
Tenebrio molitor as a Simple and Cheap Preclinical Pharmacokinetic and Toxicity Model

 , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Analysis of Vehicles
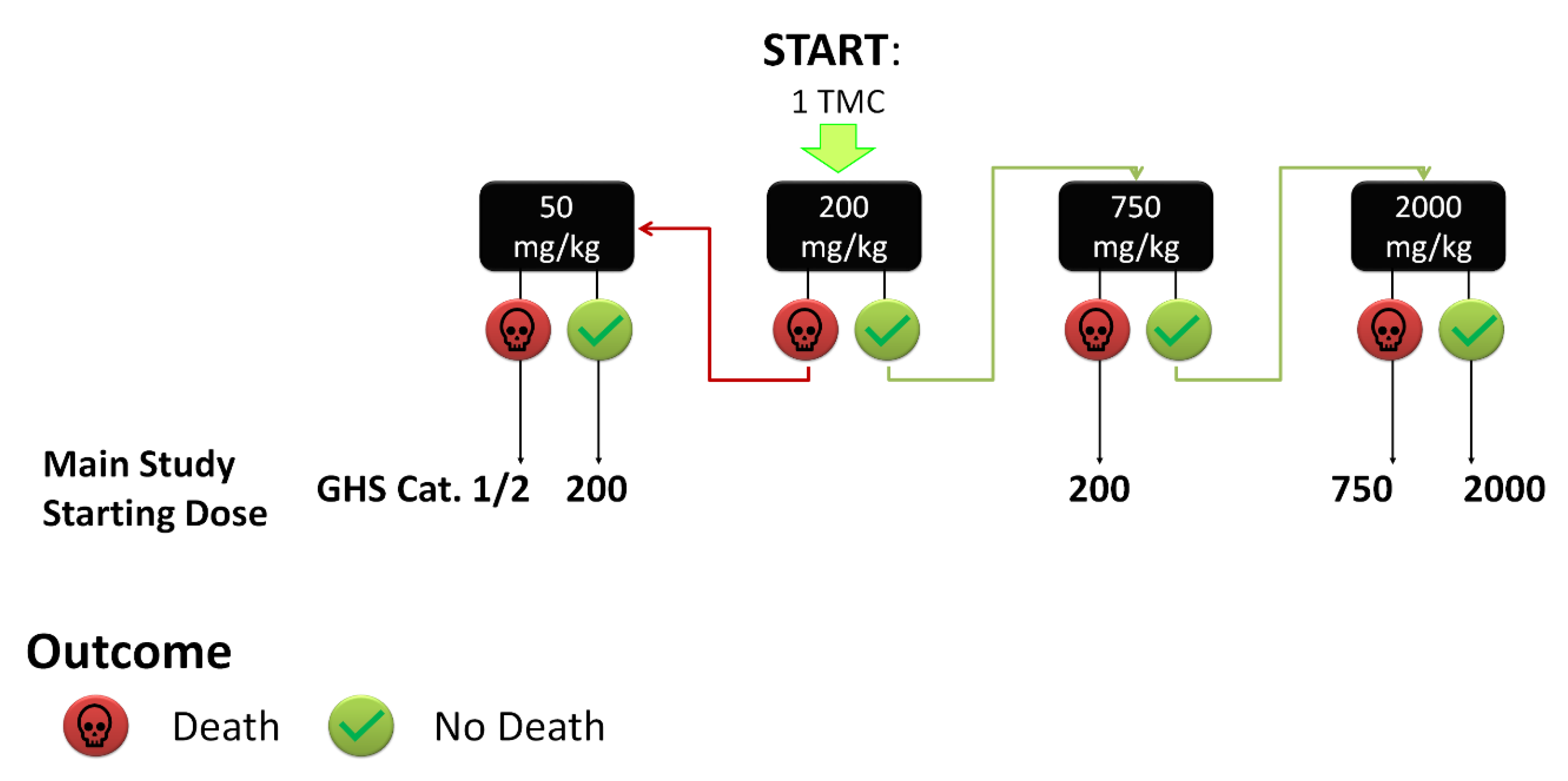
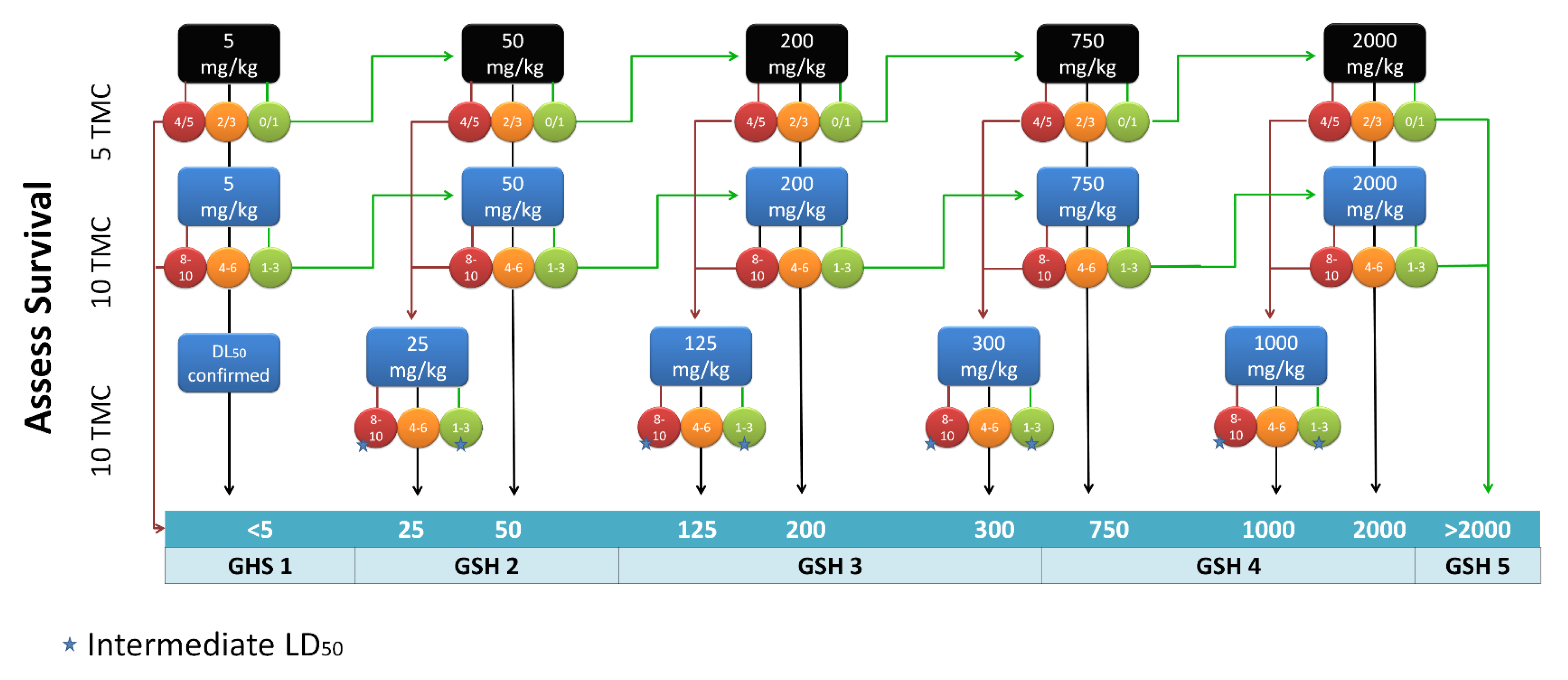
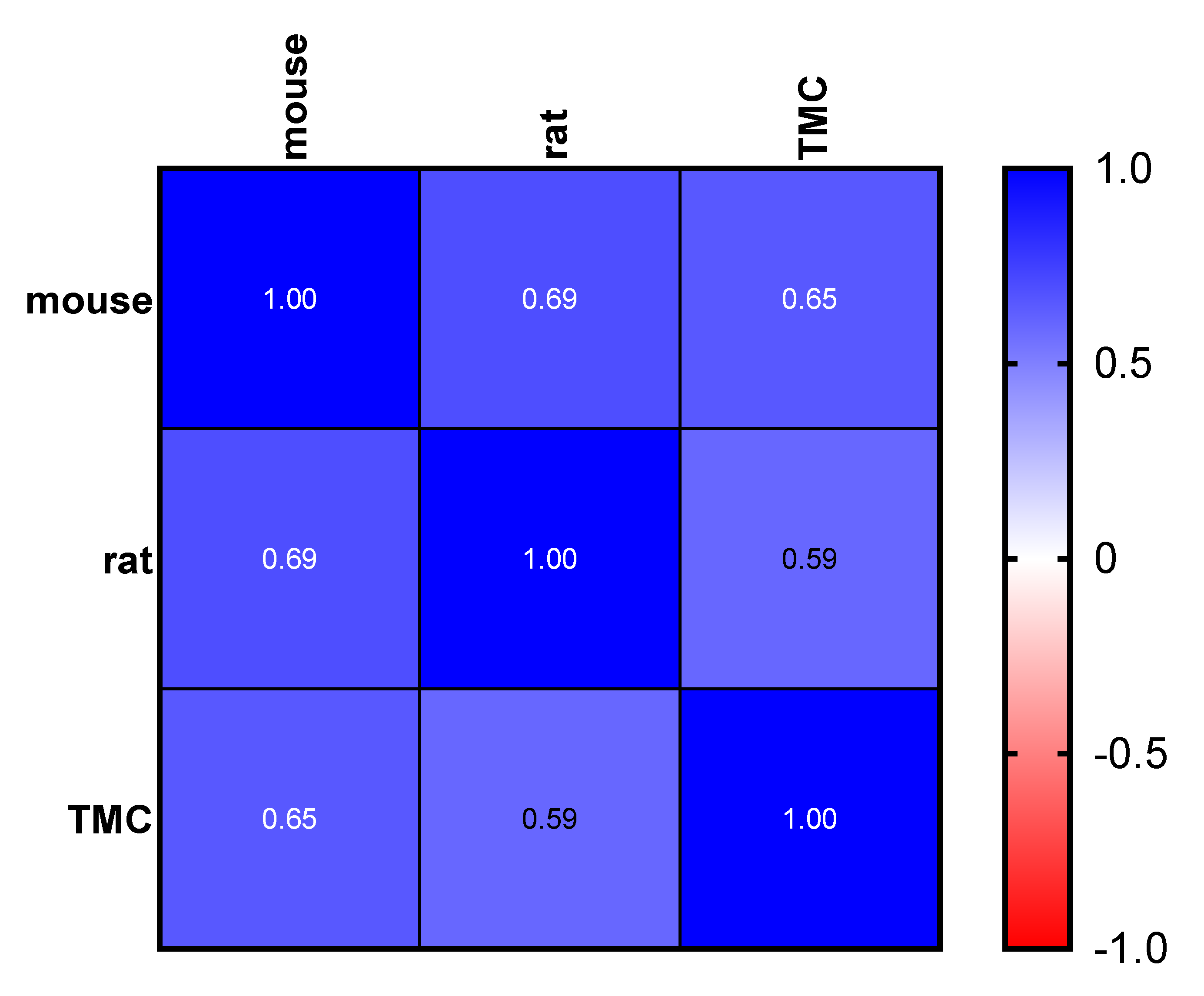
2.2. Toxicity Studies
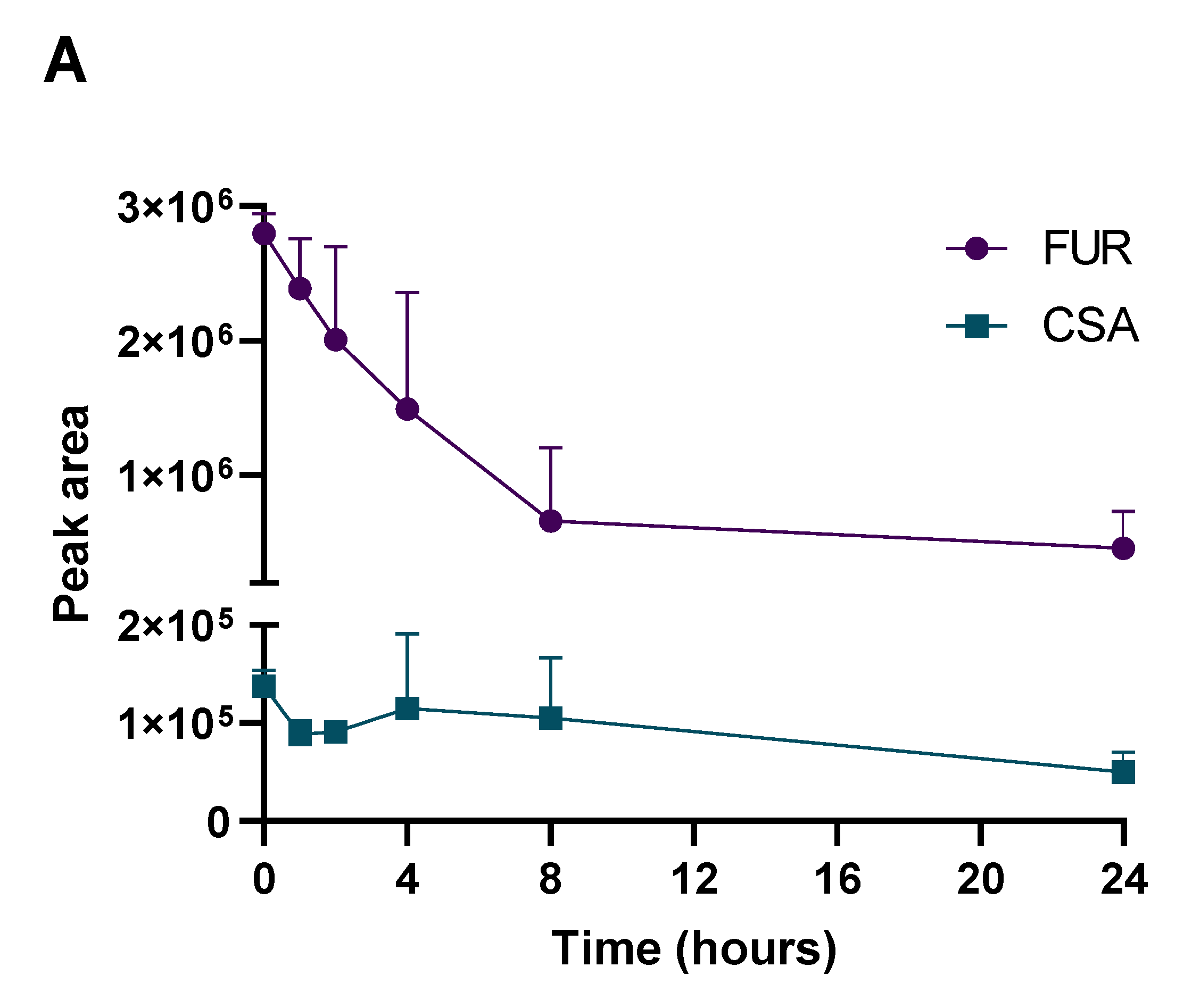
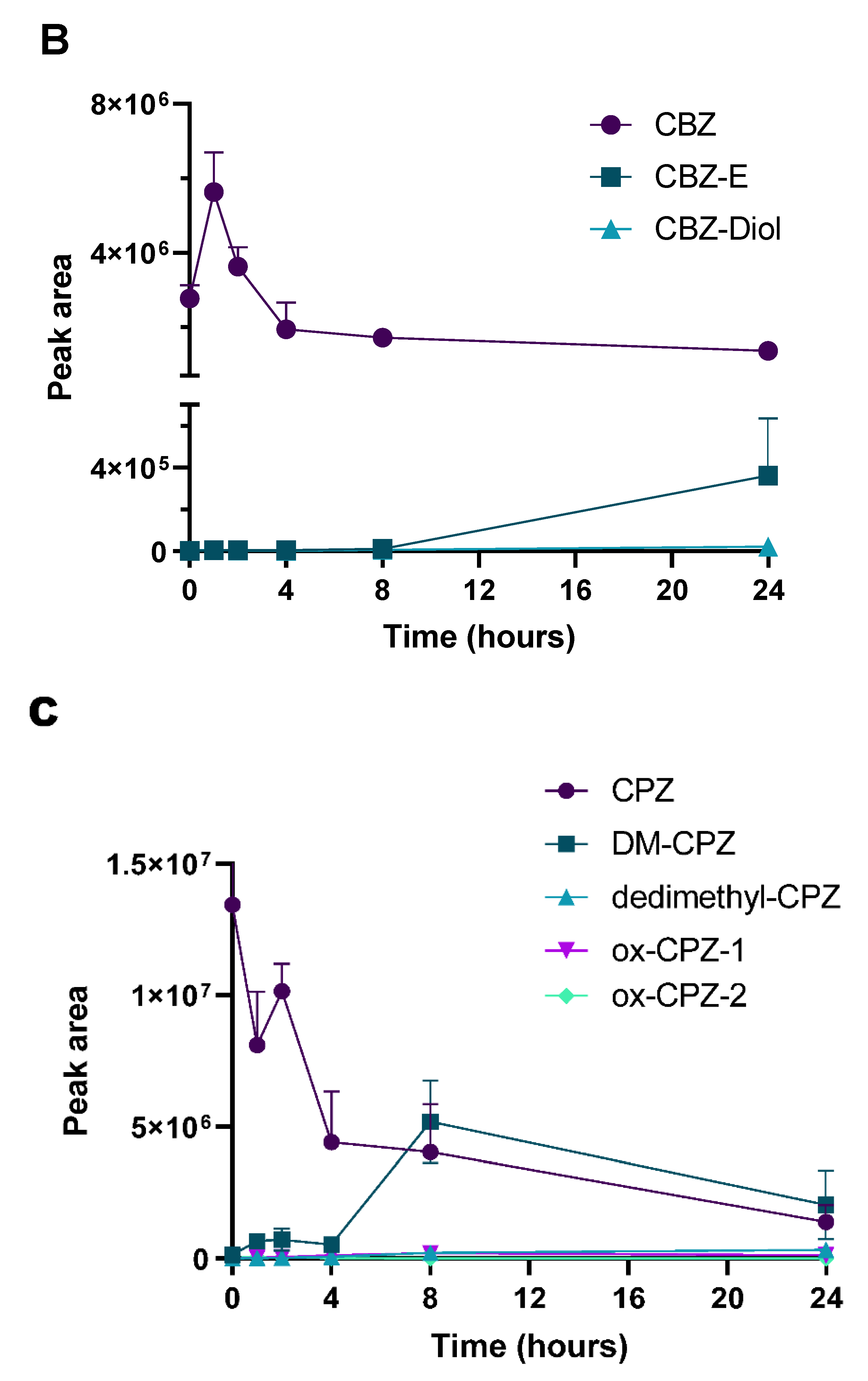
2.3. Determination of PK Parameters and Metabolic Stability
3. Materials and Methods
3.1. Materials
3.2. Animals
3.3. TMC Injection Procedure
3.4. Toxicity Testing Procedure
3.5. Metabolism Testing Procedure
3.6. In Vitro Metabolism in Human and Rat Microsomes
3.7. HPLC Analysis
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Martignoni, M.; Groothuis, G.M.M.; de Kanter, R. Species Differences between Mouse, Rat, Dog, Monkey and Human CYP-Mediated Drug Metabolism, Inhibition and Induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Tsutomu Miki Kurosawa, J. Alternative Research (3Rs) in the World, Asia and Japan. In Alternatives to Animal Testing; Springer: Singapore, 2019; pp. 33–36. [Google Scholar] [CrossRef] [Green Version]
- Katoch, S.; Patial, V. Zebrafish: An Emerging Model System to Study Liver Diseases and Related Drug Discovery. J. Appl. Toxicol. 2021, 41, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Hunt, P.R. The C. Elegans Model in Toxicity Testing. J. Appl. Toxicol. 2017, 37, 50–59. [Google Scholar] [CrossRef]
- Saeedi, B.J.; Hunter-Chang, S.; Luo, L.; Li, K.; Liu, K.H.; Robinson, B.S. Oxidative Stress Mediates End-Organ Damage in a Novel Model of Acetaminophen-Toxicity in Drosophila. Sci. Rep. 2022, 12, 19309. [Google Scholar] [CrossRef] [PubMed]
