
Interplay between Signaling Pathways and Tumor Microenvironment Components: A Paradoxical Role in Colorectal Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of the Signaling Pathways in the Carcinogenesis of CRC
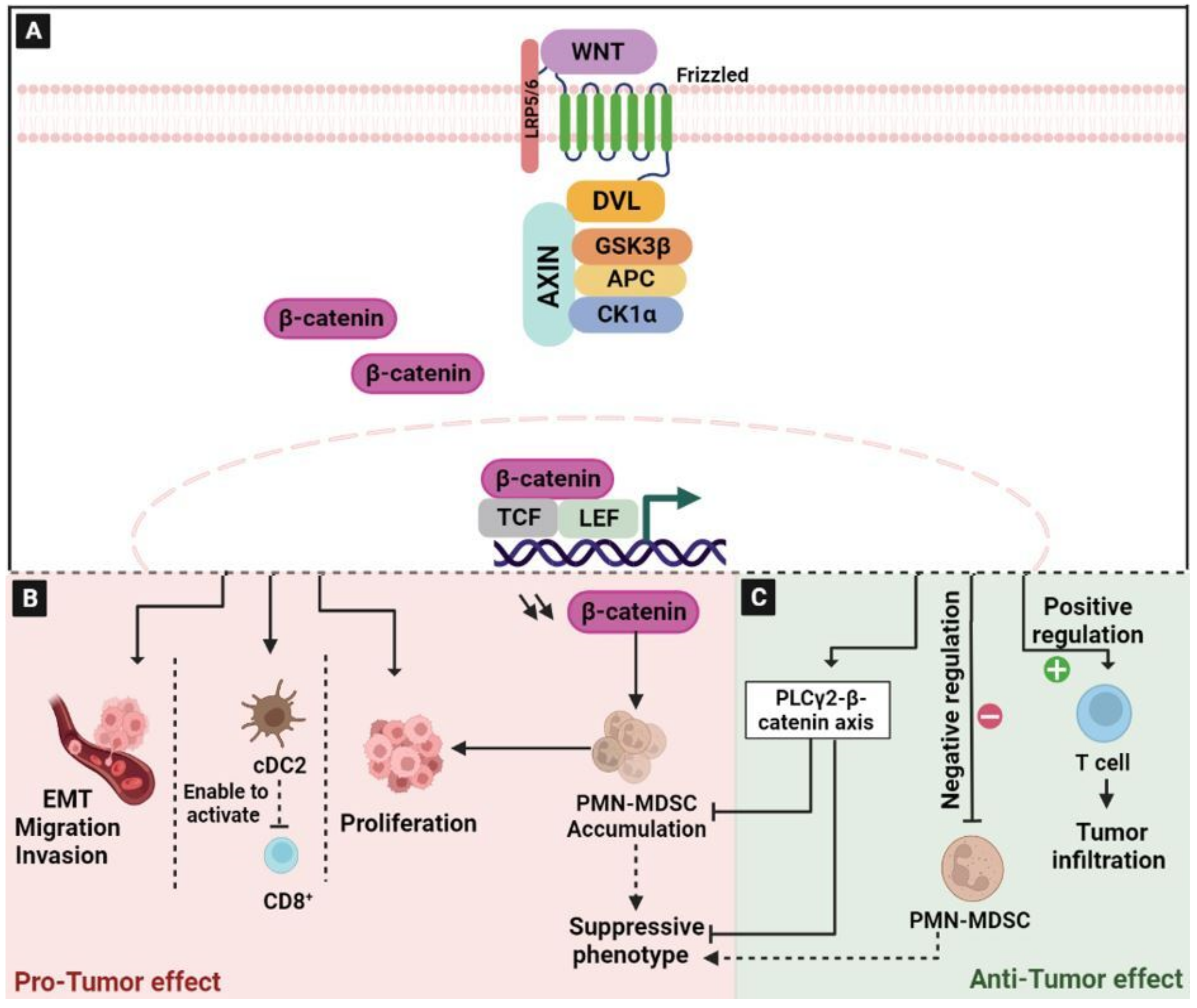
2.1. The Wingless/Integrated (Wnt)/β-Catenin Signaling Pathway
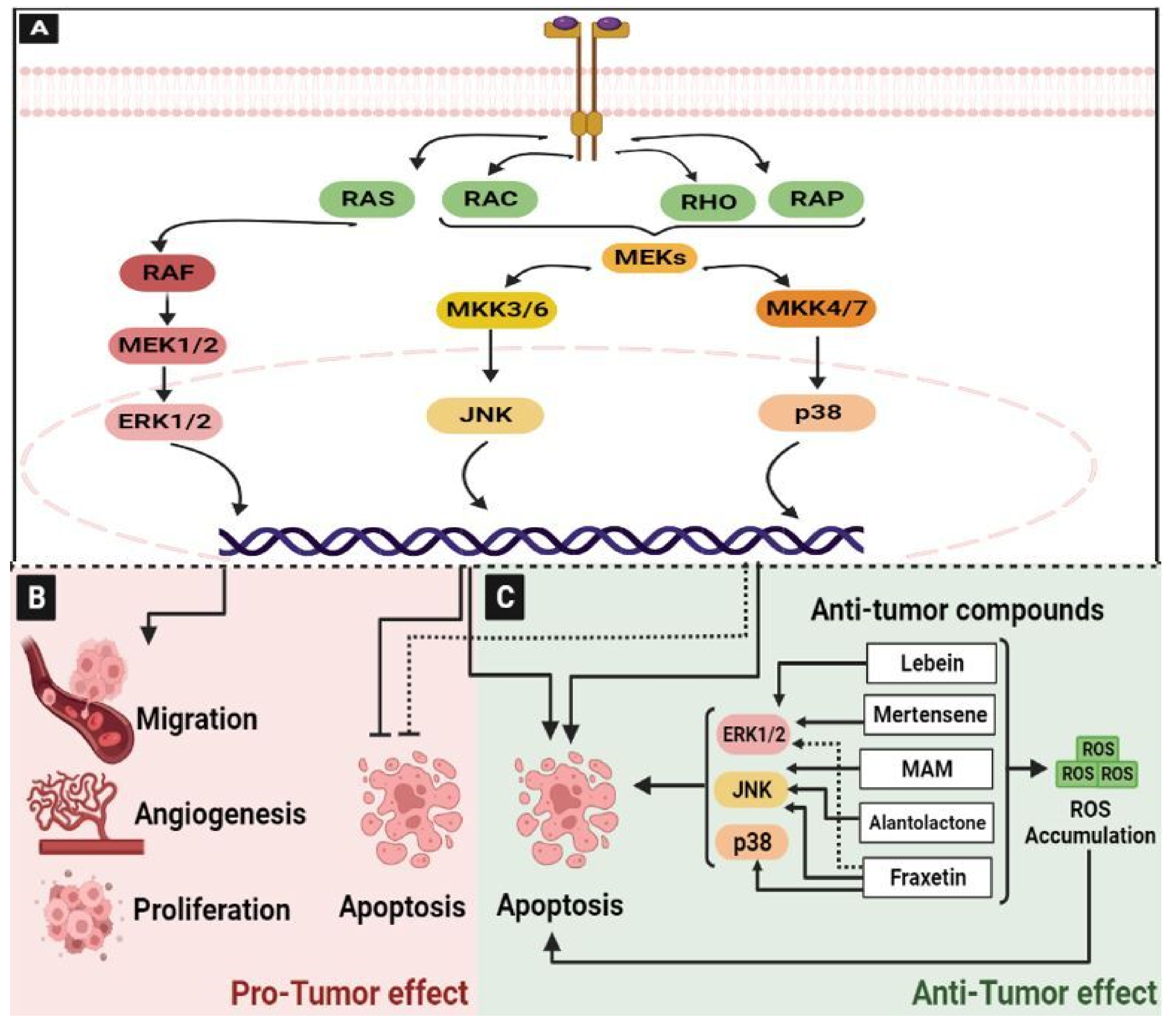
2.2. The Mitogen-Activated Protein Kinase (MAPK) Pathway
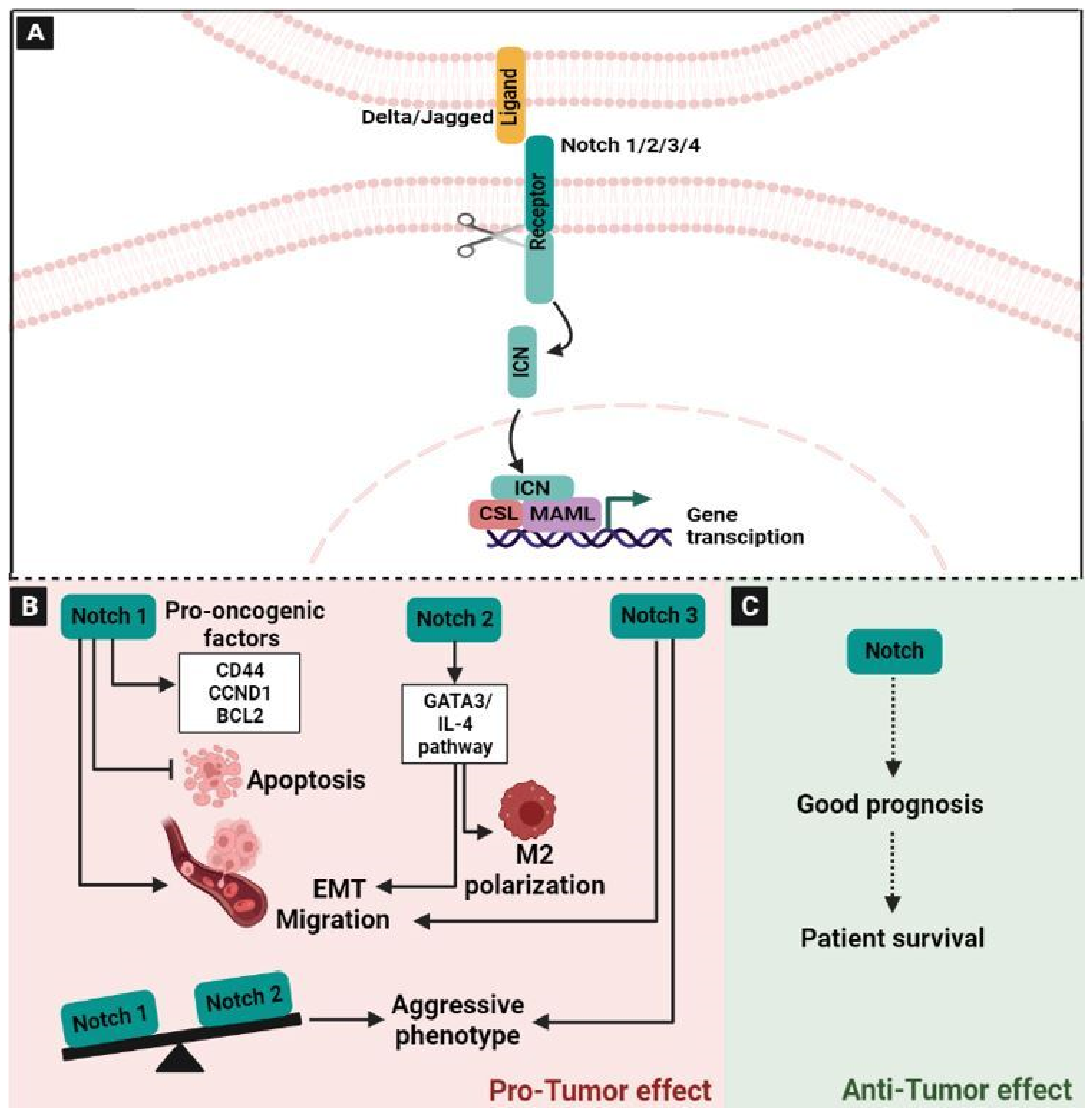
2.3. The Neurogenic Locus Notch Homolog Signaling (Notch) Pathway
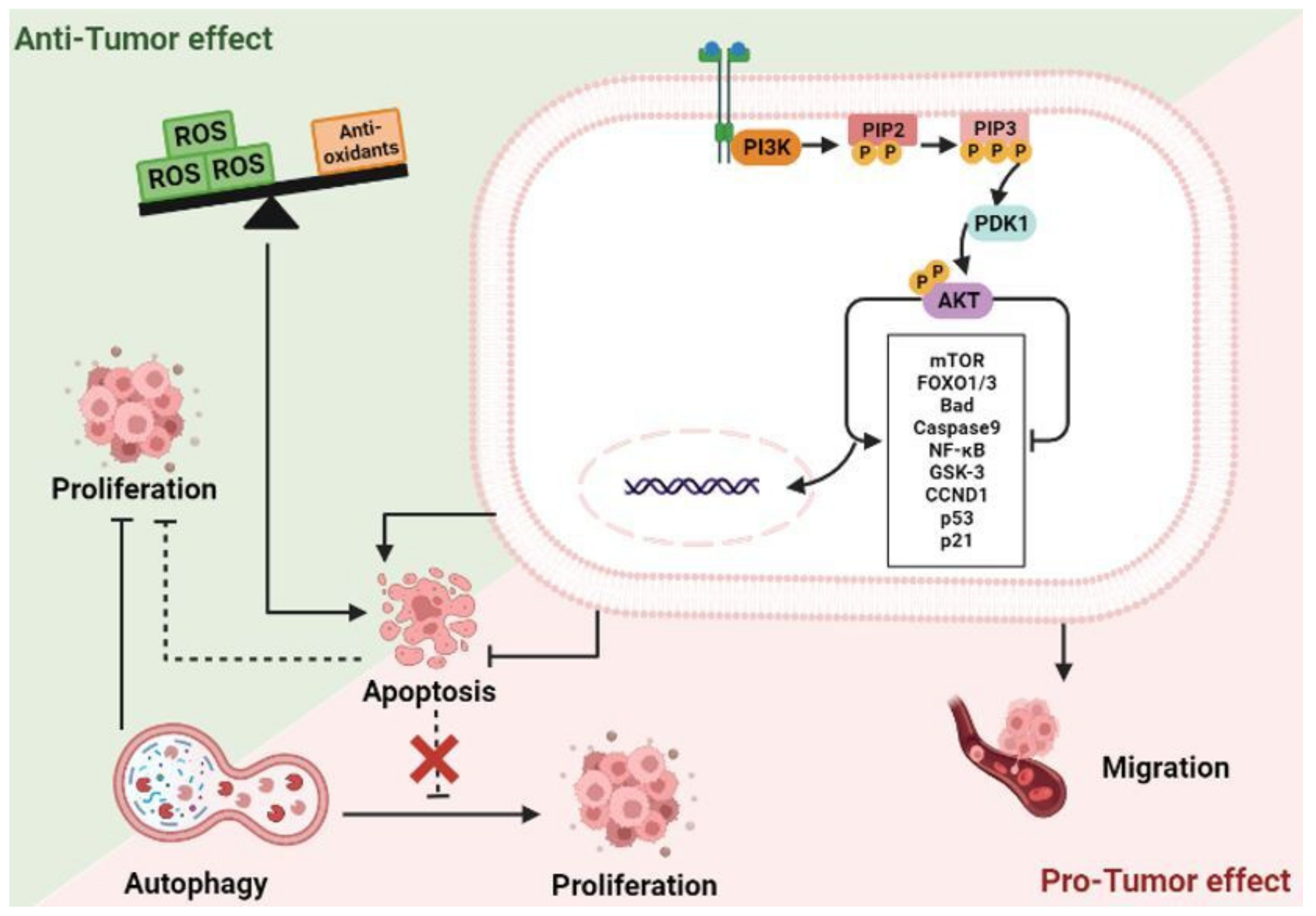
2.4. The Phosphoinositide 3-Kinase (PI3K)/Protein Kinase B (AKT) Signaling Pathway
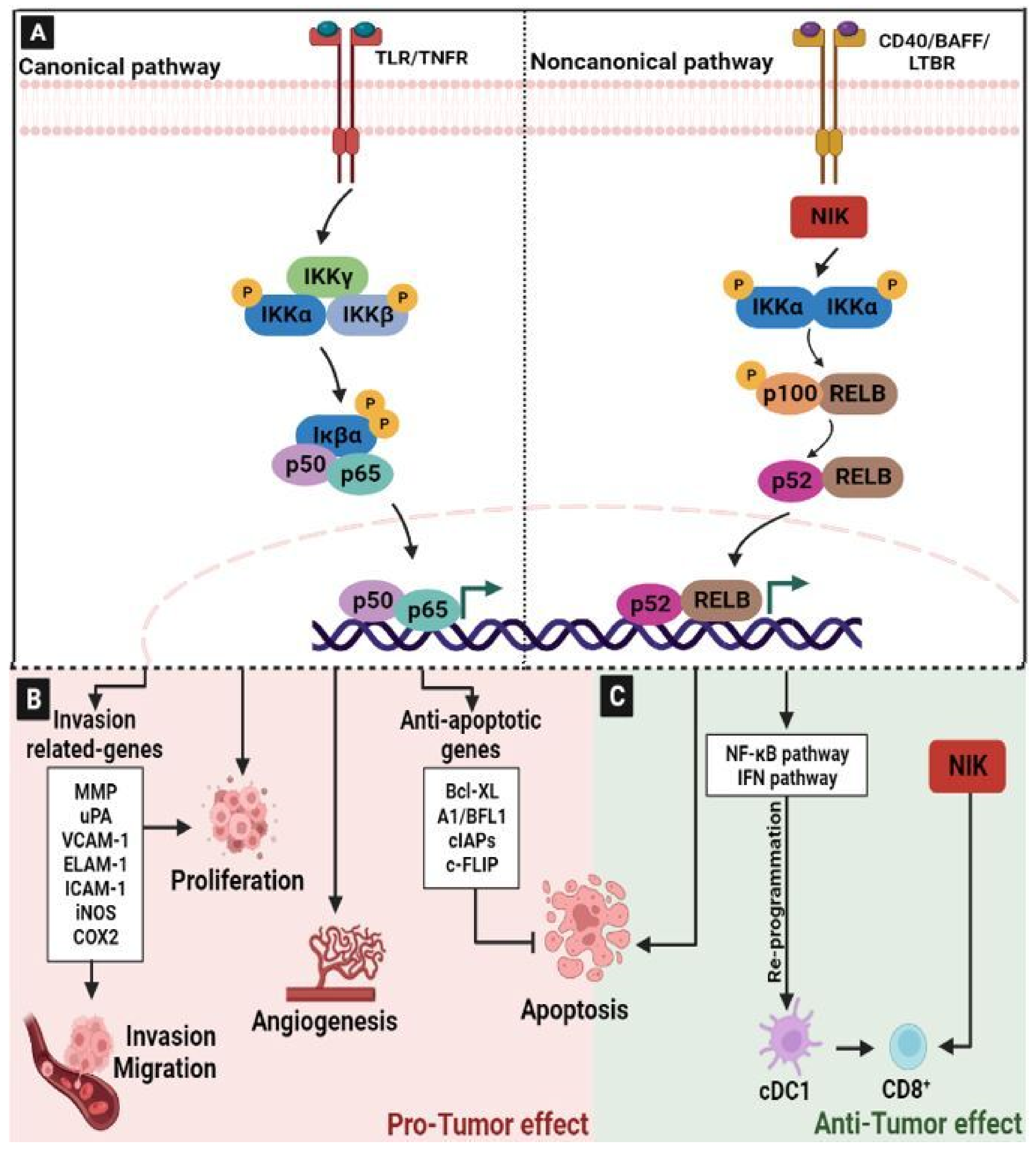
2.5. Nuclear Factor-kappaB (NF-κB) Signaling Pathway
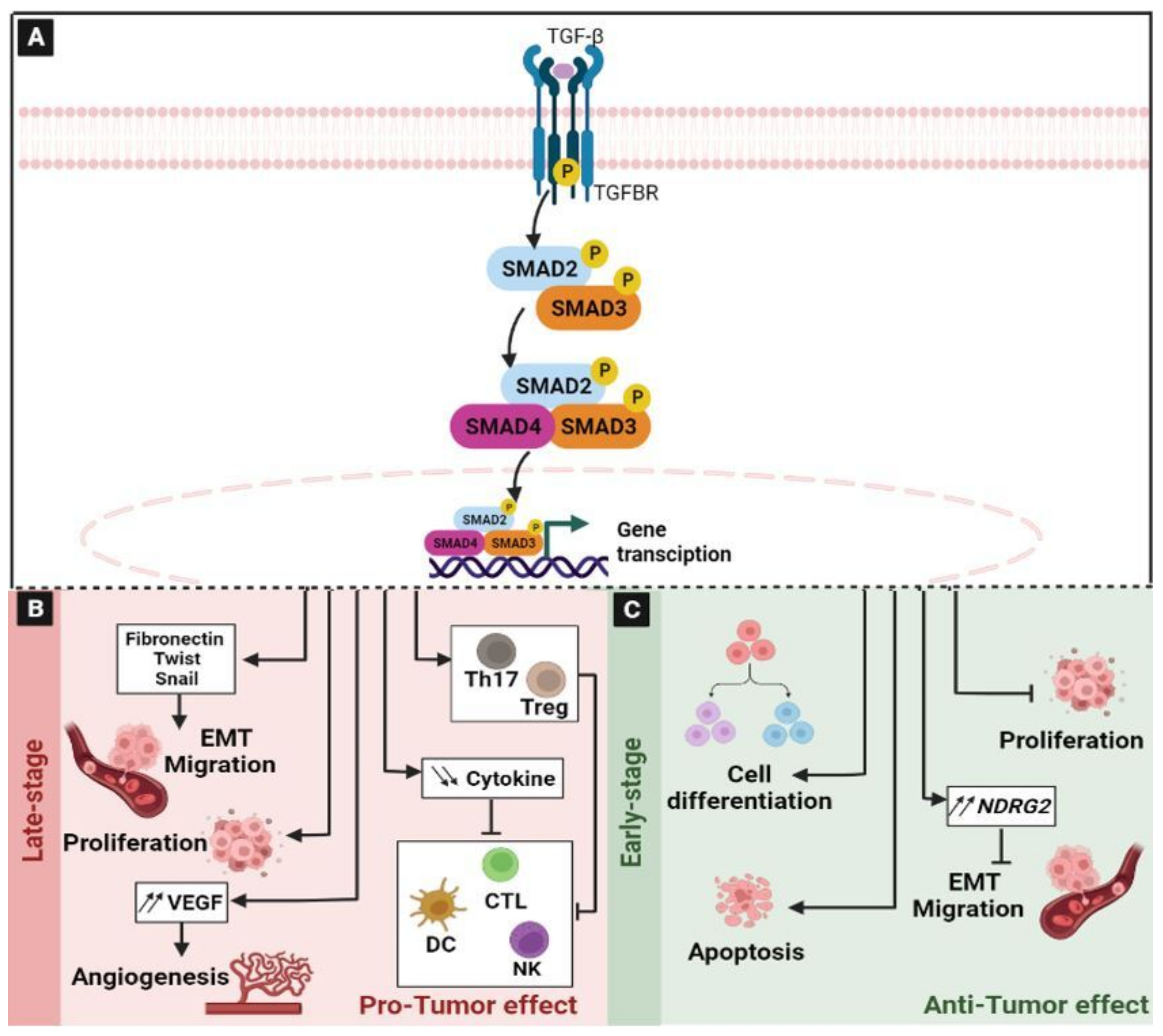
2.6. The Transforming Growth Factor-β (TGF-β) Signaling Pathway
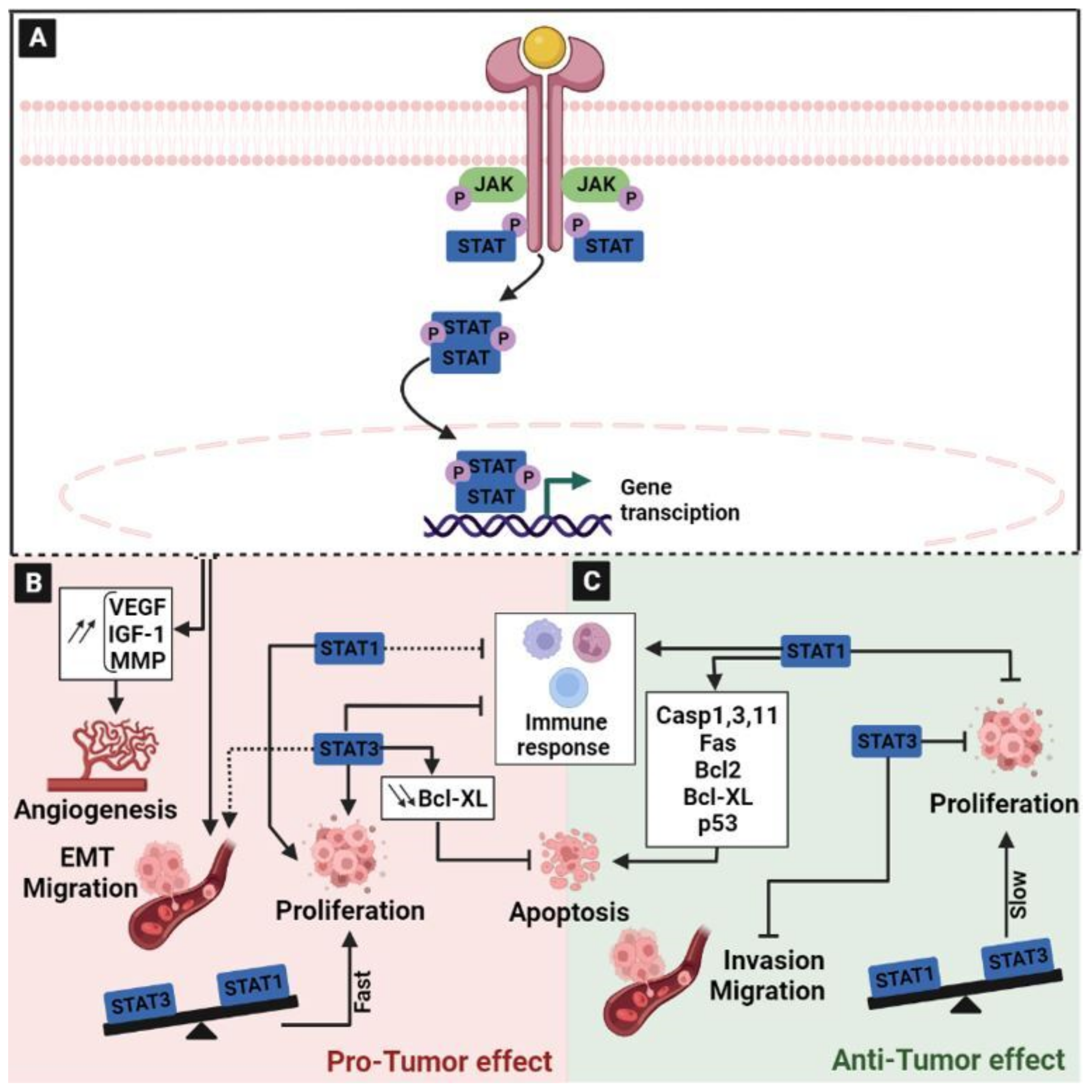
2.7. The Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) Signaling Pathway
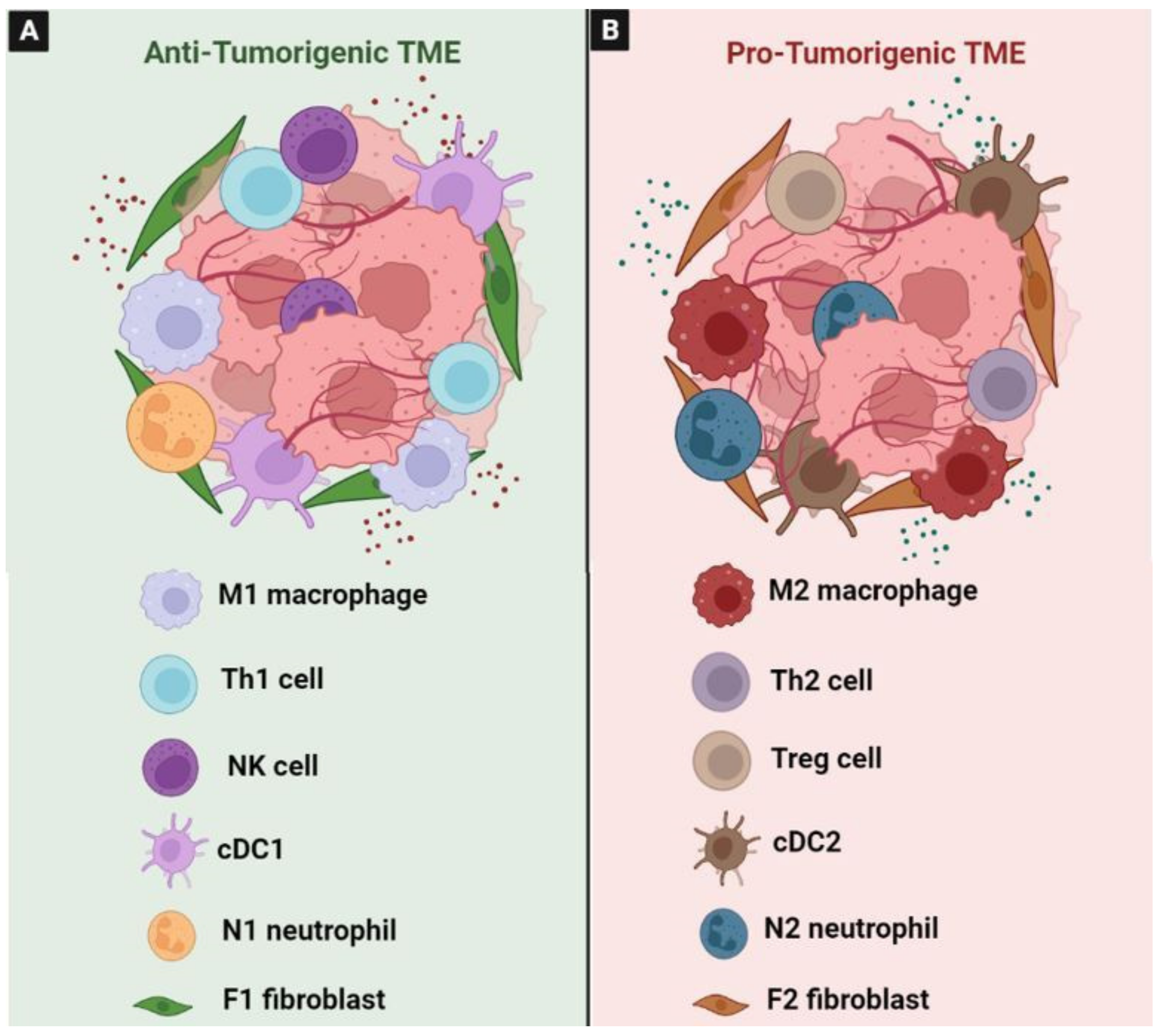
3. The Role of Tumor Microenvironment in the Carcinogenesis of CRC
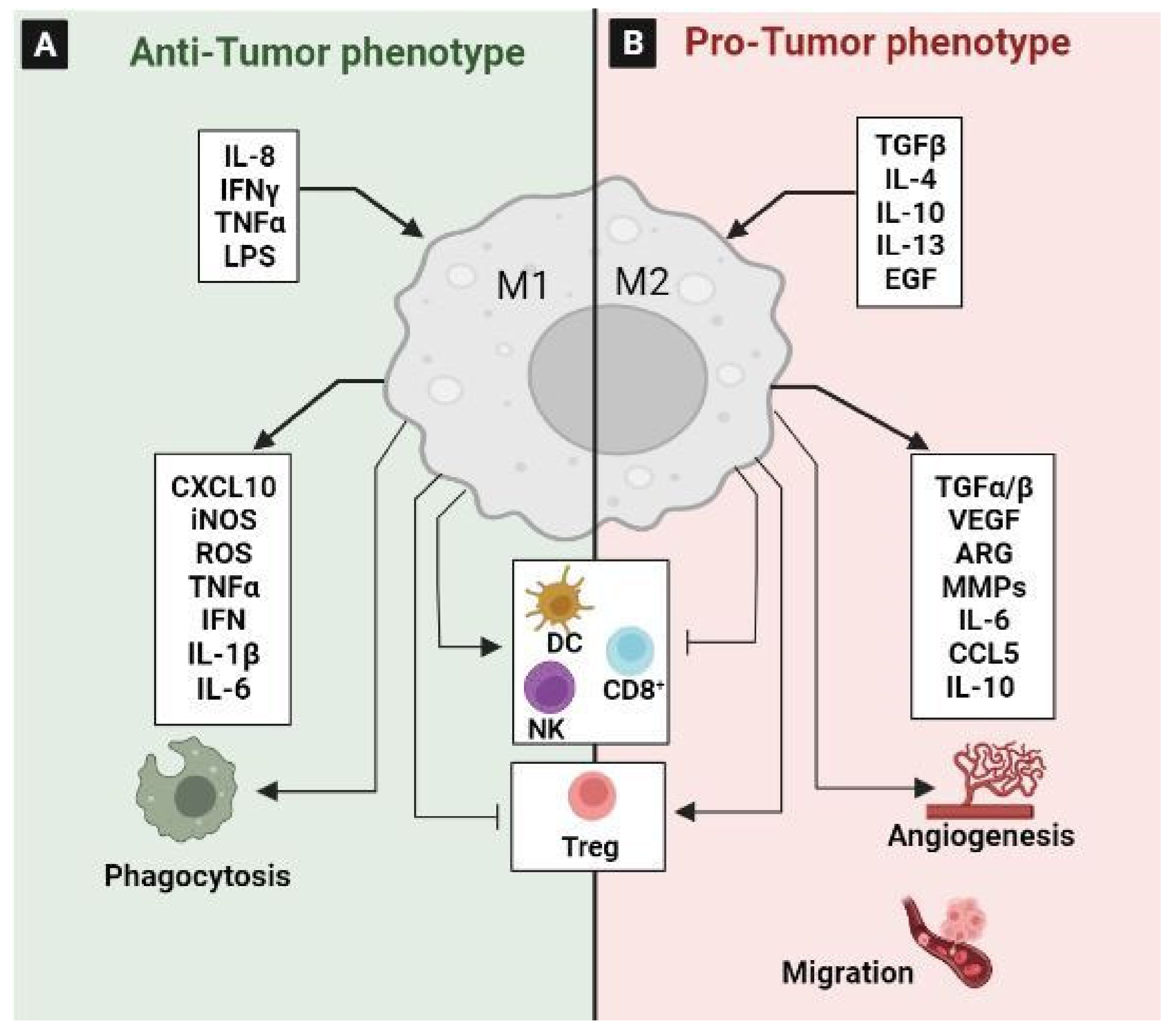
3.1. Tumor-Associated Macrophages (TAM)
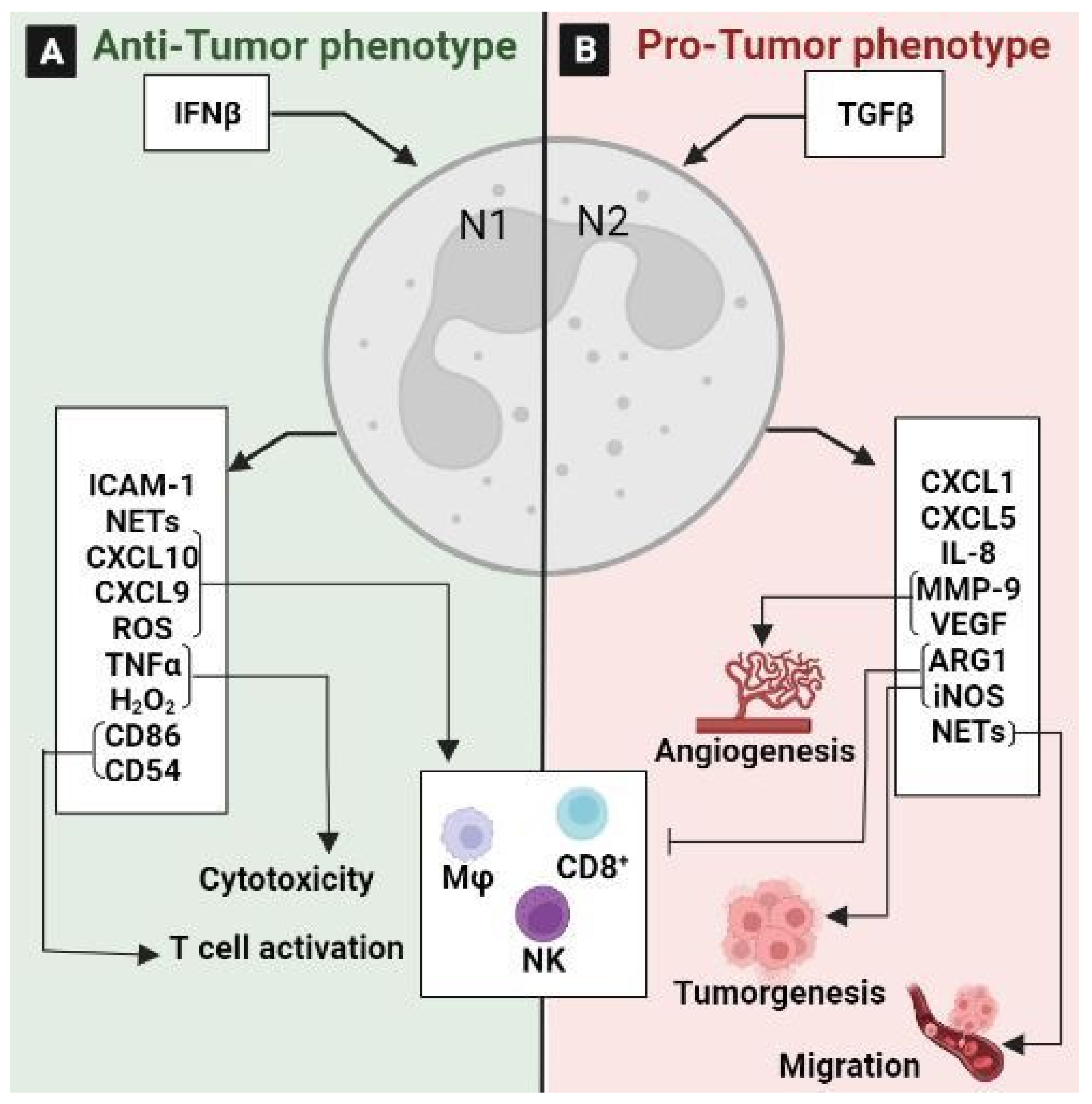
3.2. Tumor-Associated Neutrophils (TANs)
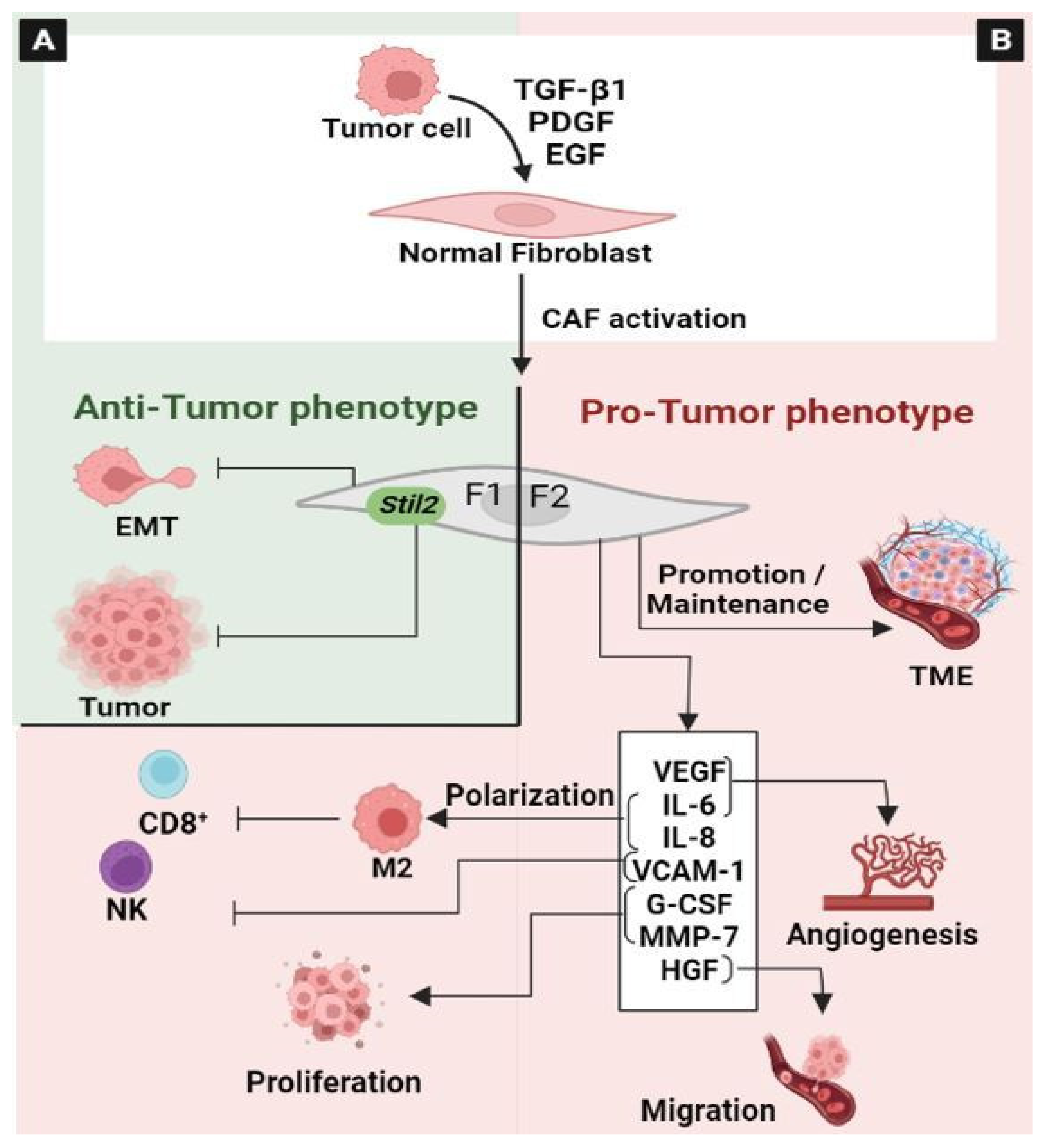
3.3. Cancer-Associated Fibroblasts (CAF)
3.4. Tumor-Infiltrating Lymphocytes (TIL)
3.5. Tumor-Associated Dendritic Cells (TADC)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murphy, N.; Ward, H.A.; Jenab, M.; Rothwell, J.A.; Boutron-Ruault, M.-C.; Carbonnel, F.; Kvaskoff, M.; Kaaks, R.; Kühn, T.; Boeing, H.; et al. Heterogeneity of Colorectal Cancer Risk Factors by Anatomical Subsite in 10 European Countries: A Multinational Cohort Study. Clin. Gastroenterol. Hepatol. 2019, 17, 1323–1331.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Global Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 6 February 2023).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Engstrand, J.; Nilsson, H.; Strömberg, C.; Jonas, E.; Freedman, J. Colorectal Cancer Liver Metastases—A Population-Based Study on Incidence, Management and Survival. BMC Cancer 2018, 18, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tieng, F.Y.F.; Baharudin, R.; Abu, N.; Mohd Yunos, R.-I.; Lee, L.-H.; Ab Mutalib, N.-S. Single Cell Transcriptome in Colorectal Cancer—Current Updates on Its Application in Metastasis, Chemoresistance and the Roles of Circulating Tumor Cells. Front. Pharm. 2020, 11, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riahi-Chebbi, I.; Souid, S.; Othman, H.; Haoues, M.; Karoui, H.; Morel, A.; Srairi-Abid, N.; Essafi, M.; Essafi-Benkhadir, K. The Phenolic Compound Kaempferol Overcomes 5-Fluorouracil Resistance in Human Resistant LS174 Colon Cancer Cells. Sci. Rep. 2019, 9, 195. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Wang, Q.; Lau, W.B.; Lau, B.; Xu, L.; Zhao, L.; Yang, H.; Feng, M.; Xuan, Y.; Yang, Y.; et al. Tumor Microenvironment: The Culprit for Ovarian Cancer Metastasis? Cancer Lett. 2016, 377, 174–182. [Google Scholar] [CrossRef]
- Ma, J.; Huang, L.; Hu, D.; Zeng, S.; Han, Y.; Shen, H. The Role of the Tumor Microbe Microenvironment in the Tumor Immune Microenvironment: Bystander, Activator, or Inhibitor? J. Exp. Clin. Cancer Res. 2021, 40, 327. [Google Scholar] [CrossRef] [PubMed]
- Peddareddigari, V.G.; Wang, D.; DuBois, R.N. The Tumor Microenvironment in Colorectal Carcinogenesis. Cancer Microenviron. 2010, 3, 149–166. [Google Scholar] [CrossRef] [Green Version]
- Virchow, R. Cellular Pathology. As Based upon Physiological and Pathological Histology. Lecture XVI—Atheromatous Affection of Arteries. 1858. Nutr. Rev. 1989, 47, 23–25. [Google Scholar] [CrossRef]
- Wang, H.; Yung, M.M.H.; Ngan, H.Y.S.; Chan, K.K.L.; Chan, D.W. The Impact of the Tumor Microenvironment on Macrophage Polarization in Cancer Metastatic Progression. Int. J. Mol. Sci. 2021, 22, 6560. [Google Scholar] [CrossRef]
- Corsale, A.M.; Di Simone, M.; Lo Presti, E.; Picone, C.; Dieli, F.; Meraviglia, S. Metabolic Changes in Tumor Microenvironment: How Could They Affect Γδ T Cells Functions? Cells 2021, 10, 2896. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Chen, C.-K.; Pan, C.-L. Cell Polarity Control by Wnt Morphogens. Dev. Biol. 2022, 487, 34–41. [Google Scholar] [CrossRef]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer Metabolism: Phenotype, Signaling and Therapeutic Targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ji, Q.; Fan, Z.; Li, Q. Cellular Signaling Pathways Implicated in Metastasis of Colorectal Cancer and the Associated Targeted Agents. Futur. Oncol. 2015, 11, 2911–2922. [Google Scholar] [CrossRef] [PubMed]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.-E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int. J. Mol. Sci. 2020, 22, 130. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [Green Version]
- Shang, S.; Hua, F.; Hu, Z.-W. The Regulation of β-Catenin Activity and Function in Cancer: Therapeutic Opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [Green Version]
- Vilchez, V.; Turcios, L.; Marti, F.; Gedaly, R. Targeting Wnt/β-Catenin Pathway in Hepatocellular Carcinoma Treatment. World J. Gastroenterol. 2016, 22, 823–832. [Google Scholar] [CrossRef]
- Polakis, P. Casein Kinase 1: A Wnt’er of Disconnect. Curr. Biol. 2002, 12, R499–R501. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-Catenin Signaling Pathway in Cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Grumolato, L.; Liu, G.; Haremaki, T.; Mungamuri, S.K.; Mong, P.; Akiri, G.; Lopez-Bergami, P.; Arita, A.; Anouar, Y.; Mlodzik, M.; et al. β-Catenin-Independent Activation of TCF1/LEF1 in Human Hematopoietic Tumor Cells through Interaction with ATF2 Transcription Factors. PLOS Genet. 2013, 9, e1003603. [Google Scholar] [CrossRef] [Green Version]
- Bu, H.; Liu, D.; Cui, J.; Cai, K.; Shen, F. Wnt/β-Catenin Signaling Pathway Is Involved in Induction of Apoptosis by Oridonin in Colon Cancer COLO205 Cells. Transl. Cancer Res. 2019, 8, 1782–1794. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e3. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-C.; Cai, D.-L.; Sun, F.; Wu, Z.-H.; Yue, B.; Zhao, S.-L.; Wu, X.-S.; Zhang, M.; Zhu, X.-W.; Peng, Z.-H.; et al. FERMT1 Mediates Epithelial-Mesenchymal Transition to Promote Colon Cancer Metastasis via Modulation of β-Catenin Transcriptional Activity. Oncogene 2017, 36, 1779–1792. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Tetsu, O.; McCormick, F. Beta-Catenin Regulates Expression of Cyclin D1 in Colon Carcinoma Cells. Nature 1999, 398, 422–426. [Google Scholar] [CrossRef]
- Neth, P.; Ciccarella, M.; Egea, V.; Hoelters, J.; Jochum, M.; Ries, C. Wnt Signaling Regulates the Invasion Capacity of Human Mesenchymal Stem Cells. Stem Cells 2006, 24, 1892–1903. [Google Scholar] [CrossRef]
