
Natural Killer Cell-Derived Extracellular Vesicles as a Promising Immunotherapeutic Strategy for Cancer: A Systematic Review
 , ,
, , 
Abstract
:1. Introduction
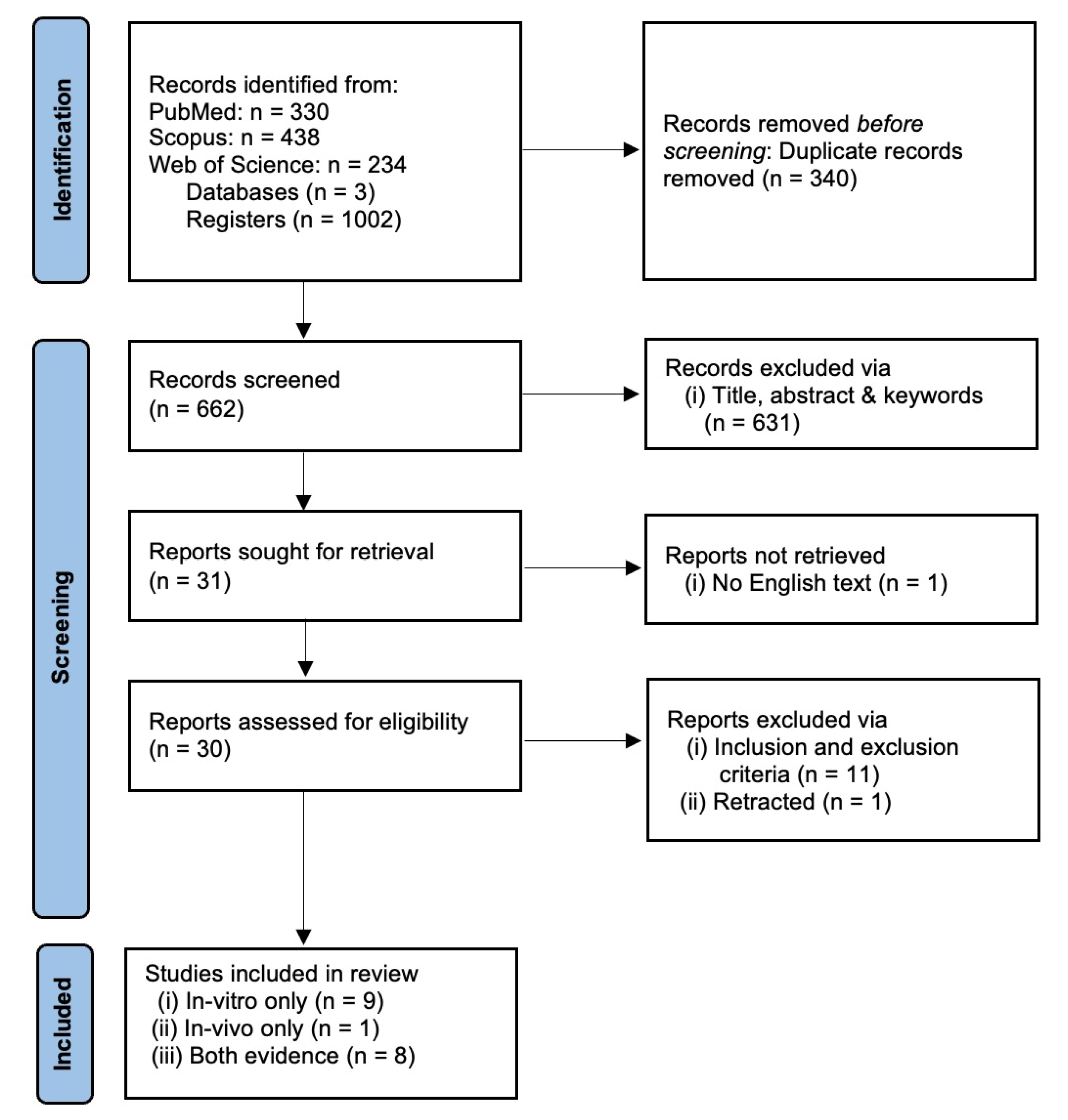
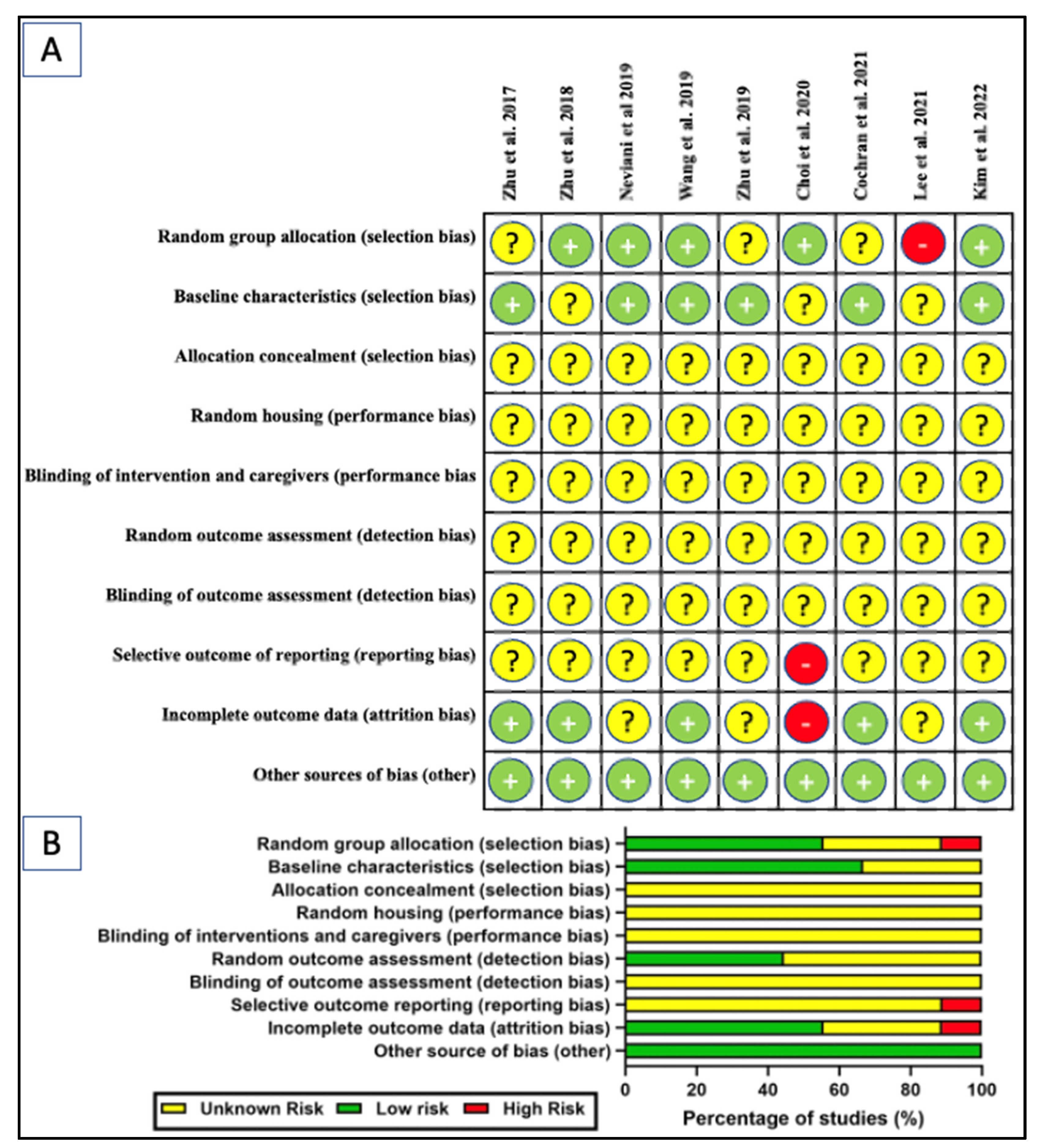
2. Methods
3. Results and Discussion
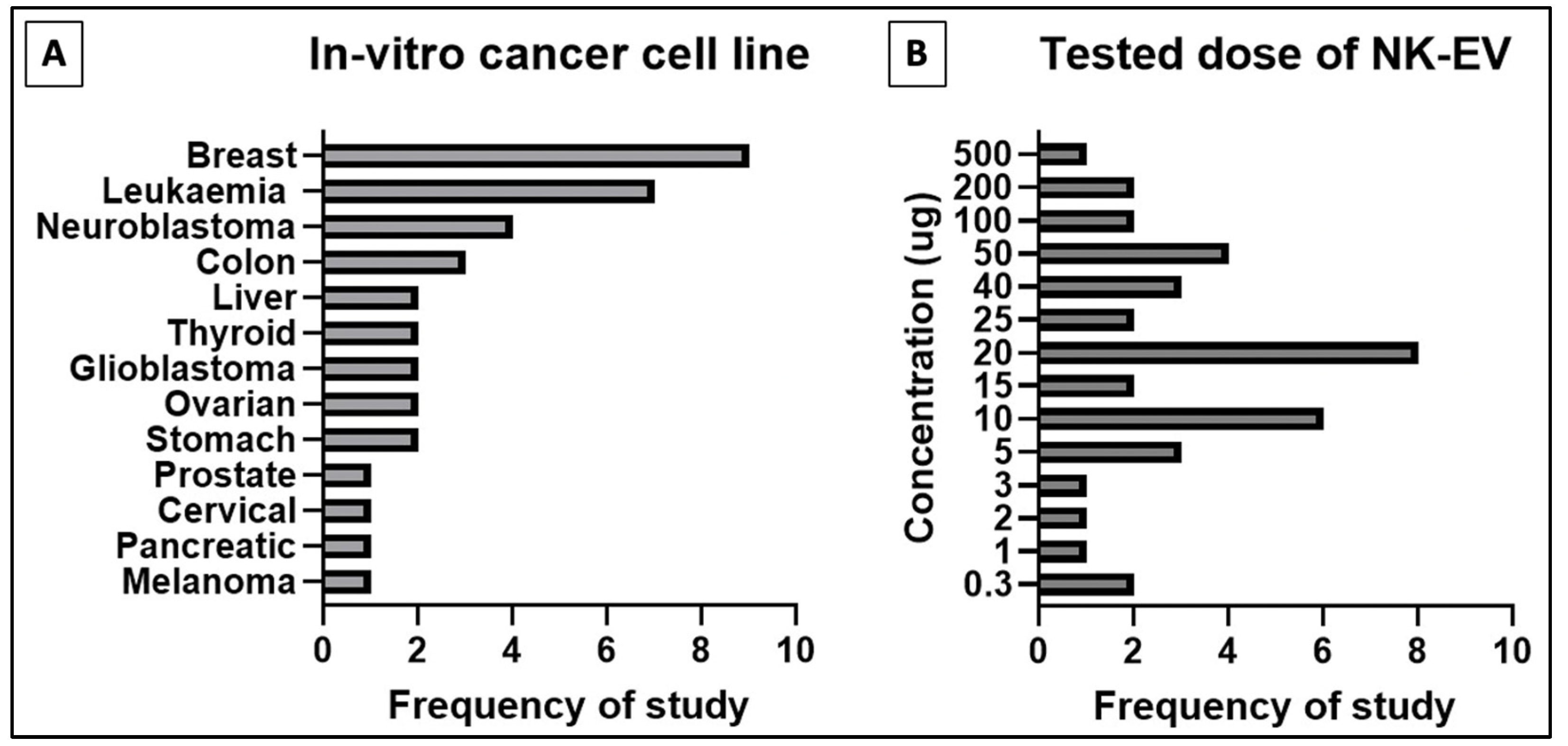
3.1. NK-EVs Express Cytotoxicity against Various Human Cancer Cell Lines
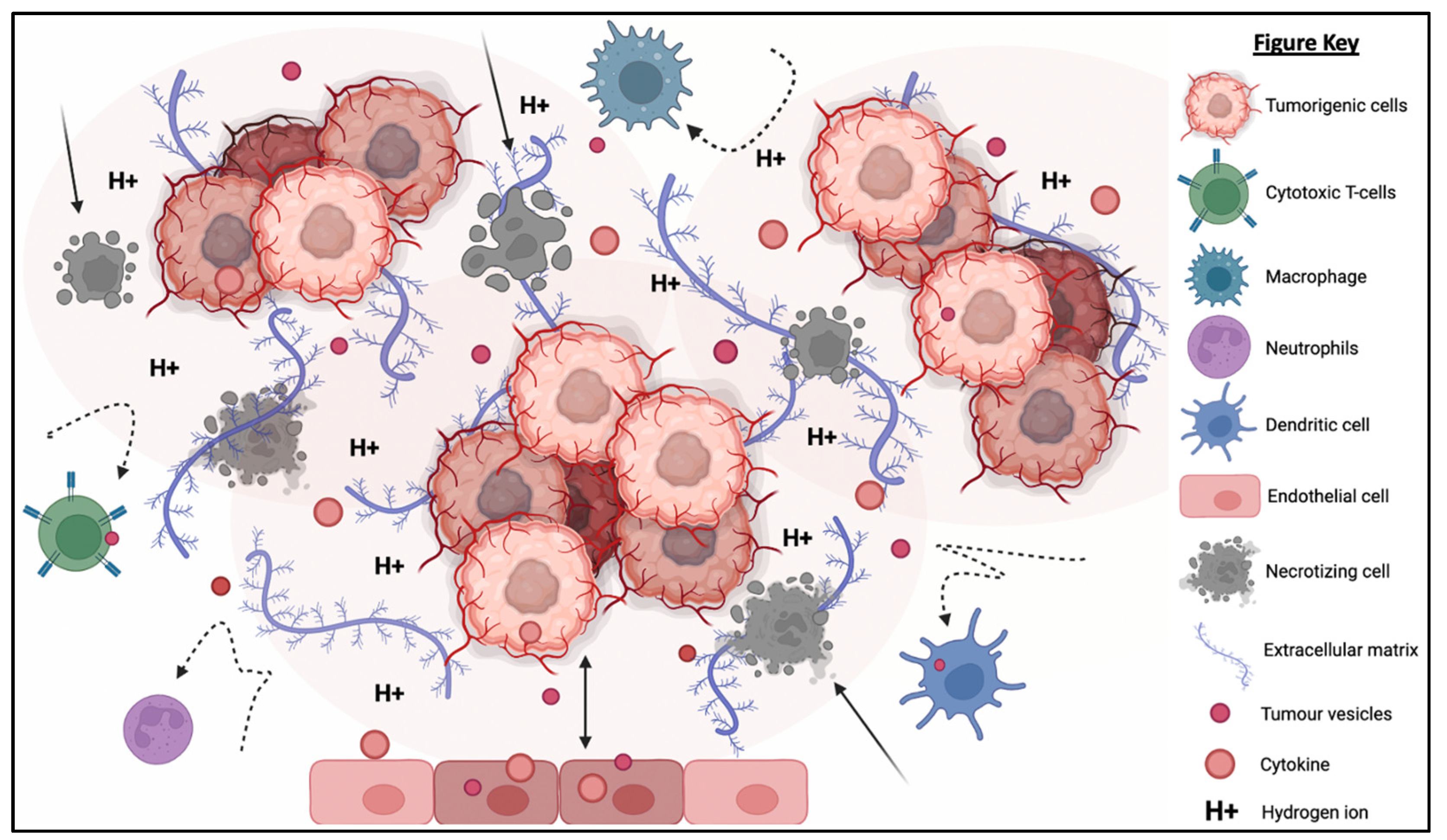
3.2. Tumor Microenvironment (TME): A Physicochemical Barrier against Immunotherapy
