
Draft Genome and Biological Characteristics of Fusarium solani and Fusarium oxysporum Causing Black Rot in Gastrodia elata
Abstract
:1. Introduction
2. Results
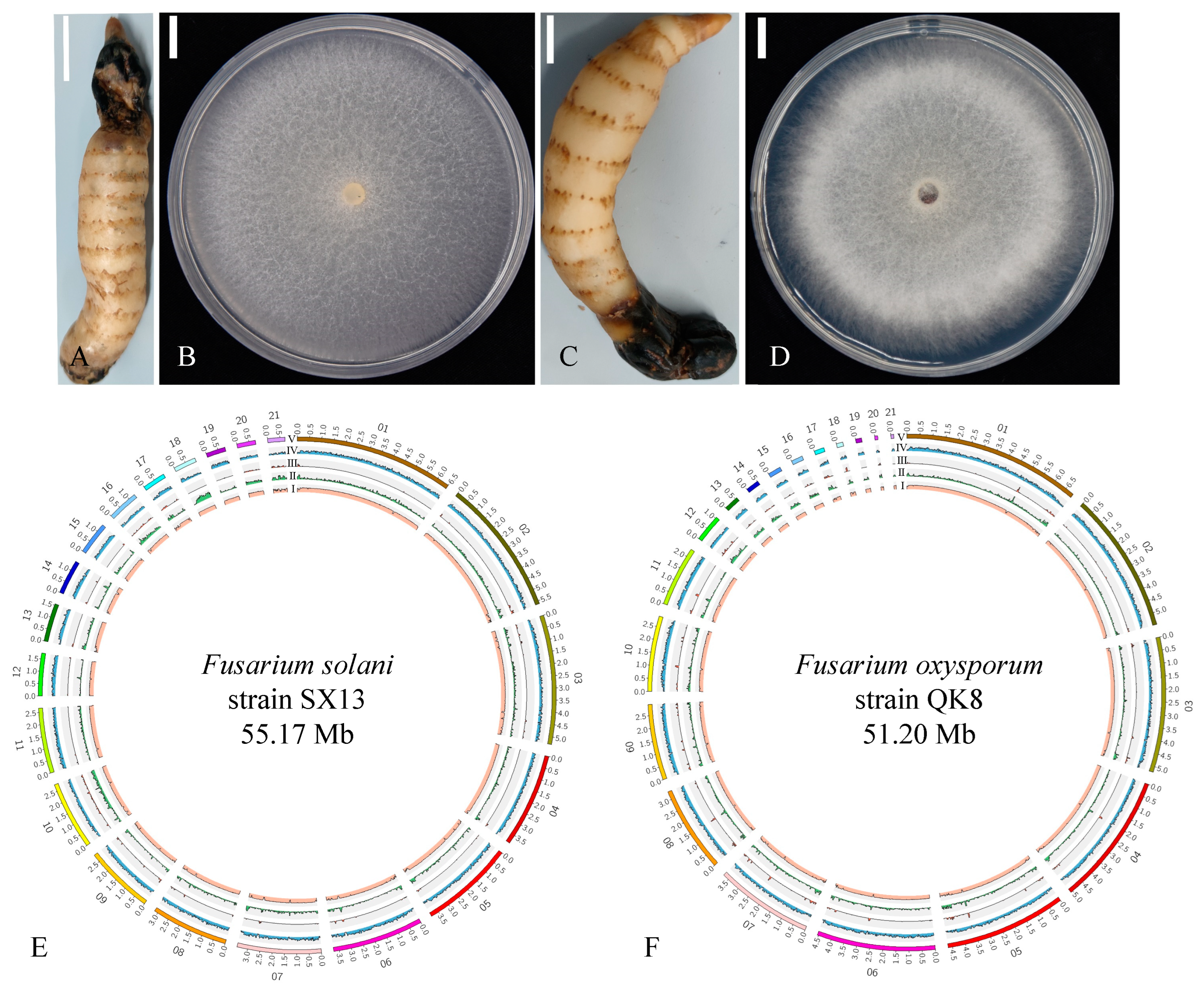
2.1. Biological Characteristics of the Fusarium Strains
2.2. Whole-Genome Sequencing
2.2.1. Genome Sequencing, Repeat Analysis, and Assembly
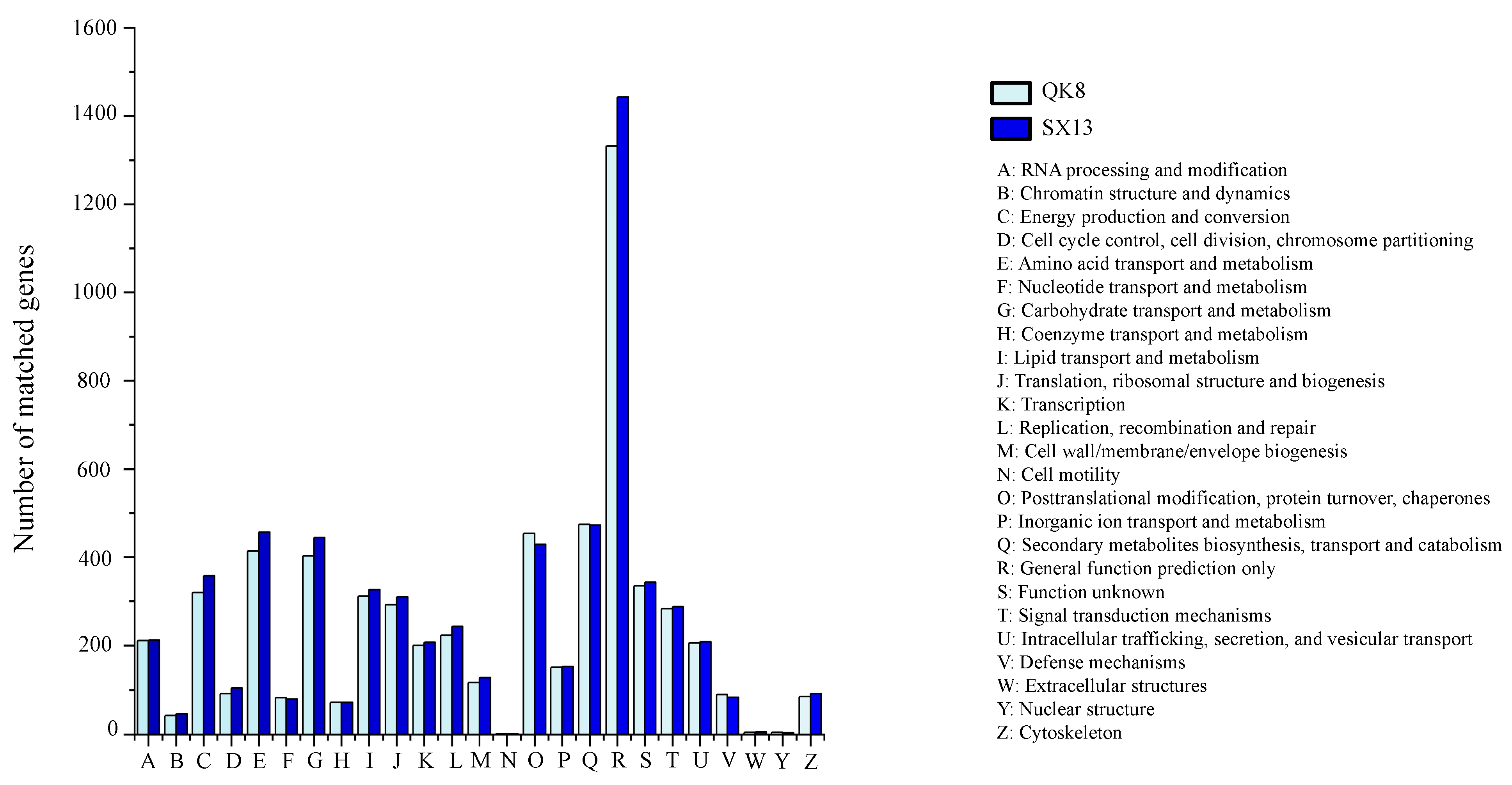
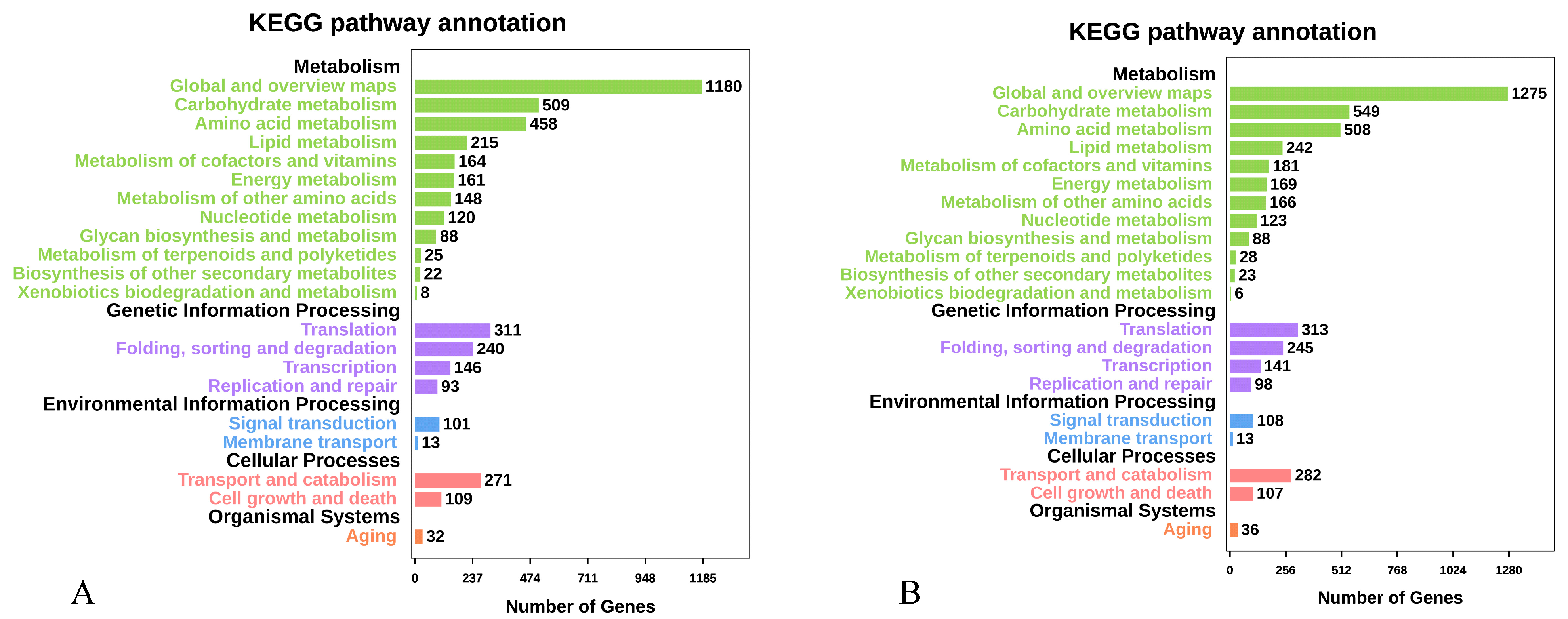
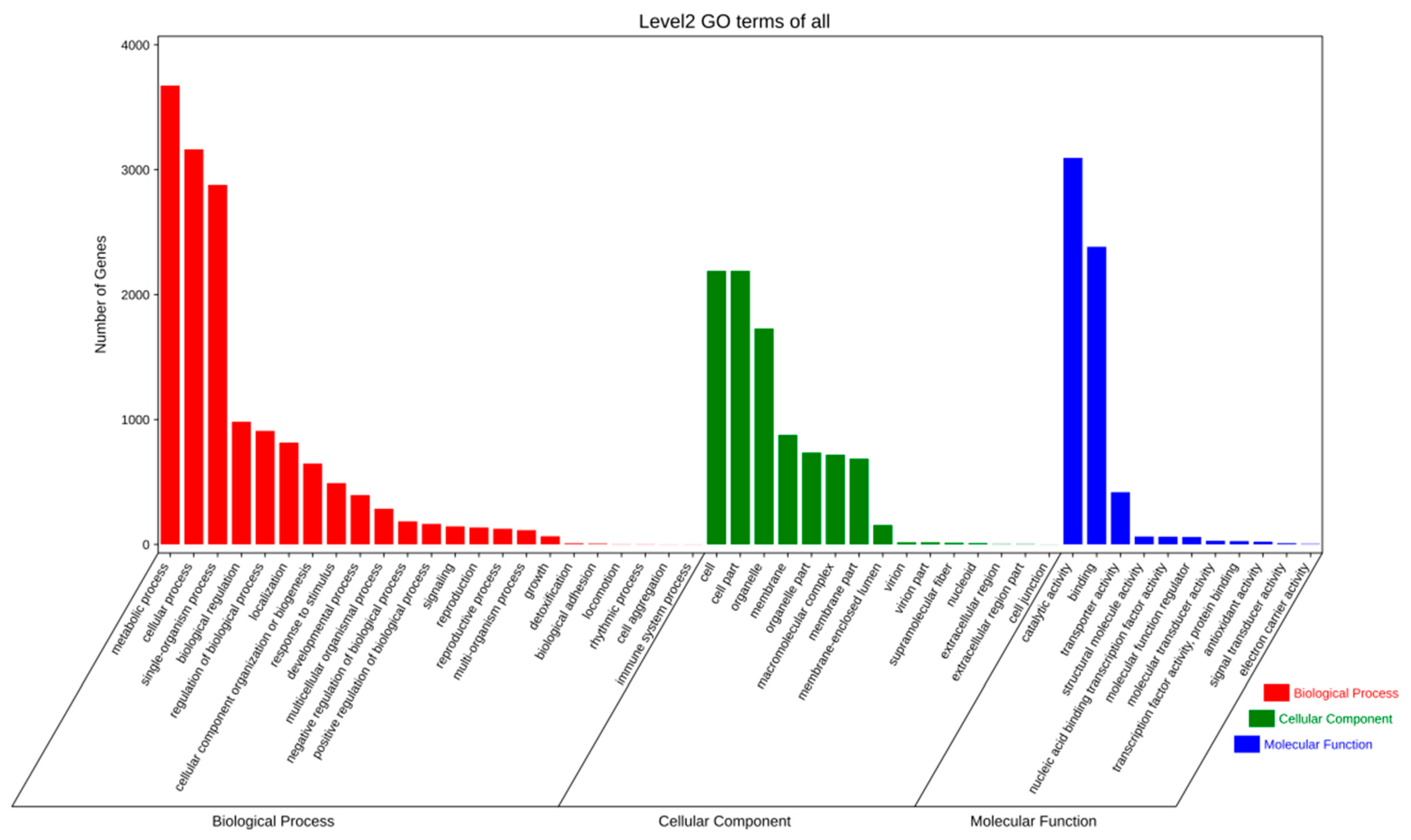
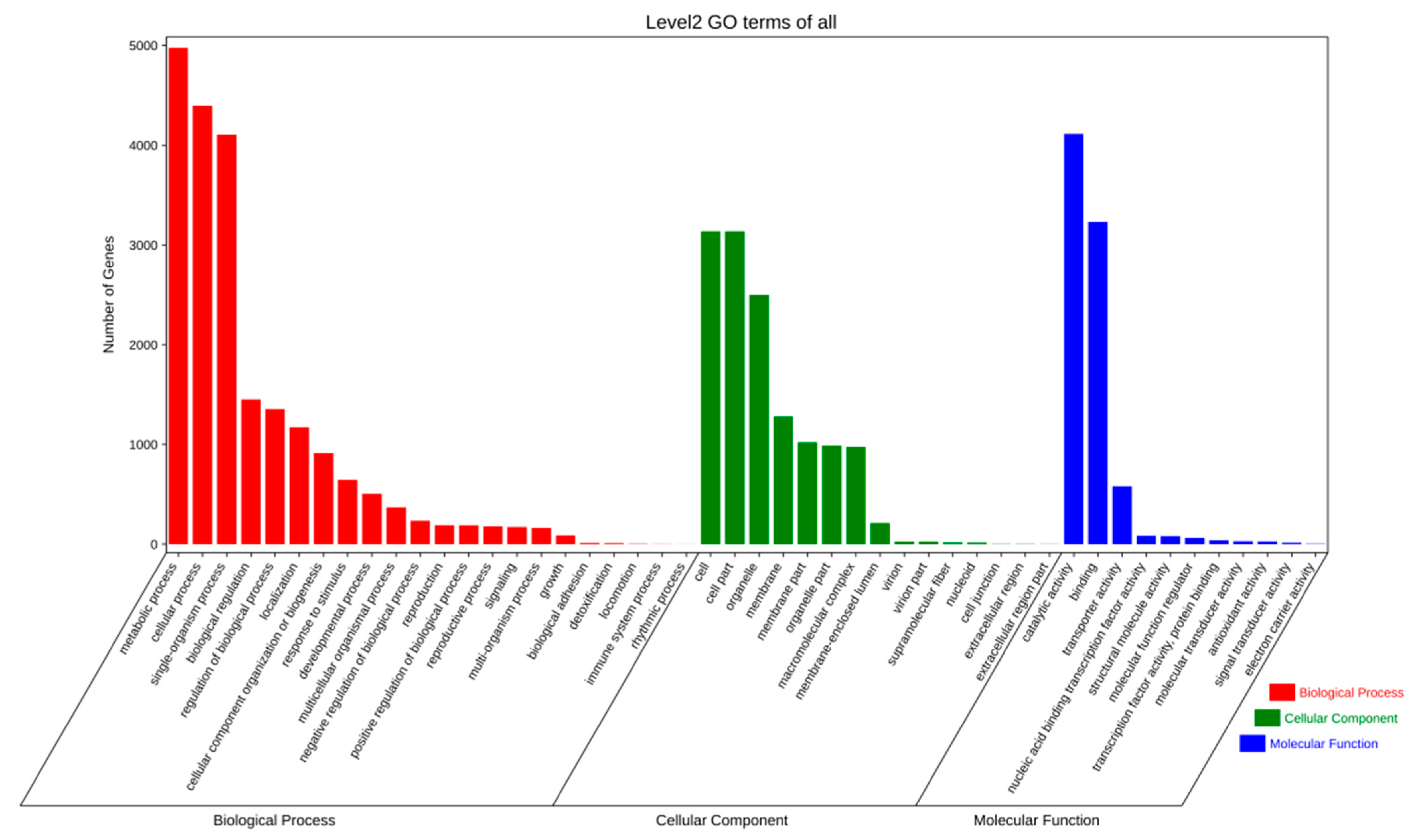
2.2.2. Gene Prediction and Annotation
2.2.3. CAZyme Annotation
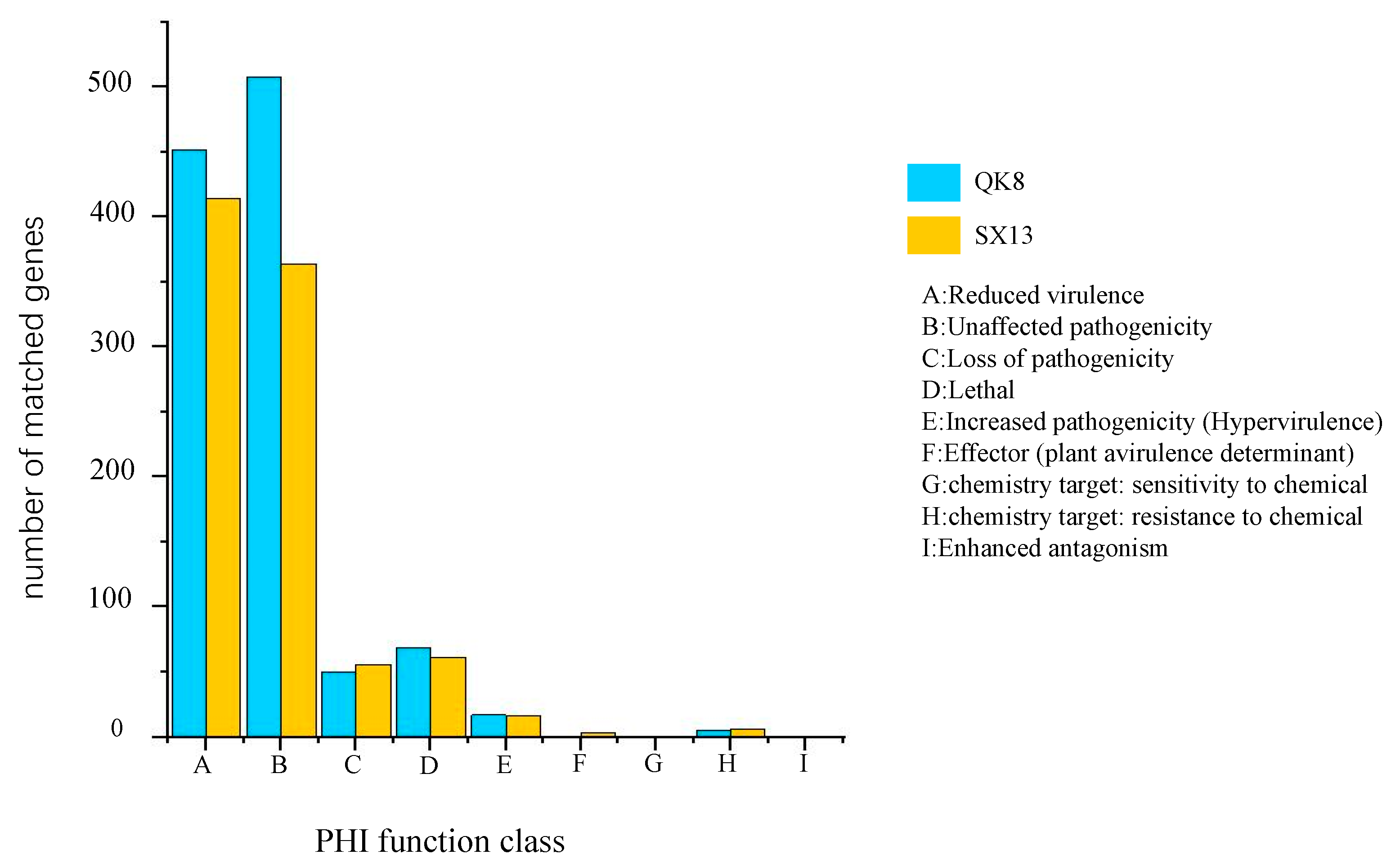
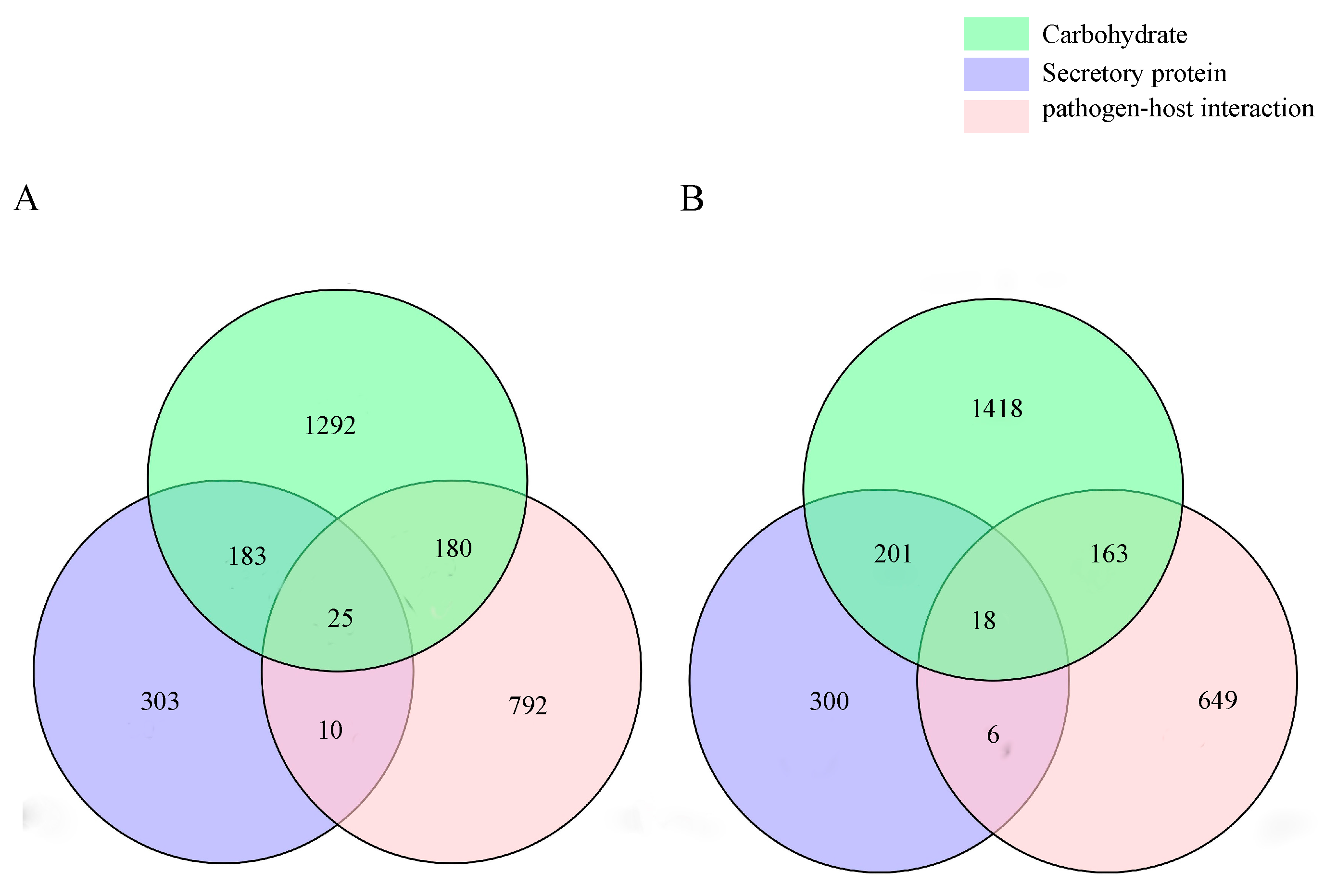
2.2.4. Pathogenicity-Related Gene Analysis
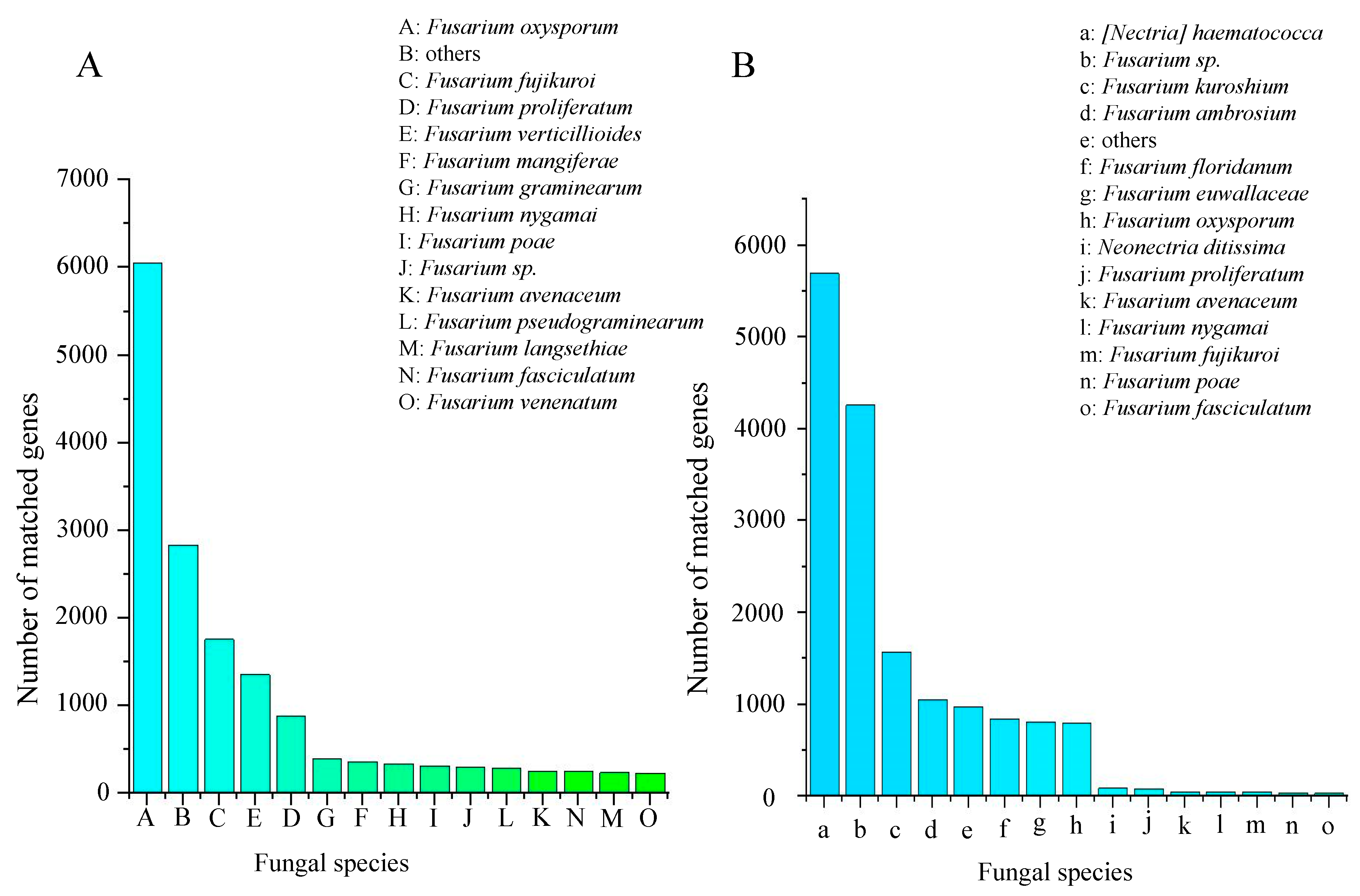
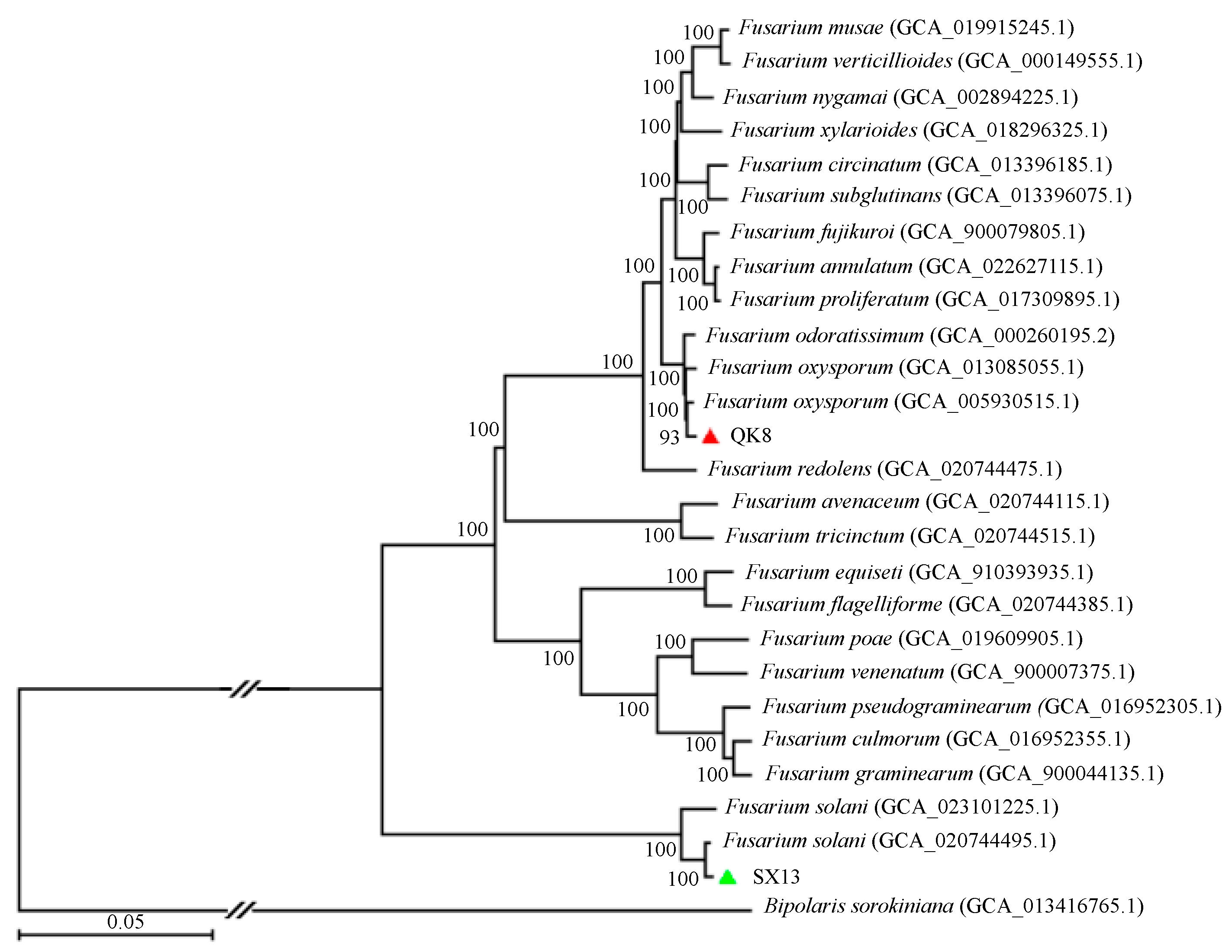
2.2.5. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Source of Pathogenic Fungi of G. elata Brown Rot
4.2. Determination of Biological Characteristics
4.3. Draft Genome Sequencing, Assembly, and Repetitive Sequences Analysis
4.4. Gene Prediction and Annotation
4.5. Analysis of Secretory and Effector Proteins
4.6. Phylogeny and Homology Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wollenweber, H.W.; Reinking, O.A. The Fusaria: Their description injurious effects and control. Paul Parey Berlin. 1935, 8, 1–135. [Google Scholar]
- Fravel, D.; Olivain, C.; Alabouvette, C. Fusarium oxysporum and its biocontrol. New Phytol. 2003, 157, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Michielse, C.B.; Rep, M. Pathogen profile update: Fusarium oxysporum. Mol. Plant Pathol. 2009, 10, 311. [Google Scholar] [CrossRef]
- Schroers, H.J.; Samuels, G.J.; Zhang, N.; Short, D.P.; Juba, J.; Geiser, D.M. Epitypification of Fusisporium (Fusarium) solani and its assignment to a common phylogenetic species in the Fusarium solani species complex. Mycologia 2016, 108, 806–819. [Google Scholar] [CrossRef] [Green Version]
- Villarino, M.; De la Lastra, E.; Basallote-Ureba, M.J.; Capote, N.; Larena, I.; Melgarejo, P.; De Cal, A. Characterization of Fusarium solani populations associated with Spanish strawberry crops. Plant Dis. 2019, 103, 1974–1982. [Google Scholar] [CrossRef]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojemann, L.M.; Nelson, W.L.; Shin, D.S.; Rowe, A.O.; Buchanan, R.A. Tian ma, an ancient Chinese herb, offers new options for the treatment of epilepsy and other conditions. Epilepsy Behav. 2006, 8, 376–383. [Google Scholar] [CrossRef]
