
Microbiota-Derived Natural Products Targeting Cancer Stem Cells: Inside the Gut Pharma Factory

 , , , and
, , , and 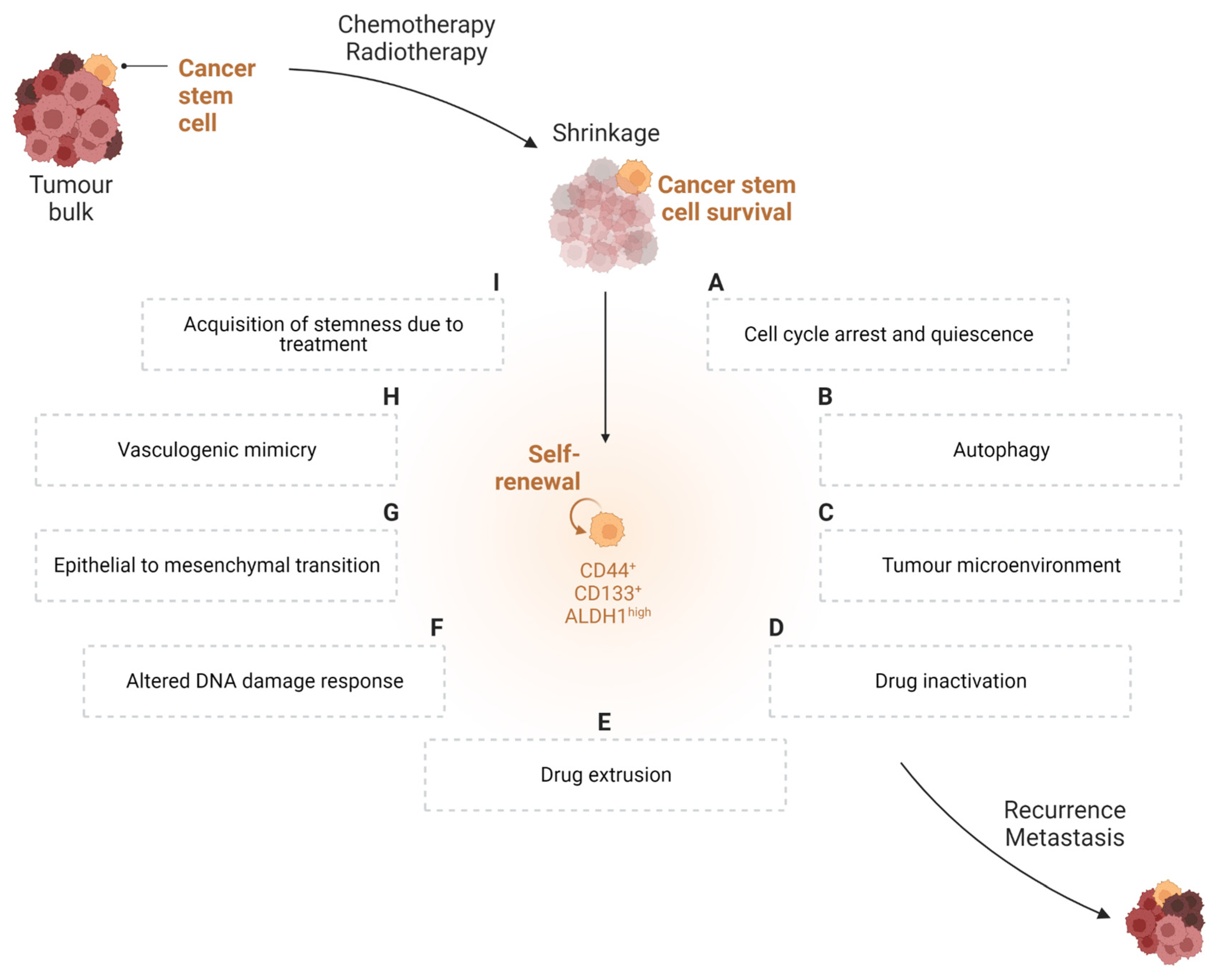{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of Cancer Stem Cells in Tumour Cell Biology
3. Therapy-Resistant Nature of Cancer Stem Cells
3.1. Cell Cycle Arrest and Quiescence
3.2. Autophagy
3.3. Tumour Microenvironment
3.4. Drug Inactivation
3.5. Drug Extrusion
3.6. Altered DNA Damage Response (DDR)
3.7. Epithelial-to-Mesenchymal Transition (EMT)
3.8. Vasculogenic Mimicry
3.9. Acquisition of Stemness Due to Treatment
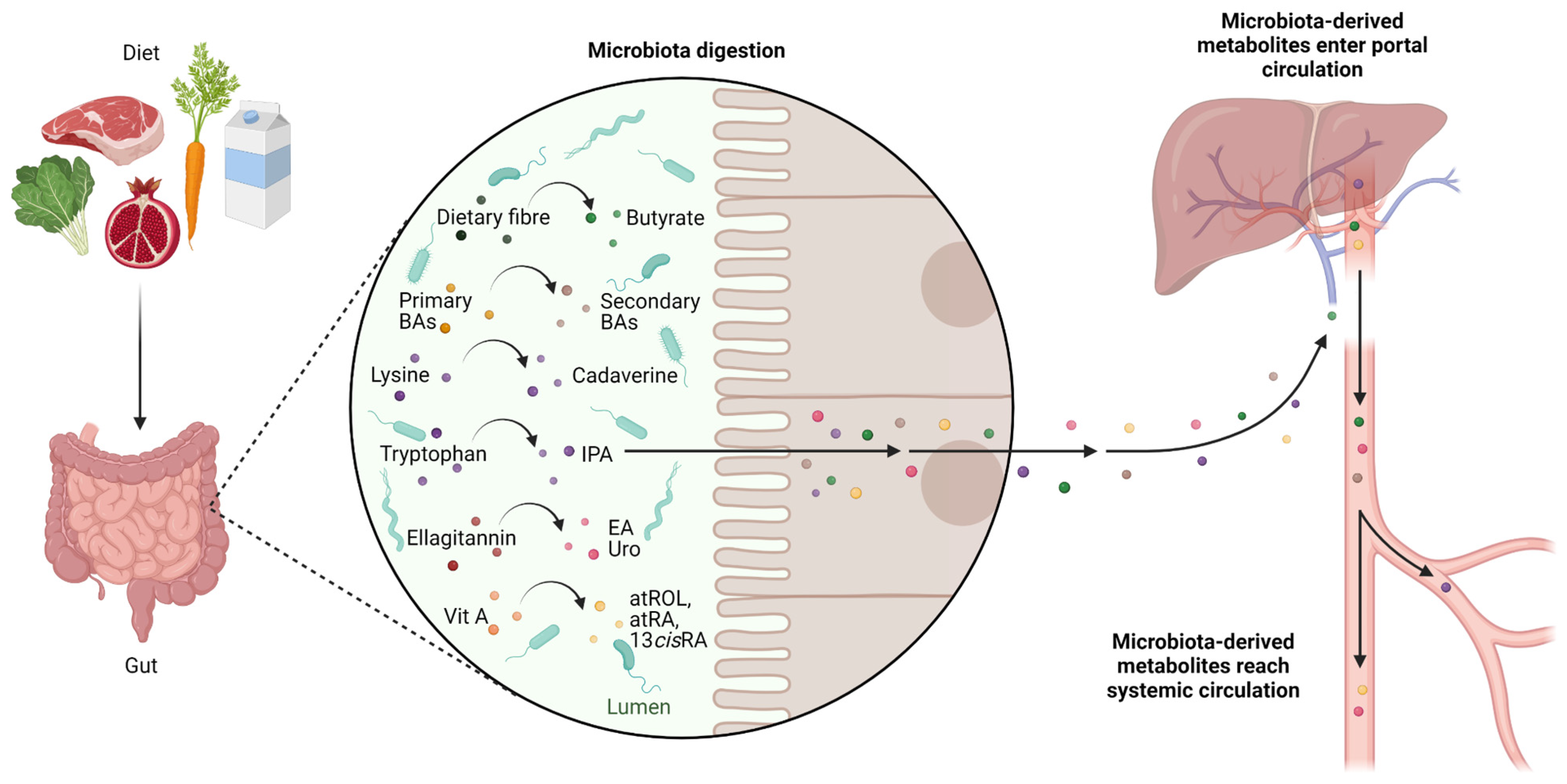
4. Role of the Gut Microbiota in Cancer
5. Microbiota-Derived Metabolites with Activity towards CSCs
5.1. Butyrate
5.2. Secondary Biliary Acids
5.3. Cadaverine and Indolepropionic Acid
5.4. Ellagic Acid and Urolithins
5.5. Retinoids
6. Conclusions
7. Data Collection
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 13cisRA | 13-cis-retinoic acid |
| 5-FU | 5-fluorouracil |
| ABC | ATP-binding cassette |
| ABCG2 | ATP binding cassette subfamily G member 2 |
| AKT | serine/threonine kinase |
| ALDH | aldehyde dehydrogenase |
| AML | acute myeloid leukaemia |
| atRA | all-trans-retinoic acid |
| atROL | all-trans-retinol |
| Bax | Bcl-2-associated X protein |
| Bcl | B-cell lymphoma 2 |
| BCRP | breast cancer resistance protein |
| BMI1 | polycomb complex protein BMI-1 |
| CFS | cell-free supernatant |
| CLDN-1 | claudin-1 |
| CMI | clomipramine |
| CQ | chloroquine |
| CSC | cancer stem cells |
| CYP26 | cytochrome P450 26A1 |
| DDR | DNA damage response |
| DIO | diet-induced obese |
| DOC or DCA | deoxycholic acid |
| DOX | doxorubicin |
| DSB | double-strand breaks |
| EA | ellagic acid |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial-to-mesenchymal transition |
| ERK | extracellular signal-regulated kinase |
| FMT | faecal microbiota transplantation |
| FXR | farnesoid X receptor |
| GIST | gastrointestinal stromal tumour |
| GLUT1 | glucose transporter 1 |
| gp130 | glycoprotein 130 |
| GPR109a | G protein-coupled receptor 109a |
| HDAC | histone deacetylase |
| SHH | sonic hedgehog |
| HIF | hypoxia-inducible factor |
| ICD | immunogenic cell death |
| IDO1 | indoleamine 2,3-dioxygenase 1 |
| IDO2 | indoleamine 2,3-dioxygenase 2 |
| IPA | indolepropionic acid |
| JAK | Janus kinase |
| LCA | lithocholic acid |
| LDC | lysine decarboxylase |
| Lgr5 | leucine-rich repeat-containing G-protein coupled receptor 5 |
| M3R | muscarinic acetylcholine receptor M3 |
| MDR | multidrug resistance |
| MET | mesenchymal-to-epithelial transition |
| MFB | metformin-butyrate |
| miRNA | micro-RNA |
| MRN | MRE11–RAD50–NBS1 protein complex |
| NLRC5 | NLR family CARD Domain Containing 5 |
| NOD/SCID | non-obese diabetic/severe combined immunodeficient |
| Notch1 | neurogenic locus notch homolog protein 1 |
| OCT3 | octamer-binding transcription factor 3 |
| OCT4 | octamer-binding transcription factor 4 |
| PKC | protein kinase C |
| RA | retinoic acid |
| SCFA | short-chain fatty acid |
| SOX2 | (sex determining region Y)-box 2 |
| STAT3 | signal transducer and activator of transcription 3 |
| TDO | tryptophan 2,3-dioxygenase |
| TLR4 | Toll-like receptor 4 |
| T-βMCA | tauro-β-muricholic acid |
| Uro | urolithin |
| VEGF | vascular-endothelial growth factor |
| VM | vasculogenic mimicry |
| Wnt | Wingless/Integrated |
References
- Baudino, T.A. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Baldo, B.A.; Pagani, M. Adverse Events to Nontargeted and Targeted Chemotherapeutic Agents: Emphasis on Hypersensitivity Responses. Immunol. Allergy Clin. N. Am. 2014, 34, 565–596, viii. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Fernandes, A.R.; Baptista, P.V. Nanoparticles as Delivery Systems in Cancer Therapy. In Applications of Targeted Nano Drugs and Delivery Systems; Elsevier: Amsterdam, The Netherlands, 2019; pp. 257–295. ISBN 9780128140291. [Google Scholar]
- Chang, J.C. Cancer Stem Cells: Role in Tumor Growth, Recurrence, Metastasis, and Treatment Resistance. Medicine 2016, 95, S20–S25. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and Expansion of Human Colon-Cancer-Initiating Cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A Human Colon Cancer Cell Capable of Initiating Tumour Growth in Immunodeficient Mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A Cell Initiating Human Acute Myeloid Leukaemia after Transplantation into SCID Mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Lin, X.; Farooqi, A.A.; Qureshi, M.Z.; Romero, M.A.; Tabassum, S.; Ismail, M. Prostate Cancer Stem Cells: Viewing Signaling Cascades at a Finer Resolution. Arch. Immunol. Ther. Exp. 2016, 64, 217–223. [Google Scholar] [CrossRef]
- Zhang, K.; Zhou, S.; Wang, L.; Wang, J.; Zou, Q.; Zhao, W.; Fu, Q.; Fang, X. Current Stem Cell Biomarkers and Their Functional Mechanisms in Prostate Cancer. Int. J. Mol. Sci. 2016, 17, 1163. [Google Scholar] [CrossRef] [Green Version]
- Hardavella, G.; George, R.; Sethi, T. Lung Cancer Stem Cells-Characteristics, Phenotype. Transl. Lung Cancer Res. 2016, 5, 272–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiler, S.; Wang, Z.; Zöller, M. Pancreatic Cancer Stem Cell Markers and Exosomes-the Incentive Push. World J. Gastroenterol. 2016, 22, 5971–6007. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Alcala, S.; Usachov, V.; Hermann, P.C.; Sainz, B., Jr. The Ever-Changing Landscape of Pancreatic Cancer Stem Cells. Pancreatology 2016, 16, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangopadhyay, S.; Nandy, A.; Hor, P.; Mukhopadhyay, A. Breast Cancer Stem Cells: A Novel Therapeutic Target. Clin. Breast Cancer 2013, 13, 7–15. [Google Scholar] [CrossRef]
- García Bueno, J.M.; Ocaña, A.; Castro-García, P.; Gil Gas, C.; Sánchez-Sánchez, F.; Poblet, E.; Serrano, R.; Calero, R.; Ramírez-Castillejo, C. An Update on the Biology of Cancer Stem Cells in Breast Cancer. Clin. Transl. Oncol. 2008, 10, 786–793. [Google Scholar] [CrossRef]
- Cherciu, I.; Bărbălan, A.; Pirici, D.; Mărgăritescu, C.; Săftoiu, A. Stem Cells, Colorectal Cancer and Cancer Stem Cell Markers Correlations. Curr. Health Sci. J. 2014, 40, 153–161. [Google Scholar] [CrossRef]
- Wilson, B.J.; Schatton, T.; Frank, M.H.; Frank, N.Y. Colorectal Cancer Stem Cells: Biology and Therapeutic Implications. Curr. Color. Cancer Rep. 2011, 7, 128–135. [Google Scholar] [CrossRef] [Green Version]
- Erhart, F.; Blauensteiner, B.; Zirkovits, G.; Printz, D.; Soukup, K.; Klingenbrunner, S.; Fischhuber, K.; Reitermaier, R.; Halfmann, A.; Lötsch, D.; et al. Gliomasphere Marker Combinatorics: Multidimensional Flow Cytometry Detects CD44+/CD133+/ITGA6+/CD36+ Signature. J. Cell. Mol. Med. 2019, 23, 281–292. [Google Scholar] [CrossRef]
- Prabavathy, D.; Swarnalatha, Y.; Ramadoss, N. Lung Cancer Stem Cells-Origin, Characteristics and Therapy. Stem Cell Investig. 2018, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Gao, L.; Xu, S.; Gong, S.; Chen, L.; Lü, S.; Chen, J.; Qiu, H.; Xu, X.; Ni, X.; et al. FISH+CD34+CD38- Cells Detected in Newly Diagnosed Acute Myeloid Leukemia Patients Can Predict the Clinical Outcome. J. Hematol. Oncol. 2013, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapasso, S.; Allegra, E. Role of CD44 as a Marker of Cancer Stem Cells in Head and Neck Cancer. Biologics 2012, 6, 379–383. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.K.; Lam, C.T.; Poon, R.T.P.; Fan, S.T. Significance of CD90+ Cancer Stem Cells in Human Liver Cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.-J.; Yin, T.; Zhu, Z.; Shi, P.-F.; Tian, Y.; Wang, C.-Y. Expression of CD44, CD24 and ESA in Pancreatic Adenocarcinoma Cell Lines Varies with Local Microenvironment. Hepatobiliary Pancreat. Dis. Int. 2011, 10, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.N.; Dakic, A.; Adams, J.M.; Nutt, S.L.; Strasser, A. Tumor Growth Need Not Be Driven by Rare Cancer Stem Cells. Science 2007, 317, 337. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.-T.; Ryu, C.J. Cancer Stem Cell Surface Markers on Normal Stem Cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Ayob, A.Z.; Ramasamy, T.S. Cancer Stem Cells as Key Drivers of Tumour Progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [Green Version]
