


TERT Extra-Telomeric Roles: Antioxidant Activity and Mitochondrial Protection
 , , , , ,
, , , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
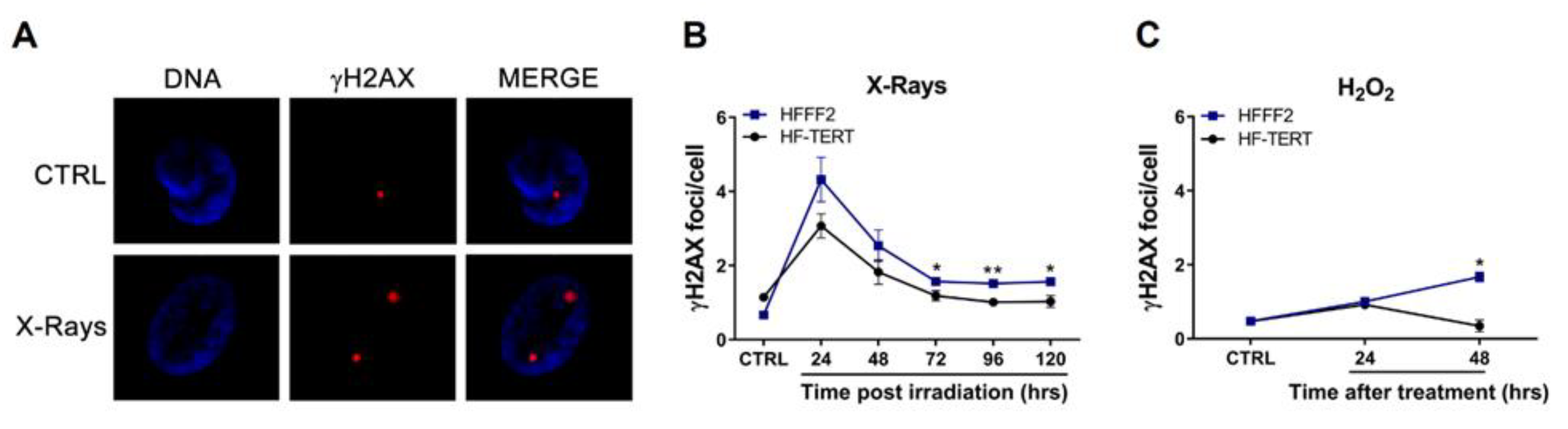
2.1. HF-TERT Cells Respond Better to Physical and Chemical Agent Treatments
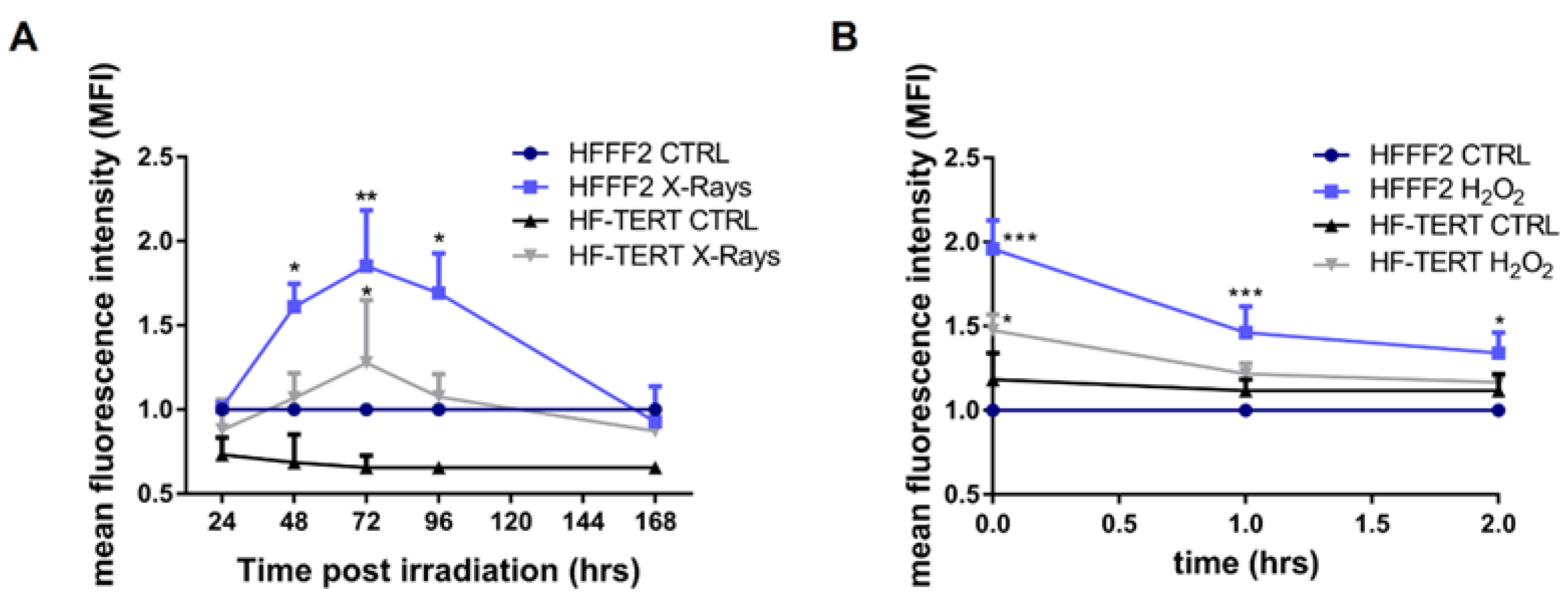
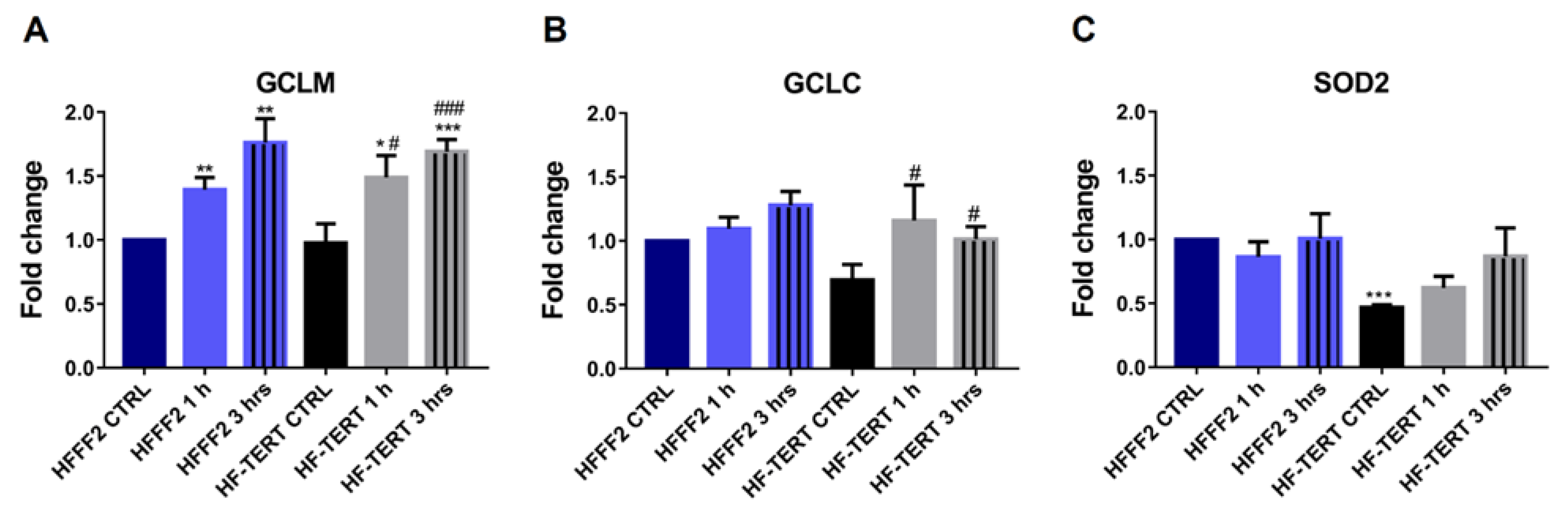
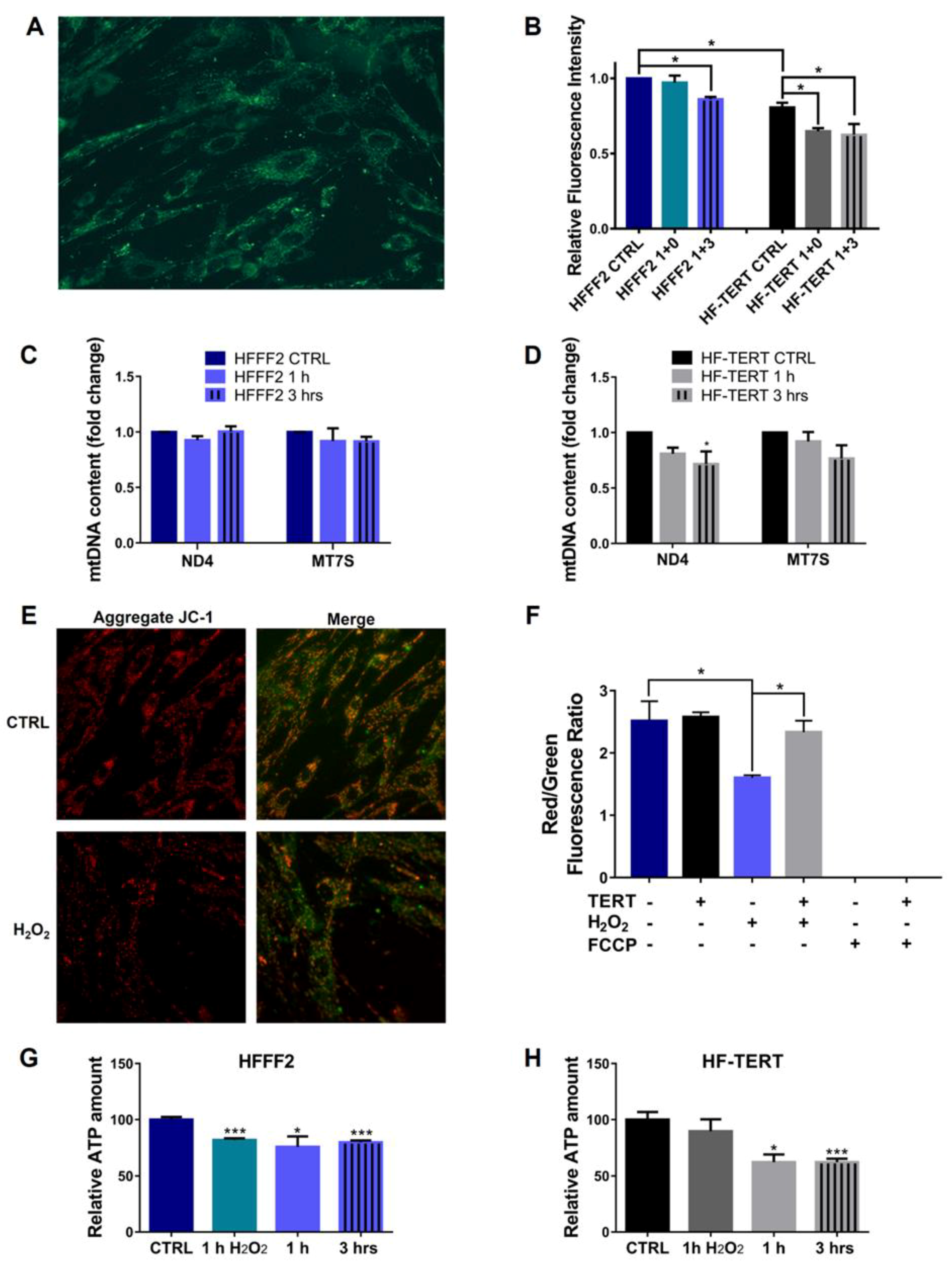
2.2. hTERT-Overexpressing Cells Display Enhanced Antioxidant Defense
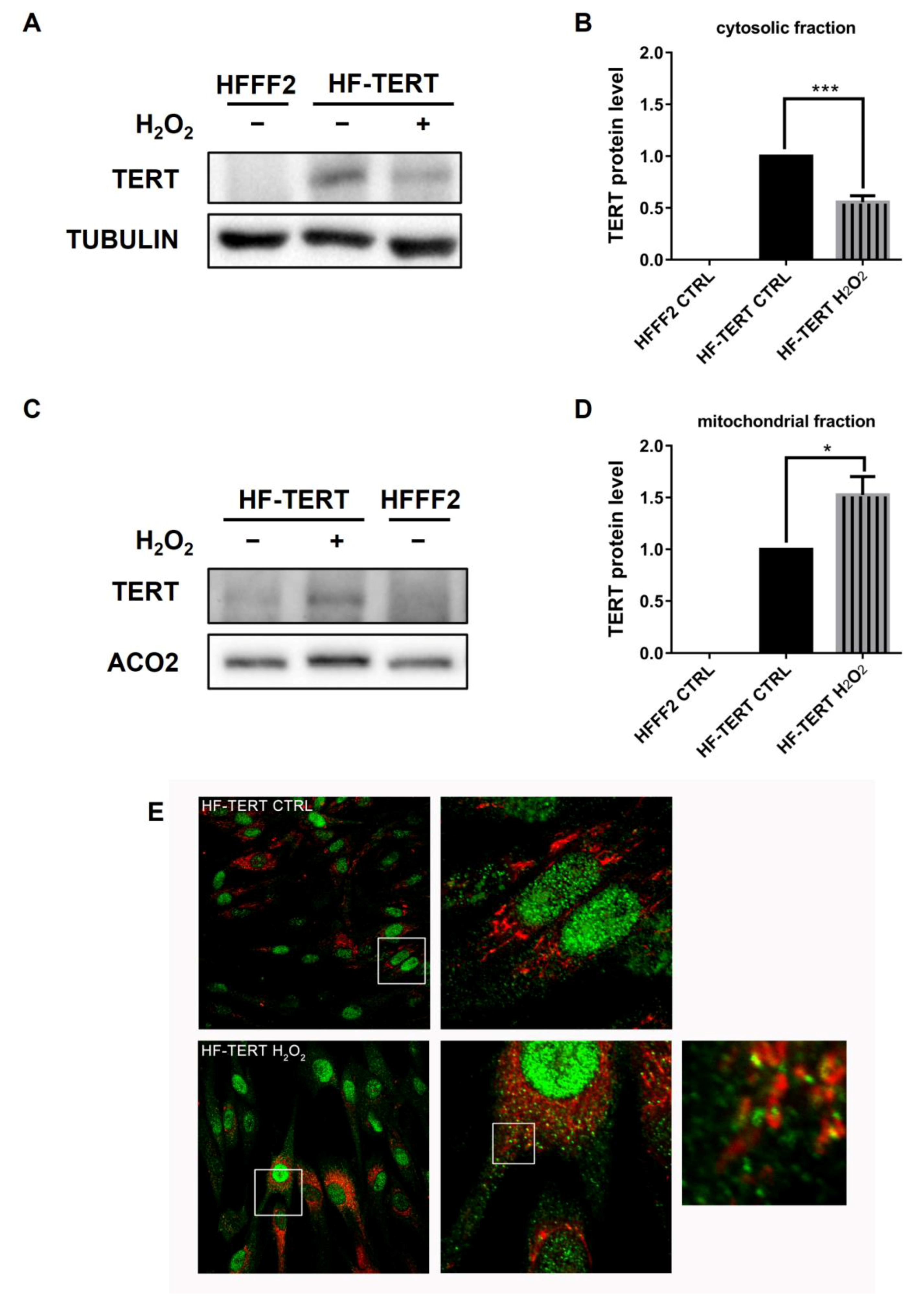
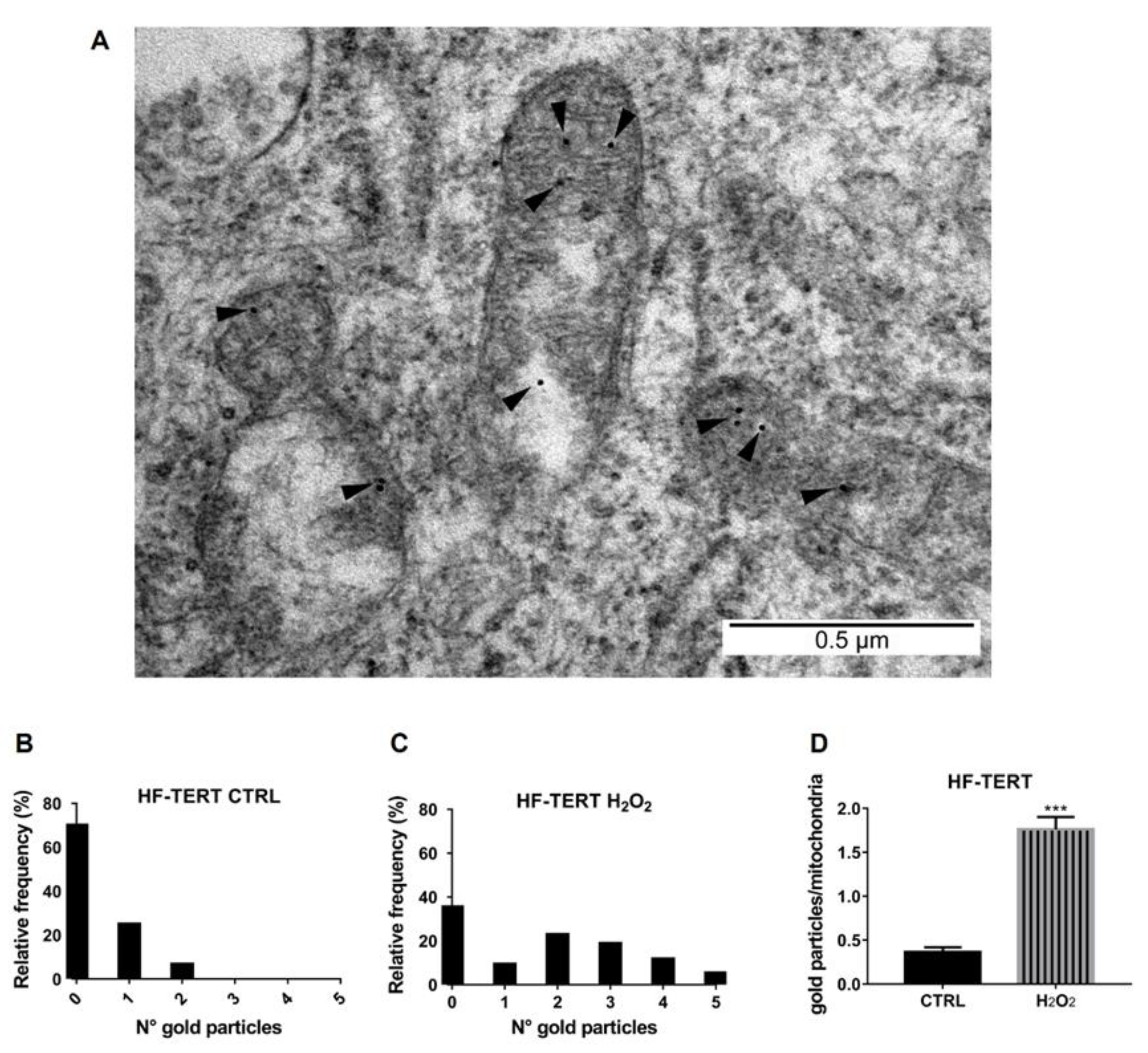
2.3. TERT Is Accumulated in Mitochondria under Oxidative Stress
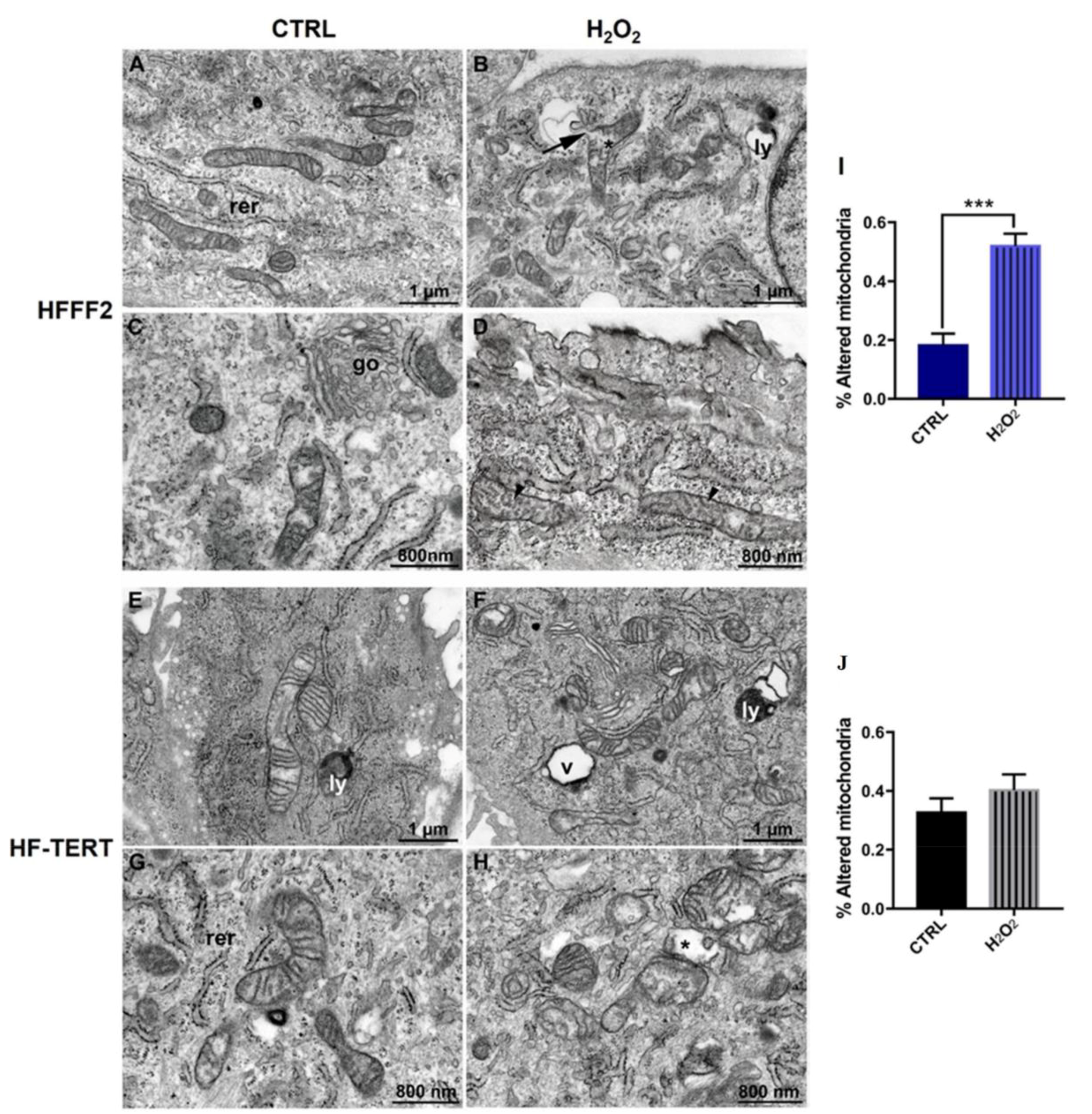
2.4. Effects of TERT on Mitochondrial Ultrastructural Features and Function
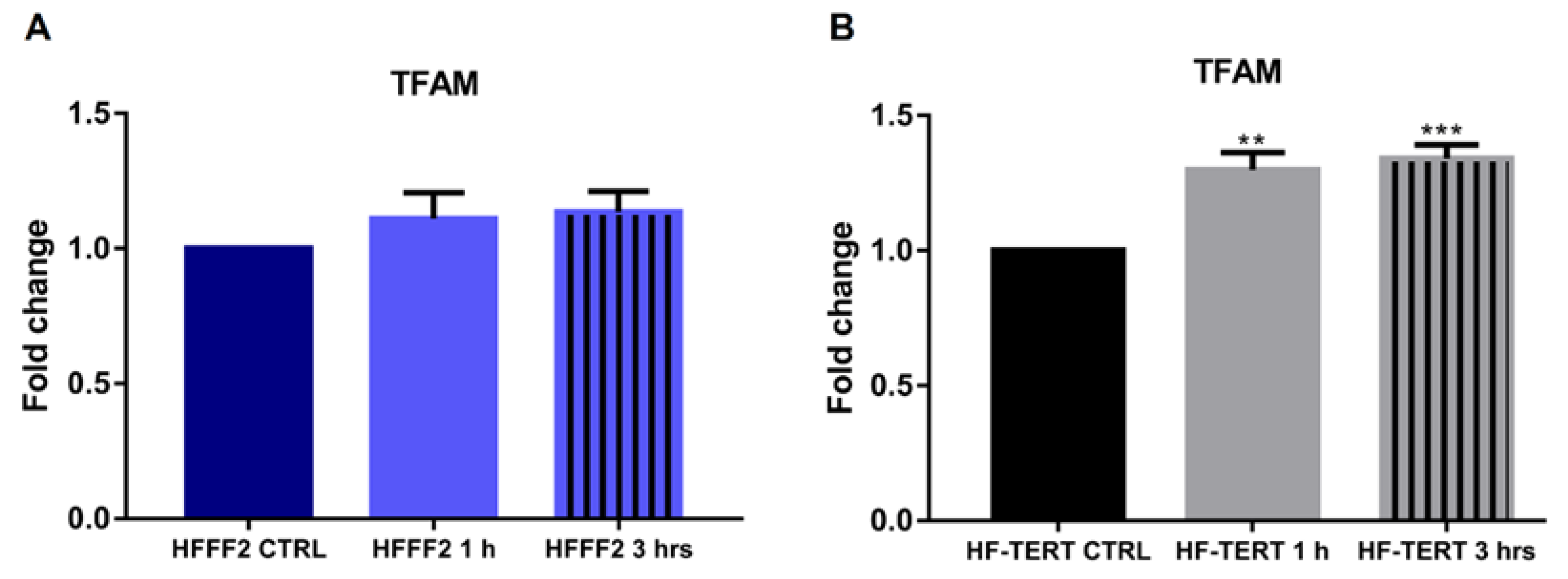
2.5. TFAM Expression Increases in HF-TERT Cells after Treatment
3. Discussion
4. Materials and Methods
4.1. Cells, Culture Condition and Retroviral Transduction
4.2. Real-time Quantitative—Telomeric Repeat Amplification Protocol Assay (RQ-TRAP)
4.3. H2O2 and X-rays Treatment
4.4. Growth Curves
4.5. Genomic and Telomeric γH2AX Immunofluorescence Staining
4.6. Collection of Chromosome Spreads and Quantitative-Fluorescence “in situ” Hybridization Analysis (Q-FISH)
4.7. Intracellular Reactive Oxygen Species (ROS) Determination
4.8. Analysis of Biogenesis and Glutathione Genes Expression
4.9. Protein Extraction from Whole Cell and Subcellular Fractions
4.10. Western Blotting
4.11. Immunofluorescence Co-Staining of Mitochondria
4.12. Immunogold at TEM
4.13. Detection of Mitochondrial Membrane Potential (ΔΨm)
4.14. ATP Content
4.15. mtDNA Copy Number and Mitochondrial Mass
4.16. Ultrastructural Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greider, C.W.; Blackburn, E.H. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 1987, 51, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, A.M. Principle of marginotomy in template synthesis of polynucleotides. Dokl. Akad. Nauk SSSR 1971, 201, 1496–1499. [Google Scholar] [PubMed]