- Zhan, H.D.; Zhou, H.Y.; Sui, Y.P.; Du, X.L.; Wang, W.H.; Dai, L.; Sui, F.; Hou, H.R.; Jiang, T.L. The rhizome of Gastrodia elata Blume–An ethnopharmacological review. J. Ethnopharmacol. 2016, 189, 361–385. [Google Scholar] [CrossRef]
- Wang, X.; Bauw, G.; Van Damme, E.J.; Peumans, W.J.; Chen, Z.L.; Van Montagu, M.; Angenon, G.; Dillen, W. Gastrodianin-like mannose-binding proteins: A novel class of plant proteins with antifungal properties. Plant J. 2001, 25, 651–661. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, M.; Yang, Z.; Li, C. Botrytis cinerea causes flower gray mold in Gastrodia elata in China. Crop Prot. 2022, 155, 105923. [Google Scholar] [CrossRef]
- Li, J.; Li, C. Fusarium solani Causing Root Rot Disease on Gastrodia elata in Shaxi, China. Plant Dis. 2022, 106, 320. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, J.; Chen, X.; Huang, M.; Shao, X.; Wang, Y. Identification of the pathogen of Gastrodia elata brown rot in Guizhou. J. Plant Pathol. 2022, 4, 699–701. [Google Scholar] [CrossRef]
- Montesinos-Matías, R.; Ordaz-Hernández, A.; Angel-Cuapio, A.; Colin-Bonifacio, Y.; Garcia-Garcia, R.E.; Ángel-Sahagún, C.A.; Arredondo-Bernal, H.C. Principal component analysis of the biological characteristics of entomopathogenic fungi in nutrient-limited and cuticle-based media. J. Basic Microbiol. 2021, 61, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Liu, T.; Guo, Z.; Zhang, L.; Mao, L.; Zhang, Y.; Jiang, H. Fumigation and contact activities of 18 plant essential oils on Villosiclava virens, the pathogenic fungus of rice false smut. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plissonneau, C.; Benevenuto, J.; Mohd-Assaad, N.; Fouché, S.; Hartmann, F.E.; Croll, D. Using population and comparative genomics to understand the genetic basis of effector-driven fungal pathogen evolution. Front. Plant Sci. 2017, 8, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Huang, L.; Huang, J.; Wang, X.; Chen, X.; Zhao, J.; Guo, J.; Zhuang, H.; Qiu, C.; Kang, Z.; et al. High genome heterozygosity and endemic genetic recombination in the wheat stripe rust fungus. Nat. Commun. 2013, 4, 2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, J.; Chen, M.; Zhong, Z.; Tang, W.; Lin, L.; Zhang, X.; Jiang, H.; Zhang, D.; Miao, C.; Wang, Z.; et al. PacBio Sequencing Reveals Transposable Element as a Key Contributor to Genomic Plasticity and Virulence Variation in Magnaporthe oryzae. Mol. Plant 2017, 10, 1465–1468. [Google Scholar] [CrossRef] [Green Version]