- Xue, J.; Yu, X.; Xue, L.; Ge, X.; Zhao, W.; Peng, W. Intrinsic β-Catenin Signaling Suppresses CD8+ T-Cell Infiltration in Colorectal Cancer. Biomed. Pharm. 2019, 115, 108921. [Google Scholar] [CrossRef]
- Zhong, Z.A.; Michalski, M.N.; Stevens, P.D.; Sall, E.A.; Williams, B.O. Regulation of Wnt Receptor Activity: Implications for Therapeutic Development in Colon Cancer. J. Biol. Chem. 2021, 296, 100782. [Google Scholar] [CrossRef]
- Famili, F.; Naber, B.A.E.; Vloemans, S.; de Haas, E.F.E.; Tiemessen, M.M.; Staal, F.J.T. Discrete Roles of Canonical and Non-Canonical Wnt Signaling in Hematopoiesis and Lymphopoiesis. Cell Death Dis. 2015, 6, e1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, L.; Mahajan, S.; Capietto, A.-H.; Yang, Z.; Zamani, A.; Ricci, B.; Bumpass, D.B.; Meyer, M.; Su, X.; Wang-Gillam, A.; et al. Dickkopf-Related Protein 1 (Dkk1) Regulates the Accumulation and Function of Myeloid Derived Suppressor Cells in Cancer. J. Exp. Med. 2016, 213, 827–840. [Google Scholar] [CrossRef] [Green Version]
- Poh, T.W.; Bradley, J.M.; Mukherjee, P.; Gendler, S.J. Lack of Muc1-Regulated Beta-Catenin Stability Results in Aberrant Expansion of CD11b+Gr1+ Myeloid Derived Suppressor Cells from the Bone Marrow. Cancer Res. 2009, 69, 3554–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capietto, A.-H.; Kim, S.; Sanford, D.E.; Linehan, D.C.; Hikida, M.; Kumosaki, T.; Novack, D.V.; Faccio, R. Down-Regulation of PLCγ2–β-Catenin Pathway Promotes Activation and Expansion of Myeloid-Derived Suppressor Cells in Cancer. J. Exp. Med. 2013, 210, 2257–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/Beta-Catenin Pathway: Modulating Anticancer Immune Response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, E.D.; Abrams, S.I. Granulocytic Myeloid-Derived Suppressor Cells as Negative Regulators of Anticancer Immunity. Front. Immunol. 2020, 11, 1963. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Asl, E.R.; Amini, M.; Najafi, S.; Mansoori, B.; Mokhtarzadeh, A.; Mohammadi, A.; Lotfinejad, P.; Bagheri, M.; Shirjang, S.; Lotfi, Z.; et al. Interplay between MAPK/ERK Signaling Pathway and MicroRNAs: A Crucial Mechanism Regulating Cancer Cell Metabolism and Tumor Progression. Life Sci. 2021, 278, 119499. [Google Scholar] [CrossRef]
- Ros, J.; Baraibar, I.; Sardo, E.; Mulet, N.; Salvà, F.; Argilés, G.; Martini, G.; Ciardiello, D.; Cuadra, J.L.; Tabernero, J.; et al. BRAF, MEK and EGFR Inhibition as Treatment Strategies in BRAF V600E Metastatic Colorectal Cancer. Ther. Adv. Med. Oncol. 2021, 13, 1758835921992974. [Google Scholar] [CrossRef]
- Yang, W.; Redpath, R.E.; Zhang, C.; Ning, N. Long Non-Coding RNA H19 Promotes the Migration and Invasion of Colon Cancer Cells via MAPK Signaling Pathway. Oncol. Lett. 2018, 16, 3365–3372. [Google Scholar] [CrossRef]
- Grossi, V.; Peserico, A.; Tezil, T.; Simone, C. P38α MAPK Pathway: A Key Factor in Colorectal Cancer Therapy and Chemoresistance. World J. Gastroenterol. 2014, 20, 9744–9758. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ung, T.T.; Nguyen, T.T.; Sah, D.K.; Park, S.Y.; Jung, Y.D. Cholic Acid Stimulates MMP-9 in Human Colon Cancer Cells via Activation of MAPK, AP-1, and NF-ΚB Activity. Int. J. Mol. Sci. 2020, 21, 3420. [Google Scholar] [CrossRef]
- Zakraoui, O.; Marcinkiewicz, C.; Aloui, Z.; Othman, H.; Grépin, R.; Haoues, M.; Essafi, M.; Srairi-Abid, N.; Gasmi, A.; Karoui, H.; et al. Lebein, a Snake Venom Disintegrin, Suppresses Human Colon Cancer Cells Proliferation and Tumor-Induced Angiogenesis through Cell Cycle Arrest, Apoptosis Induction and Inhibition of VEGF Expression: Mechanisms and Targets for Lebein in Colorectal Cancer. Mol. Carcinog. 2017, 56, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Tarhouni-Jabberi, S.; Zakraoui, O.; Ioannou, E.; Riahi-Chebbi, I.; Haoues, M.; Roussis, V.; Kharrat, R.; Essafi-Benkhadir, K. Mertensene, a Halogenated Monoterpene, Induces G2/M Cell Cycle Arrest and Caspase Dependent Apoptosis of Human Colon Adenocarcinoma HT29 Cell Line through the Modulation of ERK-1/-2, AKT and NF-ΚB Signaling. Mar. Drugs 2017, 15, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Yang, C.; Park, S.; Song, G.; Lim, W. Fraxetin Induces Cell Death in Colon Cancer Cells via Mitochondria Dysfunction and Enhances Therapeutic Effects in 5-Fluorouracil Resistant Cells. J. Cell. Biochem. 2022, 123, 469–480. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-Signaling: A Multiplexing Hub in Programmed Cell Death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Wu, X.; Gao, H.; Yu, J.; Zhao, W.; Lu, J.-J.; Wang, J.; Du, G.; Chen, X. Cytosolic Calcium Mediates RIP1/RIP3 Complex-Dependent Necroptosis through JNK Activation and Mitochondrial ROS Production in Human Colon Cancer Cells. Free Radic. Biol. Med. 2017, 108, 433–444. [Google Scholar] [CrossRef]
- Ren, Y.; Lv, C.; Zhang, J.; Zhang, B.; Yue, B.; Luo, X.; Yu, Z.; Wang, H.; Ren, J.; Wang, Z.; et al. Alantolactone Exhibits Antiproliferative and Apoptosis-Promoting Properties in Colon Cancer Model via Activation of the MAPK-JNK/c-Jun Signaling Pathway. Mol. Cell. Biochem. 2021, 476, 4387–4403. [Google Scholar] [CrossRef]
- Song, N.; Ma, J.; Hu, W.; Guo, Y.; Hui, L.; Aamer, M.; Ma, J. Lappaconitine Hydrochloride Inhibits Proliferation and Induces Apoptosis in Human Colon Cancer HCT-116 Cells via Mitochondrial and MAPK Pathway. Acta Histochem. 2021, 123, 151736. [Google Scholar] [CrossRef]
- Bray, S.J. Notch Signalling: A Simple Pathway Becomes Complex. Nat. Rev. Mol. Cell. Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- Mumm, J.S.; Kopan, R. Notch Signaling: From the outside in. Dev. Biol. 2000, 228, 151–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntziachristos, P.; Lim, J.S.; Sage, J.; Aifantis, I. From Fly Wings to Targeted Cancer Therapies: A Centennial for Notch Signaling. Cancer Cell 2014, 25, 318–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch Signaling: Simplicity in Design, Versatility in Function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, C.S.; Radtke, F. Notch as a Tumour Suppressor. Nat. Rev. Cancer 2017, 17, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Zhao, L.; Wang, M.; Zhang, R.; Cheng, L.; Qiu, L.; Tong, X.; Cai, S.; Wei, Q.; Li, Q. Novel Functional Variants in the Notch Pathway and Survival of Chinese Colorectal Cancer. Int. J. Cancer 2021, 149, 84–96. [Google Scholar] [CrossRef]
- Wang, F.; Long, J.; Li, L.; Zhao, Z.; Wei, F.; Yao, Y.; Qiu, W.; Wu, Z.; Luo, Q.; Liu, W.; et al. Mutations in the Notch Signalling Pathway Are Associated with Enhanced Anti-tumour Immunity in Colorectal Cancer. J. Cell. Mol. Med. 2020, 24, 12176–12187. [Google Scholar] [CrossRef]
- Vinson, K.E.; George, D.C.; Fender, A.W.; Bertrand, F.E.; Sigounas, G. The Notch Pathway in Colorectal Cancer. Int. J. Cancer 2016, 138, 1835–1842. [Google Scholar] [CrossRef]
