
Characterization of the Involvement of Tumour Necrosis Factor (TNF)-α-Stimulated Gene 6 (TSG-6) in Ischemic Brain Injury Caused by Middle Cerebral Artery Occlusion in Mouse

 and
and 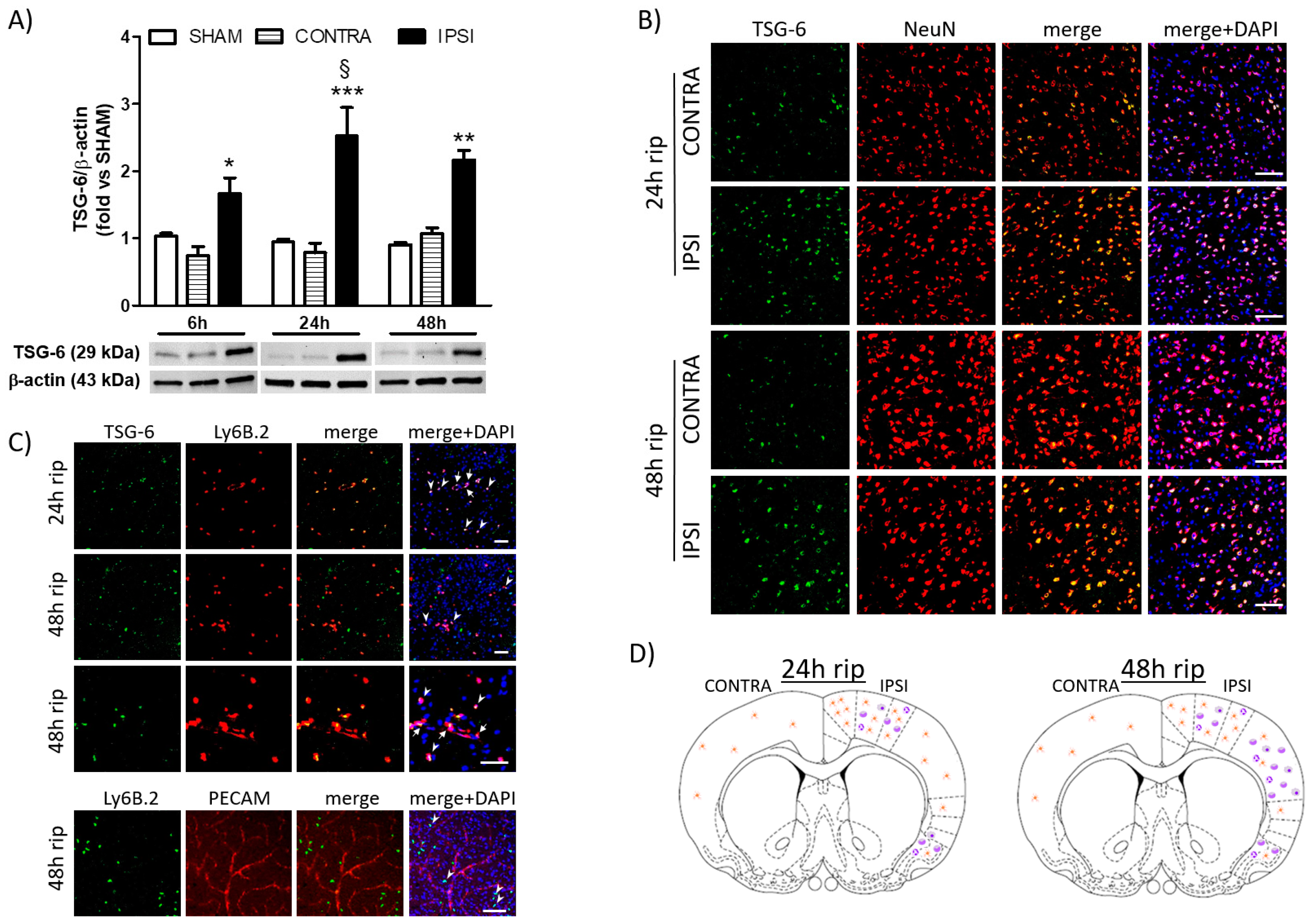{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Analysis of the Cerebral Expression of TSG-6 following Transient MCAo in Mice
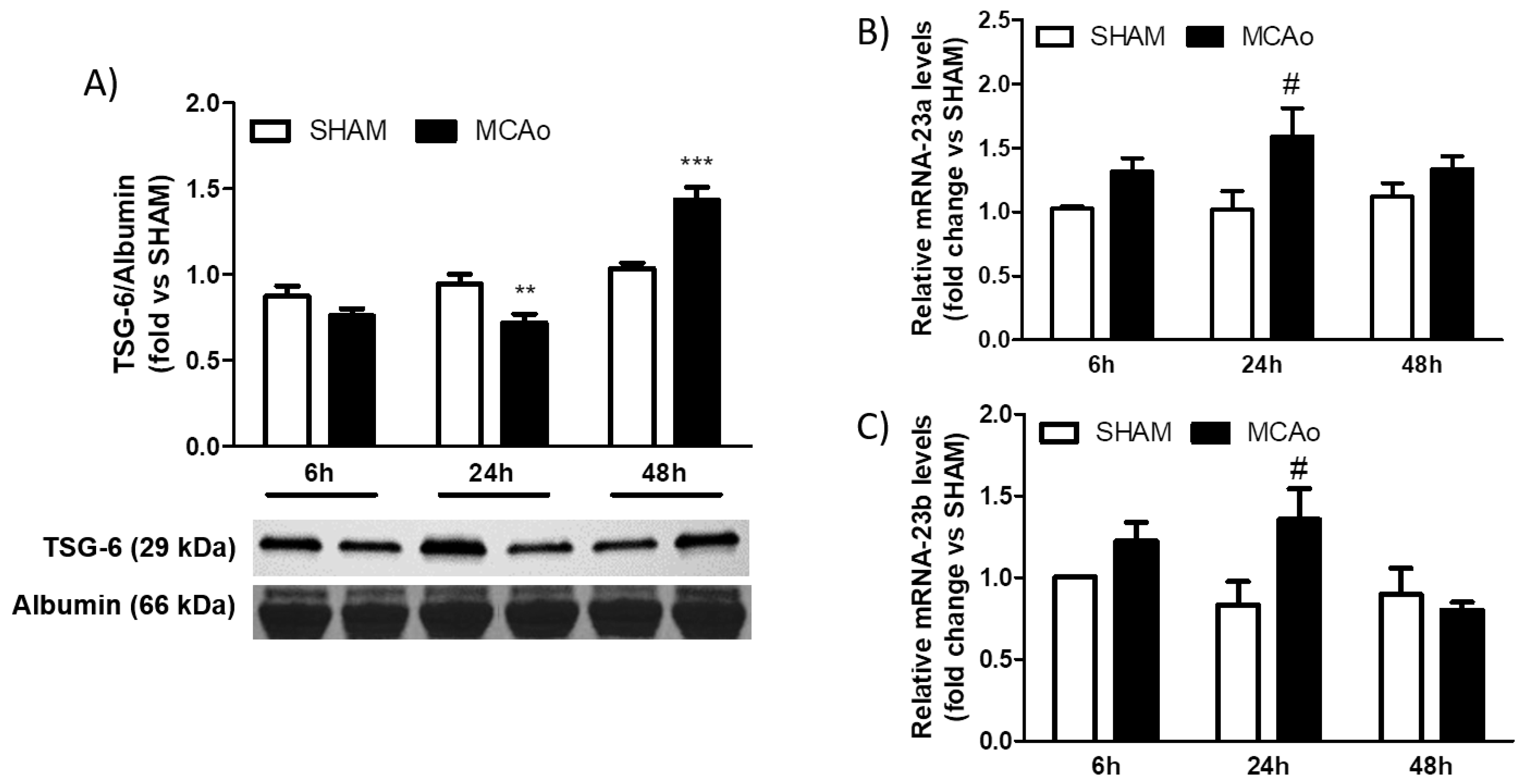
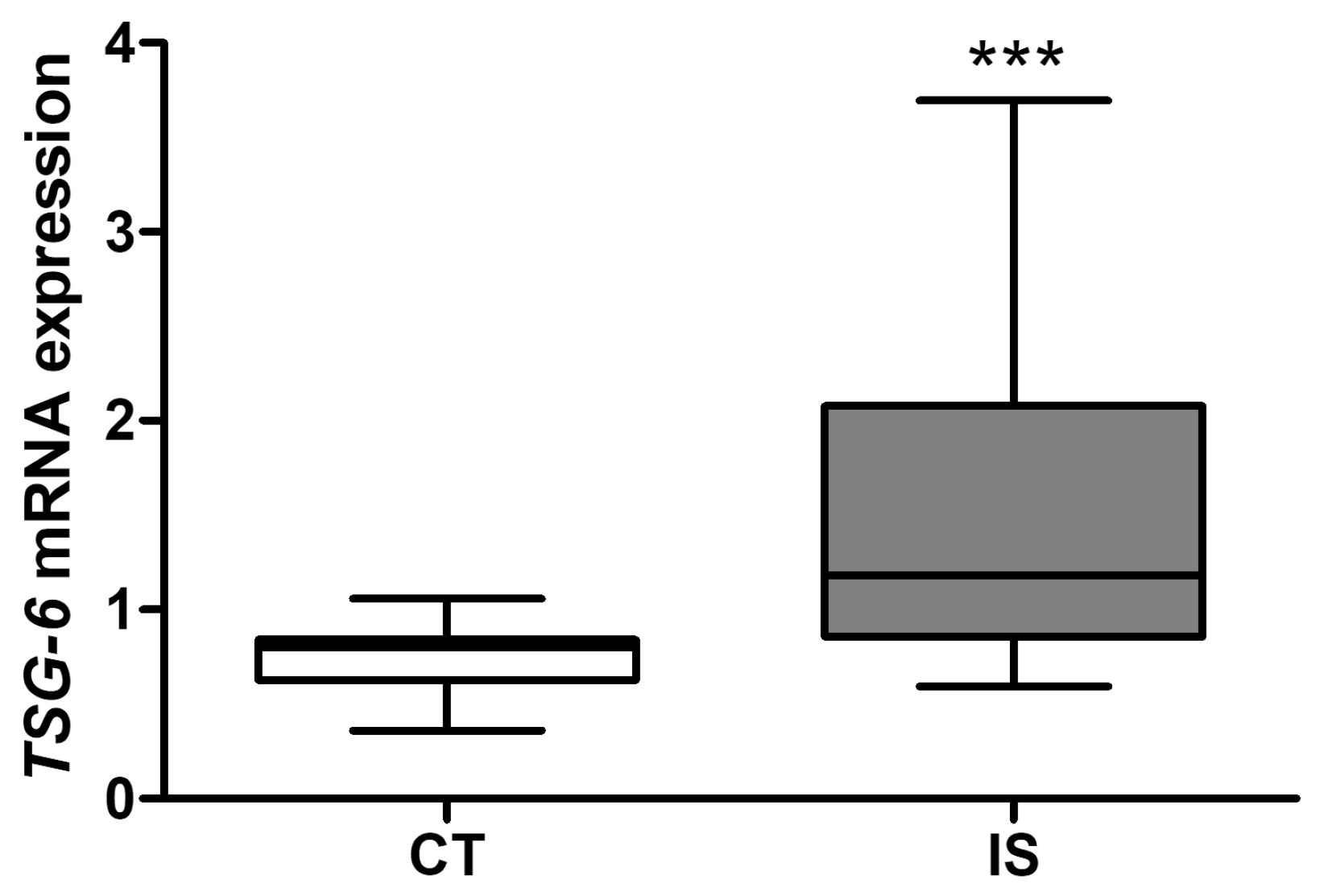
2.2. Analysis of Blood Expression of TSG-6 after Ischemic Stroke in Mice and Patients
