yao, S.; Goodpaster, J.D.; Tang, C.; Ye, Y. Tricarabrols A-C, three anti-inflammatory sesquiterpene lactone trimers featuring a methylene-tethered linkage from Carpesium faberi. Org. Chem. Front. 2020, 7, 1374–1382. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Z.; Xie, Y.; Hu, H. Antitumor activity and mechanism of costunolide and dehydrocostus lactone: Two natural sesquiterpene lactones from the Asteraceae family. Biomed. Pharmacother. 2020, 125, 109955. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Anticancer targets and signaling pathways activated by britannin and related pseudoguaianolide sesquiterpene lactones. Biomedicines 2021, 9, 1325. [Google Scholar] [CrossRef]
- Liu, X.N.; Li, H.M.; Wang, S.P.; Zhang, J.Z.; Liu, D.L. Sesquiterpene lactones of Aucklandia lappa: Pharmacology, pharmacokinetics, toxicity, and structure-activity relationship. Chin. Herb. Med. 2021, 13, 167–176. [Google Scholar] [CrossRef]
- Ma, C.; Meng, C.W.; Zhou, Q.M.; Peng, C.; Liu, F.; Zhang, J.W.; Zhou, F.; Xiong, L. New sesquiterpenoids from the stems of Dendrobium nobile and their neuroprotective activities. Fitoterapia 2019, 138, 104351. [Google Scholar] [CrossRef]
- Tang, J.J.; Huang, L.F.; Deng, J.L.; Wang, Y.M.; Guo, C.; Peng, X.N.; Liu, Z.; Gao, J.M. Cognitive enhancement and neuroprotective effects of 1,6-O,O-diacetylbritannilactone, a sesquiterpene lactone in 5xFAD Alzheimer’s disease mice model. Redox Biol. 2022, 50, 102229. [Google Scholar] [CrossRef]
- Abdelwahab, S.I.; Taha, M.M.E.; Alhazmi, H.A.; Ahsan, W.; Rehman, Z.U.; Bratty, M.A.; Makeen, H. Phytochemical profiling of costus (Saussurea lappa Clarke) root essential oil, and its antimicrobial and toxicological effects. Trop. J. Pharm. Res. 2019, 18, 2155–2160. [Google Scholar] [CrossRef]
- Sülsen, V.P.; Martino, V.S. Overview. In Sesquiterpene Lactones: Advances in Their Chemistry and Biological Aspects; Sülsen, V.P., Martino, V.S., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 3–17. [Google Scholar]
- Sosa, A.; Diaz, M.; Salvatore, A.; Bardon, A.; Borkosky, S.; Vera, N. Insecticidal effects of Vernonanthura nebularum against two economically important pest insects. Saudi J. Biol. Sci. 2019, 26, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Lajter, I.; Vasas, A.; Beni, Z.; Forgo, P.; Binder, M.; Bochkov, V.; Zupko, I.; Krupitza, G.; Frisch, R.; Kopp, B.; et al. Sesquiterpenes from Neurolaena lobata and their antiproliferative and anti-inflammatory activities. J. Nat. Prod. 2014, 77, 576–582. [Google Scholar] [CrossRef]
- Passreiter, C.M.; Wendisch, D.; Gondol, D. Sesquiterpene lactones from Neurolaena lobata. Phytochemistry 1995, 39, 133–137. [Google Scholar] [CrossRef]
- Borges-del-Castillo, J.; Manresa-Ferrero, M.T.; Rodríguez-Luis, F.; Vázquez-Bueno, P. Panama Flora. II. New Sesquiterpene Lactones from Neurolaena lobata. J. Nat. Prod. 1982, 45, 762–765. [Google Scholar] [CrossRef]