- Sonoshita, M.; Aoki, M.; Fuwa, H.; Aoki, K.; Hosogi, H.; Sakai, Y.; Hashida, H.; Takabayashi, A.; Sasaki, M.; Robine, S.; et al. Suppression of Colon Cancer Metastasis by Aes through Inhibition of Notch Signaling. Cancer Cell 2011, 19, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Rodilla, V.; Villanueva, A.; Obrador-Hevia, A.; Robert-Moreno, A.; Fernández-Majada, V.; Grilli, A.; López-Bigas, N.; Bellora, N.; Albà, M.M.; Torres, F.; et al. Jagged1 Is the Pathological Link between Wnt and Notch Pathways in Colorectal Cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 6315–6320. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, B.; Ji, Z.-Z.; Zheng, P.-S. Notch1 Regulates the Growth of Human Colon Cancers. Cancer 2010, 116, 5207–5218. [Google Scholar] [CrossRef]
- Tyagi, A.; Sharma, A.K.; Damodaran, C. A Review on Notch Signaling and Colorectal Cancer. Cells 2020, 9, 1549. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.; Zhang, Z.; Zhou, Y.; Wang, W.; Li, Y.; Zhang, H.; Dong, G.; Zhao, Q.; Ji, G. Notch1 and Notch2 Have Opposite Prognostic Effects on Patients with Colorectal Cancer. Ann. Oncol. 2011, 22, 2440–2447. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wang, S.; Sun, M.; Zhang, C.; Wei, C.; Yang, C.; Dou, R.; Liu, Q.; Xiong, B. MiR-195-5p/NOTCH2-Mediated EMT Modulates IL-4 Secretion in Colorectal Cancer to Affect M2-like TAM Polarization. J. Hematol. Oncol. 2019, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Wang, M.; Hu, H.; Huang, Q.; Chen, Y.; Wang, G. Overcoming Stemness and Chemoresistance in Colorectal Cancer through MiR-195-5p-Modulated Inhibition of Notch Signaling. Int. J. Biol. Macromol. 2018, 117, 445–453. [Google Scholar] [CrossRef]
- Serafin, V.; Persano, L.; Moserle, L.; Esposito, G.; Ghisi, M.; Curtarello, M.; Bonanno, L.; Masiero, M.; Ribatti, D.; Stürzl, M.; et al. Notch3 Signalling Promotes Tumour Growth in Colorectal Cancer. J. Pathol. 2011, 224, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Tewari, S.; Atamna, W.; Lazarova, D.L. The Notch Ligand Delta-like 1 Integrates Inputs from TGFbeta/Activin and Wnt Pathways. Exp. Cell Res. 2011, 317, 1368–1381. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Mirandola, L.; Chiriva-Internati, M.; Basile, A.; Locati, M.; Lesma, E.; Chiaramonte, R.; Platonova, N. Cancer Cells Exploit Notch Signaling to Redefine a Supportive Cytokine Milieu. Front. Immunol. 2018, 9, 1823. [Google Scholar] [CrossRef]
- Koch, U.; Radtke, F. Notch and Cancer: A Double-Edged Sword. Cell. Mol. Life Sci. 2007, 64, 2746–2762. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in Oncology: The Path Forward. Nat. Rev. Drug Discov. 2021, 20, 125–144. [Google Scholar] [CrossRef]
- Ranganathan, P.; Weaver, K.L.; Capobianco, A.J. Notch Signalling in Solid Tumours: A Little Bit of Everything but Not All the Time. Nat. Rev. Cancer 2011, 11, 338–351. [Google Scholar] [CrossRef]
- Jin, H.-Y.; Zhang, H.-Y.; Wang, X.; Xu, J.; Ding, Y. Expression and Clinical Significance of Notch Signaling Genes in Colorectal Cancer. Tumour Biol. 2012, 33, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yu, J.; Gan, J.; Song, N.; Shi, L.; Liu, J.; Zhang, Z.; Du, J. Notch1/2/3/4 Are Prognostic Biomarker and Correlated with Immune Infiltrates in Gastric Cancer. Aging 2020, 12, 2595–2609. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Li, G.; Lu, J.; Li, L. Crosstalk between CircRNAs and the PI3K/AKT Signaling Pathway in Cancer Progression. Signal Transduct. Target. Ther. 2021, 6, 400. [Google Scholar] [CrossRef]
- Narayanankutty, A. PI3K/ Akt/ MTOR Pathway as a Therapeutic Target for Colorectal Cancer: A Review of Preclinical and Clinical Evidence. Curr. Drug Targets 2019, 20, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Prossomariti, A.; Piazzi, G.; Alquati, C.; Ricciardiello, L. Are Wnt/β-Catenin and PI3K/AKT/MTORC1 Distinct Pathways in Colorectal Cancer? Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 491–506. [Google Scholar] [CrossRef]
- Nogueira, V.; Park, Y.; Chen, C.-C.; Xu, P.-Z.; Chen, M.-L.; Tonic, I.; Unterman, T.; Hay, N. Akt Determines Replicative Senescence and Oxidative or Oncogenic Premature Senescence and Sensitizes Cells to Oxidative Apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Dolado, I.; Nebreda, A.R. AKT and Oxidative Stress Team up to Kill Cancer Cells. Cancer Cell 2008, 14, 427–429. [Google Scholar] [CrossRef] [Green Version]
- Los, M.; Maddika, S.; Erb, B.; Schulze-Osthoff, K. Switching Akt: From Survival Signaling to Deadly Response. Bioessays 2009, 31, 492–495. [Google Scholar] [CrossRef] [Green Version]
- Rangel, M.; Kong, J.; Bhatt, V.; Khayati, K.; Guo, J.Y. Autophagy and Tumorigenesis. FEBS J. 2022, 289, 7177–7198. [Google Scholar] [CrossRef]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/MTOR-Mediated Autophagy for Tumor Therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef]
- Chen, T.; Yu, Q.; Xin, L.; Guo, L. Retracted: Circular RNA CircC3P1 Restrains Kidney Cancer Cell Activity by Regulating MiR-21/PTEN Axis and Inactivating PI3K/AKT and NF- k B Pathways. J. Cell. Physiol. 2020, 235, 4001–4010. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.-W. NF-ΚB in Cancer: From Innocent Bystander to Major Culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Karin, M. The Two NF-ΚB Activation Pathways and Their Role in Innate and Adaptive Immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Inducibility of κ Immunoglobulin Enhancer-Binding Protein NF-ΚB by a Posttranslational Mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Hassanzadeh, P. Colorectal Cancer and NF-ΚB Signaling Pathway. Gastroenterol. Hepatol. Bed Bench 2011, 4, 127–132. [Google Scholar]
- Soleimani, A.; Rahmani, F.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of the NF-ΚB Signaling Pathway in the Pathogenesis of Colorectal Cancer. Gene 2020, 726, 144132. [Google Scholar] [CrossRef]
- Quinn, J.A.; Bennett, L.; Patel, M.; Frixou, M.; Park, J.H.; Roseweir, A.; Horgan, P.G.; McMillan, D.C.; Edwards, J. The Relationship between Members of the Canonical NF-KB Pathway, Tumour Microenvironment and Cancer Specific Survival in Colorectal Cancer Patients. Histol. Histopathol. 2020, 35, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Hong, J. Roles of NF-ΚB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lin, A. NF-KappaB at the Crossroads of Life and Death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef]