- Olovnikov, A.M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Wellinger, R.J.; Sen, D. The DNA structures at the ends of eukaryotic chromosomes. Eur. J. Cancer 1997, 33, 735–749. [Google Scholar] [CrossRef]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J.; et al. The RNA component of human telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef]
- Cong, Y.S.; Wen, J.; Bacchetti, S. The human telomerase catalytic subunit hTERT: Organization of the gene and characterization of the promoter. Hum. Mol. Genet. 1999, 8, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Fu, D.; Collins, K. Distinct biogenesis pathways for human telomerase RNA and H/ACA small nucleolar RNAs. Mol. Cell 2003, 11, 1361–1372. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Collins, K. Control of telomerase action at human telomeres. Nat. Struct. Mol. Biol. 2015, 22, 848–852. [Google Scholar] [CrossRef] [Green Version]
- Venteicher, A.S.; Abreu, E.B.; Meng, Z.; McCann, K.E.; Terns, R.M.; Veenstra, T.D.; Terns, M.P.; Artandi, S.E. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 2009, 323, 644–648. [Google Scholar] [CrossRef] [Green Version]
- Beattie, T.L.; Zhou, W.; Robinson, M.O.; Harrington, L. Reconstitution of human telomerase activity in vitro. Curr. Biol. 1998, 8, 177–180. [Google Scholar] [CrossRef] [Green Version]
- Weinrich, S.L.; Pruzan, R.; Ma, L.; Ouellette, M.; Tesmer, V.M.; Holt, S.E.; Bodnar, A.G.; Lichtsteiner, S.; Kim, N.W.; Trager, J.B.; et al. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat. Genet. 1997, 17, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Counter, C.M.; Gupta, J.; Harley, C.B.; Leber, B.; Bacchetti, S. Telomerase Activity in Normal Leukocytes and in Hematologic Malignancies. Blood 1995, 85, 2315–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cifuentes-Rojas, C.; Shippen, D.E. Telomerase regulation. Mutat. Res. 2012, 730, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.C.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Murnane, J.P.; Sabatier, L.; Marder, B.A.; Morgan, W.F. Telomere dynamics in an immortal human cell line. EMBO J. 1994, 13, 4953. [Google Scholar] [CrossRef]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Ségal-Bendirdjian, E.; Geli, V. Non-canonical Roles of Telomerase: Unraveling the Imbroglio. Front. Cell Dev. Biol. 2019, 7, 332. [Google Scholar] [CrossRef]