- Van Kan, J.A.; Stassen, J.H.; Mosbach, A.; Van Der Lee, T.A.; Faino, L.; Farmer, A.D.; Papasotiriou, D.; Zhou, S.; Seidl, M.F.; Scalliet, G.; et al. A gapless genome sequence of the fungus Botrytis cinerea. Mol. Plant Pathol. 2017, 18, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Mesny, F.; Miyauchi, S.; Thiergart, T.; Pickel, B.; Atanasova, L.; Karlsson, M.; Hüttel, B.; Barry, K.; Haridas, S.; Hacquard, S.; et al. Genetic determinants of endophytism in the Arabidopsis root mycobiome. Nat. Commun. 2021, 12, 7227. [Google Scholar] [CrossRef]
- Kim, J.A.; Jeon, J.; Park, S.Y.; Kim, K.T.; Choi, G.; Lee, H.J.; Kim, Y.; Yang, H.S.; Yeo, J.H.; Kim, S. Genome sequence of an endophytic fungus, Fusarium solani JS-169, which has antifungal activity. Genome Announc. 2017, 5, e01071-17. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.Y.; Ma, T.; Zhao, N.; Zhang, X.; Fang, B.; Huang, L. Whole-Genome Sequencing and Comparative Genome Analysis of Fusarium solani-melongenae Causing Fusarium Root and Stem Rot in Sweetpotatoes. Microbiol. Spectr. 2022, 10, e00683-22. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yu, H.; Jia, Y.; Dong, Q.; Steinberg, C.; Alabouvette, C.; Edel-Hermann, V.; Kistler, H.; Ye, K.; Guo, L.; et al. Chromosome-scale genome assembly of Fusarium oxysporum strain Fo47, a fungal endophyte and biocontrol agent. Mol. Plant-Microbe Interact. 2020, 33, 1108–1111. [Google Scholar] [CrossRef]
- Urbaniak, C.; Massa, G.; Hummerick, M.; Khodadad, C.; Schuerger, A.; Venkateswaran, K. Draft genome sequences of two Fusarium oxysporum isolates cultured from infected Zinnia hybrida plants grown on the International Space Station. Genome Announc. 2018, 6, e00326-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xingxing, P.; Khan, R.A.A.; Yan, L.; Yuhong, Y.; Bingyan, X.; Zhenchuan, M.; Jian, L. Draft genome resource of Fusarium oxysporum f. sp. capsici, the infectious agent of pepper Fusarium wilt. Mol. Plant-Microbe Interact. 2021, 34, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Jelinski, N.A.; Broz, K.; Jonkers, W.; Ma, L.J.; Kistler, H.C. Effector gene suites in some soil isolates of Fusarium oxysporum are not sufficient predictors of vascular wilt in tomato. Phytopathology 2017, 107, 842–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.M.; Lukasiewicz, J.; Farrer, R.; van Dam, P.; Bertoldo, C.; Rep, M. Comparative genomics of Fusarium oxysporum f. sp. melonis reveals the secreted protein recognized by the Fom-2 resistance gene in melon. New Phytol. 