- Ignasiak, K.; Maxwell, A. Galleria Mellonella (Greater Wax Moth) Larvae as a Model for Antibiotic Susceptibility Testing and Acute Toxicity Trials. BMC Res. Notes 2017, 10, 428. [Google Scholar] [CrossRef] [Green Version]
- Dinh, H.; Semenec, L.; Kumar, S.S.; Short, F.L.; Cain, A.K. Microbiology’s next Top Model: Galleria in the Molecular Age. Pathog. Dis. 2021, 79, ftab006. [Google Scholar] [CrossRef]
- Desbois, A.P.; Coote, P.J. Utility of Greater Wax Moth Larva (Galleria mellonella) for Evaluating the Toxicity and Efficacy of New Antimicrobial Agents. Adv. Appl. Microbiol. 2012, 78, 25–53. [Google Scholar] [CrossRef]
- Champion, O.L.; Wagley, S.; Titball, R.W. Galleria Mellonella as a Model Host for Microbiological and Toxin Research. Virulence 2016, 7, 840–845. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Reytor, D.; García, K. Galleria Mellonella: A Model of Infection to Discern Novel Mechanisms of Pathogenesis of Non-Toxigenic Vibrio Parahaemolyticus Strains. Virulence 2018, 9, 22–24. [Google Scholar] [CrossRef]
- Tsai, C.J.Y.; Loh, J.M.S.; Proft, T. Galleria Mellonella Infection Models for the Study of Bacterial Diseases and for Antimicrobial Drug Testing. Virulence 2016, 7, 214–229. [Google Scholar] [CrossRef] [Green Version]
- Li, D.D.; Deng, L.; Hu, G.H.; Zhao, L.X.; Hu, D.D.; Jiang, Y.Y.; Wang, Y. Using Galleria Mellonella-Candida Albicans Infection Model to Evaluate Antifungal Agents. Biol. Pharm. Bull. 2013, 36, 1482–1487. [Google Scholar] [CrossRef] [Green Version]