- Choi, J.; Southworth, L.K.; Sarin, K.Y.; Venteicher, A.S.; Ma, W.; Chang, W.; Cheung, P.; Jun, S.; Artandi, M.K.; Shah, N.; et al. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet. 2008, 4, 124–138. [Google Scholar] [CrossRef] [Green Version]
- Ding, D.; Xi, P.; Zhou, J.; Wang, M.; Cong, Y.S. Human telomerase reverse transcriptase regulates MMP expression independently of telomerase activity via NF-κB-dependent transcription. FASEB J. 2013, 27, 4375–4383. [Google Scholar] [CrossRef]
- Xiang, H.; Wang, J.; Mao, Y.; Wan-Cheng Li, D.; Liu, M.; Reddy, V.N. Human telomerase accelerates growth of lens epithelial cells through regulation of the genes mediating RB/E2F pathway. Oncogene 2002, 21, 3784–3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Przyborski, S.; Cooke, M.J.; Zhang, X.; Stewart, R.; Anyfantis, G.; Atkinson, S.P.; Saretzki, G.; Armstrong, L.; Lako, M. A key role for telomerase reverse transcriptase unit in modulating human embryonic stem cell proliferation, cell cycle dynamics, and in vitro differentiation. Stem Cells 2008, 26, 850–863. [Google Scholar] [CrossRef]
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.K.; et al. Telomerase directly regulates NF-κB-dependent transcription. Nat. Cell Biol. 2012, 14, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chan, S.L.; Fu, W.; Mendoza, M.; Mattson, M.P. TERT suppresses apoptotis at a premitochondrial step by a mechanism requiring reverse transcriptase activity and 14-3-3 protein-binding ability. FASEB J. 2003, 17, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Fu, W.; Mattson, M.P. The catalytic subunit of telomerase protects neurons against amyloid beta-peptide-induced apoptosis. J. Neurochem. 