3.3. NK-EVs Show In Vivo Cytotoxicity in Tumor-Bearing Mice
3.4. NK-EVs Overcome the Chemical and Physical Barriers of TME
3.5. NK-EVs Are More Clinically Applicable than NK Cells
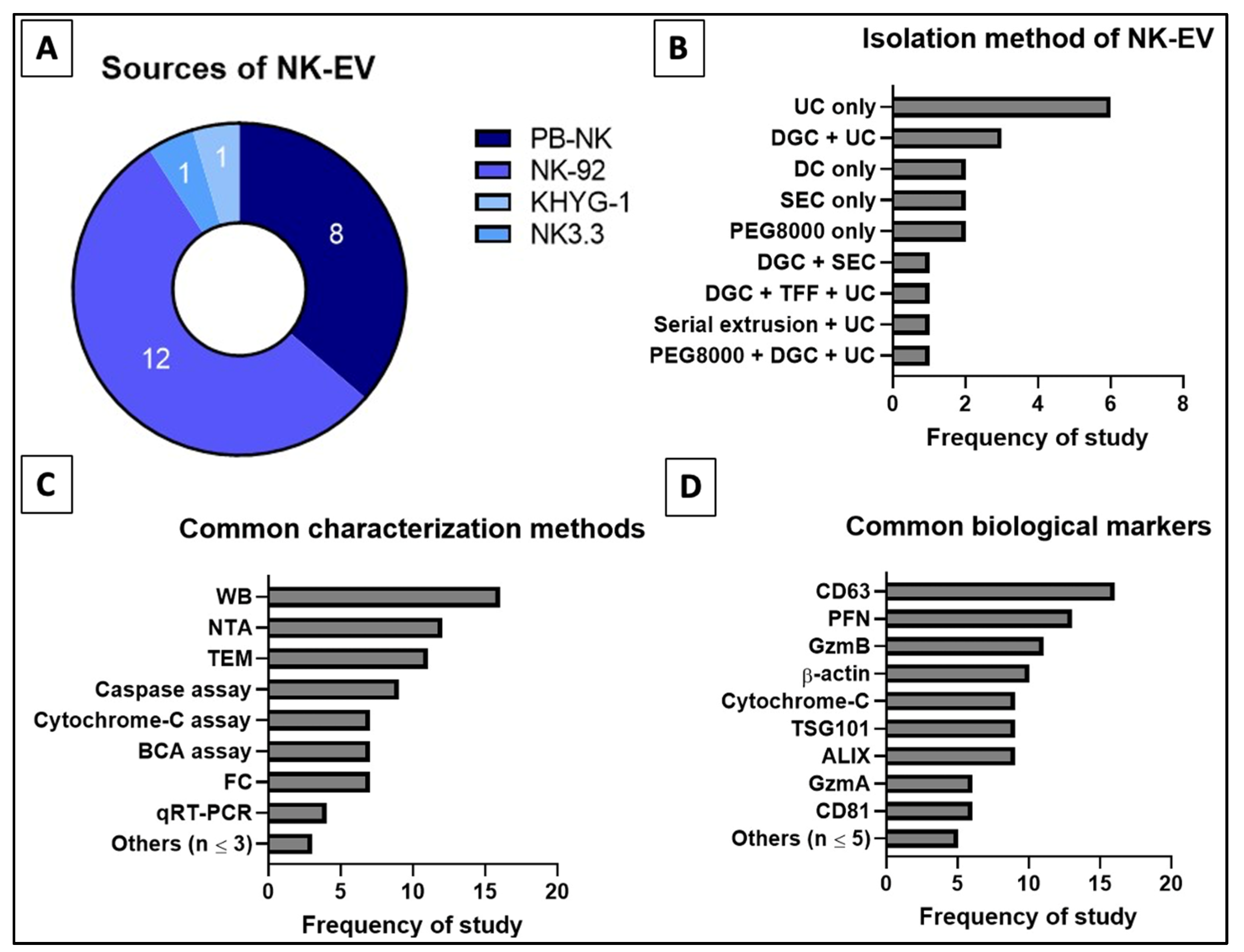
3.6. Defining Nomenclature, Isolation Technique, QA/QC Methods and Biomarkers for NK-EVs
3.7. Future Considerations of NK-EVs as a Tool for Immunotherapy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Coronavirus (COVID-19) Dashboard. 2022. Available online: https://covid19.who.int/ (accessed on 13 December 2022).