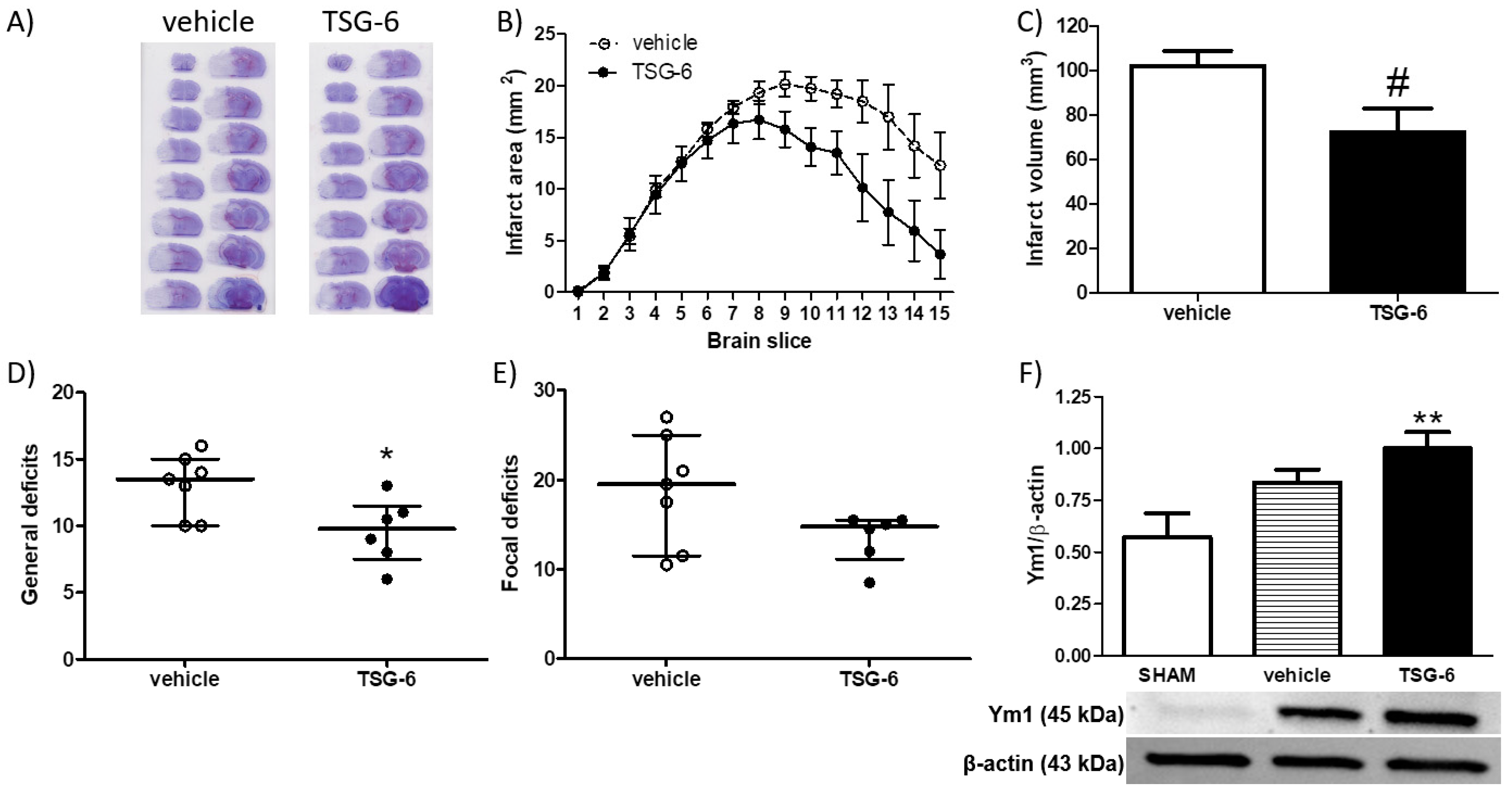
2.3. Neuroprotective Effects of Systemic Administration of TSG-6 in Mice Subjected to MCAo
3. Discussion
4. Materials and Methods
4.1. Animals
- (1)
- MCAo 6 h: mice were subjected to 1 h MCAo followed by 6 h of reperfusion;
- (2)
- SHAM 6 h: sham surgery 6 h before sacrifice;
- (3)
- MCAo 24 h: mice were subjected to 1 h MCAo followed by 24 h of reperfusion;
- (4)
- SHAM 24 h: sham surgery 24 h before sacrifice;
- (5)
- MCAo 48 h: mice were subjected to 1 h MCAo followed by 48 h of reperfusion;
- (6)
- SHAM 48 h: sham surgery 48 h before sacrifice;
- (7)
- MCAo + TSG-6: mice were subjected to 1 h MCAo followed by 48 h of reperfusion and intravenously (i.v.) injected upon reperfusion with 30 μg recombinant mouse TSG-6 (2326-TS, R & D Systems, Minneapolis, MN, USA) dissolved in 100 μL PBS;
- (8)
- MCAo + vehicle: mice were subjected to 1 h MCAo followed by 48 h of reperfusion and i.v. injected upon reperfusion with vehicle (100 μL PBS).