- Islam, F.; Gopalan, V.; Smith, R.A.; Lam, A.K.-Y. Translational Potential of Cancer Stem Cells: A Review of the Detection of Cancer Stem Cells and Their Roles in Cancer Recurrence and Cancer Treatment. Exp. Cell Res. 2015, 335, 135–147. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting Cancer Stem Cell Pathways for Cancer Therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.-M.; Zhang, J.-G.; Zhang, X.; Li, Q. Targeting Cancer Stem Cells for Reversing Therapy Resistance: Mechanism, Signaling, and Prospective Agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef]
- Caglar, H.O.; Biray Avci, C. Alterations of Cell Cycle Genes in Cancer: Unmasking the Role of Cancer Stem Cells. Mol. Biol. Rep. 2020, 47, 3065–3076. [Google Scholar] [CrossRef]
- Agudo, J.; Park, E.S.; Rose, S.A.; Alibo, E.; Sweeney, R.; Dhainaut, M.; Kobayashi, K.S.; Sachidanandam, R.; Baccarini, A.; Merad, M.; et al. Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity 2018, 48, 271–285.e5. [Google Scholar] [CrossRef] [Green Version]
- Bruschini, S.; Ciliberto, G.; Mancini, R. The Emerging Role of Cancer Cell Plasticity and Cell-Cycle Quiescence in Immune Escape. Cell Death Dis. 2020, 11, 471. [Google Scholar] [CrossRef]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massagué, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells in Solid Tumours: Accumulating Evidence and Unresolved Questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Mishra, L.; Li, S. EMT, CTCs and CSCs in Tumor Relapse and Drug-Resistance. Oncotarget 2015, 6, 10697–10711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Roy, S.; Lazar, A.J.F.; Wang, W.-L.; McAuliffe, J.C.; Reynoso, D.; McMahon, J.; Taguchi, T.; Floris, G.; Debiec-Rychter, M.; et al. Autophagy Inhibition and Antimalarials Promote Cell Death in Gastrointestinal Stromal Tumor (GIST). Proc. Natl. Acad. Sci. USA 2010, 107, 14333–14338. [Google Scholar] [CrossRef] [Green Version]
- Ojha, R.; Bhattacharyya, S.; Singh, S.K. Autophagy in Cancer Stem Cells: A Potential Link Between Chemoresistance, Recurrence, and Metastasis. Biores. Open Access 2015, 4, 97–108. [Google Scholar] [CrossRef]
- Nguyen, H.G.; Yang, J.C.; Kung, H.-J.; Shi, X.-B.; Tilki, D.; Lara, P.N.; DeVere White, R.W.; Gao, A.C.; Evans, C.P. Targeting Autophagy Overcomes Enzalutamide Resistance in Castration-Resistant Prostate Cancer Cells and Improves Therapeutic Response in a Xenograft Model. Oncogene 2014, 33, 4521–4530. [Google Scholar] [CrossRef] [Green Version]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood Flow, Oxygen and Nutrient Supply, and Metabolic Microenvironment of Human Tumors: A Review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The Role of Hypoxia in the Tumor Microenvironment and Development of Cancer Stem Cell: A Novel Approach to Developing Treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef]
- Yun, Z.; Lin, Q. Hypoxia and Regulation of Cancer Cell Stemness. Adv. Exp. Med. Biol. 2014, 772, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic Extracellular Microenvironment and Cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Andreucci, E.; Peppicelli, S.; Ruzzolini, J.; Bianchini, F.; Biagioni, A.; Papucci, L.; Magnelli, L.; Mazzanti, B.; Stecca, B.; Calorini, L. The Acidic Tumor Microenvironment Drives a Stem-like Phenotype in Melanoma Cells. J. Mol. Med. 2020, 98, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.E.; Nör, J.E. Perivascular Stem Cell Niche in Head and Neck Cancer. Cancer Lett. 2013, 338, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Zanoni, M.; Bravaccini, S.; Fabbri, F.; Arienti, C. Emerging Roles of Aldehyde Dehydrogenase Isoforms in Anti-Cancer Therapy Resistance. Front. Med. 2022, 9, 795762. [Google Scholar] [CrossRef] [PubMed]
- Croker, A.K.; Allan, A.L. Inhibition of Aldehyde Dehydrogenase (ALDH) Activity Reduces Chemotherapy and Radiation Resistance of Stem-like ALDHhiCD44+ Human Breast Cancer Cells. Breast Cancer Res. Treat. 2012, 133, 75–87. [Google Scholar] [CrossRef]
- Dylla, S.J.; Beviglia, L.; Park, I.-K.; Chartier, C.; Raval, J.; Ngan, L.; Pickell, K.; Aguilar, J.; Lazetic, S.; Smith-Berdan, S.; et al. Colorectal Cancer Stem Cells Are Enriched in Xenogeneic Tumors Following Chemotherapy. PLoS ONE 2008, 3, e2428. [Google Scholar] [CrossRef]
- Januchowski, R.; Wojtowicz, K.; Zabel, M. The Role of Aldehyde Dehydrogenase (ALDH) in Cancer Drug Resistance. Biomed. Pharmacother. 2013, 67, 669–680. [Google Scholar] [CrossRef]
- Schäfer, A.; Teufel, J.; Ringel, F.; Bettstetter, M.; Hoepner, I.; Rasper, M.; Gempt, J.; Koeritzer, J.; Schmidt-Graf, F.; Meyer, B.; et al. Aldehyde Dehydrogenase 1A1—a New Mediator of Resistance to Temozolomide in Glioblastoma. Neuro Oncol. 2012, 14, 1452–1464. [Google Scholar] [CrossRef] [Green Version]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of Breast Cancer Stem Cells Identified by Aldehyde Dehydrogenase 1 Expression with Resistance to Sequential Paclitaxel and Epirubicin-Based Chemotherapy for Breast Cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.M.S.; Wan, T.S.K.; Leung, J.C.K.; Chan, L.Y.Y.; Huang, H.; Kwong, Y.L.; Liang, R.; Leung, A.Y.H. Aldehyde Dehydrogenase Activity in Leukemic Blasts Defines a Subgroup of Acute Myeloid Leukemia with Adverse Prognosis and Superior NOD/SCID Engrafting Potential. Leukemia 2007, 21, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Houghton, J.A.; Houghton, P.J. Elucidation of Pathways of 5-Fluorouracil Metabolism in Xenografts of Human Colorectal Adenocarcinoma. Eur. J. Cancer Clin. Oncol. 1983, 19, 807–815. [Google Scholar] [CrossRef]
- Schwartz, P.M.; Moir, R.D.; Hyde, C.M.; Turek, P.J.; Handschumacher, R.E. Role of Uridine Phosphorylase in the Anabolism of 5-Fluorouracil. Biochem. Pharmacol. 1985, 34, 3585–3589. [Google Scholar] [CrossRef]
- Meijer, C.; Mulder, N.H.; Timmer-Bosscha, H.; Sluiter, W.J.; Meersma, G.J.; de Vries, E.G. Relationship of Cellular Glutathione to the Cytotoxicity and Resistance of Seven Platinum Compounds. Cancer Res. 1992, 52, 6885–6889. [Google Scholar] [PubMed]
- Wu, C.-P.; Calcagno, A.M.; Ambudkar, S.V. Reversal of ABC Drug Transporter-Mediated Multidrug Resistance in Cancer Cells: Evaluation of Current Strategies. Curr. Mol. Pharmacol. 2008, 1, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.L.; Dassa, E.; Orelle, C.; Chen, J. Structure, Function, and Evolution of Bacterial ATP-Binding Cassette Systems. Microbiol. Mol. Biol. Rev. 2008, 72, 317–364. [Google Scholar] [CrossRef] [Green Version]
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Kathawala, R.J.; Gupta, P.; Ashby, C.R., Jr.; Chen, Z.-S. The Modulation of ABC Transporter-Mediated Multidrug Resistance in Cancer: A Review of the Past Decade. Drug Resist. Updat. 2015, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Nathansen, J.; Meyer, F.; Müller, L.; Schmitz, M.; Borgmann, K.; Dubrovska, A. Beyond the Double-Strand Breaks: The Role of DNA Repair Proteins in Cancer Stem-Cell Regulation. Cancers 2021, 13, 4818. [Google Scholar] [CrossRef] [PubMed]
- Andrés-León, E.; Cases, I.; Arcas, A.; Rojas, A.M. DDRprot: A Database of DNA Damage Response-Related Proteins. Database 2016, 2016, baw123. [Google Scholar] [CrossRef] [Green Version]
- Abad, E.; Graifer, D.; Lyakhovich, A. DNA Damage Response and Resistance of Cancer Stem Cells. Cancer Lett. 2020, 474, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Anuranjani; Bala, M. Concerted Action of Nrf2-ARE Pathway, MRN Complex, HMGB1 and Inflammatory Cytokines-Implication in Modification of Radiation Damage. Redox Biol. 2014, 2, 832–846. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Wu, Q.; Huang, Z.; Guryanova, O.A.; Huang, Q.; Shou, W.; Rich, J.N.; Bao, S. L1CAM Regulates DNA Damage Checkpoint Response of Glioblastoma Stem Cells through NBS1. EMBO J. 2011, 30, 800–813. [Google Scholar] [CrossRef]
- Völker-Albert, M.; Bronkhorst, A.; Holdenrieder, S.; Imhof, A. Histone Modifications in Stem Cell Development and Their Clinical Implications. Stem Cell Rep. 2020, 15, 1196–1205. [Google Scholar] [CrossRef]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core Epithelial-to-Mesenchymal Transition Interactome Gene-Expression Signature Is Associated with Claudin-Low and Metaplastic Breast Cancer Subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Blanpain, C. Unravelling Cancer Stem Cell Potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, P.; Bonnefoi, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Becette, V.; André, S.; Piccart, M.; Campone, M.; Brain, E.; et al. A Stroma-Related Gene Signature Predicts Resistance to Neoadjuvant Chemotherapy in Breast Cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An Epithelial-Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Manna, A.; Bhattacharjee, P.; Mazumdar, M.; Saha, S.; Chakraborty, S.; Guha, D.; Adhikary, A.; Jana, D.; Gorain, M.; et al. Non-Migratory Tumorigenic Intrinsic Cancer Stem Cells Ensure Breast Cancer Metastasis by Generation of CXCR4(+) Migrating Cancer Stem Cells. Oncogene 2016, 35, 4937–4948. [Google Scholar] [CrossRef]