- Manchand, P.S.; Blount, J.F. Stereostructures of Neurolenins A and B, Novel Germacranolide Sesquiterpenes from Neurolaena lobata (L.) R.Br. J. Org. Chem. 1978, 43, 4352–4354. [Google Scholar] [CrossRef]
- Vasas, A.; Lajter, I.; Kúsz, N.; Király, S.B.; Kovács, T.; Kurtán, T.; Bózsity, N.; Nagy, N.; Schelz, Z.; Zupkó, I.; et al. Isolation, Structure Determination of Sesquiterpenes from Neurolaena lobata and Their Antiproliferative, Cell Cycle Arrest-Inducing and Anti-Invasive Properties against Human Cervical Tumor Cells. Pharmaceutics 2021, 13, 2088. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An Improved Probability for the Stereochemical Assignment of Isomeric Compounds using Quantum Chemical Calculations of NMR Shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational Prediction of 1H and 13C Chemical Shifts: A Useful Tool for Natural Product, Mechanistic, and Synthetic Organic Chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Li, W.S.; Yan, R.J.; Yu, Y.; Shi, Z.; Mándi, A.; Shen, L.; Kurtán, T.; Wu, J. Determination of the Absolute Configuration of Super-Carbon-Chain Compounds by a Combined Chemical, Spectroscopic, and Computational Approach: Gibbosols A and B. Angew. Chem. Int. Ed. 2020, 59, 13028–13036. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.P.; Sun, S.H.; Yu, Y.; Mándi, A.; Luo, J.Y.; Yang, M.H.; Kurtán, T.; Chen, W.H.; Shen, L.; Wu, J. Discovery of benthol A and its challenging stereochemical assignment: Opening up a new window for skeletal diversity of super-carbonchain compounds. Chem. Sci. 2021, 12, 10197–10206. [Google Scholar] [CrossRef] [PubMed]
- Kawka, A.; Hajdaś, G.; Kułaga, D.; Koenig, H.; Kowalczyk, I.; Pospieszny, T. Molecular structure, spectral and theoretical study of new type bile acid–sterol conjugates linked via 1,2,3-triazole ring. J. Mol. Struct. 2023, 1273, 134313. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Soldi, C.; Jones, P.B.; Olmstead, M.M.; Rita, J.; Shaw, J.T.; Tantillo, D.J. The Correct Structure of Aquatolide-Experimental Validation of a Theoretically-Predicted Structural Revision. J. Am. Chem. Soc. 2012, 134, 18550–18553. [Google Scholar] [CrossRef]
- Liu, Y.; Holt, T.A.; Kutateladze, A.; Newhouse, T.R. Stereochemical revision of xylogranatin F by GIAO and DU8+ NMR calculations. Chirality 2020, 32, 515–523. [Google Scholar] [CrossRef]
- Sarotti, A.M. In Silico Reassignment of (+)-Diplopyrone by NMR Calculations: Use of a DP4/J-DP4/DP4+/DIP Tandem to Revise Both Relative and Absolute Configuration. J. Org. Chem. 2020, 85, 11566–11570. [Google Scholar] [CrossRef]
- Zhou, T.; Zheng, A.; Zhang, W.; Lu, X.; Chen, H.; Tan, H. Concise total syntheses of two flavans and structure revision assisted by quantum NMR calculations. Org. Biomol. Chem. 2022, 20, 4096–4100. [Google Scholar] [CrossRef] [PubMed]