- Jemel, S.; Guillot, J.; Kallel, K.; Botterel, F.; Dannaoui, E. Galleria Mellonella for the Evaluation of Antifungal Efficacy against Medically Important Fungi, a Narrative Review. Microorganisms 2020, 8, 390. [Google Scholar] [CrossRef] [Green Version]
- Gizińska, M.; Staniszewska, A.; Kazek, M.; Koronkiewicz, M.; Kuryk, Ł.; Milner-Krawczyk, M.; Baran, J.; Borowiecki, P.; Staniszewska, M. Antifungal Polybrominated Proxyphylline Derivative Induces Candida Albicans Calcineurin Stress Response in Galleria Mellonella. Bioorg. Med. Chem. Lett. 2020, 30, 127545. [Google Scholar] [CrossRef]
- Antonello, R.M.; di Bella, S.; Betts, J.; la Ragione, R.; Bressan, R.; Principe, L.; Morabito, S.; Gigliucci, F.; Tozzoli, R.; Busetti, M.; et al. Zidovudine in Synergistic Combination with Fosfomycin: An in Vitro and in Vivo Evaluation against Multidrug-Resistant Enterobacterales. Int. J. Antimicrob. Agents 2021, 58, 106362. [Google Scholar] [CrossRef]
- Büyükgüzel, E.; Büyükgüzel, K. Effects of Antiviral Agent, Acyclovir, on the Biological Fitness of Galleria Mellonella (Lepidoptera: Pyralidae) Adults. J. Econ. Entomol. 2016, 109, 2090–2095. [Google Scholar] [CrossRef]
- Brai, A.; Immacolata Trivisani, C.; Vagaggini, C.; Stella, R.; Angeletti, R.; Iovenitti, G.; Francardi, V.; Dreassi, E. Proteins from Tenebrio Molitor: An Interesting Functional Ingredient and a Source of ACE Inhibitory Peptides. Food Chem. 2022, 393, 133409. [Google Scholar] [CrossRef]
- Dai, C.; Ma, H.; Luo, L.; Yin, X. Angiotensin I-Converting Enzyme (ACE) Inhibitory Peptide Derived from Tenebrio molitor (L.) Larva Protein Hydrolysate. Eur. Food Res. Technol. 2013, 236, 681–689. [Google Scholar] [CrossRef]