- Saxena, K.; Jolly, M.K.; Balamurugan, K. Hypoxia, Partial EMT and Collective Migration: Emerging Culprits in Metastasis. Transl. Oncol. 2020, 13, 100845. [Google Scholar] [CrossRef] [PubMed]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular Channel Formation by Human Melanoma Cells in Vivo and in Vitro: Vasculogenic Mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Seftor, R.E.B.; Hess, A.R.; Seftor, E.A.; Kirschmann, D.A.; Hardy, K.M.; Margaryan, N.V.; Hendrix, M.J.C. Tumor Cell Vasculogenic Mimicry: From Controversy to Therapeutic Promise. Am. J. Pathol. 2012, 181, 1115–1125. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.P.; Liao, Y.D.; Mai, D.M.; Xie, P.; Qiang, Y.Y.; Zheng, L.S.; Wang, M.Y.; Mei, Y.; Meng, D.F.; Xu, L.; et al. Tumor Vasculogenic Mimicry Predicts Poor Prognosis in Cancer Patients: A Meta-Analysis. Angiogenesis 2016, 19, 191–200. [Google Scholar] [CrossRef]
- Liu, R.; Yang, K.; Meng, C.; Zhang, Z.; Xu, Y. Vasculogenic Mimicry Is a Marker of Poor Prognosis in Prostate Cancer. Cancer Biol. Ther. 2012, 13, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Hori, A.; Shimoda, M.; Naoi, Y.; Kagara, N.; Tanei, T.; Miyake, T.; Shimazu, K.; Kim, S.J.; Noguchi, S. Vasculogenic Mimicry Is Associated with Trastuzumab Resistance of HER2-Positive Breast Cancer. Breast Cancer Res. 2019, 21, 88. [Google Scholar] [CrossRef] [Green Version]
- Murugesan, A.; Sekar, B.; Saranyan, R.; Manivannan, E.; Rajmohan, M. A Review on Cancer Stem Cells in Vasculogenic Mimicry Formation: A New Dimension for Targeted Therapy. J. Adv. Oral Res. 2021, 12, 34–41. [Google Scholar] [CrossRef]
- Bittner, M.; Meltzer, P.; Chen, Y.; Jiang, Y.; Seftor, E.; Hendrix, M.; Radmacher, M.; Simon, R.; Yakhini, Z.; Ben-Dor, A.; et al. Molecular Classification of Cutaneous Malignant Melanoma by Gene Expression Profiling. Nature 2000, 406, 536–540. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, W.; Patel, S.S.; Cong, J.; Zhang, N.; Sabbatino, F.; Liu, X.; Qi, Y.; Huang, P.; Lee, H.; et al. Blocking the Formation of Radiation-Induced Breast Cancer Stem Cells. Oncotarget 2014, 5, 3743–3755. [Google Scholar] [CrossRef] [Green Version]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-Induced Reprogramming of Breast Cancer Cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Ghisolfi, L.; Keates, A.C.; Zhang, J.; Xiang, S.; Lee, D.-K.; Li, C.J. Induction of Cancer Cell Stemness by Chemotherapy. Cell Cycle 2012, 11, 2691–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.-Y.; Tang, J.-N.; Xie, H.-X.; Du, Y.-A.; Huang, L.; Yu, P.-F.; Cheng, X.-D. 5-Fluorouracil Chemotherapy of Gastric Cancer Generates Residual Cells with Properties of Cancer Stem Cells. Int. J. Biol. Sci. 2015, 11, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Nosrati, N.; Bakovic, M.; Paliyath, G. Molecular Mechanisms and Pathways as Targets for Cancer Prevention and Progression with Dietary Compounds. Int. J. Mol. Sci. 2017, 18, 2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, T.A.; Singh, R.P. Tumor Angiogenesis—a Potential Target in Cancer Chemoprevention. Food Chem. Toxicol. 2008, 46, 1334–1345. [Google Scholar] [CrossRef]
- Liskova, A.; Kubatka, P.; Samec, M.; Zubor, P.; Mlyncek, M.; Bielik, T.; Samuel, S.M.; Zulli, A.; Kwon, T.K.; Büsselberg, D. Dietary Phytochemicals Targeting Cancer Stem Cells. Molecules 2019, 24, 899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, R.F.; Jobin, C. The Microbiome and Cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaye, K.; Li, C.G.; Chang, D.; Bhuyan, D.J. The Role of Key Gut Microbial Metabolites in the Development and Treatment of Cancer. Gut Microbes 2022, 14, 2038865. [Google Scholar] [CrossRef]
- Hsiao, W.W.L.; Metz, C.; Singh, D.P.; Roth, J. The Microbes of the Intestine: An Introduction to Their Metabolic and Signaling Capabilities. Endocrinol. Metab. Clin. N. Am. 2008, 37, 857–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pabst, O. Correlation, Consequence, and Functionality in Microbiome-Immune Interplay. Immunol. Rev. 2017, 279, 4–7. [Google Scholar] [CrossRef] [Green Version]
- Śliżewska, K.; Markowiak-Kopeć, P.; Śliżewska, W. The Role of Probiotics in Cancer Prevention. Cancers 2020, 13. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Hillman, E.T.; Lu, H.; Yao, T.; Nakatsu, C.H. Microbial Ecology along the Gastrointestinal Tract. Microbes Environ. 2017, 32, 300–313. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Liu, M.; Cao, J.; Li, X.; Fan, D.; Xia, Y.; Lu, X.; Li, J.; Ju, D.; Zhao, H. The Dynamic Interplay between the Gut Microbiota and Autoimmune Diseases. J. Immunol. Res. 2019, 2019, 7546047. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.F.; Wang, H. Environmental Exposures and Autoimmune Diseases: Contribution of Gut Microbiome. Front. Immunol. 2019, 10, 3094. [Google Scholar] [CrossRef]
- Sadrekarimi, H.; Gardanova, Z.R.; Bakhshesh, M.; Ebrahimzadeh, F.; Yaseri, A.F.; Thangavelu, L.; Hasanpoor, Z.; Zadeh, F.A.; Kahrizi, M.S. Emerging Role of Human Microbiome in Cancer Development and Response to Therapy: Special Focus on Intestinal Microflora. J. Transl. Med. 2022, 20, 301. [Google Scholar] [CrossRef] [PubMed]
- Goodman, B.; Gardner, H. The Microbiome and Cancer. J. Pathol. 2018, 244, 667–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuipers, E.J. Review Article: Exploring the Link between Helicobacter Pylori and Gastric Cancer. Aliment. Pharmacol. Ther. 1999, 13 (Suppl. S1), 3–11. [Google Scholar] [CrossRef] [PubMed]
- Wroblewski, L.E.; Peek, R.M., Jr.; Wilson, K.T. Helicobacter Pylori and Gastric Cancer: Factors That Modulate Disease Risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef] [Green Version]
- Polk, D.B.; Peek, R.M., Jr. Helicobacter Pylori: Gastric Cancer and Beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Nosho, K.; Sukawa, Y.; Adachi, Y.; Ito, M.; Mitsuhashi, K.; Kurihara, H.; Kanno, S.; Yamamoto, I.; Ishigami, K.; Igarashi, H.; et al. Association of Fusobacterium Nucleatum with Immunity and Molecular Alterations in Colorectal Cancer. World J. Gastroenterol. 2016, 22, 557–566. [Google Scholar] [CrossRef]
- Shang, F.-M.; Liu, H.-L. Fusobacterium Nucleatum and Colorectal Cancer: A Review. World J. Gastrointest. Oncol. 2018, 10, 71–81. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Heidarzadeh, S.; Jahangiri, S.; Farhood, B.; Mortezaee, K.; Khanlarkhani, N.; Negahdari, B. Fusobacterium Nucleatum and Colorectal Cancer: A Mechanistic Overview. J. Cell. Physiol. 2019, 234, 2337–2344. [Google Scholar] [CrossRef]
- Matson, V.; Chervin, C.S.; Gajewski, T.F. Cancer and the Microbiome-Influence of the Commensal Microbiota on Cancer, Immune Responses, and Immunotherapy. Gastroenterology 2021, 160, 600–613. [Google Scholar] [CrossRef]
- Bessède, E.; Staedel, C.; Acuña Amador, L.A.; Nguyen, P.H.; Chambonnier, L.; Hatakeyama, M.; Belleannée, G.; Mégraud, F.; Varon, C. Helicobacter Pylori Generates Cells with Cancer Stem Cell Properties via Epithelial–Mesenchymal Transition-like Changes. Oncogene 2013, 33, 4123–4131. [Google Scholar] [CrossRef] [Green Version]
- Cavallucci, V.; Palucci, I.; Fidaleo, M.; Mercuri, A.; Masi, L.; Emoli, V.; Bianchetti, G.; Fiori, M.E.; Bachrach, G.; Scaldaferri, F.; et al. Proinflammatory and Cancer-Promoting Pathobiont Fusobacterium Nucleatum Directly Targets Colorectal Cancer Stem Cells. Biomolecules 2022, 12, 1256. [Google Scholar] [CrossRef] [PubMed]
