
Targeting Cancer Stem Cells as the Key Driver of Carcinogenesis and Therapeutic Resistance
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Cancer Stem Cells (CSCs)
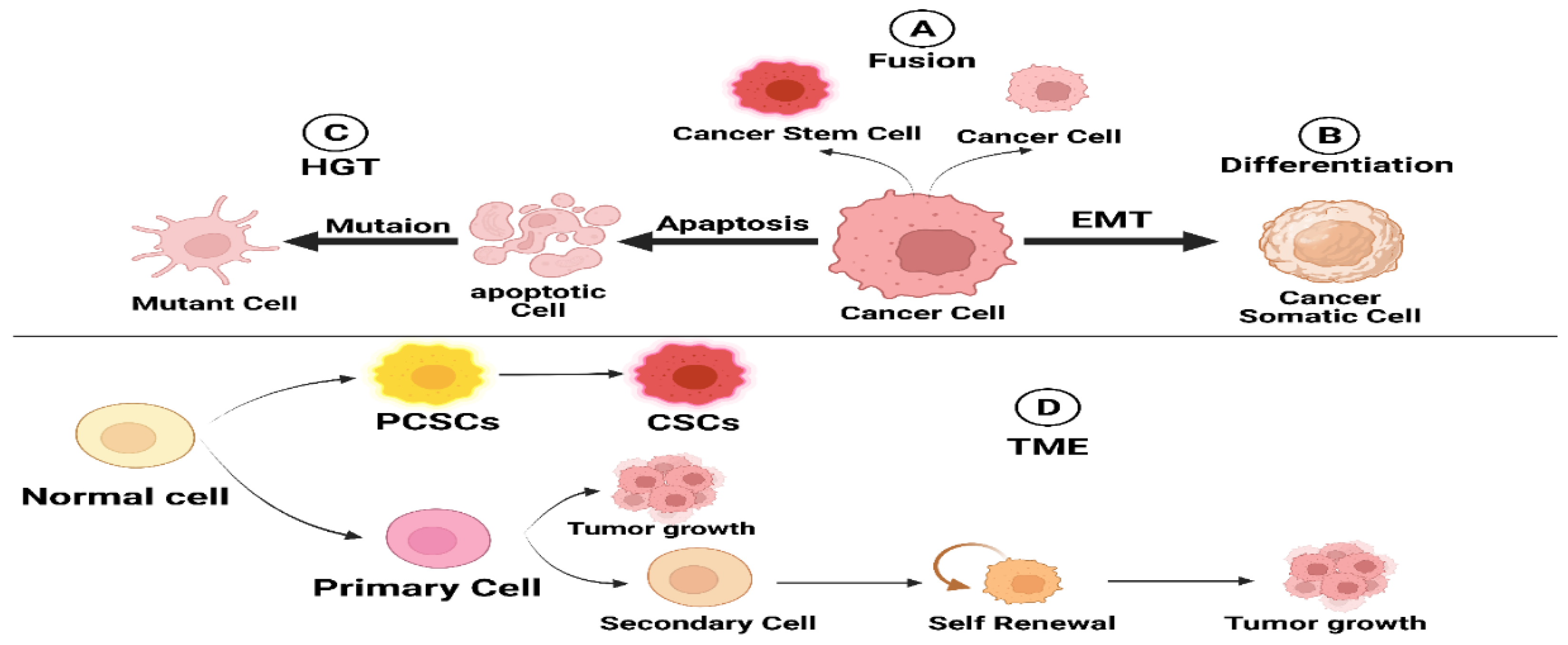
3. Origin of CSCs
3.1. Cell Fusion
3.2. Horizontal Gene Transfer