- Brai, A.; Vagaggini, C.; Pasqualini, C.; Poggialini, F.; Tarchi, F.; Francardi, V.; Dreassi, E. Use of Distillery By-Products as Tenebrio Molitor Mealworm Feed Supplement. J. Insects Food Feed. 2022, 1–14. [Google Scholar] [CrossRef]
- Brai, A.; Poggialini, F.; Trivisani, C.I.; Vagaggini, C.; Tarchi, F.; Francardi, V.; Dreassi, E. Efficient Use of Agricultural Waste to Naturally Fortify Tenebrio Molitor Mealworms and Evaluation of Their Nutraceutical Properties. J. Insects Food Feed. 2022, 1–12. [Google Scholar] [CrossRef]
- Lozoya-Pérez, N.E.; García-Carnero, L.C.; Martínez-Álvarez, J.A.; Martínez-Duncker, I.; Mora-Montes, H.M. Tenebrio Molitor as an Alternative Model to Analyze the Sporothrix Species Virulence. Infect. Drug. Resist. 2021, 14, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Gad, S.C.; Spainhour, C.B.; Shoemake, C.; Pallman, D.R.S.; Stricker-Krongrad, A.; Downing, P.A.; Seals, R.E.; Eagle, L.A.; Polhamus, K.; Daly, J. Tolerable Levels of Nonclinical Vehicles and Formulations Used in Studies by Multiple Routes in Multiple Species With Notes on Methods to Improve Utility. Int. J. Toxicol. 2016, 35, 95–178. [Google Scholar] [CrossRef] [PubMed]
- Williams, M. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 15th Edition Edited by M.J. O’Neil, Royal Society of Chemistry, Cambridge, UK ISBN 9781849736701; 2708 Pages. April 2013, $150 with 1-Year Free Access to The Merck Index Online. Drug Dev. Res. 2013, 74, 339. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Techer, W.E., Jr.; Macklim, A.W.; Szot, R.J.; Johnston, R.E.; Elion, G.B.; de Miranda, P.; Szczech, G.M. Preclinical Toxicology Studies with Acyclovir: Acute and Subchronic Tests. Fundam. Appl. Toxicol. 1983, 3, 573–578. [Google Scholar] [CrossRef]