2016, 209, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.N.; Chen, C.J.; Li, Q.Q.; Xu, F.R.; Cheng, Y.X.; Dong, X. Monitoring antifungal agents of Artemisia annua against Fusarium oxysporum and Fusarium solani, associated with Panax notoginseng root-rot disease. Molecules 2019, 24, 213. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Chen, J.; Zhao, Y.; Cai, H.; Guo, C. Bacillus subtilis HSY21 can reduce soybean root rot and inhibit the expression of genes related to the pathogenicity of Fusarium oxysporum. Pestic. Biochem. Physiol. 2021, 178, 104916. [Google Scholar] [CrossRef]
- Degani, O.; Kalman, B. Assessment of commercial fungicides against Onion (Allium cepa) basal rot disease caused by Fusarium oxysporum f. sp. cepae and Fusarium acutatum. J. Fungi. 2021, 7, 235. [Google Scholar] [CrossRef]
- Mondani, L.; Chiusa, G.; Battilani, P. Chemical and biological control of Fusarium species involved in garlic dry rot at early crop stages. Eur. J. Plant Pathol. 2021, 160, 575–587. [Google Scholar] [CrossRef]
- Tan, H.; Yu, Y.; Tang, J.; Liu, T.; Miao, R.; Huang, Z.; Martin, F.M.; Peng, W. Build Your Own Mushroom Soil: Microbiota Succession and Nutritional Accumulation in Semi-Synthetic Substratum Drive the Fructification of a Soil-Saprotrophic Morel. Front Microbiol. 2021, 24, 656656. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Liu, T.; Yu, Y.; Tang, J.; Jiang, L.; Martin, F.M.; Peng, W. Morel production related to soil microbial diversity and evenness. Microbiol. Spectr. 2021, 9, e00229-21. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Kohler, A.; Miao, R.; Liu, T.; Zhang, Q.; Zhang, B.; Jiang, L.; Wang, Y.; Xie, L.; Martin, F.M.; et al. Multi-omic analyses of exogenous nutrient bag decomposition by the black morel Morchella importuna reveal sustained carbon acquisition and transferring. Environ. Microbiol. 2019, 21, 3909–3926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Wen, T.; Zhang, H.; Zhao, M.; Penton, C.R.; Thomashow, L.S.; Shen, Q. Predicting disease occurrence with high accuracy based on soil macroecological patterns of Fusarium wilt. ISME J. 2020, 14, 2936–2950. [Google Scholar] [CrossRef]