- Marcarino, M.O.; Cicetti, S.; Zanardi, M.M.; Sarotti, A.M. A critical review on the use of DP4+ in the structural elucidation of natural products: The good, the bad and the ugly. A practical guide. Nat. Prod. Rep. 2022, 39, 58–76. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Zhou, P.J.; Jiang, H.W.; Huang, T.; He, Y.H.; Zhao, Z.Y.; Zang, Y.; Choo, Y.M.; Wang, X.; Chittiboyina, A.G.; et al. Forrestiacids A and B, Pentaterpene Inhibitors of ACL and Lipogenesis: Extending the Limits of Computational NMR Methods in the Structure Assignment of Complex Natural Products. Angew. Chem. Int. Ed. 2021, 60, 22270–22275. [Google Scholar] [CrossRef]
- Dybiec, K.; Gryff-Keller, A. Remarks on GIAO-DFT predictions of 13C chemical shifts. Magn. Reson. Chem. 2009, 47, 63–66. [Google Scholar] [CrossRef]
- Autschbach, J.; Zheng, S. Relativistic Computations of NMR Parameters from First Principles: Theory and Applications. Annu. Rep. NMR Spectrosc. 2009, 67, 1–95. [Google Scholar] [CrossRef]
- Forster, L.C.; Pierens, G.K.; Garson, M.J. Elucidation of Relative and Absolute Configurations of Highly Rearranged Diterpenoids and Evidence for a Putative Biosynthetic Intermediate from the Australian Nudibranch Goniobranchus geometricus. J. Nat. Prod. 2019, 82, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, A.G.; Reddy, D.S. High-Throughput in Silico Structure Validation and Revision of Halogenated Natural Products Is Enabled by Parametric Corrections to DFT-Computed 13C NMR Chemical Shifts and Spin−Spin Coupling Constants. J. Org. Chem. 2017, 82, 3368–3381. [Google Scholar] [CrossRef]
- Dračínský, M.; Buděšínský, M.; Warżajtis, B.; Rychlewska, U. Solution and Solid-State Effects on NMR Chemical Shifts in Sesquiterpene Lactones: NMR, X-ray, and Theoretical Methods. J. Phys. Chem. A 2012, 116, 680–688. [Google Scholar] [CrossRef]
- Li, W.S.; Mándi, A.; Liu, J.J.; Shen, L.; Kurtán, T.; Wu, J. Xylomolones A−D from the Thai Mangrove Xylocarpus moluccensis: Assignment of Absolute Stereostructures and Unveiling a Convergent Strategy for Limonoid Biosynthesis. J. Org. Chem. 2019, 84, 2596–2606. [Google Scholar] [CrossRef]
- Pierens, G.K. 1H and 13C NMR Scaling Factors for the Calculation of Chemical Shifts in Commonly Used Solvents Using Density Functional Theory. J. Comput. Chem. 2014, 35, 1388–1394. [Google Scholar] [CrossRef]
- CHESHIRE CCAT, The Chemical Shift Repository for Computed NMR Scaling Factors. Available online: https://cheshirenmr.info/index.htm (accessed on 5 October 2018).
- Bohle, F.; Grimme, S. Hydrocarbon Macrocycle Conformer Ensembles and 13C-NMR Spectra. Angew. Chem. Int. Ed. 2022, 61, e202113905. [Google Scholar] [CrossRef] [PubMed]