- Liu, T.; Liu, D.; Liu, J.; Song, J.-T.; Gao, S.-L.; Li, H.; Hu, L.-H.; Liu, B.-R. Effect of NF-ΚB Inhibitors on the Chemotherapy-Induced Apoptosis of the Colon Cancer Cell Line HT-29. Exp. Ther. Med. 2012, 4, 716–722. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Wang, G.; Chen, W.; Zhu, Z.; Liu, Y.; Huang, Z.; Huang, Y.; Du, P.; Yang, Y.; Liu, C.-Y.; et al. Co-Inhibition of BET Proteins and NF-ΚB as a Potential Therapy for Colorectal Cancer through Synergistic Inhibiting MYC and FOXM1 Expressions. Cell Death Dis. 2018, 9, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghislat, G.; Cheema, A.S.; Baudoin, E.; Verthuy, C.; Ballester, P.J.; Crozat, K.; Attaf, N.; Dong, C.; Milpied, P.; Malissen, B.; et al. NF-ΚB-Dependent IRF1 Activation Programs CDC1 Dendritic Cells to Drive Antitumor Immunity. Sci. Immunol. 2021, 6, eabg3570. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Zhou, X.; Sohn, J.H.; Zhu, L.; Jie, Z.; Yang, J.-Y.; Zheng, X.; Xie, X.; Yang, J.; Shi, Y.; et al. NF-ΚB Inducing Kinase Maintains T Cell Metabolic Fitness in Antitumor Immunity. Nat. Immunol. 2021, 22, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. The Non-Canonical NF-ΚB Pathway in Immunity and Inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Zhou, X.; Xie, X.; Chen, X.; Jie, Z.; Zou, Q.; Hu, H.; Zhu, L.; Cheng, X.; et al. Cell Intrinsic Role of NF-ΚB-Inducing Kinase in Regulating T Cell-Mediated Immune and Autoimmune Responses. Sci. Rep. 2016, 6, 22115. [Google Scholar] [CrossRef] [Green Version]
- Haque, S.; Morris, J.C. Transforming Growth Factor-β: A Therapeutic Target for Cancer. Hum. Vaccin. Immunother. 2017, 13, 1741–1750. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-Beta Signaling in Tumor Suppression and Cancer Progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef]
- Li, X.; Wu, Y.; Tian, T. TGF-β Signaling in Metastatic Colorectal Cancer (MCRC): From Underlying Mechanism to Potential Applications in Clinical Development. Int. J. Mol. Sci. 2022, 23, 14436. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.; Feng, X.-H. TGF-β Signaling in Cancer. Acta Biochim. Biophys. Sin. 2018, 50, 941–949. [Google Scholar] [CrossRef] [Green Version]
- Nakao, A.; Afrakhte, M.; Morén, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-Inducible Antagonist of TGF-Beta Signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ Signalling Pathway in Disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, N.R.; Xiang, X.; Mishra, L. TGF-β Signaling in Liver, Pancreas, and Gastrointestinal Diseases and Cancer. Gastroenterology 2021, 161, 434–452.e15. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-Z.; Li, Y.; Shao, Y.; Zeng, Y.-H.; Ren, W.-Y.; Liu, R.-X.; Zhou, L.-Y.; Hu, X.-L.; Huang, M.; He, F.; et al. TGF-Β1/PTEN/PI3K Signaling Plays a Critical Role in the Anti-Proliferation Effect of Tetrandrine in Human Colon Cancer Cells. Int. J. Oncol. 2017, 50, 1011–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iavarone, A.; Massagué, J. Repression of the CDK Activator Cdc25A and Cell-Cycle Arrest by Cytokine TGF-Beta in Cells Lacking the CDK Inhibitor P15. Nature 1997, 387, 417–422. [Google Scholar] [CrossRef]
- Lasorella, A.; Noseda, M.; Beyna, M.; Yokota, Y.; Iavarone, A. Id2 Is a Retinoblastoma Protein Target and Mediates Signalling by Myc Oncoproteins. Nature 2000, 407, 592–598. [Google Scholar] [CrossRef]
- Shen, L.; Qu, X.; Ma, Y.; Zheng, J.; Chu, D.; Liu, B.; Li, X.; Wang, M.; Xu, C.; Liu, N.; et al. Tumor Suppressor NDRG2 Tips the Balance of Oncogenic TGF-β via EMT Inhibition in Colorectal Cancer. Oncogenesis 2014, 3, e86. [Google Scholar] [CrossRef] [Green Version]
- Mishra, L.; Banker, T.; Murray, J.; Byers, S.; Thenappan, A.; He, A.R.; Shetty, K.; Johnson, L.; Reddy, E.P. Liver Stem Cells and Hepatocellular Carcinoma. Hepatology 2009, 49, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Villalba, M.; Evans, S.R.; Vidal-Vanaclocha, F.; Calvo, A. Role of TGF-β in Metastatic Colon Cancer: It Is Finally Time for Targeted Therapy. Cell Tissue Res. 2017, 370, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.L.; Blobe, G.C. Role of Transforming Growth Factor Beta in Human Cancer. J. Clin. Oncol. 2005, 23, 2078–2093. [Google Scholar] [CrossRef]
- De Caestecker, M.P.; Piek, E.; Roberts, A.B. Role of Transforming Growth Factor-Beta Signaling in Cancer. J. Natl. Cancer Inst. 2000, 92, 1388–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancino, M.; Strizzi, L.; Wechselberger, C.; Watanabe, K.; Gonzales, M.; Hamada, S.; Normanno, N.; Salomon, D.S.; Bianco, C. Regulation of Human Cripto-1 Gene Expression by TGF-Beta1 and BMP-4 in Embryonal and Colon Cancer Cells. J. Cell. Physiol. 2008, 215, 192–203. [Google Scholar] [CrossRef]
- Bianco, C.; Strizzi, L.; Normanno, N.; Khan, N.; Salomon, D.S. Cripto-1: An Oncofetal Gene with Many Faces. Curr. Top. Dev. Biol. 2005, 67, 85–133. [Google Scholar] [CrossRef]
- Langenskiöld, M.; Holmdahl, L.; Falk, P.; Angenete, E.; Ivarsson, M.-L. Increased TGF-Beta 1 Protein Expression in Patients with Advanced Colorectal Cancer. J. Surg. Oncol. 2008, 97, 409–415. [Google Scholar] [CrossRef]
- Pertovaara, L.; Kaipainen, A.; Mustonen, T.; Orpana, A.; Ferrara, N.; Saksela, O.; Alitalo, K. Vascular Endothelial Growth Factor Is Induced in Response to Transforming Growth Factor-Beta in Fibroblastic and Epithelial Cells. J. Biol. Chem. 1994, 269, 6271–6274. [Google Scholar] [CrossRef] [PubMed]
- Schwarte-Waldhoff, I.; Volpert, O.V.; Bouck, N.P.; Sipos, B.; Hahn, S.A.; Klein-Scory, S.; Lüttges, J.; Klöppel, G.; Graeven, U.; Eilert-Micus, C.; et al. Smad4/DPC4-Mediated Tumor Suppression through Suppression of Angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 9624–9629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Lee, J.; Revelo, M.; Wang, X.; Lu, S.; Dong, Z. Smad3 Is Overexpressed in Advanced Human Prostate Cancer and Necessary for Progressive Growth of Prostate Cancer Cells in Nude Mice. Clin. Cancer Res. 2007, 13, 5692–5702. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFβ in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [Green Version]
- McGeachy, M.J.; Bak-Jensen, K.S.; Chen, Y.; Tato, C.M.; Blumenschein, W.; McClanahan, T.; Cua, D.J. TGF-Beta and IL-6 Drive the Production of IL-17 and IL-10 by T Cells and Restrain T(H)-17 Cell-Mediated Pathology. Nat. Immunol. 2007, 8, 1390–1397. [Google Scholar] [CrossRef]
- Perez, L.G.; Kempski, J.; McGee, M.H.; Pelzcar, P.; Agalioti, T.; Giannou, A.; Konczalla, L.; Brockmann, L.; Wahib, R.; Xu, H.; et al. TGF-β Signaling in Th17 Cells Promotes IL-22 Production and Colitis-Associated Colon Cancer. Nat. Commun. 2020, 11, 2608. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Ménard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ Regulatory T Cells Inhibit Natural Killer Cell Functions in a Transforming Growth Factor–β–Dependent Manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.Y.; Gong, Y.; Chen, C.-M.; Zheng, Q.-D.; Chen, J.-J. Tumor-Derived TGF-Beta Reduces the Efficacy of Dendritic Cell/Tumor Fusion Vaccine. J. Immunol. 2003, 170, 3806–3811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulé, J.J.; Schwarz, S.L.; Roberts, A.B.; Sporn, M.B.; Rosenberg, S.A. Transforming Growth Factor-Beta Inhibits the in Vitro Generation of Lymphokine-Activated Killer Cells and Cytotoxic T Cells. Cancer Immunol. Immunother. 1988, 26, 95–100. [Google Scholar] [CrossRef]