- Sai, S.; Ayukawa, Y.; Gan, P.; Masuda, S.; Komatsu, K.; Shirasu, K.; Arie, T. High-quality draft genome sequence of Fusarium oxysporum f. sp. cubense strain 160527, a causal agent of Panama disease. Microbiol. Resour. Announc. 2019, 8, e00654-19. [Google Scholar] [CrossRef] [Green Version]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Aoki, T.; Ariyawansa, H.A.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Schoch, C.L.; et al. Fungal taxonomy and sequence-based nomenclature. Nat. Microbiol. 2021, 6, 540–548. [Google Scholar] [CrossRef]
- Garmendia, G.; Umpierrez-Failache, M.; Ward, T.J.; Vero, S. Development of a PCR-RFLP method based on the transcription elongation factor 1-α gene to differentiate Fusarium graminearum from other species within the Fusarium graminearum species complex. Food Microbiol. 2018, 70, 28–32. [Google Scholar] [CrossRef]
- Coleman, J.J.; Rounsley, S.D.; Rodriguez-Carres, M.; Kuo, A.; Wasmann, C.C.; Grimwood, J.; Schmutz, J.; Taga, M.; White, G.J.; Vanetten, H.D.; et al. The genome of Nectria haematococca: Contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 2009, 5, e1000618. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.J.; Van Der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Rep, M.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 18, 367–373. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ma, L.J. Deciphering Pathogenicity of Fusarium oxysporum From a Phylogenomics Perspective. Adv. Genet. 2017, 100, 179–209. [Google Scholar] [CrossRef]
- Seehausen, O.; Butlin, R.K.; Keller, I.; Wagner, C.E.; Boughman, J.W.; Hohenlohe, P.A.; Peichel, C.L.; Saetre, G.P.; Bank, C.; Widmer, A.; et al. Genomics and the origin of species. Nat. Rev. Genet. 2014, 15, 176–192. [Google Scholar] [CrossRef] [Green Version]
- Kubicek, C.P.; Starr, T.L.; Glass, N.L. Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 2014, 52, 427–451. [Google Scholar] [CrossRef] [PubMed]
- García-Maceira, F.I.; Di Pietro, A.; Huertas-González, M.D.; Ruiz-Roldán, M.C.; Roncero, M.I.G. Molecular characterization of an endopolygalacturonase from Fusarium oxysporum expressed during early stages of infection. Appl. Environ. Microbiol. 2001, 67, 2191–2196. [Google Scholar] [CrossRef] [Green Version]