- World Health Organization (WHO). Non Communicable Diseases. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases (accessed on 14 July 2022).
- Liao, G.S.; Apaya, M.K.; Shyur, L.F. Herbal medicine and acupuncture for breast cancer palliative care and adjuvant therapy. Evid. Based Complement. Alternat. Med. 2013, 2013, 437948. [Google Scholar] [CrossRef] [PubMed]
- Altun, İ.; Sonkaya, A. The Most Common Side Effects Experienced by Patients Were Receiving First Cycle of Chemotherapy. Iran. J. Public Health 2018, 47, 1218–1219. [Google Scholar] [PubMed]
- Alcindor, T.; Beauger, N. Oxaliplatin: A review in the era of molecularly targeted therapy. Curr. Oncol. 2011, 18, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrie, P.G. Cytotoxic chemotherapy: Clinical aspects. Medicine 2011, 39, 717–722. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyanaraman, B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol. 2020, 29, 101394. [Google Scholar] [CrossRef]
- Smoot, B.; Wampler, M.; Topp, K.S. Breast cancer treatments and complications: Implications for rehabilitation. Rehabilitation Oncology 2009, 27, 16–26. [Google Scholar] [CrossRef]
- Rossi, A.; Fortuna, M.C.; Caro, G.; Pranteda, G.; Garelli, V.; Pompili, U.; Carlesimo, M. Chemotherapy-induced alopecia management: Clinical experience and practical advice. J. Cosmet. Dermatol. 2017, 16, 537–541. [Google Scholar] [CrossRef]
- Frenkel, M. Refusing treatment. Oncologist 2013, 18, 634–636. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewski, W.; Dobrzyński, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef]
- Erhart, F.; Buchroithner, J.; Reitermaier, R.; Fischhuber, K.; Klingenbrunner, S.; Sloma, I.; Hibsh, D.; Kozol, R.; Efroni, S.; Ricken, G.; et al. Immunological analysis of phase II glioblastoma dendritic cell vaccine (Audencel) trial: Immune system characteristics influence outcome and Audencel up-regulates Th1-related immunovariables. Acta Neuropathol. Commun. 2018, 6, 135. [Google Scholar] [CrossRef] [Green Version]
- Margolin, K.; Morishima, C.; Velcheti, V.; Miller, J.S.; Lee, S.M.; Silk, A.W.; Holtan, S.G.; Lacroix, A.M.; Fling, S.P.; Kaiser, J.C.; et al. Phase I Trial of ALT-803, A Novel Recombinant IL15 Complex, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 5552–5561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, F.; Oliveira, R.; Cavadas, B.; Pinto, F.; Cardoso, A.P.; Castro, F.; Sousa, B.; Pinto, M.L.; Silva, A.J.; Adão, D.; et al. Hypoxia and Macrophages Act in Concert Towards a Beneficial Outcome in Colon Cancer. Cancers 2020, 12, 818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumrukcu, S.; Nguyen, T.X.; White, R.L.; Howell, G.T.; Musikanth, P. Allogeneic Natural Killer and Cytomegalovirus (CMV)-pp65 Pulsed Dendritic Cells Induced Complete Response Through 15 Months in a Patient with Recurrent Glioblastoma: A Case Study. Am. J. Case Rep. 2021, 22, e931030, Retraction in Am. J. Case Rep. 2022, 23, e937680. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I. Dendritic cells: Master regulators of the immune response. Cancer Immunol. Res. 2013, 1, 145–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, C.D.; Lenz, L.L.; Harris, R.A. A Breakthrough: Macrophage-Directed Cancer Immunotherapy. Cancer Res. 2016, 76, 513–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Oh, S.; Lee, J.H.; Kwack, K.; Choi, S.W. Natural Killer Cell Therapy: A New Treatment Paradigm for Solid Tumors. Cancers 2019, 11, 1534. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, F.; Tamiya, H. Relationship between antibody-dependent cell-mediated cytotoxicity due to anti-HTLV-1 and negative signal of major histocompatibility complex class I antigens on adult T-cell leukemia cell lines. Oncol. Res. 1998, 10, 59–67. [Google Scholar]
- Høydahl, L.S.; Frick, R.; Sandlie, I.; Løset, G.Å. Targeting the MHC Ligandome by Use of TCR-Like Antibodies. Antibodies 2019, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Nigro, C.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurdi, A.T.; Glavey, S.V.; Bezman, N.A.; Jhatakia, A.; Guerriero, J.L.; Manier, S.; Moschetta, M.; Mishima, Y.; Roccaro, A.; Detappe, A.; et al. Antibody-Dependent Cellular Phagocytosis by Macrophages is a Novel Mechanism of Action of Elotuzumab. Mol. Cancer Ther. 2018, 17, 1454–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Somiya, M.; Kuroda, S. Enhancing antibody-dependent cellular phagocytosis by Re-education of tumor-associated macrophages with resiquimod-encapsulated liposomes. Biomaterials 2021, 268, 120601. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zheng, H.; Duan, Z.; Liu, H.; Hu, D.; Bode, A.; Dong, Z.; Cao, Y. Promotion of cell proliferation and inhibition of ADCC by cancerous immunoglobulin expressed in cancer cell lines. Cell Mol Immunol. 2012, 9, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Kline, J.B.; Kennedy, R.P.; Albone, E.; Chao, Q.; Fernando, S.; McDonough, J.M.; Rybinski, K.; Wang, W.; Somers, E.B.; Schweizer, C.; et al. Tumor antigen CA125 suppresses antibody-dependent cellular cytotoxicity (ADCC) via direct antibody binding and suppressed Fc-γ receptor engagement. Oncotarget 2017, 8, 52045–52060. [Google Scholar] [CrossRef] [Green Version]
- Darwich, A.; Silvestri, A.; Benmebarek, M.R.; Mouriès, J.; Cadilha, B.; Melacarne, A.; Morelli, L.; Supino, D.; Taleb, A.; Obeck, H.; et al. Paralysis of the cytotoxic granule machinery is a new cancer immune evasion mechanism mediated by chitinase 3-like-1. J. Immunother. Cancer 2021, 9, e003224. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Shiota, G. Immune evasion by cancer stem cells. Regen. Ther. 2021, 17, 20–33. [Google Scholar] [CrossRef]
- Cullen, S.P.; Martin, S.J. Fas and TRAIL “death receptors” as initiators of inflammation: Implications for cancer. Semin. Cell Dev. Biol. 2015, 39, 26–34. [Google Scholar] [CrossRef]
- Rossin, A.; Miloro, G.; Hueber, A.O. TRAIL and FasL Functions in Cancer and Autoimmune Diseases: Towards an Increasing Complexity. Cancers 2019, 11, 639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snajdauf, M.; Havlova, K.; Vachtenheim, J., Jr.; Ozaniak, A.; Lischke, R.; Bartunkova, J.; Smrz, D.; Strizova, Z. The TRAIL in the Treatment of Human Cancer: An Update on Clinical Trials. Front. Mol. Biosci. 2021, 8, 628332. [Google Scholar] [CrossRef]
- Clark, R.; Griffiths, G.M. Lytic granules, secretory lysosomes and disease. Curr. Opin. Immunol. 2003, 15, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Krzewski, K.; Coligan, J.E. Human NK cell lytic granules and regulation of their exocytosis. Front. Immunol. 2012, 3, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabanova, A.; Zurli, V.; Baldari, C.T. Signals Controlling Lytic Granule Polarization at the Cytotoxic Immune Synapse. Front. Immunol. 2018, 9, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef] [PubMed]