- Watowich, S.S.; Wu, H.; Socolovsky, M.; Klingmuller, U.; Constantinescu, S.N.; Lodish, H.F. Cytokine Receptor Signal Transduction and the Control of Hematopoietic Cell Development. Annu. Rev. Cell Dev. Biol. 1996, 12, 91–128. [Google Scholar] [CrossRef]
- Darnell, J.E. STATs and Gene Regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Kambhampati, S.; Parmar, S.; Platanias, L.C. Jak Family of Kinases in Cancer. Cancer Metastasis Rev. 2003, 22, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Slattery, M.L.; Lundgreen, A.; Kadlubar, S.A.; Bondurant, K.L.; Wolff, R.K. JAK/STAT/SOCS-Signaling Pathway and Colon and Rectal Cancer. Mol. Carcinog. 2013, 52, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Yuan, X.; Song, J.; Chen, Y.; Tan, X.; Li, Q. Association Analyses of the JAK/STAT Signaling Pathway with the Progression and Prognosis of Colon Cancer. Oncol. Lett. 2019, 17, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Leon-Cabrera, S.; Vázquez-Sandoval, A.; Molina-Guzman, E.; Delgado-Ramirez, Y.; Delgado-Buenrostro, N.L.; Callejas, B.E.; Chirino, Y.I.; Pérez-Plasencia, C.; Rodríguez-Sosa, M.; Olguín, J.E.; et al. Deficiency in STAT1 Signaling Predisposes Gut Inflammation and Prompts Colorectal Cancer Development. Cancers 2018, 10, 341. [Google Scholar] [CrossRef] [Green Version]
- Nivarthi, H.; Gordziel, C.; Themanns, M.; Kramer, N.; Eberl, M.; Rabe, B.; Schlederer, M.; Rose-John, S.; Knösel, T.; Kenner, L.; et al. The Ratio of STAT1 to STAT3 Expression Is a Determinant of Colorectal Cancer Growth. Oncotarget 2016, 7, 51096–51106. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Jin, H.; Xu, R.; Mei, Q.; Fan, D. Triptolide Downregulates Rac1 and the JAK/STAT3 Pathway and Inhibits Colitis-Related Colon Cancer Progression. Exp. Mol. Med. 2009, 41, 717. [Google Scholar] [CrossRef] [Green Version]
- Xue, C.; Xie, J.; Zhao, D.; Lin, S.; Zhou, T.; Shi, S.; Shao, X.; Lin, Y.; Zhu, B.; Cai, X. The JAK/STAT3 Signalling Pathway Regulated Angiogenesis in an Endothelial Cell/Adipose-derived Stromal Cell Co-culture, 3D Gel Model. Cell Prolif. 2016, 50, e12307. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.; Tan, S.; Du, W.; Xu, Y.; Lin, S.; Zheng, Y.; Zou, F.; Xu, Y.; Liu, J. 3B, a Novel of Photosensitizer, Exhibited Anti-Tumor Effects via Mitochondrial Apoptosis Pathway in MCF-7 Human Breast Carcinoma Cells. Tumour Biol. 2015, 36, 5597–5606. [Google Scholar] [CrossRef] [PubMed]
- Park, K.B.; Kim, E.Y.; Chin, H.; Yoon, D.J.; Jun, K.-H. Leptin Stimulates Migration and Invasion and Maintains Cancer Stem-like Properties in Gastric Cancer Cells. Oncol. Rep. 2022, 48, 162. [Google Scholar] [CrossRef] [PubMed]
- Sakahara, M.; Okamoto, T.; Oyanagi, J.; Takano, H.; Natsume, Y.; Yamanaka, H.; Kusama, D.; Fusejima, M.; Tanaka, N.; Mori, S.; et al. IFN/STAT Signaling Controls Tumorigenesis and the Drug Response in Colorectal Cancer. Cancer Sci. 2019, 110, 1293–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodarev, N.N.; Roach, P.; Pitroda, S.P.; Golden, D.W.; Bhayani, M.; Shao, M.Y.; Darga, T.E.; Beveridge, M.G.; Sood, R.F.; Sutton, H.G.; et al. STAT1 Pathway Mediates Amplification of Metastatic Potential and Resistance to Therapy. PLoS ONE 2009, 4, e5821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-W.; Sun, Y.-M. The IL-6/JAK/STAT3 Pathway: Potential Therapeutic Strategies in Treating Colorectal Cancer (Review). Int. J. Oncol. 2014, 44, 1032–1040. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.-J.; Liu, D.-Y.; Shen, R.-R.; Xiong, Y. A Short Deletion in the DNA-Binding Domain of STAT3 Suppresses Growth and Progression of Colon Cancer Cells. Aging 2021, 13, 5185–5196. [Google Scholar] [CrossRef] [PubMed]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernández-Luna, J.L.; Nuñez, G.; et al. Constitutive Activation of Stat3 Signaling Confers Resistance to Apoptosis in Human U266 Myeloma Cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhou, L.; Xu, Y.; Yang, M.; Xu, Y.; Komaniecki, G.P.; Kosciuk, T.; Chen, X.; Lu, X.; Zou, X.; et al. A STAT3 Palmitoylation Cycle Promotes TH17 Differentiation and Colitis. Nature 2020, 586, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-Derived Suppressor Cells: Linking Inflammation and Cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.G.; et al. Inhibiting Stat3 Signaling in the Hematopoietic System Elicits Multicomponent Antitumor Immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.E.; Kitagawa, M.; Kuida, K.; Flavell, R.A.; Fu, X.Y. Activation of the STAT Signaling Pathway Can Cause Expression of Caspase 1 and Apoptosis. Mol. Cell. Biol. 1997, 17, 5328–5337. [Google Scholar] [CrossRef] [Green Version]
- Meister, N.; Shalaby, T.; von Bueren, A.O.; Rivera, P.; Patti, R.; Oehler, C.; Pruschy, M.; Grotzer, M.A. Interferon-Gamma Mediated up-Regulation of Caspase-8 Sensitizes Medulloblastoma Cells to Radio- and Chemotherapy. Eur. J. Cancer 2007, 43, 1833–1841. [Google Scholar] [CrossRef]
- Sironi, J.J.; Ouchi, T. STAT1-Induced Apoptosis Is Mediated by Caspases 2, 3, and 7. J. Biol. Chem. 2004, 279, 4066–4074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, Y.E.; Kitagawa, M.; Su, W.C.; You, Z.H.; Iwamoto, Y.; Fu, X.Y. Cell Growth Arrest and Induction of Cyclin-Dependent Kinase Inhibitor P21 WAF1/CIP1 Mediated by STAT1. Science 1996, 272, 719–722. [Google Scholar] [CrossRef]
- Stephanou, A.; Brar, B.K.; Knight, R.A.; Latchman, D.S. Opposing Actions of STAT-1 and STAT-3 on the Bcl-2 and Bcl-x Promoters. Cell Death Differ. 2000, 7, 329–330. [Google Scholar] [CrossRef] [Green Version]
- Townsend, P.A.; Scarabelli, T.M.; Davidson, S.M.; Knight, R.A.; Latchman, D.S.; Stephanou, A. STAT-1 Interacts with P53 to Enhance DNA Damage-Induced Apoptosis. J. Biol. Chem. 2004, 279, 5811–5820. [Google Scholar] [CrossRef] [Green Version]
- Tsareva, S.A.; Moriggl, R.; Corvinus, F.M.; Wiederanders, B.; Schütz, A.; Kovacic, B.; Friedrich, K. Signal Transducer and Activator of Transcription 3 Activation Promotes Invasive Growth of Colon Carcinomas through Matrix Metalloproteinase Induction. Neoplasia 2007, 9, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Zugowski, C.; Lieder, F.; Müller, A.; Gasch, J.; Corvinus, F.M.; Moriggl, R.; Friedrich, K. STAT3 Controls Matrix Metalloproteinase-1 Expression in Colon Carcinoma Cells by Both Direct and AP-1-Mediated Interaction with the MMP-1 Promoter. Biol. Chem. 2011, 392, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Musteanu, M.; Blaas, L.; Mair, M.; Schlederer, M.; Bilban, M.; Tauber, S.; Esterbauer, H.; Mueller, M.; Casanova, E.; Kenner, L.; et al. Stat3 Is a Negative Regulator of Intestinal Tumor Progression in Apc(Min) Mice. Gastroenterology 2010, 138, e1–e5. [Google Scholar] [CrossRef] [PubMed]