- Ha, N.H.; Woo, B.H.; Kim, D.J.; Ha, E.S.; Choi, J.I.; Kim, S.J.; Park, B.S.; Lee, J.H.; Park, H.R. Prolonged and Repetitive Exposure to Porphyromonas Gingivalis Increases Aggressiveness of Oral Cancer Cells by Promoting Acquisition of Cancer Stem Cell Properties. Tumour Biol. 2015, 36, 9947–9960. [Google Scholar] [CrossRef]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradere, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-Induced Gut Microbial Metabolite Promotes Liver Cancer through Senescence Secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- McIntosh, G.H.; Royle, P.J.; Playne, M.J. A Probiotic Strain of L. Acidophilus Reduces DMH-Induced Large Intestinal Tumors in Male Sprague-Dawley Rats. Nutr. Cancer 1999, 35, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Tukenmez, U.; Aktas, B.; Aslim, B.; Yavuz, S. The Relationship between the Structural Characteristics of Lactobacilli-EPS and Its Ability to Induce Apoptosis in Colon Cancer Cells in Vitro. Sci. Rep. 2019, 9, 8268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelghani, Z.; Hourani, N.; Zaidan, Z.; Dbaibo, G.; Mrad, M.; Hage-Sleiman, R. Therapeutic Applications and Biological Activities of Bacterial Bioactive Extracts. Arch. Microbiol. 2021, 203, 4755–4776. [Google Scholar] [CrossRef]
- McAllister, F.; Khan, M.A.W.; Helmink, B.; Wargo, J.A. The Tumor Microbiome in Pancreatic Cancer: Bacteria and Beyond. Cancer Cell 2019, 36, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Oliva, M.; Mulet-Margalef, N.; Ochoa-De-Olza, M.; Napoli, S.; Mas, J.; Laquente, B.; Alemany, L.; Duell, E.J.; Nuciforo, P.; Moreno, V. Tumor-Associated Microbiome: Where Do We Stand? Int. J. Mol. Sci. 2021, 22, 1446. [Google Scholar] [CrossRef] [PubMed]
- Aghamajidi, A.; Maleki Vareki, S. The Effect of the Gut Microbiota on Systemic and Anti-Tumor Immunity and Response to Systemic Therapy against Cancer. Cancers 2022, 14, 3563. [Google Scholar] [CrossRef]
- Zhou, X.; Kandalai, S.; Hossain, F.; Zheng, Q. Tumor Microbiome Metabolism: A Game Changer in Cancer Development and Therapy. Front. Oncol. 2022, 12, 933407. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.W.; Cho, W.C.S. Drug Repurposing in Cancer Therapy: Approaches and Applications; Academic Press: Cambridge, MA, USA, 2020; ISBN 9780128199039. [Google Scholar]
- Wallace, B.D.; Wang, H.; Lane, K.T.; Scott, J.E.; Orans, J.; Koo, J.S.; Venkatesh, M.; Jobin, C.; Yeh, L.-A.; Mani, S.; et al. Alleviating Cancer Drug Toxicity by Inhibiting a Bacterial Enzyme. Science 2010, 330, 831–835. [Google Scholar] [CrossRef] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The Commensal Microbiome Is Associated with Anti–PD-1 Efficacy in Metastatic Melanoma Patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [Green Version]
- West, N.R.; Powrie, F. Immunotherapy Not Working? Check Your Microbiota. Cancer Cell 2015, 28, 687–689. [Google Scholar] [CrossRef] [Green Version]
- Azad, M.A.K.; Sarker, M.; Li, T.; Yin, J. Probiotic Species in the Modulation of Gut Microbiota: An Overview. BioMed Res. Int. 2018, 2018, 9478630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindels, L.B.; Delzenne, N.M.; Cani, P.D.; Walter, J. Towards a More Comprehensive Concept for Prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 303–310. [Google Scholar] [CrossRef]
- Tsilingiri, K.; Rescigno, M. Postbiotics: What Else? Benef. Microbes 2013, 4, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Rad, A.H.; Abbasi, A.; Kafil, H.S.; Ganbarov, K. Potential Pharmaceutical and Food Applications of Postbiotics: A Review. Curr. Pharm. Biotechnol. 2020, 21, 1576–1587. [Google Scholar] [CrossRef]
- Abbasi, A.; Hajipour, N.; Hasannezhad, P.; Baghbanzadeh, A.; Aghebati-Maleki, L. Potential in Vivo Delivery Routes of Postbiotics. Crit. Rev. Food Sci. Nutr. 2022, 62, 3345–3369. [Google Scholar] [CrossRef]
- Conlon, M.A.; Bird, A.R. The Impact of Diet and Lifestyle on Gut Microbiota and Human Health. Nutrients 2014, 7, 17–44. [Google Scholar] [CrossRef]
- Sheflin, A.M.; Melby, C.L.; Carbonero, F.; Weir, T.L. Linking Dietary Patterns with Gut Microbial Composition and Function. Gut Microbes 2017, 8, 113–129. [Google Scholar] [CrossRef] [Green Version]
- Leeming, E.R.; Johnson, A.J.; Spector, T.D.; Le Roy, C.I. Effect of Diet on the Gut Microbiota: Rethinking Intervention Duration. Nutrients 2019, 11, 2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puca, F.; Fedele, M.; Rasio, D.; Battista, S. Role of Diet in Stem and Cancer Stem Cells. Int. J. Mol. Sci. 2022, 23, 8108. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The Gut Microbiota, Bacterial Metabolites and Colorectal Cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Salminen, S.; Collado, M.C.; Endo, A.; Hill, C.; Lebeer, S.; Quigley, E.M.M.; Sanders, M.E.; Shamir, R.; Swann, J.R.; Szajewska, H.; et al. The International Scientific Association of Probiotics and Prebiotics (ISAPP) Consensus Statement on the Definition and Scope of Postbiotics. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 649–667. [Google Scholar] [CrossRef]
- Patel, R.M.; Denning, P.W. Therapeutic Use of Prebiotics, Probiotics, and Postbiotics to Prevent Necrotizing Enterocolitis: What Is the Current Evidence? Clin. Perinatol. 2013, 40, 11–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Toalá, J.E.; Garcia-Varela, R.; Garcia, H.S.; Mata-Haro, V.; González-Córdova, A.F.; Vallejo-Cordoba, B.; Hernández-Mendoza, A. Postbiotics: An Evolving Term within the Functional Foods Field. Trends Food Sci. Technol. 2018, 75, 105–114. [Google Scholar] [CrossRef]
- Liu, J.; Tan, Y.; Cheng, H.; Zhang, D.; Feng, W.; Peng, C. Functions of Gut Microbiota Metabolites, Current Status and Future Perspectives. Aging Dis. 2022, 13, 1106–1126. [Google Scholar] [CrossRef]
- Zhang, L.; Song, J.; Kong, L.; Yuan, T.; Li, W.; Zhang, W.; Hou, B.; Lu, Y.; Du, G. The Strategies and Techniques of Drug Discovery from Natural Products. Pharmacol. Ther. 2020, 216, 107686. [Google Scholar] [CrossRef]
- Noori, S.M.A.; Behfar, A.; Saadat, A.; Ameri, A.; Atashi Yazdi, S.S.; Siahpoosh, A. Antimicrobial and Antioxidant Properties of Natural Postbiotics Derived from Five Lactic Acid Bacteria. Jundishapur J. Nat. Pharm. Prod. 2022. in Press. [Google Scholar] [CrossRef]
- Zin, N.M.; Abd Rashid, A.N.; Zulkhairi, N.A.; Ridzman, N.A. Isolation of Lactic Acid Bacteria from Cocoa Bean Fermentation as Potential Antibacterial Agent against ESKAPE Pathogens. Sains Malays. 2022, 51, 3401–3414. [Google Scholar]
- Ryu, S.W.; Kim, J.-S.; Oh, B.S.; Choi, W.J.; Yu, S.Y.; Bak, J.E.; Park, S.-H.; Kang, S.W.; Lee, J.; Jung, W.Y.; et al. Gut Microbiota Eubacterium Callanderi Exerts Anti-Colorectal Cancer Activity. Microbiol. Spectr. 2022, 10, e0253122. [Google Scholar] [CrossRef]
- An, J.; Ha, E.-M. Combination Therapy of Lactobacillus Plantarum Supernatant and 5-Fluouracil Increases Chemosensitivity in Colorectal Cancer Cells. J. Microbiol. Biotechnol. 2016, 26, 1490–1503. [Google Scholar] [CrossRef]
- An, J.; Ha, E.-M. Lactobacillus-Derived Metabolites Enhance the Antitumor Activity of 5-FU and Inhibit Metastatic Behavior in 5-FU-Resistant Colorectal Cancer Cells by Regulating Claudin-1 Expression. J. Microbiol. 2020, 58, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Maghsood, F.; Johari, B.; Rohani, M.; Madanchi, H.; Saltanatpour, Z.; Kadivar, M. Anti-Proliferative and Anti-Metastatic Potential of High Molecular Weight Secretory Molecules from Probiotic Lactobacillus Reuteri Cell-Free Supernatant Against Human Colon Cancer Stem-Like Cells (HT29-ShE). Int. J. Pept. Res. Ther. 2020, 26, 2619–2631. [Google Scholar] [CrossRef]
- Manson, M.M. Cancer Prevention—the Potential for Diet to Modulate Molecular Signalling. Trends Mol. Med. 2003, 9, 11–18. [Google Scholar] [CrossRef]
- Key, T.J.; Schatzkin, A.; Willett, W.C.; Allen, N.E.; Spencer, E.A.; Travis, R.C. Diet, Nutrition and the Prevention of Cancer. Public Health Nutr. 2004, 7, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Mayne, S.T.; Playdon, M.C.; Rock, C.L. Diet, Nutrition, and Cancer: Past, Present and Future. Nat. Rev. Clin. Oncol. 2016, 13, 504–515. [Google Scholar] [CrossRef]
- Bail, J.; Meneses, K.; Demark-Wahnefried, W. Nutritional Status and Diet in Cancer Prevention. Semin. Oncol. Nurs. 2016, 32, 206–214. [Google Scholar] [CrossRef]
- Chen, X.; Ding, J.; Li, H.; Carr, P.R.; Hoffmeister, M.; Brenner, H. The Power of a Healthy Lifestyle for Cancer Prevention: The Example of Colorectal Cancer. Cancer Biol. Med. 2022, 19, 1586–1597. [Google Scholar] [CrossRef]