- Pugholm, L.H.; Bæk, R.; Søndergaard, E.K.; Revenfeld, A.L.; Jørgensen, M.M.; Varming, K. Phenotyping of Leukocytes and Leukocyte-Derived Extracellular Vesicles. J. Immunol. Res. 2016, 2016, 6391264. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Xie, M.; Hun, M.; She, Z.; Li, C.; Luo, S.; Chen, X.; Wan, W.; Wen, C.; Tian, J. Natural Killer Cell-Derived Extracellular Vesicles: Novel Players in Cancer Immunotherapy. Front. Immunol. 2021, 12, 658698. [Google Scholar] [CrossRef]
- Ng, C.Y.; Kee, L.T.; Al-Masawa, M.E.; Lee, Q.H.; Subramaniam, T.; Kok, D.; Ng, M.H.; Law, J.X. Scalable Production of Extracellular Vesicles and Its Therapeutic Values: A Review. Int. J. Mol. Sci. 2022, 23, 7986. [Google Scholar] [CrossRef]
- Kee, L.T.; Ng, C.Y.; Al-Masawa, M.E.; Foo, J.B.; How, C.W.; Ng, M.H.; Law, J.X. Extracellular Vesicles in Facial Aesthetics: A Review. Int. J. Mol. Sci. 2022, 23, 6742. [Google Scholar] [CrossRef] [PubMed]
- Jong, A.Y.; Wu, C.H.; Li, J.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J. Extracell Vesicles 2017, 6, 1294368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Gangadaran, P.; Kalimuthu, S.; Oh, J.M.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Novel alternatives to extracellular vesicle-based immunotherapy—Exosome mimetics derived from natural killer cells. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. 3), S166–S179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neviani, P.; Wise, P.M.; Murtadha, M.; Liu, C.W.; Wu, C.H.; Jong, A.Y.; Seeger, R.C.; Fabbri, M. Natural Killer-Derived Exosomal miR-186 Inhibits Neuroblastoma Growth and Immune Escape Mechanisms. Cancer Res. 2019, 79, 1151–1164. [Google Scholar] [CrossRef]
- Wang, G.; Hu, W.; Chen, H.; Shou, X.; Ye, T.; Xu, Y. Cocktail Strategy Based on NK Cell-Derived Exosomes and Their Biomimetic Nanoparticles for Dual Tumor Therapy. Cancers 2019, 11, 1560. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Li, J.; Li, L.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C.; Jong, A.Y. Extracellular vesicles derived from natural killer cells use multiple cytotoxic proteins and killing mechanisms to target cancer cells. J. Extracell Vesicles 2019, 8, 1588538. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Kalimuthu, S.; Oh, J.M.; Gangadaran, P.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Enhancement of antitumor potency of extracellular vesicles derived from natural killer cells by IL-15 priming. Biomaterials 2019, 190–191, 38–50. [Google Scholar] [CrossRef]
- Choi, J.W.; Lim, S.; Kang, J.H.; Hwang, S.H.; Hwang, K.C.; Kim, S.W.; Lee, S. Proteome Analysis of Human Natural Killer Cell Derived Extracellular Vesicles for Identification of Anticancer Effectors. Molecules 2020, 25, 5216. [Google Scholar] [CrossRef]
- Aarsund, M.; Segers, F.M.; Wu, Y.; Inngjerdingen, M. Comparison of characteristics and tumor targeting properties of extracellular vesicles derived from primary NK cells or NK-cell lines stimulated with IL-15 or IL-12/15/18. Cancer Immunol. Immunother. 2022, 71, 2227–2238. [Google Scholar] [CrossRef]
- Han, D.; Wang, K.; Zhang, T.; Gao, G.C.; Xu, H. Natural killer cell-derived exosome-entrapped paclitaxel can enhance its anti-tumor effect. Eur. Rev. Med. Pharmacol Sci. 2020, 24, 5703–5713. [Google Scholar] [CrossRef]
- Cochran, A.M.; Kornbluth, J. Extracellular Vesicles From the Human Natural Killer Cell Line NK3.3 Have Broad and Potent Anti-Tumor Activity. Front. Cell Dev. Biol. 2021, 9, 698639. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Jiang, H.; Wang, K.; Liu, C.; Man, X.; Fu, Q. Hypoxia enhances the production and antitumor effect of exosomes derived from natural killer cells. Ann. Transl. Med. 2021, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Kaban, K.; Hinterleitner, C.; Zhou, Y.; Salva, E.; Kantarci, A.G.; Salih, H.R.; Märklin, M. Therapeutic Silencing of BCL-2 Using NK Cell-Derived Exosomes as a Novel Therapeutic Approach in Breast Cancer. Cancers 2021, 13, 2397. [Google Scholar] [CrossRef]
- Di Pace, A.L.; Tumino, N.; Besi, F.; Alicata, C.; Conti, L.A.; Munari, E.; Maggi, E.; Vacca, P.; Moretta, L. Characterization of Human NK Cell-Derived Exosomes: Role of DNAM1 Receptor In Exosome-Mediated Cytotoxicity Against Tumor. Cancers 2020, 12, 661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enomoto, Y.; Li, P.; Jenkins, L.M.; Anastasakis, D.; Lyons, G.C.; Hafner, M.; Leonard, W.J. Cytokine-enhanced cytolytic activity of exosomes from NK Cells. Cancer Gene Ther. 2022, 29, 734–749. [Google Scholar] [CrossRef]
- Kim, H.Y.; Min, H.K.; Song, H.W.; Yoo, A.; Lee, S.; Kim, K.P.; Park, J.O.; Choi, Y.H.; Choi, E. Delivery of human natural killer cell-derived exosomes for liver cancer therapy: An in vivo study in subcutaneous and orthotopic animal models. Drug Deliv. 2022, 29, 2897–2911. [Google Scholar] [CrossRef]
- Sun, H.; Shi, K.; Qi, K.; Kong, H.; Zhang, J.; Dai, S.; Ye, W.; Deng, T.; He, Q.; Zhou, M. Natural Killer Cell-Derived Exosomal miR-3607-3p Inhibits Pancreatic Cancer Progression by Targeting IL-26. Front. Immunol. 2019, 10, 2819. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Kalimuthu, S.; Gangadaran, P.; Oh, J.M.; Lee, H.W.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Exosomes Derived From Natural Killer Cells Exert Therapeutic Effect in Melanoma. Theranostics 2017, 7, 2732–2745. [Google Scholar] [CrossRef]
- Farcas, M.; Inngjerdingen, M. Natural killer cell-derived extracellular vesicles in cancer therapy. Scand. J. Immunol. 2020, 92, e12938. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Goldstein, D.B. In vitro assays fail to predict in vivo effects of regulatory polymorphisms. Hum. Mol. Genet. 2007, 16, 1931–1939. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Yu, L.J.; Lunardi, S.; Stamatelos, S.K.; Mack, F.; Gallo, J.M.; Birtwistle, M.R.; Walz, A.C. Predicting In Vivo Efficacy from In Vitro Data: Quantitative Systems Pharmacology Modeling for an Epigenetic Modifier Drug in Cancer. Clin. Transl. Sci. 2020, 13, 419–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.S.; Holland, A.J. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev. 2018, 32, 620–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rani, B.; Cao, Y.; Malfettone, A.; Tomuleasa, C.; Fabregat, I.; Giannelli, G. Role of the tissue microenvironment as a therapeutic target in hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 4128–4140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, A.J.; Lau, P.K.H.; Unsworth, A.S.; Loi, S.; Darcy, P.K.; Kershaw, M.H.; Slaney, C.Y. Tissue-Dependent Tumor Microenvironments and Their Impact on Immunotherapy Responses. Front. Immunol. 2018, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, X.; Zhang, H.; Hu, G. Cancer and Microenvironment Plasticity: Double-Edged Swords in Metastasis. Trend. Pharmacol. Sci. 2019, 40, 419–429. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.A.M.A.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Xue, H.; Lu, B.; Lai, M. The cancer secretome: A reservoir of biomarkers. J. Transl. Med. 2008, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Ma, L.; Zhang, D.; Gao, J.; Jin, Y.; Han, Z.; Lin, D. Tumour biomarkers—Tracing the molecular function and clinical implication. Cell Prolif. 2019, 52, e12589. [Google Scholar] [CrossRef] [Green Version]