- Gordziel, C.; Bratsch, J.; Moriggl, R.; Knösel, T.; Friedrich, K. Both STAT1 and STAT3 Are Favourable Prognostic Determinants in Colorectal Carcinoma. Br. J. Cancer 2013, 109, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Caldenhoven, E.; van Dijk, T.B.; Solari, R.; Armstrong, J.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L.; de Groot, R.P. STAT3beta, a Splice Variant of Transcription Factor STAT3, Is a Dominant Negative Regulator of Transcription. J. Biol. Chem. 1996, 271, 13221–13227. [Google Scholar] [CrossRef] [Green Version]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in Cancer: A Double Edged Sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, M.-S. STAT1 as a Key Modulator of Cell Death. Cell. Signal. 2007, 19, 454–465. [Google Scholar] [CrossRef]
- Pflügler, S.; Svinka, J.; Scharf, I.; Crncec, I.; Filipits, M.; Charoentong, P.; Tschurtschenthaler, M.; Kenner, L.; Awad, M.; Stift, J.; et al. IDO1+ Paneth Cells Promote Immune Escape of Colorectal Cancer. Commun. Biol. 2020, 3, 252. [Google Scholar] [CrossRef]
- Simpson, J.A.D.; Al-Attar, A.; Watson, N.F.S.; Scholefield, J.H.; Ilyas, M.; Durrant, L.G. Intratumoral T Cell Infiltration, MHC Class I and STAT1 as Biomarkers of Good Prognosis in Colorectal Cancer. Gut 2010, 59, 926–933. [Google Scholar] [CrossRef]
- Liang, Y.-H.; Chen, K.-H.; Tsai, J.-H.; Cheng, Y.-M.; Lee, C.-C.; Kao, C.-H.; Chan, K.-Y.; Chen, Y.-T.; Hsu, W.-L.; Yeh, K.-H. Proteasome Inhibitors Restore the STAT1 Pathway and Enhance the Expression of MHC Class I on Human Colon Cancer Cells. J. Biomed. Sci. 2021, 28, 75. [Google Scholar] [CrossRef]
- AlMusawi, S.; Ahmed, M.; Nateri, A.S. Understanding Cell-Cell Communication and Signaling in the Colorectal Cancer Microenvironment. Clin. Transl. Med. 2021, 11, e308. [Google Scholar] [CrossRef]
- Yin, Y.; Liu, B.; Cao, Y.; Yao, S.; Liu, Y.; Jin, G.; Qin, Y.; Chen, Y.; Cui, K.; Zhou, L.; et al. Colorectal Cancer-Derived Small Extracellular Vesicles Promote Tumor Immune Evasion by Upregulating PD-L1 Expression in Tumor-Associated Macrophages. Adv. Sci. 2022, 9, 2102620. [Google Scholar] [CrossRef] [PubMed]
- Keeley, T.; Costanzo-Garvey, D.L.; Cook, L.M. Unmasking the Many Faces of Tumor-Associated Neutrophils and Macrophages: Considerations for Targeting Innate Immune Cells in Cancer. Trends Cancer 2019, 5, 789–798. [Google Scholar] [CrossRef]
- Gao, L.; Zhou, Y.; Zhou, S.-X.; Yu, X.-J.; Xu, J.-M.; Zuo, L.; Luo, Y.-H.; Li, X.-A. PLD4 Promotes M1 Macrophages to Perform Antitumor Effects in Colon Cancer Cells. Oncol. Rep. 2017, 37, 408–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Zhu, Y.; Xu, W.; Xu, J.; Yang, M.; Chen, P.; Zhao, J.; Geng, L.; Gong, S. PKCα in Colon Cancer Cells Promotes M1 Macrophage Polarization via MKK3/6-P38 MAPK Pathway. Mol. Carcinog. 2018, 57, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P. The Interaction of Anticancer Therapies with Tumor-Associated Macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-Dose Irradiation Programs Macrophage Differentiation to an INOS+/M1 Phenotype That Orchestrates Effective T Cell Immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Cheung, N.K. Phagocytosis of Tumor Cells by Human Monocytes Cultured in Recombinant Macrophage Colony-Stimulating Factor. J. Exp. Med. 1990, 172, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Lian, G.; Chen, S.; Ouyang, M.; Li, F.; Chen, L.; Yang, J. Colon Cancer Cell Secretes EGF to Promote M2 Polarization of TAM Through EGFR/PI3K/AKT/MTOR Pathway. Technol. Cancer Res. Treat. 2019, 18, 1533033819849068. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Yang, C.; Wang, S.; Shi, D.; Wei, C.; Song, J.; Lin, X.; Dou, R.; Bai, J.; Xiang, Z.; et al. Wnt5a-Induced M2 Polarization of Tumor-Associated Macrophages via IL-10 Promotes Colorectal Cancer Progression. Cell Commun. Signal. 2020, 18, 51. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Dou, R.; Wei, C.; Liu, K.; Shi, D.; Zhang, C.; Liu, Q.; Wang, S.; Xiong, B. Tumor-Derived Exosomal MicroRNA-106b-5p Activates EMT-Cancer Cell and M2-Subtype TAM Interaction to Facilitate CRC Metastasis. Mol. Ther. 2021, 29, 2088–2107. [Google Scholar] [CrossRef]
- Yuan, Y.; Surui, Y.; Yaling, H.; Yuyang, F.; Min, L.; Zehua, B.; Jiwei, Z.; Yan, Q.; Xiaowei, Q.; Leyuan, Z.; et al. The Immune-Microenvironment Confers Chemoresistance of Colorectal Cancer through Macrophage-Derived IL6. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 7375–7387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Yu, S.; Zhu, C.; Guo, T.; Liu, F.; Xu, Y. HIF1α Promotes Tumor Chemoresistance via Recruiting GDF15-Producing TAMs in Colorectal Cancer. Exp. Cell Res. 2021, 398, 112394. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 Expression by Tumor-Associated Macrophages Inhibits Phagocytosis and Tumor Immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Yao, Z.; Wang, J.; Zhang, W.; Yang, Y.; Zhang, Y.; Qu, X.; Zhu, Y.; Zou, J.; Peng, S.; et al. Macrophage-Derived CCL5 Facilitates Immune Escape of Colorectal Cancer Cells via the P65/STAT3-CSN5-PD-L1 Pathway. Cell Death Differ. 2020, 27, 1765–1781. [Google Scholar] [CrossRef]
- Cao, L.; Li, T.; Ba, Y.; Chen, E.; Yang, J.; Zhang, H. Exploring Immune-Related Prognostic Signatures in the Tumor Microenvironment of Colon Cancer. Front. Genet. 2022, 13, 801484. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Min, A.K.T.; Mimura, K.; Nakajima, S.; Okayama, H.; Saito, K.; Sakamoto, W.; Fujita, S.; Endo, H.; Saito, M.; Saze, Z.; et al. Therapeutic Potential of Anti-VEGF Receptor 2 Therapy Targeting for M2-Tumor-Associated Macrophages in Colorectal Cancer. Cancer Immunol. Immunother. 2021, 70, 289–298. [Google Scholar] [CrossRef]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-Associated Neutrophils in Cancer: Going Pro. Cancers 2019, 11, 564. [Google Scholar] [CrossRef] [Green Version]
- Berry, R.S.; Xiong, M.-J.; Greenbaum, A.; Mortaji, P.; Nofchissey, R.A.; Schultz, F.; Martinez, C.; Luo, L.; Morris, K.T.; Hanson, J.A. High Levels of Tumor-Associated Neutrophils Are Associated with Improved Overall Survival in Patients with Stage II Colorectal Cancer. PLoS ONE 2017, 12, e0188799. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-Beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, F.; Liu, X.; Chen, J.; Huang, S.; Wei, W.; Zou, Y.; Liu, X.; Deng, K.; Mo, S.; Chen, J.; et al. Anti-TGF-β Attenuates Tumor Growth via Polarization of Tumor Associated Neutrophils towards an Anti-Tumor Phenotype in Colorectal Cancer. J. Cancer 2020, 11, 2580–2592. [Google Scholar] [CrossRef] [PubMed]
- Shang, A.; Gu, C.; Zhou, C.; Yang, Y.; Chen, C.; Zeng, B.; Wu, J.; Lu, W.; Wang, W.; Sun, Z.; et al. Exosomal KRAS Mutation Promotes the Formation of Tumor-Associated Neutrophil Extracellular Traps and Causes Deterioration of Colorectal Cancer by Inducing IL-8 Expression. Cell Commun. Signal. 2020, 18, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhao, R.; Cui, Y.; Zhou, Y.; Wu, X. The Dynamic Change of Neutrophil to Lymphocyte Ratio Can Predict Clinical Outcome in Stage I-III Colon Cancer. Sci. Rep. 2018, 8, 9453. [Google Scholar] [CrossRef] [Green Version]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the Tumor Microenvironment: Shield or Spear? Int. J. Mol. Sci. 2018, 19, 1532. [Google Scholar] [CrossRef] [Green Version]