- Begnini, F.; Poongavanam, V.; Atilaw, Y.; Erdelyi, M.; Schiesser, S.; Kihlberg, J. Cell Permeability of Isomeric Macrocycles: Predictions and NMR Studies. ACS Med. Chem. Lett. 2021, 12, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Brémond, É.; Savarese, M.; Su, N.Q.; Pérez-Jiménez, Á.J.; Xu, X.; Sancho-García, J.C.; Adamo, C. Benchmarking Density Functionals on Structural Parameters of Small-/Medium-Sized Organic Molecules. J. Chem. Theory Comput. 2016, 12, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melek, F.R.; Gershenzon, J.; Lee, E.; Mabry, T.J. Sesquiterpene lactones of Helianthus gracilentus. Phytochemistry 1984, 23, 2277–2279. [Google Scholar] [CrossRef]
- Liang, L.F.; Lan, L.F.; Taglialatela-Scafati, O.; Guo, Y.W. Sartrolides A-G and bissartrolide, new cembranolides from the South China Sea soft coral Sarcophyton trocheliophorum Marenzeller. Tetrahedron 2013, 69, 7381–7386. [Google Scholar] [CrossRef]
- Liang, L.F.; Kurtán, T.; Mándi, A.; Yao, L.G.; Li, J.; Lan, L.F.; Guo, Y.W. Structural, stereochemical, and bioactive studies of cembranoids from Chinese soft coral Sarcophyton trocheliophorum. Tetrahedron 2018, 74, 1933–1941. [Google Scholar] [CrossRef] [Green Version]
- Kicsák, M.; Mándi, A.; Varga, S.; Herczeg, M.; Batta, G.; Bényei, A.; Borbás, A.; Herczegh, P. Tricyclanos: Conformationally constrained nucleoside analogues with a new heterotricycle obtained from a D-ribofuranose unit. Org. Biomol. Chem. 2018, 16, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Mándi, A.; Wu, J.; Kurtán, T. TDDFT-ECD and DFT-NMR studies of thaigranatins A–E and granatumin L isolated from Xylocarpus granatum. RSC Adv. 2020, 10, 32216–32224. [Google Scholar] [CrossRef]
- Qiu, S.; de Gussem, E.; Tehrani, K.A.; Sergeyev, S.; Bultinck, P.; Herrebout, W. Stereochemistry of the Tadalafil Diastereoisomers: A Critical Assessment of Vibrational Circular Dichroism, Electronic Circular Dichroism, and Optical Rotatory Dispersion. J. Med. Chem. 2013, 56, 8903–8914. [Google Scholar] [CrossRef]
- Rablen, P.R.; Pearlman, S.A.; Finkbiner, J. A comparison of density functional methods for the estimation of proton chemical shifts with chemical accuracy. J. Phys. Chem. A 1999, 103, 7357–7363. [Google Scholar] [CrossRef]
- Jain, R.J.; Bally, T.; Rablen, P.R. Calculating accurate proton chemical shifts of organic molecules with density functional methods and modest basis sets. J. Org. Chem. 2009, 74, 4017–4023. [Google Scholar] [CrossRef] [Green Version]
- Konstantinov, I.A.; Broadbelt, L.J. Regression Formulas for Density Functional Theory Calculated 1H and 13C NMR Chemical Shifts in Toluene-d8. J. Phys. Chem. A 2011, 115, 12364–12372. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Lee, K.H.; Min, Y.D.; Choi, S.Z.; Kwon, H.C.; Cho, O.R.; Lee, K.C.; Lee, K.R. A new sesquiterpene lactone from Artemisia rubripes nakai. Arch. Pharmacal. Res. 2004, 27, 1016–1019. [Google Scholar] [CrossRef]