4.2. Surgical Procedure for MCAo in Mice
4.3. Brain Infarct and Neurological Deficits Assessment
4.4. Western Blot Analysis
4.5. Real-Time Polymerase Chain Reaction (PCR) in Mouse Plasma Samples
4.6. Immunofluorescence
4.7. Ischemic Stroke Patients and Control Subjects
4.8. Gene Expression Analysis in Human Peripheral Blood Mononuclear Cells (PBMCs)
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feigin, V.L.; Stark, B.A.; Johnson, C.O.; Roth, G.A.; Bisignano, C.; Abady, G.G.; Abbasifard, M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abedi, V.; et al. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Thomalla, G.; Boutitie, F.; Ma, H.; Koga, M.; Ringleb, P.; Schwamm, L.H.; Wu, O.; Bendszus, M.; Bladin, C.F.; Campbell, B.C.V.; et al. Intravenous alteplase for stroke with unknown time of onset guided by advanced imaging: Systematic review and meta-analysis of individual patient data. Lancet 2020, 396, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Campbell, B.C.V.; Parsons, M.W.; Churilov, L.; Levi, C.R.; Hsu, C.; Kleinig, T.J.; Wijeratne, T.; Curtze, S.; Dewey, H.M.; et al. Thrombolysis Guided by Perfusion Imaging up to 9 Hours after Onset of Stroke. N. Engl. J. Med. 2019, 380, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Dávalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with Alteplase 3 to 4.5 Hours after Acute Ischemic Stroke. N. Engl. J. Med. 2008, 359, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Shafie, M.; Yu, W. Recanalization Therapy for Acute Ischemic Stroke with Large Vessel Occlusion: Where We Are and What Comes Next? Transl. Stroke Res. 2021, 12, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Amantea, D.; Greco, R. Neuroprotection Following Stroke. In Comprehensive Pharmacology; Elsevier: Amsterdam, The Netherlands, 2022; pp. 64–90. ISBN 9780128012383. [Google Scholar]
- Haupt, M.; Gerner, S.T.; Bähr, M.; Doeppner, T.R. Quest for Quality in Translational Stroke Research—A New Dawn for Neuroprotection? Int. J. Mol. Sci. 2022, 23, 5381. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Amantea, D.; Greco, R.; Micieli, G.; Bagetta, G. Paradigm Shift to Neuroimmunomodulation for Translational Neuroprotection in Stroke. Front. Neurosci. 2018, 12, 241. [Google Scholar] [CrossRef]
- Endres, M.; Moro, M.A.; Nolte, C.H.; Dames, C.; Buckwalter, M.S.; Meisel, A. Immune Pathways in Etiology, Acute Phase, and Chronic Sequelae of Ischemic Stroke. Circ. Res. 2022, 130, 1167–1186. [Google Scholar] [CrossRef]
- Frank, D.; Zlotnik, A.; Boyko, M.; Gruenbaum, B.F. The Development of Novel Drug Treatments for Stroke Patients: A Review. Int. J. Mol. Sci. 2022, 23, 5796. [Google Scholar] [CrossRef]
- Iadecola, C.; Buckwalter, M.S.; Anrather, J. Immune responses to stroke: Mechanisms, modulation, and therapeutic potential. J. Clin. Investig. 2020, 130, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Amantea, D.; La Russa, D.; Frisina, M.; Giordano, F.; Di Santo, C.; Panno, M.L.; Pignataro, G.; Bagetta, G. Ischemic Preconditioning Modulates the Peripheral Innate Immune System to Promote Anti-Inflammatory and Protective Responses in Mice Subjected to Focal Cerebral Ischemia. Front. Immunol. 2022, 13, 825834. [Google Scholar] [CrossRef]
- La Russa, D.; Di Santo, C.; Lizasoain, I.; Moraga, A.; Bagetta, G.; Amantea, D. Tumor necrosis factor (TNF)-α-stimulated gene 6 (TSG-6): A promising immunomodulatory target in acute neurodegenerative diseases. Int. J. Mol. Sci. 2022, 24, 1162. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.G.; Maier, R.; Lotz, M.; Lee, S.; Klampfer, L.; Lee, T.H.; Vilcek, J. TSG-6: A TNF-, IL-1-, and LPS-inducible secreted glycoprotein associated with arthritis. J. Immunol. 1993, 151, 6593–6601. [Google Scholar] [CrossRef]
- Klampfer, L.; Lee, T.H.; Hsu, W.; Vilcek, J.; Chen-Kiang, S. NF-IL6 and AP-1 cooperatively modulate the activation of the TSG-6 gene by tumor necrosis factor alpha and interleukin-1. Mol. Cell. Biol. 1994, 14, 6561–6569. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Wisniewski, H.G.; Vilcek, J. A novel secretory tumor necrosis factor-inducible protein (TSG-6) is a member of the family of hyaluronate binding proteins, closely related to the adhesion receptor CD44. J. Cell Biol. 1992, 116, 545–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdani, M.; Johnson, P.Y.; Potter-Perigo, S.; Nagy, N.; Day, A.J.; Bollyky, P.L.; Wight, T.N. Hyaluronan and Hyaluronan-Binding Proteins Accumulate in Both Human Type 1 Diabetic Islets and Lymphoid Tissues and Associate with Inflammatory Cells in Insulitis. Diabetes 2014, 63, 2727–2743. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.; McGrouther, D.; Day, A.; Milner, C.; Bayat, A. Characterization of hyaluronan and TSG-6 in skin scarring: Differential distribution in keloid scars, normal scars and unscarred skin. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 317–327. [Google Scholar] [CrossRef]