- Painter, W.P.; Holman, W.; Bush, J.A.; Almazedi, F.; Malik, H.; Eraut, N.C.J.E.; Morin, M.J.; Szewczyk, L.J.; Painter, G.R. Human Safety, Tolerability, and Pharmacokinetics of Molnupiravir, a Novel Broad-Spectrum Oral Antiviral Agent with Activity against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65, e02428-20. [Google Scholar] [CrossRef]
- Nelson, E.B.; Montes, M.; Goldstein, M. Effectiveness of Methyrapone in the Treatment of Acetaminophen Toxicity in Mice. Toxicology 1980, 17, 73–81. [Google Scholar] [CrossRef]
- Coman, L.; Păunescu, H.; Ghiță, C.I.V.; Țincu, R.C.; Vasile, S.; Cinteza, D.; Fulga, I.; Coman, O.A. Paracetamol-Induced Hypothermia in Rodents: A Review on Pharmacodynamics. Processes 2022, 10, 687. [Google Scholar] [CrossRef]
- Bjørge, J.D.; Overgaard, J.; Malte, H.; Gianotten, N.; Heckmann, L.H. Role of Temperature on Growth and Metabolic Rate in the Tenebrionid Beetles Alphitobius Diaperinus and Tenebrio Molitor. J. Insect. Physiol. 2018, 107, 89–96. [Google Scholar] [CrossRef]
- Ashley, C.; Dunleavy, A.; Cunningham, J. The Renal Drug Handbook: The Ultimate Prescribing Guide for Renal Practitioners, 5th ed.; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar] [CrossRef]
- Boles Ponto, L.L.; Schoenwald, R.D. Furosemide (Frusemide) A Pharmacokinetic/Pharmacodynamic Review (Part I). Clin. Pharmacokinet. 2012, 18, 381–408. [Google Scholar] [CrossRef]