- Kartikasari, A.E.R.; Huertas, C.S.; Mitchell, A.; Plebanski, M. Tumor-Induced Inflammatory Cytokines and the Emerging Diagnostic Devices for Cancer Detection and Prognosis. Front. Oncol. 2021, 11, 692142. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- El-Fattah Ibrahim, S.A.; Abudu, A.; Johnson, E.; Aftab, N.; Conrad, S.; Fluck, M. Correction: The role of AP-1 in self-sufficient proliferation and migration of cancer cells and its potential impact on an autocrine/paracrine loop. Oncotarget 2019, 10, 799, Erratum for: Oncotarget 2018, 9, 34259–34278. [Google Scholar] [CrossRef] [PubMed]
- Qiao, F.; Pan, P.; Yan, J.; Sun, J.; Zong, Y.; Wu, Z.; Lu, X.; Chen, N.; Mi, R.; Ma, Y.; et al. Role of tumor-derived extracellular vesicles in cancer progression and their clinical applications (Review). Int. J. Oncol. 2019, 54, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro Franco, P.I.; Rodrigues, A.P.; de Menezes, L.B.; Pacheco Miguel, M. Tumor microenvironment components: Allies of cancer progression. Pathol. Res. Pract. 2020, 216, 152729. [Google Scholar] [CrossRef]
- Li, P.; Lu, M.; Shi, J.; Hua, L.; Gong, Z.; Li, Q.; Shultz, L.D.; Ren, G. Dual roles of neutrophils in metastatic colonization are governed by the host NK cell status. Nat. Commun. 2020, 11, 4387. [Google Scholar] [CrossRef]
- Nowak, M.; Klink, M. The Role of Tumor-Associated Macrophages in the Progression and Chemoresistance of Ovarian Cancer. Cells 2020, 9, 1299. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T.P. The Role of Macrophages in Cancer Development and Therapy. Cancers 2021, 13, 1946. [Google Scholar] [CrossRef] [PubMed]
- Hodge, G.; Barnawi, J.; Jurisevic, C.; Moffat, D.; Holmes, M.; Reynolds, P.N.; Jersmann, H.; Hodge, S. Lung cancer is associated with decreased expression of perforin, granzyme B and interferon (IFN)-γ by infiltrating lung tissue T cells, natural killer (NK) T-like and NK cells. Clin. Exp. Immunol. 2014, 178, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Huang, T.; Chen, X.; Li, W.; Yang, X.; Zhang, W.; Li, M.; Gao, R. Uncovering the interplay between pH receptors and immune cells: Potential drug targets (Review). Oncol. Rep. 2021, 46, 228. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Schlößer, H.A.; Wang, Z.; Qin, J.; Li, J.; Popp, F.; Popp, M.C.; Alakus, H.; Chon, S.H.; Hansen, H.P.; et al. Tumor-Derived Extracellular Vesicles Inhibit Natural Killer Cell Function in Pancreatic Cancer. Cancers 2019, 11, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Cai, S.; Li, M.; Salma, K.I.; Zhou, X.; Han, F.; Chen, J.; Huyan, T. Tumor-Derived Extracellular Vesicles: Their Role in Immune Cells and Immunotherapy. Int. J. Nanomed. 2021, 16, 5395–5409. [Google Scholar] [CrossRef]
- Nakase, I.; Ueno, N.; Matsuzawa, M.; Noguchi, K.; Hirano, M.; Omura, M.; Takenaka, T.; Sugiyama, A.; Bailey Kobayashi, N.; Hashimoto, T.; et al. Environmental pH stress influences cellular secretion and uptake of extracellular vesicles. FEBS Open Biol. 2021, 11, 753–767. [Google Scholar] [CrossRef]
- Honary, S.; Zahir, F. Effect of Zeta Potential on the Properties of Nano-Drug Delivery Systems—A Review (Part 1). Trop. J. Pharm. Res. 2013, 12, 255–264. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.A.; Gu, N.Y.; Jeong, S.Y.; Byeon, J.S.; Jeong, D.U.; Ouh, I.O.; Lee, Y.H.; Hyun, B.H. Canine Natural Killer Cell-Derived Exosomes Exhibit Antitumor Activity in a Mouse Model of Canine Mammary Tumor. Biomed. Res. Int. 2021, 2021, 6690704. [Google Scholar] [CrossRef]
- Kim, Y.I.; Ahn, B.C.; Ronald, J.A.; Katzenberg, R.; Singh, A.; Paulmurugan, R.; Ray, S.; Gambhir, S.S.; Hofmann, L.V. Intratumoral versus intravenous gene therapy using a transcriptionally targeted viral vector in an orthotopic hepatocellular carcinoma rat model. J. Vasc. Interv. Radiol. 2012, 23, 704–711. [Google Scholar] [CrossRef] [Green Version]
- Baniel, C.C.; Sumiec, E.G.; Hank, J.A.; Bates, A.M.; Erbe, A.K.; Pieper, A.A.; Hoefges, A.G.; Patel, R.B.; Rakhmilevich, A.L.; Morris, Z.S.; et al. Intratumoral injection reduces toxicity and antibody-mediated neutralization of immunocytokine in a mouse melanoma model. J. Immunother. Cancer 2020, 8, e001262. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Rovers, M.M.; de Vries, R.B.; Leenaars, M.; Ritskes-Hoitinga, M.; Langendam, M.W. SYRCLE’s risk of bias tool for animal studies. BMC Med. Res. Methodol. 2014, 14, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dommelen, S.M.; Vader, P.; Lakhal, S.; Kooijmans, S.A.; van Solinge, W.W.; Wood, M.J.; Schiffelers, R.M. Microvesicles and exosomes: Opportunities for cell-derived membrane vesicles in drug delivery. J. Control Release 2012, 161, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Papoutsakis, E.T. Extracellular vesicles: Exosomes, microparticles, their parts, and their targets to enable their biomanufacturing and clinical applications. Curr. Opin. Biotechnol. 2019, 60, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev. 2006, 214, 73–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, S.P.; Foulds, G.A.; Imrie, H.; Reeder, S.; McArdle, S.E.B.; Khan, M.; Pockley, A.G. Phenotype and Function of Activated Natural Killer Cells From Patients With Prostate Cancer: Patient-Dependent Responses to Priming and IL-2 Activation. Front. Immunol. 2019, 9, 3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A.; et al. Immune surveillance properties of human NK cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef] [Green Version]
- Shoae-Hassani, A.; Hamidieh, A.A.; Behfar, M.; Mohseni, R.; Mortazavi-Tabatabaei, S.A.; Asgharzadeh, S. NK Cell-derived Exosomes From NK Cells Previously Exposed to Neuroblastoma Cells Augment the Antitumor Activity of Cytokine-activated NK Cells. J. Immunother. 2017, 40, 265–276. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Bluman, E.M.; Porter, M.M.; Mrózek, E.; Cooper, M.A.; VanDeusen, J.B.; Frankel, S.R.; Stock, W.; Caligiuri, M.A. Potential mechanisms of human natural killer cell expansion in vivo during low-dose IL-2 therapy. J. Clin. Invest. 2000, 106, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Okayama, T.; Sakamoto, N.; Ideno, M.; Oka, K.; Enoki, T.; Mineno, J.; Yoshida, N.; Katada, K.; Kamada, K.; et al. Phase I clinical trial of adoptive transfer of expanded natural killer cells in combination with IgG1 antibody in patients with gastric or colorectal cancer. Int. J. Cancer 2018, 142, 2599–2609. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martinez, D.; Allende-Vega, N.; Orecchioni, S.; Talarico, G.; Cornillon, A.; Vo, D.N.; Rene, C.; Lu, Z.Y.; Krzywinska, E.; Anel, A.; et al. Expansion of allogeneic NK cells with efficient antibody-dependent cell cytotoxicity against multiple tumors. Theranostics 2018, 8, 3856–3869. [Google Scholar] [CrossRef]
- Manuel, J. Low cost tissue culture technology for the regeneration of some economically important plants for developing countries. Int. J. Agric. Environ. Biotechnol. 2013, 6, 703–711. [Google Scholar]