- Ali, R.; Staub, H.; Coccodrilli, G., Jr.; Schanbacher, L. Nutritional Significance of Dietary Fiber: Effect on Nutrient Bioavailability and Selected Gastrointestinal Functions. J. Agric. Food Chem. 1981, 29, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.M. CODEX-Aligned Dietary Fiber Definitions Help to Bridge the “Fiber Gap”. Nutr. J. 2014, 13, 34. [Google Scholar] [CrossRef] [Green Version]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The Role of Short-Chain Fatty Acids in the Interplay between Diet, Gut Microbiota, and Host Energy Metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aune, D.; Chan, D.S.M.; Lau, R.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Dietary Fibre, Whole Grains, and Risk of Colorectal Cancer: Systematic Review and Dose-Response Meta-Analysis of Prospective Studies. BMJ 2011, 343, d6617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunzmann, A.T.; Coleman, H.G.; Huang, W.-Y.; Kitahara, C.M.; Cantwell, M.M.; Berndt, S.I. Dietary Fiber Intake and Risk of Colorectal Cancer and Incident and Recurrent Adenoma in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Am. J. Clin. Nutr. 2015, 102, 881–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masrul, M.; Nindrea, R.D. Dietary Fibre Protective against Colorectal Cancer Patients in Asia: A Meta-Analysis. Open Access Maced. J. Med. Sci. 2019, 7, 1723–1727. [Google Scholar] [CrossRef] [Green Version]
- Comalada, M.; Bailón, E.; de Haro, O.; Lara-Villoslada, F.; Xaus, J.; Zarzuelo, A.; Gálvez, J. The Effects of Short-Chain Fatty Acids on Colon Epithelial Proliferation and Survival Depend on the Cellular Phenotype. J. Cancer Res. Clin. Oncol. 2006, 132, 487–497. [Google Scholar] [CrossRef]
- Gibson, P.R.; Moeller, I.; Kagelari, O.; Folino, M.; Young, G.P. Contrasting Effects of Butyrate on the Expression of Phenotypic Markers of Differentiation in Neoplastic and Non-Neoplastic Colonic Epithelial Cells in Vitro. J. Gastroenterol. Hepatol. 1992, 7, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.R.; Rosella, O.; Wilson, A.J.; Mariadason, J.M.; Rickard, K.; Byron, K.; Barkla, D.H. Colonic Epithelial Cell Activation and the Paradoxical Effects of Butyrate. Carcinogenesis 1999, 20, 539–544. [Google Scholar] [CrossRef] [Green Version]
- Lupton, J.R. Microbial Degradation Products Influence Colon Cancer Risk: The Butyrate Controversy. J. Nutr. 2004, 134, 479–482. [Google Scholar] [CrossRef] [Green Version]
- Geng, H.-W.; Yin, F.-Y.; Zhang, Z.-F.; Gong, X.; Yang, Y. Butyrate Suppresses Glucose Metabolism of Colorectal Cancer Cells via GPR109a-AKT Signaling Pathway and Enhances Chemotherapy. Front. Mol. Biosci. 2021, 8, 634874. [Google Scholar] [CrossRef]
- Li, Q.; Ding, C.; Meng, T.; Lu, W.; Liu, W.; Hao, H.; Cao, L. Butyrate Suppresses Motility of Colorectal Cancer Cells via Deactivating Akt/ERK Signaling in Histone Deacetylase Dependent Manner. J. Pharmacol. Sci. 2017, 135, 148–155. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvi, P.S.; Cowles, R.A. Butyrate and the Intestinal Epithelium: Modulation of Proliferation and Inflammation in Homeostasis and Disease. Cells 2021, 10, 1775. [Google Scholar] [CrossRef] [PubMed]
- Kaiko, G.E.; Ryu, S.H.; Koues, O.I.; Collins, P.L.; Solnica-Krezel, L.; Pearce, E.J.; Pearce, E.L.; Oltz, E.M.; Stappenbeck, T.S. The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell 2016, 165, 1708–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-M.; Lee, M.; Lee, J.; Kim, S.W.; Moon, H.-G.; Noh, D.-Y.; Han, W. Enhanced Anti-Tumor Activity and Cytotoxic Effect on Cancer Stem Cell Population of Metformin-Butyrate Compared with Metformin HCl in Breast Cancer. Oncotarget 2016, 7, 38500–38512. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Kwon, J.; Shin, H.-J.; Moon, S.M.; Kim, S.B.; Shin, U.S.; Han, Y.-H.; Kim, Y. Butyrate Enhances the Efficacy of Radiotherapy via FOXO3A in Colorectal Cancer Patient-derived Organoids. Int. J. Oncol. 2020, 57, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.-J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-Fat Diet Enhances Stemness and Tumorigenicity of Intestinal Progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Staels, B.; Fonseca, V.A. Bile Acids and Metabolic Regulation: Mechanisms and Clinical Responses to Bile Acid Sequestration. Diabetes Care 2009, 32 (Suppl. S2), S237–S245. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, A.F. The Continuing Importance of Bile Acids in Liver and Intestinal Disease. Arch. Intern. Med. 1999, 159, 2647–2658. [Google Scholar] [CrossRef] [PubMed]
- Phelan, J.P.; Reen, F.J.; Caparros-Martin, J.A.; O’Connor, R.; O’Gara, F. Rethinking the Bile Acid/Gut Microbiome Axis in Cancer. Oncotarget 2017, 8, 115736–115747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Qian, L. Research on Gut Microbiota-Derived Secondary Bile Acids in Cancer Progression. Integr. Cancer Ther. 2022, 21. [Google Scholar] [CrossRef] [PubMed]
- Bayerdörffer, E.; Mannes, G.A.; Ochsenkühn, T.; Dirschedl, P.; Wiebecke, B.; Paumgartner, G. Unconjugated Secondary Bile Acids in the Serum of Patients with Colorectal Adenomas. Gut 1995, 36, 268–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farhana, L.; Nangia-Makker, P.; Arbit, E.; Shango, K.; Sarkar, S.; Mahmud, H.; Hadden, T.; Yu, Y.; Majumdar, A.P.N. Bile Acid: A Potential Inducer of Colon Cancer Stem Cells. Stem Cell Res. Ther. 2016, 7, 181. [Google Scholar] [CrossRef] [Green Version]
- Fu, T.; Coulter, S.; Yoshihara, E.; Oh, T.G.; Fang, S.; Cayabyab, F.; Zhu, Q.; Zhang, T.; Leblanc, M.; Liu, S.; et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 2019, 176, 1098–1112.e18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Osaka, T.; Tsuneda, S. Bacterial Metabolites Directly Modulate Farnesoid X Receptor Activity. Nutr. Metab. 2015, 12, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovács, T.; Mikó, E.; Vida, A.; Sebő, É.; Toth, J.; Csonka, T.; Boratkó, A.; Ujlaki, G.; Lente, G.; Kovács, P.; et al. Cadaverine, a Metabolite of the Microbiome, Reduces Breast Cancer Aggressiveness through Trace Amino Acid Receptors. Sci. Rep. 2019, 9, 1300. [Google Scholar] [CrossRef] [Green Version]
- Ball, H.J.; Jusof, F.F.; Bakmiwewa, S.M.; Hunt, N.H.; Yuasa, H.J. Tryptophan-Catabolizing Enzymes—Party of Three. Front. Immunol. 2014, 5, 485. [Google Scholar] [CrossRef] [Green Version]
- Tennoune, N.; Andriamihaja, M.; Blachier, F. Production of Indole and Indole-Related Compounds by the Intestinal Microbiota and Consequences for the Host: The Good, the Bad, and the Ugly. Microorganisms 2022, 10, 930. [Google Scholar] [CrossRef]
- Sakurai, K.; Amano, S.; Enomoto, K.; Kashio, M.; Saito, Y.; Sakamoto, A.; Matsuo, S.; Suzuki, M.; Kitajima, A.; Hirano, T.; et al. Study of indoleamine 2,3-dioxygenase expression in patients with breast cancer. Gan Kagaku Ryoho. Cancer Chemother. 2005, 32, 1546–1549. [Google Scholar]
- Liu, Q.; Zhai, J.; Kong, X.; Wang, X.; Wang, Z.; Fang, Y.; Wang, J. Comprehensive Analysis of the Expressionand Prognosis for TDO2 in Breast Cancer. Mol. Ther. Oncolytics 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Sári, Z.; Mikó, E.; Kovács, T.; Jankó, L.; Csonka, T.; Lente, G.; Sebő, É.; Tóth, J.; Tóth, D.; Árkosy, P.; et al. Indolepropionic Acid, a Metabolite of the Microbiome, Has Cytostatic Properties in Breast Cancer by Activating AHR and PXR Receptors and Inducing Oxidative Stress. Cancers 2020, 12, 2411. [Google Scholar] [CrossRef]
- Wyatt, M.; Greathouse, K.L. Targeting Dietary and Microbial Tryptophan-Indole Metabolism as Therapeutic Approaches to Colon Cancer. Nutrients 2021, 13, 1189. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, Y.; Wang, M. Bioactive Substances of Plant Origin. In Handbook of Food Chemistry; Cheung, P.C.K., Mehta, B.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 967–1008. ISBN 9783642366055. [Google Scholar]
- Samtiya, M.; Aluko, R.E.; Dhewa, T.; Moreno-Rojas, J.M. Potential Health Benefits of Plant Food-Derived Bioactive Components: An Overview. Foods 2021, 10, 839. [Google Scholar] [CrossRef]
- Bié, J.; Sepodes, B.; Fernandes, P.C.B.; Ribeiro, M.H.L. Polyphenols in Health and Disease: Gut Microbiota, Bioaccessibility, and Bioavailability. Compounds 2023, 3, 40–72. [Google Scholar] [CrossRef]
- Park, J.-H.; Choi, J.W.; Ju, E.J.; Pae, A.N.; Park, K.D. Antioxidant and Anti-Inflammatory Activities of a Natural Compound, Shizukahenriol, through Nrf2 Activation. Molecules 2015, 20, 15989–16003. [Google Scholar] [CrossRef] [Green Version]