- Jewell, J.S.; Szarek, W.A. The light-induced addition of 1,3-dioxolan to unsaturated carbohydrates. Tetrahedron Lett. 1969, 10, 43–46. [Google Scholar] [CrossRef]
- Hough, L.; Otte, B. Furanoid Vinyl Ethers. Chem. Commun. 1966, 173–174. [Google Scholar] [CrossRef]
- Miyoshi, T.; Miyairi, N.; Aoki, H.; Kohsaka, M.; Sakai, H.I.; Imanaka, H. Bicyclomycin, a new antibiotic I. Taxonomy, isolation and characterization. J. Antibiot. 1972, 25, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Domínguez, E.; Romo, J. Mexicanin—I. A new sesquiterpene lactone related to tenulin. Tetrahedron 1963, 19, 1415–1421. [Google Scholar] [CrossRef]
- Li, J.; Tang, H.; Kurtán, T.; Mándi, A.; Zhuang, C.L.; Su, L.; Zheng, G.L.; Zhang, W. Swinhoeisterols from the South China Sea Sponge Theonella swinhoei. J. Nat. Prod. 2018, 81, 1645–1650. [Google Scholar] [CrossRef]
- Kobayashi, S.; Lu, C.; Hoye, T.R.; Hillmyer, M.A. Controlled Polymerization of a Cyclic Diene Prepared from the Ring-Closing Metathesis of a Naturally Occurring Monoterpene. J. Am. Chem. Soc. 2009, 131, 7960–7961. [Google Scholar] [CrossRef]
- Nishida, T.; Satoh, K.; Kamigaito, M. Biobased polymers via radical homopolymerization and copolymerization of a series of terpenoid derived conjugated dienes with exo-methylene and 6-membered ring. Molecules 2020, 25, 5890. [Google Scholar] [CrossRef]
- Raghavendra, S.; Tadiparthi, K.; Yadav, J.S. Total syntheses of Prelactone V and Prelactone B. Carbohydr. Res. 2017, 442, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Doboszewski, B.; Herdewijn, P. Carbohydrate chiral-pool approach to four enantiomerically pure 2-naphthylmethyl 3-hydroxy-2-methylbutanoates. Tetrahedron 2008, 64, 5551–5562. [Google Scholar] [CrossRef]
- Ivanova, N.A.; Valiullina, Z.R.; Shitikova, O.V.; Miftakhov, M.S. Reaction of methyl-4-methylene-2,3-O-isopropylidene-β-D-ribofuranoside with N-bromosuccinimide in aqueous tetrahydrofurane. Russ. J. Org. Chem. 2007, 43, 742–746. [Google Scholar] [CrossRef]
- Kohn, H.; Abuzar, S.; Korp, J.D.; Zektzer, A.S.; Martin, G.E. Structural studies of bicyclomycin. J. Heterocyclic. Chem. 1988, 25, 1511–1517. [Google Scholar] [CrossRef]
- Błoszyk, E.; Dudek, A.; Kosturkiewicz, Z.; Rychłewska, U.; Daniewski, W.M.; Gumulka, M.; Nawrot, J.; Buděšínský, M.; Vašíčková, S.; Holub, M. Sesquiterpene lactones of Cephalophora aromatica (HOOK.) SCHRADER and their deterrent activity. The stereostructure of geigerinin. Collect. Czech. Chem. Commun. 1989, 54, 1903–1918. [Google Scholar] [CrossRef]
- Grimblat, N.; Gavín, J.A.; Daranas, A.H.; Sarotti, A.M. Combining the Power of J Coupling and DP4 Analysis on Stereochemical Assignments: The J-DP4 Methods. Org. Lett. 2019, 21, 4003–4007. [Google Scholar] [CrossRef] [PubMed]