- WIRTH, P.J.; BETTIS, C.J.; NELSON, W.L. Microsomal Metabolism of Furosemide Evidence for the Nature of the Reactive Intermediate Involved in Covalent Binding. Mol. Pharmacol. 1976, 12, 759–768. [Google Scholar]
- Williams, D.P.; Antoine, D.J.; Butler, P.J.; Jones, R.; Randle, L.; Payne, A.; Howard, M.; Gardner, I.; Blagg, J.; Park, B.K. The Metabolism and Toxicity of Furosemide in the Wistar Rat and CD-1 Mouse: A Chemical and Biochemical Definition of the Toxicophore. J. Pharmacol. Exp. Ther. 2007, 322, 1208–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Lin, H.; Pan, Y.; Sun, Y.; Wang, Y.; Qiao, J.Q.; Lian, H.Z.; Xu, C. xiang Determination of Chlorpromazine and Its Metabolites in Animal-Derived Foods Using QuEChERS-Based Extraction, EMR-Lipid Cleanup, and UHPLC-Q-Orbitrap MS Analysis. Food Chem. 2023, 403, 134298. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Li, G.Y.; Li, L.; Song, Q.S.; Stanley, D.; Wei, S.J.; Zhu, J.Y. Genome-Wide and Expression-Profiling Analyses of the Cytochrome P450 Genes in Tenebrionidea. Arch Insect. Biochem. Physiol. 2022, 111, e21954. [Google Scholar] [CrossRef] [PubMed]
- Yeung, P.K.F.; Hubbard, J.W.; Korchinski, E.D.; Midha, K.K. Pharmacokinetics of Chlorpromazine and Key Metabolites. Eur. J. Clin. Pharmacol. 1993, 45, 563–569. [Google Scholar] [CrossRef]
- Tabunoki, H.; Dittmer, N.T.; Gorman, M.J.; Kanost, M.R. Development of a New Method for Collecting Hemolymph and Measuring Phenoloxidase Activity in Tribolium Castaneum. BMC Res. Notes 2019, 12, 7. [Google Scholar] [CrossRef]