- Augsten, M. Cancer-Associated Fibroblasts as Another Polarized Cell Type of the Tumor Microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Flaberg, E.; Markasz, L.; Petranyi, G.; Stuber, G.; Dicso, F.; Alchihabi, N.; Oláh, È.; Csízy, I.; Józsa, T.; Andrén, O.; et al. High-Throughput Live-Cell Imaging Reveals Differential Inhibition of Tumor Cell Prolif.eration by Human Fibroblasts. Int. J. Cancer 2011, 128, 2793–2802. [Google Scholar] [CrossRef]
- Chang, P.-H.; Hwang-Verslues, W.W.; Chang, Y.-C.; Chen, C.-C.; Hsiao, M.; Jeng, Y.-M.; Chang, K.-J.; Lee, E.Y.-H.P.; Shew, J.-Y.; Lee, W.-H. Activation of Robo1 Signaling of Breast Cancer Cells by Slit2 from Stromal Fibroblast Restrains Tumorigenesis via Blocking PI3K/Akt/β-Catenin Pathway. Cancer Res. 2012, 72, 4652–4661. [Google Scholar] [CrossRef] [Green Version]
- Green, J.L.; La, J.; Yum, K.W.; Desai, P.; Rodewald, L.-W.; Zhang, X.; Leblanc, M.; Nusse, R.; Lewis, M.T.; Wahl, G.M. Paracrine Wnt Signaling Both Promotes and Inhibits Human Breast Tumor Growth. Proc. Natl. Acad. Sci. USA 2013, 110, 6991–6996. [Google Scholar] [CrossRef] [Green Version]
- Mosa, M.H.; Michels, B.E.; Menche, C.; Nicolas, A.M.; Darvishi, T.; Greten, F.R.; Farin, H.F. A Wnt-Induced Phenotypic Switch in Cancer-Associated Fibroblasts Inhibits EMT in Colorectal Cancer. Cancer Res. 2020, 80, 5569–5582. [Google Scholar] [CrossRef] [PubMed]
- Unterleuthner, D.; Neuhold, P.; Schwarz, K.; Janker, L.; Neuditschko, B.; Nivarthi, H.; Crncec, I.; Kramer, N.; Unger, C.; Hengstschläger, M.; et al. Cancer-Associated Fibroblast-Derived WNT2 Increases Tumor Angiogenesis in Colon Cancer. Angiogenesis 2020, 23, 159–177. [Google Scholar] [CrossRef] [Green Version]
- Kramer, N.; Schmöllerl, J.; Unger, C.; Nivarthi, H.; Rudisch, A.; Unterleuthner, D.; Scherzer, M.; Riedl, A.; Artaker, M.; Crncec, I.; et al. Autocrine WNT2 Signaling in Fibroblasts Promotes Colorectal Cancer Progression. Oncogene 2017, 36, 5460–5472. [Google Scholar] [CrossRef] [PubMed]
- Stuelten, C.H.; Busch, J.I.; Tang, B.; Flanders, K.C.; Oshima, A.; Sutton, E.; Karpova, T.S.; Roberts, A.B.; Wakefield, L.M.; Niederhuber, J.E. Transient Tumor-Fibroblast Interactions Increase Tumor Cell Malignancy by a TGF-β Mediated Mechanism in a Mouse Xenograft Model of Breast Cancer. PLoS ONE 2010, 5, e9832. [Google Scholar] [CrossRef] [PubMed]
- Koliaraki, V.; Pallangyo, C.K.; Greten, F.R.; Kollias, G. Mesenchymal Cells in Colon Cancer. Gastroenterology 2017, 152, 964–979. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, G.J. Regulation of Heterogeneous Cancer-Associated Fibroblasts: The Molecular Pathology of Activated Signaling Pathways. J. Exp. Clin. Cancer Res. 2020, 39, 112. [Google Scholar] [CrossRef]
- Takahashi, H.; Sakakura, K.; Kudo, T.; Toyoda, M.; Kaira, K.; Oyama, T.; Chikamatsu, K. Cancer-Associated Fibroblasts Promote an Immunosuppressive Microenvironment through the Induction and Accumulation of Protumoral Macrophages. Oncotarget 2016, 8, 8633–8647. [Google Scholar] [CrossRef] [Green Version]
- Stadler, M.; Pudelko, K.; Biermeier, A.; Walterskirchen, N.; Gaigneaux, A.; Weindorfer, C.; Harrer, N.; Klett, H.; Hengstschläger, M.; Schüler, J.; et al. Stromal Fibroblasts Shape the Myeloid Phenotype in Normal Colon and Colorectal Cancer and Induce CD163 and CCL2 Expression in Macrophages. Cancer Lett. 2021, 520, 184–200. [Google Scholar] [CrossRef]
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-Associated Fibroblasts Enhance Tumor-Associated Macrophages Enrichment and Suppress NK Cells Function in Colorectal Cancer. Cell Death Dis. 2019, 10, 273. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Maroto, N.G.; Garcia-Vicién, G.; Polcaro, G.; Bañuls, M.; Albert, N.; Villanueva, A.; Molleví, D.G. The Blockade of Tumoral IL1β-Mediated Signaling in Normal Colonic Fibroblasts Sensitizes Tumor Cells to Chemotherapy and Prevents Inflammatory CAF Activation. Int. J. Mol. Sci. 2021, 22, 4960. [Google Scholar] [CrossRef]
- Bai, Y.-P.; Shang, K.; Chen, H.; Ding, F.; Wang, Z.; Liang, C.; Xu, Y.; Sun, M.-H.; LI, Y.-Y. FGF-1/-3/FGFR4 Signaling in Cancer-Associated Fibroblasts Promotes Tumor Progression in Colon Cancer through Erk and MMP-7. Cancer Sci. 2015, 106, 1278–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heichler, C.; Scheibe, K.; Schmied, A.; Geppert, C.I.; Schmid, B.; Wirtz, S.; Thoma, O.-M.; Kramer, V.; Waldner, M.J.; Büttner, C.; et al. STAT3 Activation through IL-6/IL-11 in Cancer-Associated Fibroblasts Promotes Colorectal Tumour Development and Correlates with Poor Prognosis. Gut 2020, 69, 1269–1282. [Google Scholar] [CrossRef]
- Ferrari, N.; Ranftl, R.; Chicherova, I.; Slaven, N.D.; Moeendarbary, E.; Farrugia, A.J.; Lam, M.; Semiannikova, M.; Westergaard, M.C.W.; Tchou, J.; et al. Dickkopf-3 Links HSF1 and YAP/TAZ Signalling to Control Aggressive Behaviours in Cancer-Associated Fibroblasts. Nat. Commun. 2019, 10, 130. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Approaches to Treat Immune Hot, Altered and Cold Tumours with Combination Immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, H. Focus on TILs: Prognostic Significance of Tumor Infiltrating Lymphocytes in Human Colorectal Cancer. Cancer Immun. 2007, 7, 4. [Google Scholar] [PubMed]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.-H.; Pagès, F.; et al. Clinical Impact of Different Classes of Infiltrating T Cytotoxic and Helper Cells (Th1, Th2, Treg, Th17) in Patients with Colorectal Cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Qi, Q.; Pan, Y.; Zhou, Q.; Wu, Y.; Zhuang, J.; Xu, J.; Pan, M.; Han, S. Single-Cell Analysis Reveals Characterization of Infiltrating T Cells in Moderately Differentiated Colorectal Cancer. Front. Immunol. 2021, 11, 620196. [Google Scholar] [CrossRef]
- Toor, S.M.; Murshed, K.; Al-Dhaheri, M.; Khawar, M.; Abu Nada, M.; Elkord, E. Immune Checkpoints in Circulating and Tumor-Infiltrating CD4+ T Cell Subsets in Colorectal Cancer Patients. Front. Immunol. 2019, 10, 2936. [Google Scholar] [CrossRef]
- Akeus, P.; Langenes, V.; Kristensen, J.; von Mentzer, A.; Sparwasser, T.; Raghavan, S.; Quiding-Järbrink, M. Treg-Cell Depletion Promotes Chemokine Production and Accumulation of CXCR3(+) Conventional T Cells in Intestinal Tumors. Eur. J. Immunol. 2015, 45, 1654–1666. [Google Scholar] [CrossRef]
- Disis, M.L. Immune Regulation of Cancer. J. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef]