3.3. Dedifferentiation in Cancer Cells
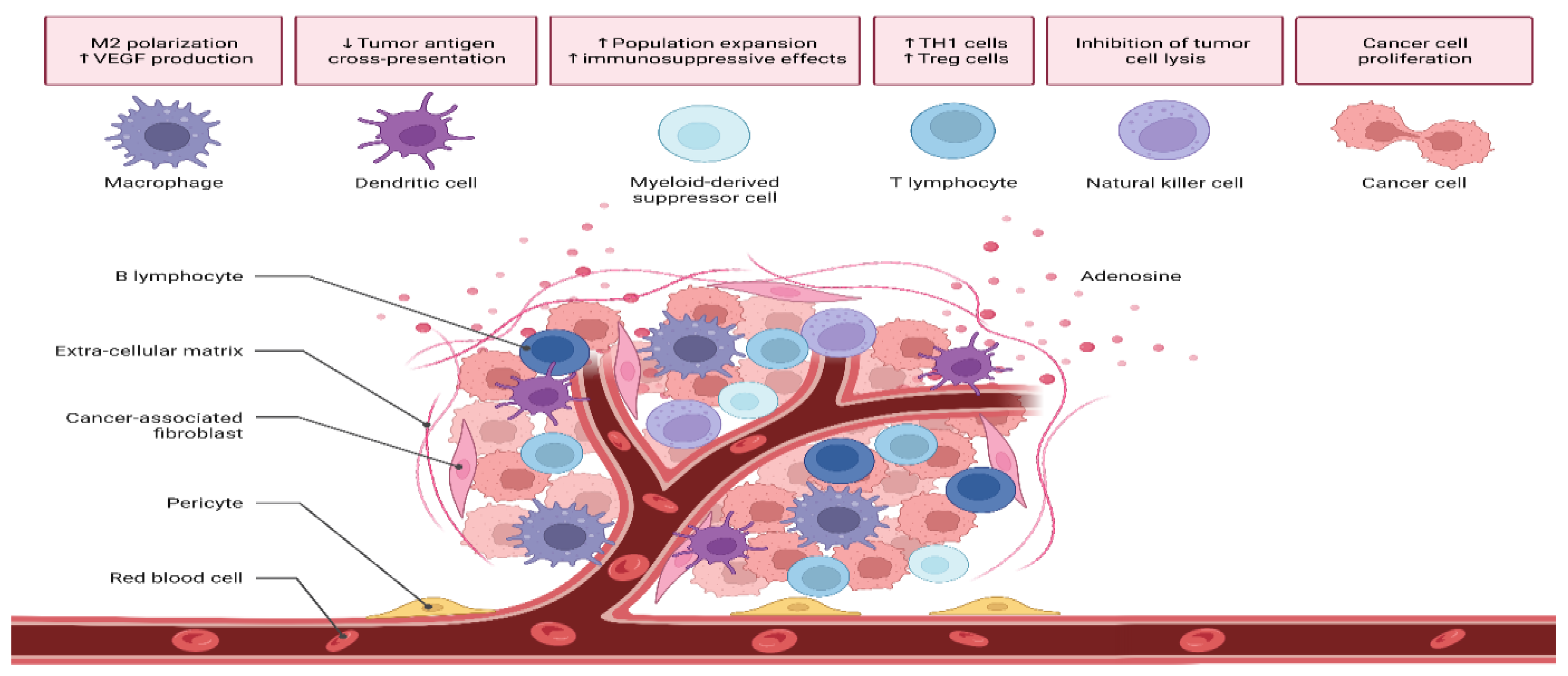
3.4. Tumor Microenvironment (TME)
4. Features of CSCs
4.1. Autophagy
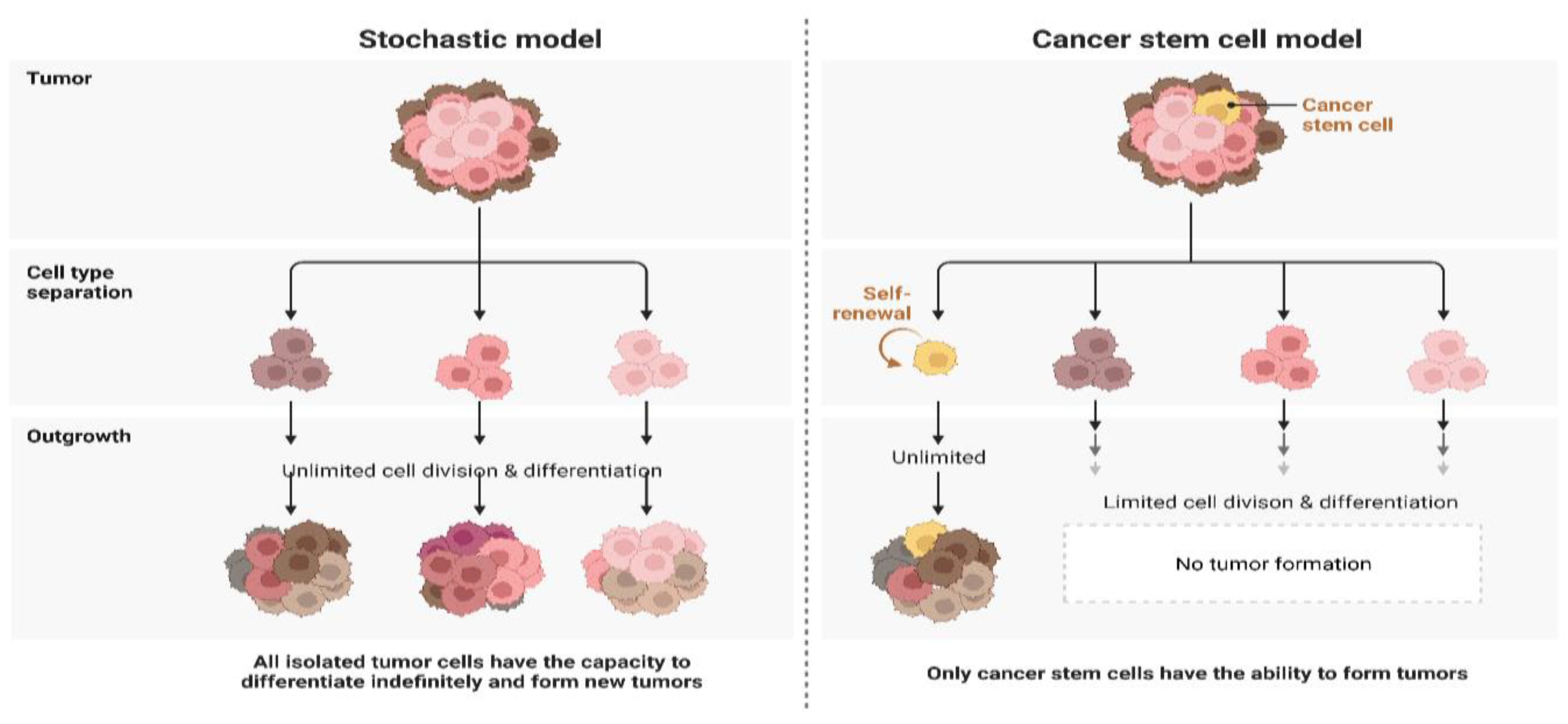
4.2. Self-Renewal, Differentiation, and Tumor Recurrence
4.3. Induction of Angiogenesis