- Ospina-Giraldo, M.D.; Griffith, J.G.; Laird, E.W.; Mingora, C. The CAZyome of Phytophthora spp.: A comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus Phytophthora. Bmc Genom. 2010, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.S.; Xie, X.L.; Liang, G.; Gong, F.; Wang, Y.; Wei, X.Q.; Wang, Q.; Ji, Z.L.; Chen, Q.X. The GH18 family of chitinases: Their domain architectures, functions and evolutions. Glycobiology 2012, 22, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, K.V.; Haitjema, C.H.; Henske, J.K.; Gilmore, S.P.; Borges-Rivera, D.; Lipzen, A.; Brewer, H.M.; Purvine, S.O.; Wright, A.T.; O’Malley, M.A.; et al. Early-branching gut fungi possess a large, comprehensive array of biomass-degrading enzymes. Science 2016, 351, 1192–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruninger, R.J.; Nguyen, T.T.; Reid, I.D.; Yanke, J.L.; Wang, P.; Abbott, D.W.; Tsang, A.; McAllister, T. Application of transcriptomics to compare the carbohydrate active enzymes that are expressed by diverse genera of anaerobic fungi to degrade plant cell wall carbohydrates. Front. Microbiol. 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Z.; Li, Y.; Jiang, Y.; Xu, J.; Li, J.; Luo, L. Genome Sequence Analysis of the Fungal Pathogen Fusarium graminearum Using Oxford Nanopore Technology. J. Fungi. 2021, 7, 699. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Shin, Y.K.; Lee, S.W.; Wimonmuang, K.; Kang, K.B.; Lee, Y.S.; Yun, S.H. FgPKS7 is an essential player in mating-type-mediated regulatory pathway required for completing sexual cycle in Fusarium graminearum. Environ. Microbiol. 2021, 23, 1972–1990. [Google Scholar] [CrossRef]
- Kwon, J.H.; Choi, O.; Kang, B.; Lee, Y.; Park, J.; Kang, D.W.; Han, I.; Park, E.J.; Kim, J. Identification of Neocosmospora ipomoeae causing tomato stem rot in Korea. Australas. Plant Dis. Notes 2017, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, A.; Ghosh, S.; Sahoo, D.; Jha, G. Fungal effectors, the double edge sword of phytopathogens. Curr. Genet. 2021, 67, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Liu, H.; Li, Z.; Ke, X.; Dou, D.; Gao, X.; Song, N.; Dai, Q.; Wu, Y.; Huang, L.; et al. Genome sequence of Valsa canker pathogens uncovers a potential adaptation of colonization of woody bark. New Phytol. 2015, 208, 1202–1216. [Google Scholar] [CrossRef]