- Mándi, A.; Kurtán, T. Applications of OR/ECD/VCD to the structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 889–918. [Google Scholar] [CrossRef]
- Superchi, S.; Scafato, P.; Górecki, M.; Pescitelli, G. Configuration Determination by Quantum Mechanical Calculation of Chiroptical Spectra: Basics and Applications to Fungal Metabolites. Curr. Med. Chem. 2018, 25, 287–320. [Google Scholar] [CrossRef] [PubMed]
- De, B.C.; Zhang, W.; Yang, C.; Mándi, A.; Huang, C.; Zhang, L.; Liu, W.; Ruszczycky, M.W.; Zhu, Y.; Ma, M.; et al. Flavin-enabled reductive and oxidative epoxide ring opening reactions. Nat. Commun. 2022, 13, 4896. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.; Handy, N. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Pecul, M.; Marchesan, D.; Ruud, K.; Coriani, S. Polarizable continuum model study of solvent effects on electronic circular dichroism parameters. J. Chem. Phys. 2005, 122, 024106. [Google Scholar] [CrossRef]
- Szabó, K.E.; Kun, S.; Mándi, A.; Kurtán, T.; Somsák, L. Glucopyranosylidene-Spiro-Thiazolinones: Synthetic Studies and Determination of Absolute Configuration by TDDFT-ECD Calculations. Molecules 2017, 22, 1760. [Google Scholar] [CrossRef] [Green Version]
- MacroModel. Schrödinger LLC. 2015. Available online: http://www.schrodinger.com/MacroModel (accessed on 10 January 2023).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Stephens, P.J.; Harada, N. ECD cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Varetto, U. MOLEKEL, 5.4; Swiss National Supercomputing Centre: Manno, Switzerland, 2009. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Reutelingsperger, C. Flow cytometry of apoptotic cell death. J. Immunol. Methods 2000, 243, 167–190. [Google Scholar] [CrossRef] [PubMed]
- Latif, D.A.; Gonda, T.; Vágvölgyi, M.; Kúsz, N.; Kulmány, Á.; Ocsovszki, I.; Zomborszki, P.Z.; Zupkó, I.; Hunyadi, A. Synthesis and In Vitro Antitumor Activity of Naringenin Oxime and Oxime Ether Derivatives. Int. J. Mol. Sci. 2019, 20, 2184. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.Y.; Schelz, Z.; Tóth, B.; Vasas, A.; Ocsovszki, I.; Chang, F.R.; Hohmann, J.; Zupkó, I.; Wang, H.C. Investigation of natural phenanthrenes and the antiproliferative potential of juncusol in cervical cancer cell lines. Phytomedicine 2019, 58, 152770. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; Kameritsch, P.; Wallner, S.; Pohl, U.; Pogoda, K. The carboxyl tail of Cx43 augments p38 mediated cell migration in a gap junction-independent manner. Eur. J. Cell Biol. 2010, 89, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Xu, D.X.; Mándi, A.; Kurtán, T.; Li, T.J.; Schulz, B.; Zhang, W. Structure, Absolute Configuration, and Conformational Study of 12-Membered Macrolides from the Fungus Dendrodochium sp. Associated with the Sea Cucumber Holothuria nobilis Selenka. J. Org. Chem. 2013, 78, 7030–7047. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1H | 13C |