- Zhang, S.; He, H.; Day, A.J.; Tseng, S.C.G. Constitutive Expression of Inter-α-inhibitor (IαI) Family Proteins and Tumor Necrosis Factor-stimulated Gene-6 (TSG-6) by Human Amniotic Membrane Epithelial and Stromal Cells Supporting Formation of the Heavy Chain-Hyaluronan (HC-HA) Complex. J. Biol. Chem. 2012, 287, 12433–12444. [Google Scholar] [CrossRef] [Green Version]
- Coulson-Thomas, V.J.; Lauer, M.E.; Soleman, S.; Zhao, C.; Hascall, V.C.; Day, A.J.; Fawcett, J.W. Tumor Necrosis Factor-stimulated Gene-6 (TSG-6) Is Constitutively Expressed in Adult Central Nervous System (CNS) and Associated with Astrocyte-mediated Glial Scar Formation following Spinal Cord Injury. J. Biol. Chem. 2016, 291, 19939–19952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, C.M.; Day, A.J. TSG-6: A multifunctional protein associated with inflammation. J. Cell Sci. 2003, 116, 1863–1873. [Google Scholar] [CrossRef] [Green Version]
- Day, A.J.; Milner, C.M. TSG-6: A multifunctional protein with anti-inflammatory and tissue-protective properties. Matrix Biol. 2019, 78–79, 60–83. [Google Scholar] [CrossRef] [Green Version]
- Lesley, J.; Gál, I.; Mahoney, D.J.; Cordell, M.R.; Rugg, M.S.; Hyman, R.; Day, A.J.; Mikecz, K. TSG-6 Modulates the Interaction between Hyaluronan and Cell Surface CD44. J. Biol. Chem. 2004, 279, 25745–25754. [Google Scholar] [CrossRef] [Green Version]
- Kota, D.J.; Wiggins, L.L.; Yoon, N.; Lee, R.H. TSG-6 Produced by hMSCs Delays the Onset of Autoimmune Diabetes by Suppressing Th1 Development and Enhancing Tolerogenicity. Diabetes 2013, 62, 2048–2058. [Google Scholar] [CrossRef] [Green Version]
- Baranova, N.S.; Nilebäck, E.; Haller, F.M.; Briggs, D.C.; Svedhem, S.; Day, A.J.; Richter, R.P. The Inflammation-associated Protein TSG-6 Cross-links Hyaluronan via Hyaluronan-induced TSG-6 Oligomers. J. Biol. Chem. 2011, 286, 25675–25686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.; Lee, R.H.; Bazhanov, N.; Oh, J.Y.; Prockop, D.J. Anti-inflammatory protein TSG-6 secreted by activated MSCs attenuates zymosan-induced mouse peritonitis by decreasing TLR2/NF-κB signaling in resident macrophages. Blood 2011, 118, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Baranova, N.S.; Inforzato, A.; Briggs, D.C.; Tilakaratna, V.; Enghild, J.J.; Thakar, D.; Milner, C.M.; Day, A.J.; Richter, R.P. Incorporation of Pentraxin 3 into Hyaluronan Matrices Is Tightly Regulated and Promotes Matrix Cross-linking. J. Biol. Chem. 2014, 289, 30481–30498. [Google Scholar] [CrossRef] [Green Version]
- Baranova, N.S.; Foulcer, S.J.; Briggs, D.C.; Tilakaratna, V.; Enghild, J.J.; Milner, C.M.; Day, A.J.; Richter, R.P. Inter-α-inhibitor Impairs TSG-6-induced Hyaluronan Cross-linking. J. Biol. Chem. 2013, 288, 29642–29653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulson-Thomas, V.J.; Gesteira, T.F.; Hascall, V.; Kao, W. Umbilical Cord Mesenchymal Stem Cells Suppress Host Rejection. J. Biol. Chem. 2014, 289, 23465–23481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauer, M.E.; Loftis, J.; de la Motte, C.; Hascall, V.C. Analysis of the Heavy-Chain Modification and TSG-6 Activity in Pathological Hyaluronan Matrices. Methods Mol. Biol. 2015, 1229, 543–548. [Google Scholar]
- Liao, Z.; Wang, W.; Deng, W.; Zhang, Y.; Song, A.; Deng, S.; Zhao, H.; Zhang, S.; Li, Z. Human Umbilical Cord Mesenchymal Stem Cells-Secreted TSG-6 Is Anti-Inflammatory and Promote Tissue Repair After Spinal Cord Injury. ASN Neuro 2021, 13, 175909142110106. [Google Scholar] [CrossRef]
- Tang, B.; Song, M.; Xie, X.; Le, D.; Tu, Q.; Wu, X.; Chen, M. Tumor Necrosis Factor-stimulated Gene-6 (TSG-6) Secreted by BMSCs Regulates Activated Astrocytes by Inhibiting NF-κB Signaling Pathway to Ameliorate Blood Brain Barrier Damage After Intracerebral Hemorrhage. Neurochem. Res. 2021, 46, 2387–2402. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liu, W.; Yin, J.; Chen, Y.; Guo, S.; Fan, H.; Li, X.; Zhang, X.; He, X.; Duan, C. TSG-6 attenuates inflammation-induced brain injury via modulation of microglial polarization in SAH rats through the SOCS3/STAT3 pathway. J. Neuroinflamm. 2018, 15, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, J.; Shetty, A.K.; Hattiangady, B.; Kim, D.-K.; Foraker, J.E.; Nishida, H.; Prockop, D.J. Administration of TSG-6 improves memory after traumatic brain injury in mice. Neurobiol. Dis. 2013, 59, 86–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roura, S.; Monguió-Tortajada, M.; Munizaga-Larroudé, M.; Clos-Sansalvador, M.; Franquesa, M.; Rosell, A.; Borràs, F.E. Potential of Extracellular Vesicle-Associated TSG-6 from Adipose Mesenchymal Stromal Cells in Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 6761. [Google Scholar] [CrossRef]