- Sica, A.; Larghi, P.; Mancino, A.; Rubino, L.; Porta, C.; Totaro, M.G.; Rimoldi, M.; Biswas, S.K.; Allavena, P.; Mantovani, A. Macrophage Polarization in Tumour Progression. Semin. Cancer Biol. 2008, 18, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.; Chen, Y.; Wang, S.; Yu, L.; Shen, Y.; Zhong, H.; Yang, Y. Exosomes from Heat-stressed Tumour Cells Inhibit Tumour Growth by Converting Regulatory T Cells to Th17 Cells via IL-6. Immunology 2018, 154, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Yaguchi, T.; Kawakami, Y. Cancer-Induced Heterogeneous Immunosuppressive Tumor Microenvironments and Their Personalized Modulation. Int. Immunol. 2016, 28, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Li, Y.; Zhang, J.; Zhang, B. PD-L1 Expression Increased by IFN-γ via JAK2-STAT1 Signaling and Predicts a Poor Survival in Colorectal Cancer. Oncol. Lett. 2020, 20, 1127–1134. [Google Scholar] [CrossRef]
- Kikuchi, T.; Mimura, K.; Okayama, H.; Nakayama, Y.; Saito, K.; Yamada, L.; Endo, E.; Sakamoto, W.; Fujita, S.; Endo, H.; et al. A Subset of Patients with MSS/MSI-Low-Colorectal.l Cancer Showed Increased CD8(+) TILs Together with up-Regulated IFN-γ. Oncol. Lett. 2019, 18, 5977–5985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Klement, J.D.; Ibrahim, M.L.; Xiao, W.; Redd, P.S.; Nayak-Kapoor, A.; Zhou, G.; Liu, K. Type I Interferon Suppresses Tumor Growth through Activating the STAT3-Granzyme B Pathway in Tumor-Infiltrating Cytotoxic T Lymphocytes. J. Immunother. Cancer 2019, 7, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legitimo, A.; Consolini, R.; Failli, A.; Orsini, G.; Spisni, R. Dendritic Cell Defects in the Colorectal Cancer. Hum. Vaccines Immunother. 2014, 10, 3224–3235. [Google Scholar] [CrossRef] [Green Version]
- Kießler, M.; Plesca, I.; Sommer, U.; Wehner, R.; Wilczkowski, F.; Müller, L.; Tunger, A.; Lai, X.; Rentsch, A.; Peuker, K.; et al. Tumor-Infiltrating Plasmacytoid Dendritic Cells Are Associated with Survival in Human Colon Cancer. J. Immunother. Cancer 2021, 9, e001813. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [Green Version]
- Schlitzer, A.; Sivakamasundari, V.; Chen, J.; Sumatoh, H.R.B.; Schreuder, J.; Lum, J.; Malleret, B.; Zhang, S.; Larbi, A.; Zolezzi, F.; et al. Identification of CDC1- and CDC2-Committed DC Progenitors Reveals Early Lineage Priming at the Common DC Progenitor Stage in the Bone Marrow. Nat. Immunol. 2015, 16, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e29. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Cueto, F.J.; del Fresno, C.; Brandi, P.; Combes, A.J.; Hernández-García, E.; Sánchez-Paulete, A.R.; Enamorado, M.; Bromley, C.P.; Gomez, M.J.; Conde-Garrosa, R.; et al. DNGR-1 Limits Flt3L-Mediated Antitumor Immunity by Restraining Tumor-Infiltrating Type I Conventional Dendritic Cells. J. Immunother. Cancer 2021, 9, e002054. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yu, S.; Chan, E.R.; Chen, K.-Y.; Liu, C.; Che, D.; Awadallah, A.; Myers, J.; Askew, D.; Huang, A.Y.; et al. Notch-Regulated Dendritic Cells Restrain Inflammation-Associated Colorectal Carcinogenesis. Cancer Immunol. Res. 2021, 9, 348–361. [Google Scholar] [CrossRef]
- Krishnaswamy, J.K.; Gowthaman, U.; Zhang, B.; Mattsson, J.; Szeponik, L.; Liu, D.; Wu, R.; White, T.; Calabro, S.; Xu, L.; et al. Migratory CD11b+ Conventional Dendritic Cells Induce T Follicular Helper Cell–Dependent Antibody Responses. Sci. Immunol. 2017, 2, eaam9169. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Ruhland, M.K.; Kersten, K.; Tsui, J.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell 2019, 177, 556–571.e16. [Google Scholar] [CrossRef]
- Kim, C.W.; Kim, K.-D.; Lee, H.K. The Role of Dendritic Cells in Tumor Microenvironments and Their Uses as Therapeutic Targets. BMB Rep. 2021, 54, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Poropatich, K.; Dominguez, D.; Chan, W.-C.; Andrade, J.; Zha, Y.; Wray, B.; Miska, J.; Qin, L.; Cole, L.; Coates, S.; et al. OX40+ Plasmacytoid Dendritic Cells in the Tumor Microenvironment Promote Antitumor Immunity. J. Clin. Investig. 2020, 130, 3528–3542. [Google Scholar] [CrossRef]
- Sisirak, V.; Vey, N.; Goutagny, N.; Renaudineau, S.; Malfroy, M.; Thys, S.; Treilleux, I.; Labidi-Galy, S.I.; Bachelot, T.; Dezutter-Dambuyant, C.; et al. Breast Cancer-Derived Transforming Growth Factor-β and Tumor Necrosis Factor-α Compromise Interferon-α Production by Tumor-Associated Plasmacytoid Dendritic Cells. Int. J. Cancer 2013, 133, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Ling, Z.; Shao, L.; Liu, X.; Cheng, Y.; Yan, C.; Mei, Y.; Ji, F.; Liu, X. Regulatory T Cells and Plasmacytoid Dendritic Cells Within the Tumor Microenvironment in Gastric Cancer Are Correlated with Gastric Microbiota Dysbiosis: A Preliminary Study. Front. Immunol. 2019, 10, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Yang, M.; Wang, Y.-H.; Lande, R.; Gregorio, J.; Perng, O.A.; Qin, X.-F.; Liu, Y.-J.; Gilliet, M. Plasmacytoid Dendritic Cells Prime IL-10–Producing T Regulatory Cells by Inducible Costimulator Ligand. J. Exp. Med. 2007, 204, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-M.; Liu, X.-S.; Lin, X.-K.; Yu, H.; Sun, J.-Y.; Liu, X.-K.; Chen, C.; Jin, H.-L.; Zhang, G.-E.; Shi, X.-X.; et al. Role of Plasmacytoid Dendritic Cells and Inducible Costimulator-Positive Regulatory T Cells in the Immunosuppression Microenvironment of Gastric Cancer. Cancer Sci. 2014, 105, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Gregorio, J.; Wang, Y.-H.; Ito, T.; Meller, S.; Hanabuchi, S.; Anderson, S.; Atkinson, N.; Ramirez, P.T.; Liu, Y.-J.; et al. Plasmacytoid Dendritic Cells Promote Immunosuppression in Ovarian Cancer via ICOS Costimulation of Foxp3+ T-Regulatory Cells. Cancer Res. 2012, 72, 5240–5249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fucikova, J.; Palova-Jelinkova, L.; Bartunkova, J.; Spisek, R. Induction of Tolerance and Immunity by Dendritic Cells: Mechanisms and Clinical Applications. Front. Immunol. 2019, 10, 2393. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Ren, J.; ten Dijke, P. Targeting TGFβ Signal Transduction for Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 8. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben Hamouda, S.; Essafi-Benkhadir, K. Interplay between Signaling Pathways and Tumor Microenvironment Components: A Paradoxical Role in Colorectal Cancer. Int. J. Mol. Sci. 2023, 24, 5600. https://doi.org/10.3390/ijms24065600
Ben Hamouda S, Essafi-Benkhadir K. Interplay between Signaling Pathways and Tumor Microenvironment Components: A Paradoxical Role in Colorectal Cancer. International Journal of Molecular Sciences. 2023; 24(6):5600. https://doi.org/10.3390/ijms24065600
Chicago/Turabian StyleBen Hamouda, Sonia, and Khadija Essafi-Benkhadir. 2023. "Interplay between Signaling Pathways and Tumor Microenvironment Components: A Paradoxical Role in Colorectal Cancer" International Journal of Molecular Sciences 24, no. 6: 5600. https://doi.org/10.3390/ijms24065600