2000, 75, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Indran, I.R.; Hande, M.P.; Pervaiz, S. hTERT overexpression alleviates intracellular ROS production, improves mitochondrial function, and inhibits ROS-mediated apoptosis in cancer cells. Cancer Res. 2011, 71, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Martens, A.; Schmid, B.; Akintola, O.; Saretzki, G. Telomerase Does Not Improve DNA Repair in Mitochondria upon Stress but Increases MnSOD Protein under Serum-Free Conditions. Int. J. Mol. Sci. 2019, 21, 27. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Kondo, S.; Tanaka, Y.; Haqqi, T.; Barna, B.P.; Cowell, J.K. Inhibition of telomerase increases the susceptibility of human malignant glioblastoma cells to cisplatin-induced apoptosis. Oncogene 1998, 16, 2243–2248. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, A.; Saretzki, G.; Holm, P.S.; Tiemann, F.; Lorenz, M.; Emrich, T.; Harley, C.B.; von Zglinicki, T. Ribozyme Cleavage of Telomerase mRNA Sensitizes Breast Epithelial Cells to Inhibitors of Topoisomerase1. Cancer Res. 2001, 61, 3053–3061. [Google Scholar]
- Sharma, G.G.; Gupta, A.; Wang, H.; Scherthan, H.; Dhar, S.; Gandhi, V.; Iliakis, G.; Shay, J.W.; Young, C.S.H.; Pandita, T.K. hTERT associates with human telomeres and enhances genomic stability and DNA repair. Oncogene 2003, 22, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Mattiussi, M.; Tilman, G.; Lenglez, S.; Decottignies, A. Human telomerase represses ROS-dependent cellular responses to Tumor Necrosis Factor-α without affecting NF-κB activation. Cell Signal. 2012, 24, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Hoffmann, J.; Rahman, S.; Zeiher, A.M.; Dimmeler, S. Regulation of telomerase activity and anti-apoptotic function by protein-protein interaction and phosphorylation. FEBS Lett. 2003, 536, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.H.; Meyer, J.N.; Skorvaga, M.; Annab, L.A.; Van Houten, B. Mitochondrial hTERT exacerbates free-radical-mediated mtDNA damage. Aging Cell 2004, 3, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Dröse, S.; Büchner, N.; Jakob, S.; Altschmied, J.; Goy, C.; Spyridopoulos, I.; Zeiher, A.M.; Brandt, U.; Dimmeler, S. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Naamati, A.; Regev-Rudzki, N.; Galperin, S.; Lill, R.; Pines, O. Dual targeting of Nfs1 and discovery of its novel processing enzyme, Icp55. J. Biol. Chem. 2009, 284, 30200–30208. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Passos, J.F.; Birket, M.J.; Beckmann, T.; Brings, S.; Peters, H.; Birch-Machin, M.A.; von Zglinicki, T.; Saretzki, G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J. Cell Sci. 2008, 121, 1046–1053. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.H.; Meyer, J.N.; Van Houten, B. Mitochondrial localization of telomerase as a determinant for hydrogen peroxide-induced mitochondrial DNA damage and apoptosis. Hum. Mol. Genet. 2006, 15, 1757–1768. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [Green Version]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef] [Green Version]
- Petersen, S.; Saretzki, G.; Von Zglinicki, T. Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp. Cell Res. 1998, 239, 152–160. [Google Scholar] [CrossRef]
- Opresko, P.L.; Fan, J.; Danzy, S.; Wilson, D.M.; Bohr, V.A. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005, 33, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Coluzzi, E.; Buonsante, R.; Leone, S.; Asmar, A.J.; Miller, K.L.; Cimini, D.; Sgura, A. Transient ALT activation protects human primary cells from chromosome instability induced by low chronic oxidative stress. Sci. Rep. 2017, 7, 43309. [Google Scholar] [CrossRef] [PubMed]
- Coluzzi, E.; Leone, S.; Sgura, A. Oxidative Stress Induces Telomere Dysfunction and Senescence by Replication Fork Arrest. Cells 2019, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, H.; Smogorzewska, A.; De Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- De Vitis, M.; Berardinelli, F.; Coluzzi, E.; Marinaccio, J.; O’sullivan, R.J.; Sgura, A. X-rays Activate Telomeric Homologous Recombination Mediated Repair in Primary Cells. Cells 2019, 8, 708. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J. Mitochondrial Antioxidants and the Maintenance of Cellular Hydrogen Peroxide Levels. Oxid. Med. Cell Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, Y.; Johansson, E.; Schneider, S.N.; Shertzer, H.G.; Nebert, D.W.; Dalton, T.P. Interaction between the catalytic and modifier subunits of glutamate-cysteine ligase. Biochem. Pharmacol. 2007, 74, 372–381. [Google Scholar] [CrossRef]
- Murata, S.; Minami, Y.; Minami, M.; Chiba, T.; Tanaka, K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001, 2, 1133–1138. [Google Scholar] [CrossRef]
- Lee, J.H.; Khadka, P.; Baek, S.H.; Chung, I.K. CHIP promotes human telomerase reverse transcriptase degradation and negatively regulates telomerase activity. J. Biol. Chem. 2010, 285, 42033–42045. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio-Protocol. 2019, 9, e3128. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Rimessi, A.; de Marchi, E.; Suski, J.M.; Bononi, A.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. ATP synthesis and storage. Purinergic Signal. 2012, 8, 343–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, M.A.; Clayton, D.A. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science 1991, 252, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.P.; Clayton, D.A. Purification and characterization of human mitochondrial transcription factor 1. Mol. Cell Biol. 1988, 8, 3496–3509. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Maynard, S.; Bayne, A.C.V.; Sykora, P.; Tian, J.; de Souza-Pinto, N.C.; Croteau, D.L.; Bohr, V.A. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair 2010, 9, 1080–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Zglinicki, T.; Pilger, R.; Sitte, N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med. 2000, 28, 64–74. [Google Scholar] [CrossRef]