- Lin, Q.; Zhao, S.; Zhou, L.; Fang, X.; Fu, Y.; Huang, Z. Mesenchymal stem cells transplantation suppresses inflammatory responses in global cerebral ischemia: Contribution of TNF-α-induced protein 6. Acta Pharmacol. Sin. 2013, 34, 784–792. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Liu, Y.; Yan, K.; Chen, L.; Chen, X.-R.; Li, P.; Chen, F.-F.; Jiang, X.-D. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J. Neuroinflamm. 2013, 10, 106. [Google Scholar] [CrossRef] [Green Version]
- Al’Qteishat, A. Changes in hyaluronan production and metabolism following ischaemic stroke in man. Brain 2006, 129, 2158–2176. [Google Scholar] [CrossRef]
- Qu, Y.; Yang, F.; Meng, F.; Chen, X.; Zhang, Q.; Yu, T.; Wen, S.; Pan, Y. Plasma Concentration of Tumor Necrosis Factor-Stimulated Gene-6 as a Novel Diagnostic and 3-Month Prognostic Indicator in Non-Cardioembolic Acute Ischemic Stroke. Front. Immunol. 2022, 13, 713379. [Google Scholar] [CrossRef]
- Lin, Q.; Lin, S.; Lv, Y.; Zhou, L.; Fu, Y.; Fang, X.; Chen, F.; Huang, Z. Suppression of inflammatory damage to the brain after global cerebral ischemia by transplanted mesenchymal stem cells via secretion of TSG-6. Neurol. Asia 2016, 21, 113–122. [Google Scholar]
- Jung, H.-S.; Jeong, S.-Y.; Yang, J.; Kim, S.-D.; Zhang, B.; Yoo, H.S.; Song, S.U.; Jeon, M.-S.; Song, Y.S. Neuroprotective effect of mesenchymal stem cell through complement component 3 downregulation after transient focal cerebral ischemia in mice. Neurosci. Lett. 2016, 633, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Bárdos, T.; Kamath, R.V.; Mikecz, K.; Glant, T.T. Anti-Inflammatory and Chondroprotective Effect of TSG-6 (Tumor Necrosis Factor-α-Stimulated Gene-6) in Murine Models of Experimental Arthritis. Am. J. Pathol. 2001, 159, 1711–1721. [Google Scholar] [CrossRef]
- Getting, S.J.; Mahoney, D.J.; Cao, T.; Rugg, M.S.; Fries, E.; Milner, C.M.; Perretti, M.; Day, A.J. The Link Module from Human TSG-6 Inhibits Neutrophil Migration in a Hyaluronan- and Inter-α-inhibitor-independent Manner. J. Biol. Chem. 2002, 277, 51068–51076. [Google Scholar] [CrossRef] [Green Version]
- Bertling, F.; Bendix, I.; Drommelschmidt, K.; Wisniewski, H.G.; Felderhoff-Mueser, U.; Keller, M.; Prager, S. Tumor necrosis factor-inducible gene 6 protein: A novel neuroprotective factor against inflammation-induced developmental brain injury. Exp. Neurol. 2016, 279, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, M.J.; Damodarasamy, M.; Pathan, J.L.; Chan, C.K.; Spiekerman, C.; Wight, T.N.; Banks, W.A.; Day, A.J.; Vernon, R.B.; Keene, C.D. Increased Hyaluronan and TSG-6 in Association with Neuropathologic Changes of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 67, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhan, Y.; Xu, L.; Feuerstein, G.Z.; Wang, X. Use of Suppression Subtractive Hybridization for Differential Gene Expression in Stroke. Stroke 2001, 32, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, L.; Wang, H.; Zhan, Y.; Puré, E.; Feuerstein, G.Z. CD44 deficiency in mice protects brain from cerebral ischemia injury. J. Neurochem. 2002, 83, 1172–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Song, M.; Xie, X.; Chen, Z.; Gao, Z.; Wu, X.; Huang, R.; Chen, M. BMSCs Regulate Astrocytes through TSG-6 to Protect the Blood-Brain Barrier after Subarachnoid Hemorrhage. Mediat. Inflamm. 2021, 2021, 5522291. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhu, J.; Hang, C.-H.; Wang, Y.-H. The Potassium SK Channel Activator NS309 Protects Against Experimental Traumatic Brain Injury Through Anti-Inflammatory and Immunomodulatory Mechanisms. Front. Pharmacol. 2019, 10, 1432. [Google Scholar] [CrossRef]