- Diniz do Nascimento, L.; Barbosa de Moraes, A.A.; Santana da Costa, K.; Pereira Galúcio, J.M.; Taube, P.S.; Costa, C.M.L.; Neves Cruz, J.; de Aguiar Andrade, E.H.; Guerreiro de Faria, L.J. Bioactive Natural Compounds and Antioxidant Activity of Essential Oils from Spice Plants: New Findings and Potential Applications. Biomolecules 2020, 10, 988. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Martins-Gomes, C.; Souto, E.B.; Schäfer, J.; Santos, J.A.; Bunzel, M.; Nunes, F.M. Thymus Zygis Subsp. Zygis an Endemic Portuguese Plant: Phytochemical Profiling, Antioxidant, Anti-Proliferative and Anti-Inflammatory Activities. Antioxidants 2020, 9, 482. [Google Scholar] [CrossRef] [PubMed]
- Abraão, A.S.; Fernandes, N.; Silva, A.M.; Domínguez-Perles, R.; Barros, A. Prunus lusitanica L. Fruits as a Novel Source of Bioactive Compounds with Antioxidant Potential: Exploring the Unknown. Antioxidants 2022, 11, 1738. [Google Scholar] [CrossRef]
- Rivas-Chacón, L.d.M.; Yanes-Díaz, J.; de Lucas, B.; Riestra-Ayora, J.I.; Madrid-García, R.; Sanz-Fernández, R.; Sánchez-Rodríguez, C. Cocoa Polyphenol Extract Inhibits Cellular Senescence via Modulation of SIRT1 and SIRT3 in Auditory Cells. Nutrients 2023, 15, 544. [Google Scholar] [CrossRef]
- Tomas-Hernandez, S.; Garcia-Vallvé, S.; Pujadas, G.; Valls, C.; Ojeda-Montes, M.J.; Gimeno, A.; Cereto-Massagué, A.; Roca-Martinez, J.; Suárez, M.; Arola, L.; et al. Anti-Inflammatory and Proapoptotic Properties of the Natural Compound o-Orsellinaldehyde. J. Agric. Food Chem. 2018, 66, 10952–10963. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Deng, W.; Wu, L.; Chen, S.; Zheng, Z.; Song, H. Anti-Inflammatory Effects of Polyphenols from Plum (Prunus Salicina Lindl) on RAW264.7 Macrophages Induced by Monosodium Urate and Potential Mechanisms. Foods 2023, 12, 254. [Google Scholar] [CrossRef]
- Zhou, Z.; He, W.; Tian, H.; Zhan, P.; Liu, J. Thyme (Thymus Vulgaris L.) Polyphenols Ameliorate DSS-Induced Ulcerative Colitis of Mice by Mitigating Intestinal Barrier Damage, Regulating Gut Microbiota, and Suppressing TLR4/NF-ΚB-NLRP3 Inflammasome Pathways. Food Funct. 2023, 14, 1113–1132. [Google Scholar] [CrossRef] [PubMed]
- Artusa, V.; Ciaramelli, C.; D’Aloia, A.; Facchini, F.A.; Gotri, N.; Bruno, A.; Costa, B.; Palmioli, A.; Airoldi, C.; Peri, F. Green and Roasted Coffee Extracts Inhibit Interferon-β Release in LPS-Stimulated Human Macrophages. Front. Pharmacol. 2022, 13, 806010. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Shang, J.; Zeng, H.; Wang, X.; Fang, M.; Xu, L.; Liu, X.; Wu, K.; Gong, Z.; Yang, Q. Hepatoprotective Effects of Rosmarinic Acid on Ovalbumin-Induced Intestinal Food Allergy Mouse Model. Molecules 2023, 28, 788. [Google Scholar] [CrossRef]
- Neves, B.R.O.; de Freitas, S.; Borelli, P.; Rogero, M.M.; Fock, R.A. Delphinidin-3-O-Glucoside in Vitro Suppresses NF-ΚB and Changes the Secretome of Mesenchymal Stem Cells Affecting Macrophage Activation. Nutrition 2023, 105, 111853. [Google Scholar] [CrossRef]
- Daskalova, E.; Delchev, S.; Peeva, Y.; Vladimirova-Kitova, L.; Kratchanova, M.; Kratchanov, C.; Denev, P. Antiatherogenic and Cardioprotective Effects of Black Chokeberry (Aronia Melanocarpa) Juice in Aging Rats. Evid. Based Complement. Altern. Med. 2015, 2015, 717439. [Google Scholar] [CrossRef] [Green Version]
- Humeniuk, E.; Adamczuk, G.; Kubik, J.; Adamczuk, K.; Józefczyk, A.; Korga-Plewko, A. Cardioprotective Effect of Centaurea Castriferrei Borbás & Waisb Extract against Doxorubicin-Induced Cardiotoxicity in H9c2 Cells. Molecules 2023, 28, 420. [Google Scholar] [CrossRef]
- Li, L.; Ma, H.; Zhang, Y.; Jiang, H.; Xia, B.; Sberi, H.A.; Elhefny, M.A.; Lokman, M.S.; Kassab, R.B. Protocatechuic Acid Reverses Myocardial Infarction Mediated by β-Adrenergic Agonist via Regulation of Nrf2/HO-1 Pathway, Inflammatory, Apoptotic, and Fibrotic Events. J. Biochem. Mol. Toxicol. 2023, e23270. [Google Scholar] [CrossRef]
- D’Aloia, A.; Molteni, L.; Gullo, F.; Bresciani, E.; Artusa, V.; Rizzi, L.; Ceriani, M.; Meanti, R.; Lecchi, M.; Coco, S.; et al. Palmitoylethanolamide Modulation of Microglia Activation: Characterization of Mechanisms of Action and Implication for Its Neuroprotective Effects. Int. J. Mol. Sci. 2021, 22, 3054. [Google Scholar] [CrossRef]
- Samani, P.; Costa, S.; Cai, S. Neuroprotective Effects of Blueberries through Inhibition on Cholinesterase, Tyrosinase, Cyclooxygenase-2, and Amyloidogenesis. Nutraceuticals 2023, 3, 39–57. [Google Scholar] [CrossRef]
- An, L.; Li, M.; Zou, C.; Wang, K.; Zhang, W.; Huang, X.; Wang, Y. Walnut Polyphenols and the Active Metabolite Urolithin A Improve Oxidative Damage in SH-SY5Y Cells by up-Regulating PKA/CREB/BDNF Signaling. Food Funct. 2023. [Google Scholar] [CrossRef]
- Laghezza Masci, V.; Bernini, R.; Villanova, N.; Clemente, M.; Cicaloni, V.; Tinti, L.; Salvini, L.; Taddei, A.R.; Tiezzi, A.; Ovidi, E. In Vitro Anti-Proliferative and Apoptotic Effects of Hydroxytyrosyl Oleate on SH-SY5Y Human Neuroblastoma Cells. Int. J. Mol. Sci. 2022, 23, 12348. [Google Scholar] [CrossRef] [PubMed]
- Luz, J.R.D.d.; López, J.A.; Ferreira, M.P.; de Sousa, R.M.; Silva, S.V.e.; Almeida, M. das G.; Araujo-Silva, G. In Vitro Antithrombotic, Antitumor and Antiangiogenic Activities of Green Tea Polyphenols and Its Main Constituent Epigallocatechin-3-Gallate. Processes 2022, 11, 76. [Google Scholar] [CrossRef]
- Cuciniello, R.; Di Meo, F.; Sulli, M.; Demurtas, O.C.; Tanori, M.; Mancuso, M.; Villano, C.; Aversano, R.; Carputo, D.; Baldi, A.; et al. Aglianico Grape Seed Semi-Polar Extract Exerts Anticancer Effects by Modulating MDM2 Expression and Metabolic Pathways. Cells 2023, 12, 210. [Google Scholar] [CrossRef] [PubMed]
- Foti, P.; Ballistreri, G.; Timpanaro, N.; Rapisarda, P.; Romeo, F.V. Prebiotic Effects of Citrus Pectic Oligosaccharides. Nat. Prod. Res. 2022, 36, 3173–3176. [Google Scholar] [CrossRef]
- Nisa, S.; Bibi, Y.; Masood, S.; Ali, A.; Alam, S.; Sabir, M.; Qayyum, A.; Ahmed, W.; Alharthi, S.; Santali, E.Y.; et al. Isolation, Characterization and Anticancer Activity of Two Bioactive Compounds from Arisaema Flavum (Forssk.) Schott. Molecules 2022, 27, 7932. [Google Scholar] [CrossRef] [PubMed]
- NK, T.; MP, R. Biologically Active Naneoicglycolate of Aristolochia Littoralis Parodi Seed Extract with Anti-Bacterial Activity Induces Cytotoxicity and Apoptosis in A431 Human Skin Cancer Cell Line. Indian J. Nat. Prod. Resour. (IJNPR) [Former. Nat. Prod. Radiance (NPR)] 2022, 13, 301–309. [Google Scholar] [CrossRef]
- Ibrahim, A.; Siswandono, S.; Prajogo, B.E.W. Anticancer Activity of Peronema Canescens Jack Leaves Extracts against Human Cells: HT-29 and HeLa in Vitro. Res. J. Pharm. Technol. Raipur 2022, 15, 4739–4745. [Google Scholar] [CrossRef]
- Molina, L.; Williams, D.E.; Andersen, R.J.; Golsteyn, R.M. Isolation of a Natural Product with Anti-Mitotic Activity from a Toxic Canadian Prairie Plant. Heliyon 2021, 7, e07131. [Google Scholar] [CrossRef]
- Gallo, C.; Dallaglio, K.; Bassani, B.; Rossi, T.; Rossello, A.; Noonan, D.M.; D’Uva, G.; Bruno, A.; Albini, A. Hop Derived Flavonoid Xanthohumol Inhibits Endothelial Cell Functions via AMPK Activation. Oncotarget 2016, 7, 59917–59931. [Google Scholar] [CrossRef] [Green Version]
- Baci, D.; Gallazzi, M.; Cascini, C.; Tramacere, M.; De Stefano, D.; Bruno, A.; Noonan, D.M.; Albini, A. Downregulation of Pro-Inflammatory and Pro-Angiogenic Pathways in Prostate Cancer Cells by a Polyphenol-Rich Extract from Olive Mill Wastewater. Int. J. Mol. Sci. 2019, 20, 307. [Google Scholar] [CrossRef] [Green Version]
- Grynberg, N.F.; Carvalho, M.G.; Velandia, J.R.; Oliveira, M.C.; Moreira, I.C.; Braz-Filho, R.; Echevarria, A. DNA Topoisomerase Inhibitors: Biflavonoids from Ouratea Species. Braz. J. Med. Biol. Res. 2002, 35, 819–822. [Google Scholar] [CrossRef] [Green Version]