- Brai, A.; Riva, V.; Clementi, L.; Falsitta, L.; Zamperini, C.; Sinigiani, V.; Festuccia, C.; Sabetta, S.; Aiello, D.; Roselli, C.; et al. Targeting DDX3X Helicase Activity with BA103 Shows Promising Therapeutic Effects in Preclinical Glioblastoma Models. Cancers 2021, 13, 5569. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vehicles | CAS # | Survival % | |
|---|---|---|---|
| 1 µL | 2 µL | ||
| Saline | 7647-14-5 | 100 | 100 |
| DMSO | 67-68-5 | 100 | 0 |
| DMSO: Saline (1:1) | - | 100 | 100 |
| DMSO: Saline (2:1) | - | 100 | 83.4 |
| Acetone | 67-64-1 | 100 | 16.6 |
| EtOH | 64-17-5 | 100 | 50 |
| Drug | Class | LD50 (mg/kg) | ||||
|---|---|---|---|---|---|---|
| Mice | Rats | Tenebrio molitor | ||||
| ip | iv | ip | iv | |||
| Acyclovir | Antivirals | 1000 | NR | NR | NR | >2000 |
| Amoxicillin | Antibiotics | 3590 | NR | 2870 | NR | 750 |
| Carbamazepine | Anticonvulsants | 114 | NR | 158 | NR | 125 |
| Chloramphenicol | Antibiotics | 171 | NR | 300 | ||
| Chlorpromazine | Neuroleptics | 14 | 106 | 137 | NR | 300 |
| Cimetidine | Antiulcer | 306 | 150 | 328 | 326 | 750 |
| Cloxacillin | Antibiotics | 1660 | 750 | |||
| Codeine | Analgesic | 60 | 54 | 100 | 75 | 50 |
| Diazepam | Neuroleptics | 37 | 25 | 46.5 | 32 | 50 |
| Diphenhydramine | Antihistamines | 56 | NR | NR | 42 | 50 |
| Furosemide | Loop diuretic | 308 | 800 | 800 | 300 | |
| Ketoprofen | Anti-inflammatory | 300 | 500 | 80 | 350 | 500 |
| Mepivacaine | Local anesthetic | 135 | 35 | NR | 30 | 125 |
| Metoprolol | Antihypertensives | >200 | 62 | NR | 71.9 | 500 |
| Molnupiravir | Antivirals | NR | NR | NR | NR | >2000 |