|---|---|---|
| 1a | 2.72 dd (11.0, 5.9) | 48.6 |
| 1b | 2.07 m | |
| 2 | 4.75 td (9.8, 5.9) | 69.2 |
| 3 | 5.25 d (9.8) | 133.9 |
| 4 | - | 135.1 |
| 5a | 2.76 dd (14.3, 5.1) | 44.0 |
| 5b | 2.32 dd (14.3, 1.6) | |
| 6 | 5.76 brd (5.1) | 70.8 |
| 7 | 2.93 d (7.9) | 52.9 |
| 8 | 5.50 t (9.9, 7.9) | 75.3 |
| 9 | 4.98 brd (9.9) | 129.3 |
| 10 | - | 142.7 |
| 11 | - | 136.4 |
| 12 | - | 169.4 |
| 13a | 6.30 d (3.2) | 121.2 |
| 13b | 5.60 d (3.2) | |
| 14 | 1.79 s | 18.7 |
| 15 | 1.53 s | 20.0 |
| 8-iVal | ||
| 1′ | - | 171.8 |
| 2′ | 2.15–2.17 m (2H) | 43.3 |
| 3′ | 2.05 m | 25.4 |
| 4′ | 0.92 d (6.2) | 22.3 |
| 5′ | 0.90 d (6.3) | 22.3 |
| NMR Level//DFT Optimization Level | MAE (Isomer 1; Isomer 2; Isomer 3, Isomer 4) | sDP4+ (Isomer 1; Isomer 2; Isomer 3, Isomer 4) |
|---|---|---|
| mPW1PW91/6-311+G(2d,p)//B3LYP/6-31+G(d,p) | 2.32; 2.10; 2.57; 2.49 | 5.28%; 93.00%; 0.25%; 1.47% |
| mPW1PW91/6-311+G(2d,p) SMD/CHCl3//B3LYP/6-31+G(d,p) | 2.06; 2.07; 2.16; 2.34 | 55.91%; 30.41%; 12.46%; 1.22% |
| mPW1PW91/6-311+G(2d,p) SMD/CHCl3//mPW1PW91/6-311+G(2d,p) SMD/CHCl3 | 2.15; 1.89; 2.38; 2.41 | 3.40%; 96.26%; 0.19%; 0.16% |
| NMR Level//DFT Optimization Level | MAE [(8R)-Epimer; (8S)-Epimer] | sDP4+ [(8R)-Epimer; (8S)-Epimer] |
|---|---|---|
| mPW1PW91/6-31G(d)//M06-2X/6-31G(d) | 1.63; 1.68 | (69.35%; 30.65%) |
| mPW1PW91/6-31G(d) SMD/CHCl3//M06-2X/6-31G(d) | 1.40; 1.54 | (93.58%; 6.42%) |
| M06/6-31G(d)//B3LYP/6-31+G(d,p) | 1.68; 2.01 | (99.13%; 0.87%) |
| OPBE0/6-31G(d)//B3LYP/6-31+G(d,p) | 1.93; 2.35 | (99.79%; 0.21%) |
| NMR Level//DFT Optimization Level | MAE (Isomer 1; Isomer 2; Isomer 3, Isomer 4) | sDP4+ (Isomer 1; Isomer 2; Isomer 3, Isomer 4) |
|---|---|---|
| mPW1PW91/6-31G(d) SMD/CHCl3//M06-2X/6-31G(d) | 2.42; 2.59; 2.62; 2.85 | (90.66% 3.14% 5.16% 1.04%) |
| M06/6-31G(d)//B3LYP/6-31+G(d,p) | 3.12; 2.83; 3.19; 2.76 | (2.54%; 69.71%; 9.58%; 18.17%) |
| Numbering | δExp (ppm) | δCalc (ppm) | Δδ (ppm) |
|---|---|---|---|
| C-1 | 79.28 | 78.78 | 0.50 |
| C-2 | 32.17 | 31.26 | 0.91 |
| C-3 | 35.34 | 36.89 | 1.55 |
| C-4 | 146.48 | 152.97 | 6.49 |
| C-5 | 56.15 | 56.06 | 0.09 |
| C-6 | 67.25 | 64.33 | 2.92 |
| C-7 | 49.59 | 48.55 | 1.04 |
| C-8 | 18.43 | 19.65 | 1.22 |
| C-9 | 36.55 | 35.90 | 0.65 |
| C-10 | 41.93 | 44.33 | 2.40 |
| C-11 | 26.26 | 27.81 | 1.55 |
| C-12 | 21.32 | 19.60 | 1.72 |
| C-13 | 16.44 | 15.72 | 0.72 |
| C-14 | 11.82 | 10.57 | 1.25 |
| C-15 | 108.03 | 105.99 | 2.04 |
| MAE | N/A | N/A | 1.67 |
| Numbering | δExp (ppm) | δCalc (ppm) | Δδ (ppm) |
|---|---|---|---|
| C-1 | 79.28 | 77.99 | 1.29 |
| C-2 | 32.17 | 31.72 | 0.45 |
| C-3 | 35.34 | 36.68 | 1.34 |
| C-4 | 146.48 | 152.90 | 6.42 |
| C-5 | 56.15 | 55.65 | 0.50 |
| C-6 | 67.25 | 64.65 | 2.60 |