- Sossah, F.L.; Liu, Z.; Yang, C.; Okorley, B.A.; Sun, L.; Fu, Y.; Li, Y. Genome sequencing of Cladobotryum protrusum provides insights into the evolution and pathogenic mechanisms of the cobweb disease pathogen on cultivated mushroom. Genes 2019, 10, 124. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Wang, J.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 274, 578–579. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Schatz, M.C.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.L.; Chen, Y.; Xie, S.Q.; Chen, K.N.; Wang, Y.; Han, Y.; Luo, F.; Xie, Z. MECAT: Fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat. Methods 2017, 14, 1072–1074. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, Q.; Earl, A.M.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Ye, C.; Hill, C.M.; Wu, S.; Ruan, J.; Ma, Z.S. DBG2OLC: Efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 2016, 6, 31900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.; Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 51, 59. [Google Scholar] [CrossRef] [Green Version]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Sherlock, G.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Rutherford, K.; Irvine, A.; Pedro, H.; Pant, R.; Sadanadan, V.; Khamari, L.; Billal, S.; Hammond-Kosack, K.E.; et al. PHI-base: A new interface and further additions for the multi-species pathogen–host interactions database. Nucleic Acids Res. 2017, 45, D604–D610. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; Heijne, G.V.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Kroh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kall, L.; Krogh, A.; Sonnhammer, E.L. Advantages of combined transmembrane topology and signal peptide prediction—The Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emanuelsson, O.; Nielsen, H.; Brunak, S.; Von Heijne, G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000, 300, 1005–1016. [Google Scholar] [CrossRef] [Green Version]
- Pierleoni, A.; Martelli, P.L.; Casadio, R. PredGPI: A GPI-anchor predictor. BMC Bioinform. 2008, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 564, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A.; Aberer, A.J.; Goll, C.; Smith, S.A.; Berger, S.A.; Izquierdo-Carrasco, F. RAxML-Light: A tool for computing terabyte phylogenies. Bioinformatics 2012, 28, 2064–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scaffold Characteristics | QK8 | SX13 |
|---|---|---|
| Total counts of contig sequences | 21 | 21 |
| N50 Length (bp) | 4,657,513 | 3,724,189 |
| N90 Length (bp) | 2,323,049 | 1,334,751 |