- Rani, V.; BC, R.; GS, M.; Deshpande, S.; Venkatesan, J.; Appana Dalavi, P.; Prabhu, A. Cytotoxic and Apoptotic Efficacy of Alkanna Tinctoria on Glioma Cells. Nat. Prod. Res. 2022, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sigstedt, S.C.; Hooten, C.J.; Callewaert, M.C.; Jenkins, A.R.; Romero, A.E.; Pullin, M.J.; Kornienko, A.; Lowrey, T.K.; Van Slambrouck, S.; Steelant, W.F.A. Evaluation of Aqueous Extracts of Taraxacum Officinale on Growth and Invasion of Breast and Prostate Cancer Cells. Int. J. Oncol. 2008, 32, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Matić, I.Z.; Aljancić, I.; Vajs, V.; Jadranin, M.; Gligorijević, N.; Milosavljević, S.; Juranić, Z.D. Cancer-Suppressive Potential of Extracts of Endemic Plant Helichrysum Zivojinii: Effects on Cell Migration, Invasion and Angiogenesis. Nat. Prod. Commun. 2013, 8, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Albini, A.; Festa, M.M.G.; Ring, N.; Baci, D.; Rehman, M.; Finzi, G.; Sessa, F.; Zacchigna, S.; Bruno, A.; Noonan, D.M. A Polyphenol-Rich Extract of Olive Mill Wastewater Enhances Cancer Chemotherapy Effects, While Mitigating Cardiac Toxicity. Front. Pharmacol. 2021, 12, 694762. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, N.; Calabrone, L.; Gutmańska, K.; Macrì, N.; Cerrito, M.G.; Ricotta, R.; Pelosi, G.; Bruno, A.; Noonan, D.M.; Albini, A. An Olive Oil Mill Wastewater Extract Improves Chemotherapeutic Activity Against Breast Cancer Cells While Protecting From Cardiotoxicity. Front. Cardiovasc. Med. 2022, 9, 867867. [Google Scholar] [CrossRef]
- Li, M.; Zhang, H.; Hu, X.; Liu, Y.; Liu, Y.; Song, M.; Wu, R.; Wu, J. Isolation of a New Polysaccharide from Dandelion Leaves and Evaluation of Its Antioxidant, Antibacterial, and Anticancer Activities. Molecules 2022, 27, 7641. [Google Scholar] [CrossRef]
- Efferth, T. Stem Cells, Cancer Stem-like Cells, and Natural Products. Planta Med. 2012, 78, 935–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moselhy, J.; Srinivasan, S.; Ankem, M.K.; Damodaran, C. Natural Products That Target Cancer Stem Cells. Anticancer Res. 2015, 35, 5773–5788. [Google Scholar]
- Pistollato, F.; Giampieri, F.; Battino, M. The Use of Plant-Derived Bioactive Compounds to Target Cancer Stem Cells and Modulate Tumor Microenvironment. Food Chem. Toxicol. 2015, 75, 58–70. [Google Scholar] [CrossRef]
- Scarpa, E.-S.; Ninfali, P. Phytochemicals as Innovative Therapeutic Tools against Cancer Stem Cells. Int. J. Mol. Sci. 2015, 16, 15727–15742. [Google Scholar] [CrossRef] [Green Version]
- Taylor, W.F.; Jabbarzadeh, E. The Use of Natural Products to Target Cancer Stem Cells. Am. J. Cancer Res. 2017, 7, 1588–1605. [Google Scholar]
- Palermo, R.; Ghirga, F.; Piccioni, M.G.; Bernardi, F.; Zhdanovskaya, N.; Infante, P.; Mori, M. Natural Products Inspired Modulators of Cancer Stem Cells-Specific Signaling Pathways Notch and Hedgehog. Curr. Pharm. Des. 2018, 24, 4251–4269. [Google Scholar] [CrossRef]
- Das, P.K.; Zahan, T.; Abdur Rakib, M.; Khanam, J.A.; Pillai, S.; Islam, F. Natural Compounds Targeting Cancer Stem Cells: A Promising Resource for Chemotherapy. Anticancer Agents Med. Chem. 2019, 19, 1796–1808. [Google Scholar] [CrossRef]
- Ganesan, K.; Jayachandran, M.; Xu, B. Diet-Derived Phytochemicals Targeting Colon Cancer Stem Cells and Microbiota in Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 3976. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Saha, S.K.; Rahman, M.S.; Uddin, M.J.; Uddin, M.S.; Pang, M.-G.; Rhim, H.; Cho, S.-G. Molecular Insights Into Therapeutic Potential of Autophagy Modulation by Natural Products for Cancer Stem Cells. Front. Cell Dev. Biol. 2020, 8, 283. [Google Scholar] [CrossRef]
- Gairola, K.; Gururani, S.; Bahuguna, A.; Garia, V.; Pujari, R.; Dubey, S.K. Natural Products Targeting Cancer Stem Cells: Implications for Cancer Chemoprevention and Therapeutics. J. Food Biochem. 2021, 45, e13772. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Saraff, M.; Gahtori, R.; Negi, N.; Tripathi, S.K.; Kumar, J.; Kumar, S.; Aldhayan, S.H.; Dhanasekaran, S.; Abomughaid, M.M.; et al. Phytomedicines Targeting Cancer Stem Cells: Therapeutic Opportunities and Prospects for Pharmaceutical Development. Pharmaceuticals 2021, 14, 676. [Google Scholar] [CrossRef] [PubMed]
- Meerson, A.; Khatib, S.; Mahajna, J. Natural Products Targeting Cancer Stem Cells for Augmenting Cancer Therapeutics. Int. J. Mol. Sci. 2021, 22, 13044. [Google Scholar] [CrossRef]
- Hashem, S.; Ali, T.A.; Akhtar, S.; Nisar, S.; Sageena, G.; Ali, S.; Al-Mannai, S.; Therachiyil, L.; Mir, R.; Elfaki, I.; et al. Targeting Cancer Signaling Pathways by Natural Products: Exploring Promising Anti-Cancer Agents. Biomed. Pharmacother. 2022, 150, 113054. [Google Scholar] [CrossRef]
- Singh, D.; Pandey, H.; Singh, V. Natural Products That Target Cancer Stem Cells. In Handbook of Research on Natural Products and Their Bioactive Compounds as Cancer Therapeutics; IGI Global: Hershey, PA, USA, 2022; pp. 169–186. [Google Scholar]
- Bonuccelli, G.; Sotgia, F.; Lisanti, M.P. Identification of Natural Products and FDA-Approved Drugs for Targeting Cancer Stem Cell (CSC) Propagation. Aging 2022, 14, 9466–9483. [Google Scholar] [CrossRef]
- Kawabata, K.; Yoshioka, Y.; Terao, J. Role of Intestinal Microbiota in the Bioavailability and Physiological Functions of Dietary Polyphenols. Molecules 2019, 24, 370. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, K.; Sugiyama, M.; Mukai, T. Adhesion Properties of Lactic Acid Bacteria on Intestinal Mucin. Microorganisms 2016, 4, 34. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, J.-H.; Kim, S.-L.; Deng, H.-Y.; Lee, D.; Kim, C.S.; Yun, B.-S.; Lee, D.-S. Catechol Derived from Aronia Juice through Lactic Acid Bacteria Fermentation Inhibits Breast Cancer Stem Cell Formation via Modulation Stat3/IL-6 Signaling Pathway. Mol. Carcinog. 2018, 57, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Cerdá, B.; Tomás-Barberán, F.A.; Espín, J.C. Metabolism of Antioxidant and Chemopreventive Ellagitannins from Strawberries, Raspberries, Walnuts, and Oak-Aged Wine in Humans: Identification of Biomarkers and Individual Variability. J. Agric. Food Chem. 2005, 53, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Seeram, N.P.; Henning, S.M.; Zhang, Y.; Suchard, M.; Li, Z.; Heber, D. Pomegranate Juice Ellagitannin Metabolites Are Present in Human Plasma and Some Persist in Urine for up to 48 Hours. J. Nutr. 2006, 136, 2481–2485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés-Martín, A.; Selma, M.V.; Tomás-Barberán, F.A.; González-Sarrías, A.; Espín, J.C. Where to Look into the Puzzle of Polyphenols and Health? The Postbiotics and Gut Microbiota Associated with Human Metabotypes. Mol. Nutr. Food Res. 2020, 64, e1900952. [Google Scholar] [CrossRef]
- Núñez-Sánchez, M.Á.; Karmokar, A.; González-Sarrías, A.; García-Villalba, R.; Tomás-Barberán, F.A.; García-Conesa, M.T.; Brown, K.; Espín, J.C. In Vivo Relevant Mixed Urolithins and Ellagic Acid Inhibit Phenotypic and Molecular Colon Cancer Stem Cell Features: A New Potentiality for Ellagitannin Metabolites against Cancer. Food Chem. Toxicol. 2016, 92, 8–16. [Google Scholar] [CrossRef]
- González-Sarrías, A.; Miguel, V.; Merino, G.; Lucas, R.; Morales, J.C.; Tomás-Barberán, F.; Alvarez, A.I.; Espín, J.C. The Gut Microbiota Ellagic Acid-Derived Metabolite Urolithin A and Its Sulfate Conjugate Are Substrates for the Drug Efflux Transporter Breast Cancer Resistance Protein (ABCG2/BCRP). J. Agric. Food Chem. 2013, 61, 4352–4359. [Google Scholar] [CrossRef] [PubMed]
- González-Sarrías, A.; Tomé-Carneiro, J.; Bellesia, A.; Tomás-Barberán, F.A.; Espín, J.C. The Ellagic Acid-Derived Gut Microbiota Metabolite, Urolithin A, Potentiates the Anticancer Effects of 5-Fluorouracil Chemotherapy on Human Colon Cancer Cells. Food Funct. 2015, 6, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Cañestro, C.; Catchen, J.M.; Rodríguez-Marí, A.; Yokoi, H.; Postlethwait, J.H. Consequences of Lineage-Specific Gene Loss on Functional Evolution of Surviving Paralogs: ALDH1A and Retinoic Acid Signaling in Vertebrate Genomes. PLoS Genet. 2009, 5, e1000496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezquita, B.; Mezquita, C. Two Opposing Faces of Retinoic Acid: Induction of Stemness or Induction of Differentiation Depending on Cell-Type. Biomolecules 2019, 9, 567. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.-H.; Gudas, L.J. Retinoids, Retinoic Acid Receptors, and Cancer. Annu. Rev. Pathol. 2011, 6, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Hunsu, V.O.; Facey, C.O.B.; Fields, J.Z.; Boman, B.M. Retinoids as Chemo-Preventive and Molecular-Targeted Anti-Cancer Therapies. Int. J. Mol. Sci. 2021, 22, 7731. [Google Scholar] [CrossRef]
- Bouriez, D.; Giraud, J.; Gronnier, C.; Varon, C. Efficiency of All-Trans Retinoic Acid on Gastric Cancer: A Narrative Literature Review. Int. J. Mol. Sci. 2018, 19, 3388. [Google Scholar] [CrossRef] [Green Version]