| C-7 | 49.59 | 48.49 | 1.10 |
| C-8 | 18.43 | 19.48 | 1.05 |
| C-9 | 36.55 | 35.92 | 0.63 |
| C-10 | 41.93 | 44.70 | 2.77 |
| C-11 | 26.26 | 28.01 | 1.75 |
| C-12 | 21.32 | 19.80 | 1.52 |
| C-13 | 16.44 | 15.27 | 1.17 |
| C-14 | 11.82 | 11.13 | 0.69 |
| C-15 | 108.03 | 104.29 | 3.74 |
| MAE | N/A | N/A | 1.80 |
| NMR Level//DFT Optimization Level | MAE [(8R)-Epimer; (8S)-Epimer] | sDP4+ [(8R)-Epimer; (8S)-Epimer] |
|---|---|---|
| mPW1PW91/6-311+G(2d,p)//ωB97XD/6-31+G(d,p) | 1.82; 1.87 | (66.38%; 33.62%) |
| mPW1PW91/6-311+G(2d,p) SMD/CHCl3//ωB97XD/6-31+G(d,p) SMD/CHCl3 | 1.60; 1.91 | (99.26%; 0.74%) |
| NMR Level//DFT Optimization Level | MAE (Isomer 1; Isomer 2; Isomer 3, Isomer 4) |
|---|---|
| mPW1PW91/6-311+G(2d,p)//ωB97XD/6-31+G(d,p) | 2.33; 2.48; 2.58; 3.11 |
| mPW1PW91/6-311+G(2d,p) SMD/CHCl3//ωB97XD/6-31+G(d,p) SMD/CHCl3 | 2.26; 2.42; 2.48; 3.23 |
| Coupling Atoms | JExp (Hz) | Ji1 (Hz) | Ji2 (Hz) | ΔJi1 | ΔJi2 |
|---|---|---|---|---|---|
| 1aH-2H | 5.90 | 6.62 | 0.93 | 0.72 | 4.97 |
| 2H-3H | 9.80 | 10.08 | 6.51 | 0.28 | 3.29 |
| 5aH-6H | 5.10 | 6.58 | 2.82 | 1.48 | 2.28 |
| 7H-8H | 7.90 | 4.33 | 3.28 | 3.57 | 4.62 |
| 8H-9H | 9.90 | 7.36 | 8.73 | 2.54 | 1.17 |
| MAE | 1.72 | 3.27 |
| Compound | IC50 Values (µM) [95% CI] | ||||
|---|---|---|---|---|---|
| HeLa | C33A | SiHa | NIH-3T3 | MRC-5 | |
| 1 | 16.62 [14.55–18.99] | 4.43 [3.62–5.43] | 2.82 [2.40–3.32] | 13.05 [11.44–14.90] | 14.82 [13.90–15.80] |
| cisplatin | 12.14 [10.18–14.46] | 5.85 [5.37–6.38] | 4.29 [3.72–4.95] | 5.49 [4.76–6.35] | 5.17 [3.88–6.89] |
| SI for 1 NIH-3T3/cell line | 0.79 | 2.95 | 4.63 | ||
| SI for 1 MRC-5/cell line | 0.89 | 3.35 | 5.26 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, T.; Lajter, I.; Kúsz, N.; Schelz, Z.; Bózsity-Faragó, N.; Borbás, A.; Zupkó, I.; Krupitza, G.; Frisch, R.; Hohmann, J.; et al. Isolation and NMR Scaling Factors for the Structure Determination of Lobatolide H, a Flexible Sesquiterpene from Neurolaena lobata . Int. J. Mol. Sci. 2023, 24, 5841. https://doi.org/10.3390/ijms24065841
Kovács T, Lajter I, Kúsz N, Schelz Z, Bózsity-Faragó N, Borbás A, Zupkó I, Krupitza G, Frisch R, Hohmann J, et al. Isolation and NMR Scaling Factors for the Structure Determination of Lobatolide H, a Flexible Sesquiterpene from Neurolaena lobata . International Journal of Molecular Sciences. 2023; 24(6):5841. https://doi.org/10.3390/ijms24065841
Chicago/Turabian StyleKovács, Tibor, Ildikó Lajter, Norbert Kúsz, Zsuzsanna Schelz, Noémi Bózsity-Faragó, Anikó Borbás, István Zupkó, Georg Krupitza, Richard Frisch, Judit Hohmann, and et al. 2023. "Isolation and NMR Scaling Factors for the Structure Determination of Lobatolide H, a Flexible Sesquiterpene from Neurolaena lobata " International Journal of Molecular Sciences 24, no. 6: 5841. https://doi.org/10.3390/ijms24065841