- Watanabe, R.; Watanabe, H.; Takahashi, Y.; Kojima, M.; Konii, H.; Watanabe, K.; Shirai, R.; Sato, K.; Matsuyama, T.; Ishibashi-Ueda, H.; et al. Atheroprotective Effects of Tumor Necrosis Factor–Stimulated Gene-6. JACC Basic Transl. Sci. 2016, 1, 494–509. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Wang, X.; Shi, Z.; Yu, C.; Li, M.; Chen, L.; Jia, Q.; Liang, G. Tumor necrosis factor-stimulated gene-6-a new serum identification marker to identify severe and symptomatic carotid artery stenosis. Pathol.-Res. Pract. 2022, 232, 153838. [Google Scholar] [CrossRef]
- Li, X.; Liu, W.; Li, R.; Guo, S.; Fan, H.; Wei, B.; Zhang, X.; He, X.; Duan, C. TSG-6 Attenuates Oxidative Stress-Induced Early Brain Injury in Subarachnoid Hemorrhage Partly by the HO-1 and Nox2 Pathways. J. Stroke Cerebrovasc. Dis. 2020, 29, 104986. [Google Scholar] [CrossRef]
- Maina, V.; Cotena, A.; Doni, A.; Nebuloni, M.; Pasqualini, F.; Milner, C.M.; Day, A.J.; Mantovani, A.; Garlanda, C. Coregulation in human leukocytes of the long pentraxin PTX3 and TSG-6. J. Leukoc. Biol. 2009, 86, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, D.; Xu, L.; Dong, L.; Zheng, J.; Lin, Y.; Huang, J.; Zhang, Y.; Tao, Y.; Zang, X.; et al. Cell–cell contact with proinflammatory macrophages enhances the immunotherapeutic effect of mesenchymal stem cells in two abortion models. Cell Mol. Immunol. 2019, 16, 908–920. [Google Scholar] [CrossRef]
- Mittal, M.; Tiruppathi, C.; Nepal, S.; Zhao, Y.-Y.; Grzych, D.; Soni, D.; Prockop, D.J.; Malik, A.B. TNFα-stimulated gene-6 (TSG6) activates macrophage phenotype transition to prevent inflammatory lung injury. Proc. Natl. Acad. Sci. USA 2016, 50, E8151–E8158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Li, X.; Shi, Z.; Wu, P.; Fu, J.; Tang, J.; Qing, L. Exosomes from LPS-preconditioned bone marrow MSCs accelerated peripheral nerve regeneration via M2 macrophage polarization: Involvement of TSG-6/NF-κB/NLRP3 signaling pathway. Exp. Neurol. 2022, 356, 114139. [Google Scholar] [CrossRef] [PubMed]
- Bartosh, T.J.; Ylöstalo, J.H.; Mohammadipoor, A.; Bazhanov, N.; Coble, K.; Claypool, K.; Lee, R.H.; Choi, H.; Prockop, D.J. Aggregation of human mesenchymal stromal cells (MSCs) into 3D spheroids enhances their antiinflammatory properties. Proc. Natl. Acad. Sci. USA 2010, 107, 13724–13729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Jiang, D.; Sindrilaru, A.; Stegemann, A.; Schatz, S.; Treiber, N.; Rojewski, M.; Schrezenmeier, H.; Beken, S.V.; Wlaschek, M.; et al. TSG-6 Released from Intradermally Injected Mesenchymal Stem Cells Accelerates Wound Healing and Reduces Tissue Fibrosis in Murine Full-Thickness Skin Wounds. J. Invest. Dermatol. 2014, 2, 526–537. [Google Scholar] [CrossRef] [Green Version]
- Greco, R.; Mangione, A.S.; Amantea, D.; Bagetta, G.; Nappi, G.; Tassorelli, C. IkappaB-alpha expression following transient focal cerebral ischemia is modulated by nitric oxide. Brain Res. 2011, 1372, 145–151. [Google Scholar] [CrossRef]
- Deng, W.; Mandeville, E.; Terasaki, Y.; Li, W.; Holder, J.; Chuang, A.T.; Ning, M.; Arai, K.; Lo, E.H.; Xing, C. Transcriptomic characterization of microglia activation in a rat model of ischemic stroke. J. Cereb. Blood Flow Metab. 2020, 40, S34–S48. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, Y.; Li, F.; Zhang, Z.; Cui, L.; He, H.; Yan, X.; He, W.; Sun, H.; Feng, Z.; et al. Neuronal chemokine-like-factor 1 (CKLF1) up-regulation promotes M1 polarization of microglia in rat brain after stroke. Acta Pharmacol. Sin. 2022, 43, 1217–1230. [Google Scholar] [CrossRef]
- Gaire, B.P.; Song, M.-R.; Choi, J.W. Sphingosine 1-phosphate receptor subtype 3 (S1P3) contributes to brain injury after transient focal cerebral ischemia via modulating microglial activation and their M1 polarization. J. Neuroinflamm. 2018, 15, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.; Wang, J.; Wen, L.; Sun, M.; Liu, H.; Gao, Y. Remote Limb Ischemic Postconditioning Protects against Ischemic Stroke via Modulating Microglia/Macrophage Polarization in Mice. J. Immunol. Res. 2021, 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Dai, Z.; Cao, Y.; Wang, L. Caspase-1 inhibition mediates neuroprotection in experimental stroke by polarizing M2 microglia/macrophage and suppressing NF-κB activation. Biochem. Biophys. Res. Commun. 2019, 513, 479–485. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, D.; Li, X.; Jiang, Y.; Wang, C.; Zhang, Y.; Kong, Q.; Tian, C.; Dai, Y.; Zhao, W.; et al. Excess salt intake promotes M1 microglia polarization via a p38/MAPK/AR-dependent pathway after cerebral ischemia in mice. Int. Immunopharmacol. 2020, 81, 106176. [Google Scholar] [CrossRef]