- Daniszewski, M.; Crombie, D.E.; Henderson, R.; Liang, H.H.; Wong, R.C.B.; Hewitt, A.W.; Pébay, A. Automated Cell Culture Systems and Their Applications to Human Pluripotent Stem Cell Studies. SLAS Technol. 2018, 23, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, M.N.F.B.; Yazid, M.D.; Yunus, M.H.M.; Chowdhury, S.R.; Lokanathan, Y.; Idrus, R.B.H.; Ng, A.M.H.; Law, J.X. Large-Scale Expansion of Human Mesenchymal Stem Cells. Stem Cells Int. 2020, 2020, 9529465. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Li, Y.J.; Hu, X.B.; Huang, S.; Xiang, D.X. Preservation of small extracellular vesicles for functional analysis and therapeutic applications: A comparative evaluation of storage conditions. Drug Deliv. 2021, 28, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Gelibter, S.; Marostica, G.; Mandelli, A.; Siciliani, S.; Podini, P.; Finardi, A.; Furlan, R. The impact of storage on extracellular vesicles: A systematic study. J. Extracell Vesicles 2022, 11, e12162. [Google Scholar] [CrossRef]
- Sivanantham, A.; Jin, Y. Impact of Storage Conditions on EV Integrity/Surface Markers and Cargos. Life 2022, 12, 697. [Google Scholar] [CrossRef]
- Palay, S.L.; Palade, G.E. The fine structure of neurons. J. Biophys. Biochem. Cytol. 1955, 1, 69–88. [Google Scholar] [CrossRef]
- Rashed, M.H.; Bayraktar, E.; Helal, G.K.; Abd-Ellah, M.F.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From Garbage Bins to Promising Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 538. [Google Scholar] [CrossRef] [Green Version]
- Vidal, M. Exosomes: Revisiting their role as “garbage bags”. Traffic 2019, 20, 815–828. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Koh, B.; Tan, K.L.; Chan, H.H.; Daniel Looi, Q.H.; Lim, M.N.; How, C.W.; Law, J.X.; Foo, J.B. A Simple Benchtop Filtration Method to Isolate Small Extracellular Vesicles from Human Mesenchymal Stem Cells. J. Vis. Exp. 2022, 184, e64106. [Google Scholar] [CrossRef] [PubMed]
- Al-Masawa, M.E.; Alshawsh, M.A.; Ng, C.Y.; Ng, A.M.H.; Foo, J.B.; Vijakumaran, U.; Subramaniam, R.; Ghani, N.A.A.; Witwer, K.W.; Law, J.X. Efficacy and safety of small extracellular vesicle interventions in wound healing and skin regeneration: A systematic review and meta-analysis of animal studies. Theranostics 2022, 12, 6455–6508. [Google Scholar] [CrossRef] [PubMed]
- van Reis, R.; Leonard, L.C.; Hsu, C.C.; Builder, S.E. Industrial scale harvest of proteins from mammalian cell culture by tangential flow filtration. Biotechnol. Bioeng. 1991, 38, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Busatto, S.; Vilanilam, G.; Ticer, T.; Lin, W.L.; Dickson, D.W.; Shapiro, S.; Bergese, P.; Wolfram, J. Tangential Flow Filtration for Highly Efficient Concentration of Extracellular Vesicles from Large Volumes of Fluid. Cells 2018, 7, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, D.C.; Yung, B.C.; Bergamaschi, C.; Chowdhury, B.; Bear, J.; Stellas, D.; Morales-Kastresana, A.; Jones, J.C.; Felber, B.K.; Chen, X.; et al. Scalable, cGMP-compatible purification of extracellular vesicles carrying bioactive human heterodimeric IL-15/lactadherin complexes. J. Extracell Vesicles 2018, 7, 1442088. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin–Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Kazemi, N.Y.; Gendrot, B.; Berishvili, E.; Markovic, S.N.; Cohen, M. The Role and Clinical Interest of Extracellular Vesicles in Pregnancy and Ovarian Cancer. Biomedicines 2021, 9, 1257. [Google Scholar] [CrossRef]
- Witwer, K.W.; Théry, C. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J. Extracell Vesicles 2019, 8, 1648167. [Google Scholar] [CrossRef]
- Ashammakhi, N.; Ahadian, S.; Darabi, M.A.; El Tahchi, M.; Lee, J.; Suthiwanich, K.; Sheikhi, A.; Dokmeci, M.R.; Oklu, R.; Khademhosseini, A. Minimally Invasive and Regenerative Therapeutics. Adv. Mater. 2019, 31, e1804041. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.; Faiz, O.; Davis, R.; Almoudaris, A.; Vincent, C. Surgical complications and their impact on patients’ psychosocial well-being: A systematic review and meta-analysis. BMJ Open 2016, 6, e007224. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Clymer, J.W.; Po-Han Chen, B.; Sadeghirad, B.; Ferko, N.C.; Cameron, C.G.; Hinoul, P. Prolonged operative duration is associated with complications: A systematic review and meta-analysis. J. Surg. Res. 2018, 229, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Sultana, A.; Zare, M.; Thomas, V.; Kumar, T.S.; Ramakrishna, S. Nano-based drug delivery systems: Conventional drug delivery routes, recent developments and future prospects. Med. Drug Discov. 2022, 15, 100134. [Google Scholar] [CrossRef]
- Jin, J.F.; Zhu, L.L.; Chen, M.; Xu, H.M.; Wang, H.F.; Feng, X.Q.; Zhu, X.P.; Zhou, Q. The optimal choice of medication administration route regarding intravenous, intramuscular, and subcutaneous injection. Patient Prefer. Adherence 2015, 9, 923–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, W.J.; Bhowmick, T.; Gross, A.; Vanschooneveld, T.C.; Weinstein, M.P. Using higher doses to compensate for tubing residuals in extended-infusion piperacillin-tazobactam. Ann. Pharmacother. 2013, 47, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Bolla, B.; Buxani, Y.; Wong, R.; Jones, L.; Dube, M. Understanding IV antimicrobial drug losses: The importance of flushing infusion administration sets. JAC Antimicrob. Resist. 2020, 2, dlaa061. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.L.; Zhang, M.; Tang, Y.L.; Liang, X.H. Cancer cell dormancy: Mechanisms and implications of cancer recurrence and metastasis. OncoTarget. Ther. 2017, 10, 5219–5228. [Google Scholar] [CrossRef] [Green Version]
- Gomis, R.R.; Gawrzak, S. Tumor cell dormancy. Mol. Oncol. 2017, 11, 62–78. [Google Scholar] [CrossRef] [Green Version]
- Becker, P.S.; Suck, G.; Nowakowska, P.; Ullrich, E.; Seifried, E.; Bader, P.; Tonn, T.; Seidl, C. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol. Immunother. 2016, 65, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Bt Hj Idrus, R.; Abas, A.; Ab Rahim, F.; Saim, A.B. Clinical Translation of Cell Therapy, Tissue Engineering, and Regenerative Medicine Product in Malaysia and Its Regulatory Policy. Tissue Eng. Part A 2015, 21, 2812–2816. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Type of NK-EV | Dose of NK-EV | Tested Cancer Cell Lines | Key Findings |
|---|---|---|---|---|
| Jong et al., 2017 [44] | NK-EVs from PB-NK of healthy donors | 20 or 40 µg | Human acute lymphoblastic leukemia (NALM-6; SUPB15), neuroblastoma (CHLA-136; CHLA-255) and breast cancer (MCF-7) | NK-EVs increased apoptosis of all tumor cell lines in a time- and dose-dependent manner. |
| Zhu et al., 2017 [60] | NK-EXO from human NK cell line (NK92-MI) | 5 or 20 µg | Mouse melanoma (B16F10); Human gastric carcinoma (SNU484) and colon cancer (HCT-5) | All cell lines showed lower cell viability in a time and dose-dependent manner. NK-EXO did not illicit any response in healthy cells. NK-EXO induced cancer cell apoptosis through PFN and Gzm as well as activation of Fas/FasL pathway. |