- Karsy, M.; Albert, L.; Tobias, M.E.; Murali, R.; Jhanwar-Uniyal, M. All-Trans Retinoic Acid Modulates Cancer Stem Cells of Glioblastoma Multiforme in an MAPK-Dependent Manner. Anticancer Res. 2010, 30, 4915–4920. [Google Scholar]
- Lim, Y.C.; Kang, H.J.; Kim, Y.S.; Choi, E.C. All-Trans-Retinoic Acid Inhibits Growth of Head and Neck Cancer Stem Cells by Suppression of Wnt/β-Catenin Pathway. Eur. J. Cancer 2012, 48, 3310–3318. [Google Scholar] [CrossRef] [PubMed]
- Li, R.-J.; Ying, X.; Zhang, Y.; Ju, R.-J.; Wang, X.-X.; Yao, H.-J.; Men, Y.; Tian, W.; Yu, Y.; Zhang, L.; et al. All-Trans Retinoic Acid Stealth Liposomes Prevent the Relapse of Breast Cancer Arising from the Cancer Stem Cells. J. Control. Release 2011, 149, 281–291. [Google Scholar] [CrossRef]
- Sun, R.; Liu, Y.; Li, S.-Y.; Shen, S.; Du, X.-J.; Xu, C.-F.; Cao, Z.-T.; Bao, Y.; Zhu, Y.-H.; Li, Y.-P.; et al. Co-Delivery of All-Trans-Retinoic Acid and Doxorubicin for Cancer Therapy with Synergistic Inhibition of Cancer Stem Cells. Biomaterials 2015, 37, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Wang, L.; Huang, H.; Li, X.; Wang, P.; Mi, K.; Cheng, J.; Liu, H.; Gu, C.; Huang, L.; et al. All-Trans Retinoic Acid Reduces Cancer Stem Cell-like Cell-Mediated Resistance to Gefitinib in NSCLC Adenocarcinoma Cells. BMC Cancer 2020, 20, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berardi, D.E.; Ariza Bareño, L.; Amigo, N.; Cañonero, L.; Pelagatti, M.d.L.N.; Motter, A.N.; Taruselli, M.A.; Díaz Bessone, M.I.; Cirigliano, S.M.; Edelstein, A.; et al. All-Trans Retinoic Acid and Protein Kinase C α/Β1 Inhibitor Combined Treatment Targets Cancer Stem Cells and Impairs Breast Tumor Progression. Sci. Rep. 2021, 11, 6044. [Google Scholar] [CrossRef]
- MacDonagh, L.; Santiago, R.M.; Gray, S.G.; Breen, E.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Barr, M.P. Exploitation of the Vitamin A/Retinoic Acid Axis Depletes ALDH1-Positive Cancer Stem Cells and Re-Sensitises Resistant Non-Small Cell Lung Cancer Cells to Cisplatin. Transl. Oncol. 2021, 14, 101025. [Google Scholar] [CrossRef]
- Bonakdar, M.; Czuba, L.C.; Han, G.; Zhong, G.; Luong, H.; Isoherrannen, N.; Vaishnava, S. Gut Commensals Expand Vitamin A Metabolic Capacity of the Mammalian Host. Cell Host Microbe 2022, 30, 1084–1092.e5. [Google Scholar] [CrossRef] [PubMed]
- Ubago-Guisado, E.; Rodríguez-Barranco, M.; Ching-López, A.; Petrova, D.; Molina-Montes, E.; Amiano, P.; Barricarte-Gurrea, A.; Chirlaque, M.-D.; Agudo, A.; Sánchez, M.-J. Evidence Update on the Relationship between Diet and the Most Common Cancers from the European Prospective Investigation into Cancer and Nutrition (EPIC) Study: A Systematic Review. Nutrients 2021, 13, 3582. [Google Scholar] [CrossRef]
- Kamal, N.; Ilowefah, M.A.; Hilles, A.R.; Anua, N.A.; Awin, T.; Alshwyeh, H.A.; Aldosary, S.K.; Jambocus, N.G.S.; Alosaimi, A.A.; Rahman, A.; et al. Genesis and Mechanism of Some Cancer Types and an Overview on the Role of Diet and Nutrition in Cancer Prevention. Molecules 2022, 27, 1794. [Google Scholar] [CrossRef]
- Bamia, C.; Lagiou, P.; Buckland, G.; Grioni, S.; Agnoli, C.; Taylor, A.J.; Dahm, C.C.; Overvad, K.; Olsen, A.; Tjønneland, A.; et al. Mediterranean Diet and Colorectal Cancer Risk: Results from a European Cohort. Eur. J. Epidemiol. 2013, 28, 317–328. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Hoffmann, G. Adherence to Mediterranean Diet and Risk of Cancer: A Systematic Review and Meta-Analysis of Observational Studies. Int. J. Cancer 2014, 135, 1884–1897. [Google Scholar] [CrossRef]
- Rosato, V.; Guercio, V.; Bosetti, C.; Negri, E.; Serraino, D.; Giacosa, A.; Montella, M.; La Vecchia, C.; Tavani, A. Mediterranean Diet and Colorectal Cancer Risk: A Pooled Analysis of Three Italian Case-Control Studies. Br. J. Cancer 2016, 115, 862–865. [Google Scholar] [CrossRef] [Green Version]
- Praud, D.; Bertuccio, P.; Bosetti, C.; Turati, F.; Ferraroni, M.; La Vecchia, C. Adherence to the Mediterranean Diet and Gastric Cancer Risk in Italy. Int. J. Cancer 2014, 134, 2935–2941. [Google Scholar] [CrossRef] [Green Version]
- Bosetti, C.; Turati, F.; Dal Pont, A.; Ferraroni, M.; Polesel, J.; Negri, E.; Serraino, D.; Talamini, R.; La Vecchia, C.; Zeegers, M.P. The Role of Mediterranean Diet on the Risk of Pancreatic Cancer. Br. J. Cancer 2013, 109, 1360–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demetriou, C.A.; Hadjisavvas, A.; Loizidou, M.A.; Loucaides, G.; Neophytou, I.; Sieri, S.; Kakouri, E.; Middleton, N.; Vineis, P.; Kyriacou, K. The Mediterranean Dietary Pattern and Breast Cancer Risk in Greek-Cypriot Women: A Case-Control Study. BMC Cancer 2012, 12, 113. [Google Scholar] [CrossRef] [Green Version]
- Turati, F.; Carioli, G.; Bravi, F.; Ferraroni, M.; Serraino, D.; Montella, M.; Giacosa, A.; Toffolutti, F.; Negri, E.; Levi, F.; et al. Mediterranean Diet and Breast Cancer Risk. Nutrients 2018, 10, 326. [Google Scholar] [CrossRef] [Green Version]
- Laudisio, D.; Castellucci, B.; Barrea, L.; Pugliese, G.; Savastano, S.; Colao, A.; Muscogiuri, G. Mediterranean Diet and Breast Cancer Risk: A Narrative Review. Minerva Endocrinol. 2021, 46, 441–452. [Google Scholar] [CrossRef]
- Turati, F.; Bravi, F.; Polesel, J.; Bosetti, C.; Negri, E.; Garavello, W.; Taborelli, M.; Serraino, D.; Libra, M.; Montella, M.; et al. Adherence to the Mediterranean Diet and Nasopharyngeal Cancer Risk in Italy. Cancer Causes Control 2017, 28, 89–95. [Google Scholar] [CrossRef]
- Fortes, C.; Forastiere, F.; Farchi, S.; Mallone, S.; Trequattrinni, T.; Anatra, F.; Schmid, G.; Perucci, C.A. The Protective Effect of the Mediterranean Diet on Lung Cancer. Nutr. Cancer 2003, 46, 30–37. [Google Scholar] [CrossRef]
- Itsiopoulos, C.; Hodge, A.; Kaimakamis, M. Can the Mediterranean Diet Prevent Prostate Cancer? Mol. Nutr. Food Res. 2009, 53, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Bravi, F.; Spei, M.-E.; Polesel, J.; Di Maso, M.; Montella, M.; Ferraroni, M.; Serraino, D.; Libra, M.; Negri, E.; La Vecchia, C.; et al. Mediterranean Diet and Bladder Cancer Risk in Italy. Nutrients 2018, 10, 1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botero, L.E.; Delgado-Serrano, L.; Cepeda Hernandez, M.L.; Del Portillo Obando, P.; Zambrano Eder, M.M. The Human Microbiota: The Role of Microbial Communities in Health and Disease. Acta Biológica Colomb. 2016, 21, 5–15. [Google Scholar]
- Karkman, A.; Lehtimäki, J.; Ruokolainen, L. The Ecology of Human Microbiota: Dynamics and Diversity in Health and Disease. Ann. N. Y. Acad. Sci. 2017, 1399, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yao, M.; Lv, L.; Ling, Z.; Li, L. The Human Microbiota in Health and Disease. Engineering 2017, 3, 71–82. [Google Scholar] [CrossRef]
- Tungland, B. Human Microbiota in Health and Disease: From Pathogenesis to Therapy; Academic Press: Cambridge, MA, USA, 2018; ISBN 9780128146507. [Google Scholar]
- Martínez, J.E.; Vargas, A.; Pérez-Sánchez, T.; Encío, I.J.; Cabello-Olmo, M.; Barajas, M. Human Microbiota Network: Unveiling Potential Crosstalk between the Different Microbiota Ecosystems and Their Role in Health and Disease. Nutrients 2021, 13, 2905. [Google Scholar] [CrossRef]
- Artemev, A.; Naik, S.; Pougno, A.; Honnavar, P.; Shanbhag, N.M. The Association of Microbiome Dysbiosis With Colorectal Cancer. Cureus 2022, 14, e22156. [Google Scholar] [CrossRef] [PubMed]
- Fong, W.; Li, Q.; Yu, J. Gut Microbiota Modulation: A Novel Strategy for Prevention and Treatment of Colorectal Cancer. Oncogene 2020, 39, 4925–4943. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut Microbiota Functions: Metabolism of Nutrients and Other Food Components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Artusa, V.; Calabrone, L.; Mortara, L.; Peri, F.; Bruno, A. Microbiota-Derived Natural Products Targeting Cancer Stem Cells: Inside the Gut Pharma Factory. Int. J. Mol. Sci. 2023, 24, 4997. https://doi.org/10.3390/ijms24054997
Artusa V, Calabrone L, Mortara L, Peri F, Bruno A. Microbiota-Derived Natural Products Targeting Cancer Stem Cells: Inside the Gut Pharma Factory. International Journal of Molecular Sciences. 2023; 24(5):4997. https://doi.org/10.3390/ijms24054997
Chicago/Turabian StyleArtusa, Valentina, Luana Calabrone, Lorenzo Mortara, Francesco Peri, and Antonino Bruno. 2023. "Microbiota-Derived Natural Products Targeting Cancer Stem Cells: Inside the Gut Pharma Factory" International Journal of Molecular Sciences 24, no. 5: 4997. https://doi.org/10.3390/ijms24054997