- Jha, K.A.; Pentecost, M.; Lenin, R.; Gentry, J.; Klaic, L.; Del Mar, N.; Reiner, A.; Yang, C.H.; Pfeffer, L.M.; Sohl, N.; et al. TSG-6 in conditioned media from adipose mesenchymal stem cells protects against visual deficits in mild traumatic brain injury model through neurovascular modulation. Stem Cell Res. Ther. 2019, 10, 318. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zeng, R.; Wang, Y.; Huang, W.; Hu, B.; Zhu, G.; Zhang, R.; Li, F.; Han, J.; Li, Y. Mesenchymal stem cells enhance microglia M2 polarization and attenuate neuroinflammation through TSG-6. Brain Res. 2019, 1724, 146422. [Google Scholar] [CrossRef] [PubMed]
- Armogida, M.; Spalloni, A.; Amantea, D.; Nutini, M.; Petrelli, F.; Longone, P.; Bagetta, G.; Nisticò, R.; Mercuri, N.B. The protective role of catalase against cerebral ischemia in vitro and in vivo. Int. J. Immunopathol. Pharmacol. 2011, 24, 735–747. [Google Scholar] [CrossRef]
- Tettamanti, M.; Beretta, S.; Pignataro, G.; Fumagalli, S.; Perego, C.; Sironi, L.; Pedata, F.; Amantea, D.; Bacigalluppi, M. Multi-center Translational Trial of Remote Ischemic Conditioning in Acute Ischemic Stroke (TRICS). Protocol of a multi-center, parallel group, randomized, preclinical trial in female and male rat and mouse from the Italian Stroke Organization (ISO) Basic. Br. Med. J. Open Sci. 2020, 4, e100063. [Google Scholar]
- Petrelli, F.; Muzzi, M.; Chiarugi, A.; Bagetta, G.; Amantea, D. Poly(ADP-ribose) polymerase is not involved in the neuroprotection exerted by azithromycin against ischemic stroke in mice. Eur. J. Pharmacol. 2016, 791, 518–522. [Google Scholar] [CrossRef]
- Certo, M.; Endo, Y.; Ohta, K.; Sakurada, S.; Bagetta, G.; Amantea, D. Activation of RXR/PPARγ underlies neuroprotection by bexarotene in ischemic stroke. Pharmacol. Res. 2015, 102, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Villa, P.; Parrella, S.; Zangari, R.; Zanier, E.R.; Gesuete, R.; Stravalaci, M.; Fumagalli, S.; Ottria, R.; Reina, J.J.; et al. Targeting Mannose-Binding Lectin Confers Long-Lasting Protection With a Surprisingly Wide Therapeutic Window in Cerebral Ischemia. Circulation 2012, 126, 1484–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paxinos, G.; Franklin, K.B.J. Paxinos and Franklin’s The Mouse Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2012; ISBN 9780128161609. [Google Scholar]
- La Russa, D.; Frisina, M.; Secondo, A.; Bagetta, G.; Amantea, D. Modulation of Cerebral Store-operated Calcium Entry-regulatory Factor (SARAF) and Peripheral Orai1 Following Focal Cerebral Ischemia and Preconditioning in Mice. Neuroscience 2020, 441, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Amantea, D.; Certo, M.; Petrelli, F.; Tassorelli, C.; Micieli, G.; Corasaniti, M.T.; Puccetti, P.; Fallarino, F.; Bagetta, G. Azithromycin protects mice against ischemic stroke injury by promoting macrophage transition towards M2 phenotype. Exp. Neurol. 2016, 275, 116–125. [Google Scholar] [CrossRef]
- Greco, R.; Demartini, C.; Zanaboni, A.; Tumelero, E.; Candeloro, E.; Persico, A.; Morotti, A.; Amantea, D.; Tassorelli, C. Characterization of CB2 receptor expression in peripheral blood monocytes of acute ischemic stroke patients. Transl. Stroke Res. 2021, 12, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Demartini, C.; Zanaboni, A.M.; Tumelero, E.; Persico, A.; Candeloro, E.; Morotti, A.; Amantea, D.; Tassorelli, C. CD163 as a potential biomarker of monocyte activation in ischemic stroke patients. Int. J. Mol. Sci. 2021, 22, 6712. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Santo, C.; La Russa, D.; Greco, R.; Persico, A.; Zanaboni, A.M.; Bagetta, G.; Amantea, D. Characterization of the Involvement of Tumour Necrosis Factor (TNF)-α-Stimulated Gene 6 (TSG-6) in Ischemic Brain Injury Caused by Middle Cerebral Artery Occlusion in Mouse. Int. J. Mol. Sci. 2023, 24, 5800. https://doi.org/10.3390/ijms24065800
Di Santo C, La Russa D, Greco R, Persico A, Zanaboni AM, Bagetta G, Amantea D. Characterization of the Involvement of Tumour Necrosis Factor (TNF)-α-Stimulated Gene 6 (TSG-6) in Ischemic Brain Injury Caused by Middle Cerebral Artery Occlusion in Mouse. International Journal of Molecular Sciences. 2023; 24(6):5800. https://doi.org/10.3390/ijms24065800
Chicago/Turabian StyleDi Santo, Chiara, Daniele La Russa, Rosaria Greco, Alessandra Persico, Anna Maria Zanaboni, Giacinto Bagetta, and Diana Amantea. 2023. "Characterization of the Involvement of Tumour Necrosis Factor (TNF)-α-Stimulated Gene 6 (TSG-6) in Ischemic Brain Injury Caused by Middle Cerebral Artery Occlusion in Mouse" International Journal of Molecular Sciences 24, no. 6: 5800. https://doi.org/10.3390/ijms24065800