| Longest Length (bp) | 6,849,339 | 6,503,946 |
| Shortest Length (bp) | 106,531 | 704,098 |
| tRNA | 282 | 275 |
| rRNA | 63 | 70 |
| Average Length (bp) | 2,438,320 | 2,627,238 |
| Genome coverage (%) | 98.04 | 97.91 |
| GC content (%) | 47.21 | 50.61 |
| Total Size (bp) | 51,204,719 | 55,171,989 |
| Gene Characteristics | ||
| Number of genes | 15,917 | 16,650 |
| Exon average length (bp) | 1557 | 1598 |
| Genome GC percent (%) | 47.21% | 50.61 |
| Exon Gene GC percent (%) | 51.43 | 54.91 |
| Total Size (bp) | 24,778,044 | 26,605,002 |
| Annotation Database | Number of Genes | |
|---|---|---|
| QK8 | SX13 | |
| Carbohydrate-active Enzymes Database (CAZy) | 1680 | 1800 |
| Kyoto Encyclopedia of Genes and Genomes (KEGG) | 15,595 | 15,861 |
| Eukaryotic Orthologous Groups (KOG) | 7045 | 7395 |
| Gene Ontology (GO) | 5540 | 7513 |
| Cytochrome P450 monooxygenase (P450) | 156 | 152 |
| Pathogen–Host Interactions Database (PHI) | 1007 | 836 |
| NCBI Non-Redundant Protein Sequence Database (NR) | 15,877 | 16,384 |
| Protein families database (Pfam) | 11,770 | 12,267 |
| ID | Uncharacterized Proteins | Hypothetical Proteins | Functional Genes | Total |
|---|---|---|---|---|
| QK8 | 2678 | 8377 | 4819 | 15,874 |
| SX13 | 186 | 13,119 | 3079 | 16,384 |
| Isolate | PLs | AAs | GHs | GTs | CEs | CBMs | Total |
|---|---|---|---|---|---|---|---|
| QK8 | 41 | 198 | 649 | 405 | 140 | 247 | 1680 |
| SX13 | 43 | 263 | 653 | 444 | 138 | 259 | 1800 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; He, K.; Zhang, Q.; Wu, X.; Li, Z.; Pan, X.; Wang, Y.; Li, C.; Zhang, M. Draft Genome and Biological Characteristics of Fusarium solani and Fusarium oxysporum Causing Black Rot in Gastrodia elata. Int. J. Mol. Sci. 2023, 24, 4545. https://doi.org/10.3390/ijms24054545
Li J, He K, Zhang Q, Wu X, Li Z, Pan X, Wang Y, Li C, Zhang M. Draft Genome and Biological Characteristics of Fusarium solani and Fusarium oxysporum Causing Black Rot in Gastrodia elata. International Journal of Molecular Sciences. 2023; 24(5):4545. https://doi.org/10.3390/ijms24054545
Chicago/Turabian StyleLi, Jinshao, Ke He, Qian Zhang, Xiaoyi Wu, Zhong Li, Xuejun Pan, Yong Wang, Cheng Li, and Manman Zhang. 2023. "Draft Genome and Biological Characteristics of Fusarium solani and Fusarium oxysporum Causing Black Rot in Gastrodia elata" International Journal of Molecular Sciences 24, no. 5: 4545. https://doi.org/10.3390/ijms24054545