| Zhu et al., 2018 [45] | NK-EXO and NK-EM from human NK cell line (NK92-MI) | 10, 20 or 30 µg/mL | Human glioblastoma (D54), breast carcinoma (MDA-MB-231), anaplastic thyroid cancer (CAL-62) and hepatic carcinoma (HepG2) | NK-EM showed greater anti-tumor properties compared to NK-EXO. Both treatment groups reduced BLI signal intensity for all tested tumor cell lines in a time and dose-dependent manner. |
| Neviani et al., 2019 [46] | miR-186 enriched NK-EVs from PB-NK of healthy donors | A series of 2-fold dilutions starting from 4 × 1011 particles/mL | Human MYCN-amplified (CHLA-136 and LAN-5) and non-amplified (CHLA-255) neuroblastoma | IL-15 treated NK cells and its exosomes successfully halted growth of neuroblastoma cell lines. Inactivation of NK cells via TGFβ1 did not affect the functions of the secreted exosomes despite downregulation of cytotoxic proteins. miR-186 delivery via NK-EXO reduced expression of tumor escape oncogenes, i.e., MYCN, AURKA, TGFβ1R and TGFβ2R. |
| Sun et al., 2019 [59] | miR-3607-3p enriched NK-EVs from PB-NK of healthy donors | − | Human pancreatic cancer (MIA PaCa-2 and PANC-1) | NK-EVs inhibited growth, migration and invasive properties of both cancer cell lines. |
| Wang et al., 2019 [47] | NK-EXO and NN/NK-EXO from PB-NK of healthy donors | 10, 20 or 40 µg | Human breast cancer (MDA-MB-231) and neuroblastoma (CHLA-255) | NK-EXOs reduced the viability of both tumor cell lines in a dose-dependent manner. NN/NK-EXOs showed higher levels of tumor cytotoxicity compared to NK-EXOs. |
| Wu et al., 2019 [48] | NK-EVs from PB-NK of healthy donors and human NK cell line (NK92-MI) | 40 µg/100 µL | Human acute lymphoblastic leukemia (SUPB15) and neuroblastoma (CHLA255) | NK-EVs were cytotoxic towards both cancer cell lines. PFN, GzmA, GzmB and GNLY were found to work collectively to induce tumor cytotoxicity. NK-EVs were able to enter caspase-dependent and independent pathways, highlighting the flexibility of NK-EVs to access multiple cytotoxic pathways. |
| Zhu et al., 2019 [49] | NK-EVs from human NK cell line (NK-92MI) with or without IL-15 treatment | 5, 10 or 15 µg | Human breast cancer (MDA-MB-231), anaplastic thyroid cancer (CAL-62) and glioblastoma (U87/MG) | Both NK-EVIL-15 and NK-EVs displayed dose- and time-dependent cytotoxicity against all cancer cell lines. NK-EVIL-15 had greater cytotoxic effect compared to NK-EVs. |
| Choi et al., 2020 [50] | NK-EVs from PB-NK of healthy donors | 2, 5, 10 or 20 µg | Human hepatocarcinoma (HEPG2), colon cancer (SW-620), stomach cancer (MKN-74), breast cancer (MCF-7) and brain cancer (T98G). | NK-EVs were cytotoxicity against all cancer cell lines. |
| Di Pace et al., 2020 [56] | NK-EXO from IL-2 or IL-15 stimulated PB-NK of healthy donors | 5, 20 or 50 µg/100 µL | Human childhood B acute lymphoblastic leukemia (NALM-18) and erythroleukemia (K562) | NK-EXOs exerted cytotoxicity against both cancer cell lines in a dose-dependent manner. |
| Han et al., 2020 [52] | NK-EXO and PTX-NK-EXO from human NK cell line (NK92-MI) | 40 µg/mL of NK-EXO or 15 µg/mL of PTX-NK-EXO | Human breast cancer (MCF-7) | NK-EXOs were as cytotoxic as PTX against the breast cancer cell line. NK-EXOs showed great potential as cancer drug carriers with PTX-NK-EXOs showed the highest tumor cytotoxicity. |
| Cochran et al., 2021 [53] | NK-EVs from human NK cell lines (NK3.3 and NK92-MI) | 1, 10, 25, 50 or 100 μg/mL | Human T cell leukemia (K562 and JURKAT) and breast cancer (HEK293, MCF-7 and MDA-MB-231) | NK-EVs showed cytotoxicity against both cancer cell lines in a time- and dose-dependent manner. NK-EVs did not trigger apoptosis in normal cells, i.e., HEK293 and PB- and CB-derived lymphocytes. |
| Enomoto et al., 2021 [57] | NK-EV from human NK cell line (NK92-MI) | 0.3, 1 or 3 μg | Human T cell leukemia (K562 and JURKAT), lung (A549) and cervical cancer (HELA) | NK-EVs exerted time- and dose-dependent cytotoxic towards all the cancer cell lines albeit the dosage is much lower compared to other studies. |
| Jiang et al., 2021 [54] | NK92-EXO from human NK cell lines (NK92-MI and NK92-hIL-15) cultured in either normal or hypoxic condition | 25 or 50 μg/mL | Human breast cancer (MCF-7) and ovarian cancer (A2780) | NK cells in hypoxic culture (24 and 48 h) doubled its EV production. Hypoxic and normoxic NK-EVs demonstrated similar degree of cytotoxic towards the cancer cell lines. |
| Kaban et al., 2021 [55] | NK-EXO from human NK cell line (NK92-MI) overexpressing BCL-2 siRNAs | 200 µg/mL | Human T cell leukemia (K562 and MEC1) and breast cancer (HEK293T, SKBR3, MCF-7, MCF-10A, T-47D and MDA-MB-231) | Transduced cells produced EVs enriched with BCL-2 siRNAs. Modified NK-EVs showed greater cytotoxicity against the breast cancer cell lines but have no effect on the normal cells. |
| Aarsund et al., 2022 [51] | NK-EVs from IL-15 or IL-12/15/18-stimulated PB-NK of healthy donors and human NK cell line (NK92 and KHYG-1) | 20 µg | Human colon cancer (HCT116 and HCT-15), prostate cancer (DU145 and PC3), breast cancer (SK-BR-3 and T-4D7), ovarian cancer (OVCAR-3), leukemia (KHYG-1), melanoma (WM9) and glioblastoma (U87). | NK-EVs isolated from IL-15 and IL-12/15/18-stimulated NK cells and NK-92 cells were able to kill the cancer cells in 2D and spheroid cultures. KHYG-1 EVs showed no tumor cytotoxicity in both in vitro models. |
| Kim et al., 2022 [58] | NK-EXO from human NK cell line (NK-92) | 10, 20, 50, 100, 200 or 500 µg | Human hepatocellular carcinoma (Hep3B, HepG2 and Huh7) | NK-EXOs had a cytotoxicity towards Hep3B compared to HepG2 and Huh7 cells in a dose-dependent but not time-dependent manner. |
| Study | Type of Cancer and Animal Model (n = Sample Size per Group) | Dosage and Method of Administration | Key Findings |
|---|---|---|---|
| Zhu et al., 2017 [60] | B16F10/effluc cells (1 × 105 cells/100 µL) were subcutaneously injected into the right thigh of pathogen-free 6-week-old female C57BL/6 mice (n = 6) | 20 µg/100 µL of human NK-EXO via IT | Tumor was effectively reduced (3.5 folds) after 2 to 5 days. In vivo and ex vivo BLI confirmed reduced signal intensity and reduced tumor mass in the treatment group. |
| Zhu et al., 2018 [45] | D54 cells (5 × 106 cells/100 µL) were subcutaneously injected into pathogen-free 6-week-old female BALB/c mice (n = 5) | 100 µg/150 µL (via IV) and 30 µg/50 µL (via IT) of human NK-EM, 3 times at intervals of 3 days | Both treatments reduced the tumor mass with IT route showed greater reduction. |
| Neviani et al., 2019 [46] | CHLA-136-Fluc cells (1 × 106 cells) were intra-renally injected into left kidney of 4- to 8-week-old female and male NSG mice (n = 5 − 10) | 1 mg/kg/d of miR-186 enriched human NK-EVs via IV 3 times per week | BLI signal intensity of tumor cells and weight of kidneys were decreased in the animals received miR-186-enriched NK-EVs compared to the control group. The treated animals showed improved survival rate. |