- Masutomi, K.; Possemato, R.; Wong, J.M.Y.; Currier, J.L.; Tothova, Z.; Manola, J.B.; Ganesan, S.; Lansdorp, P.M.; Collins, K.; Hahn, W.C. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc. Natl. Acad. Sci. USA 2005, 102, 8222–8227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, M.; Yamada, O.; Hideshima, T.; Yanagisawa, T.; Yokoi, K.; Fujisawa, K.; Eto, Y.; Yamada, H.; Anderson, K.C. TNFα induces rapid activation and nuclear translocation of telomerase in human lymphocytes. Biochem. Biophys. Res. Commun. 2004, 316, 528–532. [Google Scholar] [CrossRef]
- Allen, R.G.; Tresini, M. Oxidative stress and gene regulation. Free Radic. Biol. Med. 2000, 28, 463–499. [Google Scholar] [CrossRef]
- Bubici, C.; Papa, S.; Dean, K.; Franzoso, G. Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: Molecular basis and biological significance. Oncogene 2006, 25, 6731–6748. [Google Scholar] [CrossRef] [Green Version]
- Seimiya, H.; Sawada, H.; Muramatsu, Y.; Shimizu, M.; Ohko, K.; Yamane, K.; Tsuruo, T. Involvement of 14-3-3 proteins in nuclear localization of telomerase. EMBO J. 2000, 19, 2652–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büchner, N.; Zschauer, T.C.; Lukosz, M.; Altschmied, J.; Haendeler, J. Downregulation of mitochondrial telomerase reverse transcriptase induced by H2O2 is Src kinase dependent. Exp. Gerontol. 2010, 45, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Stierhof, Y.D.; Schwarz, H.; Frank, H. Transverse sectioning of plastic-embedded immunolabeled cryosections: Morphology and permeability to protein A-colloidal gold complexes. J. Ultrastruct. Mol. Struct. Res. 1986, 97, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G. Fine Structure Immunocytochemistry; Springer: Berlin/Heidelberg, Germany, 1993. [Google Scholar]
- Counter, C.M.; Meyerson, M.; Eaton, E.N.; Ellisen, L.W.; Caddle, S.D.; Haber, D.A.; Weinberg, R.A. Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene 1998, 16, 1217–1222. [Google Scholar] [CrossRef] [Green Version]
- Wege, H.; Chui, M.S.; Le, H.T.; Tran, J.M.; Zern, M.A. SYBR Green real-time telomeric repeat amplification protocol for the rapid quantification of telomerase activity. Nucleic Acids Res. 2003, 31, e3. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.W.; Wu, F. Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP). Nucleic Acids Res. 1997, 25, 2595–2597. [Google Scholar] [CrossRef] [Green Version]