| Nimesulide | Antipyretic analgesic | NR | NR | 300 | NR | 50 |
| Paracetamol | Antipyretic analgesic | 340 | NR | NR | NR | 125 |
| Salicylic acid | Antipyretic analgesic | 300 | 184 | 157 | NR | 250 |
| Sulfadiazine | Antibiotics | 750 | 180 | 446 | 880 | 300 |
| Trimethoprim | Antibiotics | 400 | 132 | 500 | NR | 300 |
| T ½ (h) a | LogP a | LogS a | Protein Binding (%) a | Vd (L/kg) a | Stability % Rats Microsomes | Stability % Human Microsomes | |
|---|---|---|---|---|---|---|---|
| Furosemide | 0.5–2 | 2.03 | −3.66 | 91–99 | 0.07–0.2 | 95.1 | 96.90 |
| Carbamazepine | 5 | 2.77 | −3.20 | 70–80 | 0.8–1.9 | 64.47 | 77.54 |
| Chlorpromazine | 8–35 | 5.41 | −5.01 | 95–98 | 7–20 | 77.99 | 86.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brai, A.; Poggialini, F.; Vagaggini, C.; Pasqualini, C.; Simoni, S.; Francardi, V.; Dreassi, E. Tenebrio molitor as a Simple and Cheap Preclinical Pharmacokinetic and Toxicity Model. Int. J. Mol. Sci. 2023, 24, 2296. https://doi.org/10.3390/ijms24032296
Brai A, Poggialini F, Vagaggini C, Pasqualini C, Simoni S, Francardi V, Dreassi E. Tenebrio molitor as a Simple and Cheap Preclinical Pharmacokinetic and Toxicity Model. International Journal of Molecular Sciences. 2023; 24(3):2296. https://doi.org/10.3390/ijms24032296
Chicago/Turabian StyleBrai, Annalaura, Federica Poggialini, Chiara Vagaggini, Claudia Pasqualini, Sauro Simoni, Valeria Francardi, and Elena Dreassi. 2023. "Tenebrio molitor as a Simple and Cheap Preclinical Pharmacokinetic and Toxicity Model" International Journal of Molecular Sciences 24, no. 3: 2296. https://doi.org/10.3390/ijms24032296