| Wang et al., 2019 [47] | CHLA-255-luc cells (1 × 107 cells/500 µL) were injected via IV into specific pathogen-free, 6-week-old female NOD/SCID mice (n = 3) | 100 µg of human NK-EXO with or without NN let-7a loaded polyamidoamine dendrimer via IV route | NN/NK-EXOs showed better homing efficacy and greater suppression of tumor growth compared to the NK-EXOs. |
| Zhu et al., 2019 [49] | U87/MG/F cells were administered into specific pathogen-free, 6-week-old female BALB/c nude mice (n = 15) | 50 µg of human NK-EVs or 50 µg of human NK-EVsIL-15 via IV for 5 times at intervals of 2 days | Both treatments significantly reduced BLI signals and tumor weight compared to the control group. NK-EVsIL-15 were significantly more effective compared to NK-EVs. Both treatments did not elicit toxic response in tumor-bearing animal model. |
| Choi et al., 2020 [50] | MCF-7 cells (3 × 106 cells) were injected subcutaneously into the right flank of 5-week-old, female athymic nude mice (n = 4) | 50 µg/100 µL of human NK-EVs via IV 3 times weekly | The tumor dimension and mass reduced significantly compared to the control group after 2 weeks. |
| Cochran et al., 2021 [53] | GFP-expressing MDA-MB-231 cells (2 × 106 cells) were injected into the 4th mammary fat pads of female athymic nude mice (n = 4–5) | 50 µg of human NK3.3-EVs via IT, 7 times at intervals of 3 to 4 days | The NK3.3-EV treated animals showed a higher number of dead cells both histologically and in TUNEL assay. |
| Kim et al., 2022 [58] | Hep3B cells (1 × 107 cells/100 µL) was subcutaneously (right back) or (2 × 106 cells/50 µL) orthotopically (liver) xenografted into 6-week-old male BALB/c nude mice (n = 5) | 50, 100, 200 or 500 µg of human NK-EXO via IV 6 times at intervals of 2 days | NK-EXO exerted migratory and targeting ability to inhibit tumor growth in dose-dependent manner in both subcutaneous and orthotopic animal models. |
| Lee et al., 2021 [90] | Canine REM134 cells (1 × 104 cells) xenografted into mammary fat pad of BALB/c nude mice via IT route (n = n/a) | 100 µg of canine NK-EXO via IT once and IV twice per week for 6 weeks | NK-EXO suppressed tumor growth and reduced the expression of tumor-associated markers. |
| Study | Isolation Method | Size Range or mean (±SEM) (nm) | Size Peak or Mode (nm) | Methods of Assessment | Identification Markers | Functional Markers |
|---|---|---|---|---|---|---|
| Jong et al., 2017 [44] | PEG8000 | 155 ± 5.9 | 120 ± 6.4 | Caspase assay, NTA, TEM, WB | CD63, FN, GNLY, GzmA, GzmB, PFN | CD56, Cytochrome-C, Rab5A |
| Zhu et al., 2018 [45] | a Serial extrusion + UC or b UC + DGC | a 100–150 b 100–120 | a 99.2 ± 21.5 b 118 ± 33.1 | Caspase assay, Cellular uptake assay, Inhibition assay, NTA, TEM, WB | ALIX, β-actin, CD63, Cytochrome-C, Fas, GM-130 | AKT, ERK, PFN, p-AKT, p-ERK |
| Neviani et al., 2019 [46] | SEC | 122.2 ± 1.3 | 92.5 ± 1.2 | NTA, TEM, WB | ALIX, β-actin, CANX, CD81, FN, HSP70, TSG101 | AURKA, GzmA, GzmB, MYCN, PFN, TGFBR1, TGFBR2 |
| Wang et al., 2019 [47] | DC | 100 | − | BCA assay, DLS, NTA, TEM, WB | ALIX, CD63, TSG101 | CD47, CXCR4, Cytochrome-C |
| Wu et al., 2019 [48] | PEG8000 | − | − | Caspase assay, Cytochrome-C assay, ELISA, WB | β-actin, Cytochrome-C FasL, GzmA, GzmB, GNLY, PFN | − |
| Zhu et al., 2019 [49] | SEC + DGC | NK-EVs: 106.9 ± 21.6 NK-EVsIL-15:118.2 ± 20.3 | − | BLI, Caspase assay, FC, MTT assay, NTA, TEM, WB | ALIX, β-actin, Calnexin, CD63, Cytochrome-C, GM-130 | Membrane-FasL, Cytoplasm-FasL, PFN, GzmB |
| Choi et al., 2020 [50] | DGC + UC | − | − | 2-DE proteome analysis, Antibody blocking assay, BCA assay, WB | CD40L, CD49, CD51, CD63, Integrin α1, Integrin α3, Integrin β1, L-selectin | Apo A-IV, Apo E, β-actin, DR4, DR5, DNAM-1, Fas, FasL, FGB, FGG, FN, HSP90 α/β, IFN-γ, IL-6, L-plastin, NKG2D, NKP44, NKP46, TRAIL, TNF-α, VCP |
| Aarsund et al., 2022 [51] | SEC | 60–125 | − | BCA assay, LC-MS/MS, NTA, TEM, WB | CD63, CD81, FasL, GzmB, PFN, TSG101 | DNAM-1, NKG2D, NKP46, NKP30 |
| Han et al., 2020 [52] | UC | 80–110 | − | DLS, HPLC, TEM, qRT-PCR, WB | ALIX, CD63, TSG101 | Bax, Bcl-2, β-actin, Cas-3, |
| Cochran et al., 2021 [53] | PEG8000 + DGC + UC | 188.6 ± 2.7 | 133.4 ± 8.0 | BCA assay, Caspase assay, GO, LC-MS/MS, qPCR, NTA, TEM, WB | ALIX, Annexin V, β-actin, CD9, CD63, HSP70, HSP90, LAMP1, NKLAM, TSG101 | DNAM1, GNLY, GzmA, GzmB, ICAM1, MHC-I, MHC-II, PFN, VCAM1 |
| Jiang et al., 2021 [54] | UC | 50–200 | 205.6 ± 29.65 | BCA assay, FC, TEM, WB, Wound healing assay | CD63, FasL, GAPDH, GzmB, PFN, TSG101 | − |
| Kaban et al., 2021 [55] | UC | 115.8–128.9 | − | Caspase assay, FC, NTA, TEM, Immunogold staining, qRT-PCR | CD56, CD63 | 7-AAD, Annexin V, Bcl-2, Cas 3/7, Cas-9, Cytochrome-C, TMRE |
| Di Pace et al., 2020 [56] | DC | 135.9 ± 0.5 | 88 ± 1.3 | Bradford assay, Caspase assay, Cellular uptake assay, ELISA, FC, NTA, WB | CANX, CD63, CD81, TSG101 | CD3, CD16, CD19, CD56, CD69, DNAM1, GzmA, GzmB, IFN-γ, LFA-1, NKP44, NKG2D, PFN, PD-1 |
| Enomoto et al., 2021 [57] | UC | − | 148.2 | BCA assay, FC, Migration assay, miRNA profiling (small RNA-seq), MS, NTA, GO, qRT-PCR, WB | α-tubulin, β-actin, CD63, CD81, Cytochrome-C | CD226, FasL, GNLY, GzmA, GzmB, GzmH, PFN, TRAIL |
| Kim et al., 2022 [58] | UC + DC + TFF | a 106.1 ± 71.5 b 128.5 ± 33.3 | − | Caspase assay, Cellular uptake assay, a DLS, LDH assay, b NTA, TEM, WB | ALIX, CD63, CD81, GzmB, PFN, TRAIL, FasL | β-actin, Cas3, Cas7, Cas8, Cas9, Cytochrome-C, PARP, p-AKT, p-ERK1/2 |
| Sun et al., 2019 [59] | UC | 50–200 | − | FC, Migration and invasion assay, SEM, qRT-PCR | ACTIN, CD63, TSG101 | IL-26, mir-3607-3p |
| Zhu et al., 2017 [60] | DGC + UC | 100–150 | − | BCA assay, Caspase assay, ELISA, FC, TEM, WB | ALIX, CD63, β-actin, GM-130 | Annexin V, Cytochrome-C, FasL, PFN, p38, TNF-α |
| Lee et al., 2021 [90] | UC | 136.6 ± 9.4 | − | NTA, WB | ALIX, CD63, CD81, HSP70, TSG101, GzmB, PFN | Bax, Bcl-xL, Bmi-1, CD133, IL-1β, IL-6, MDR, MMP-3, p53, PCNA, TNF-α, VEGF |
| Pros | Cons | |
|---|---|---|
| NK CELL |
|
|
| NK-EV |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, A.M.L.; Cheah, J.M.; Lokanathan, Y.; Ng, M.H.; Law, J.X. Natural Killer Cell-Derived Extracellular Vesicles as a Promising Immunotherapeutic Strategy for Cancer: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 4026. https://doi.org/10.3390/ijms24044026
Chan AML, Cheah JM, Lokanathan Y, Ng MH, Law JX. Natural Killer Cell-Derived Extracellular Vesicles as a Promising Immunotherapeutic Strategy for Cancer: A Systematic Review. International Journal of Molecular Sciences. 2023; 24(4):4026. https://doi.org/10.3390/ijms24044026
Chicago/Turabian StyleChan, Alvin Man Lung, Jin Min Cheah, Yogeswaran Lokanathan, Min Hwei Ng, and Jia Xian Law. 2023. "Natural Killer Cell-Derived Extracellular Vesicles as a Promising Immunotherapeutic Strategy for Cancer: A Systematic Review" International Journal of Molecular Sciences 24, no. 4: 4026. https://doi.org/10.3390/ijms24044026