- Durante, M.; Furusawa, Y.; Gotoh, E. A simple method for simultaneous interphase-metaphase chromosome analysis in biodosimetry. Int. J. Radiat. Biol. 1998, 74, 457–462. [Google Scholar] [CrossRef]
- Berardinelli, F.; Antoccia, A.; Buonsante, R.; Gerardi, S.; Cherubini, R.; De Nadal, V.; Tanzarella, C.; Sgura, A. The role of telomere length modulation in delayed chromosome instability induced by ionizing radiation in human primary fibroblasts. Environ. Mol. Mutagen. 2013, 54, 172–179. [Google Scholar] [CrossRef]
- Colasuonno, F.; Niceforo, A.; Marioli, C.; Fracassi, A.; Stregapede, F.; Massey, K.; Tartaglia, M.; Bertini, E.; Compagnucci, C.; Moreno, S. Mitochondrial and Peroxisomal Alterations Contribute to Energy Dysmetabolism in Riboflavin Transporter Deficiency. Oxid. Med. Cell Longev. 2020, 2020, 6821247. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marinaccio, J.; Micheli, E.; Udroiu, I.; Di Nottia, M.; Carrozzo, R.; Baranzini, N.; Grimaldi, A.; Leone, S.; Moreno, S.; Muzzi, M.; et al. TERT Extra-Telomeric Roles: Antioxidant Activity and Mitochondrial Protection. Int. J. Mol. Sci. 2023, 24, 4450. https://doi.org/10.3390/ijms24054450
Marinaccio J, Micheli E, Udroiu I, Di Nottia M, Carrozzo R, Baranzini N, Grimaldi A, Leone S, Moreno S, Muzzi M, et al. TERT Extra-Telomeric Roles: Antioxidant Activity and Mitochondrial Protection. International Journal of Molecular Sciences. 2023; 24(5):4450. https://doi.org/10.3390/ijms24054450
Chicago/Turabian StyleMarinaccio, Jessica, Emanuela Micheli, Ion Udroiu, Michela Di Nottia, Rosalba Carrozzo, Nicolò Baranzini, Annalisa Grimaldi, Stefano Leone, Sandra Moreno, Maurizio Muzzi, and et al. 2023. "TERT Extra-Telomeric Roles: Antioxidant Activity and Mitochondrial Protection" International Journal of Molecular Sciences 24, no. 5: 4450. https://doi.org/10.3390/ijms24054450