4.4. CSCs Promote Metastasis
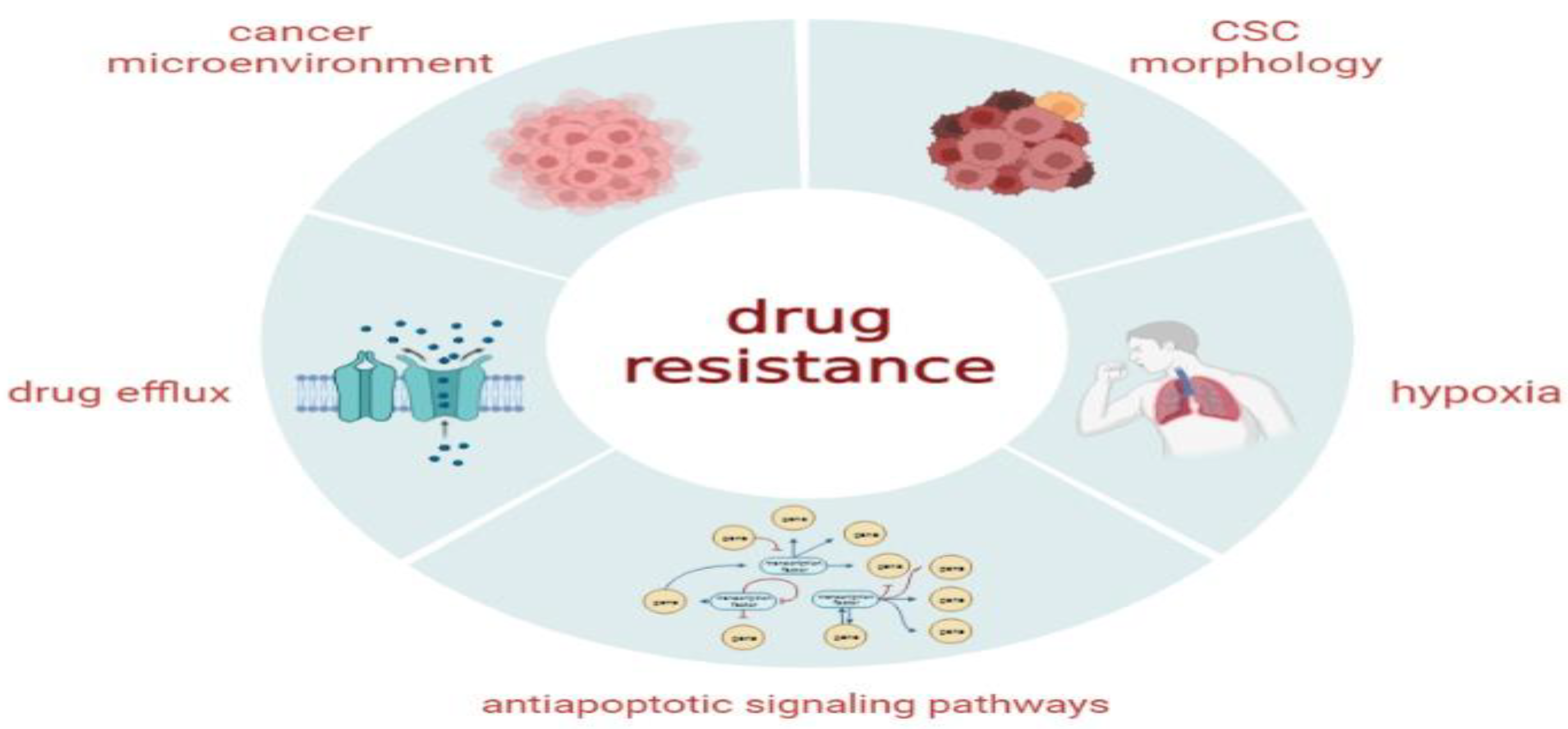
4.5. Radiation and Chemoresistance
5. Isolation Techniques of CSCs
5.1. Isolation with Surface Markers
5.2. Side Population Assay (SP)
5.3. Label-Retaining Methods (Lipophilic Dyes)
5.4. Tumorigenicity
5.5. Aldehyde Dehydrogenase Assay
5.6. Spheroid Formation Assay
5.7. Stemness Gene Expression and Transcriptional Factors
6. Signaling Pathways Governing CSCs’ Behavior
6.1. Wnt Signaling Pathway in CSCs
6.2. Hedgehog (Hh) Signaling
6.3. Notch Signaling
7. Novel Therapeutic Approaches for Targeting CSCs
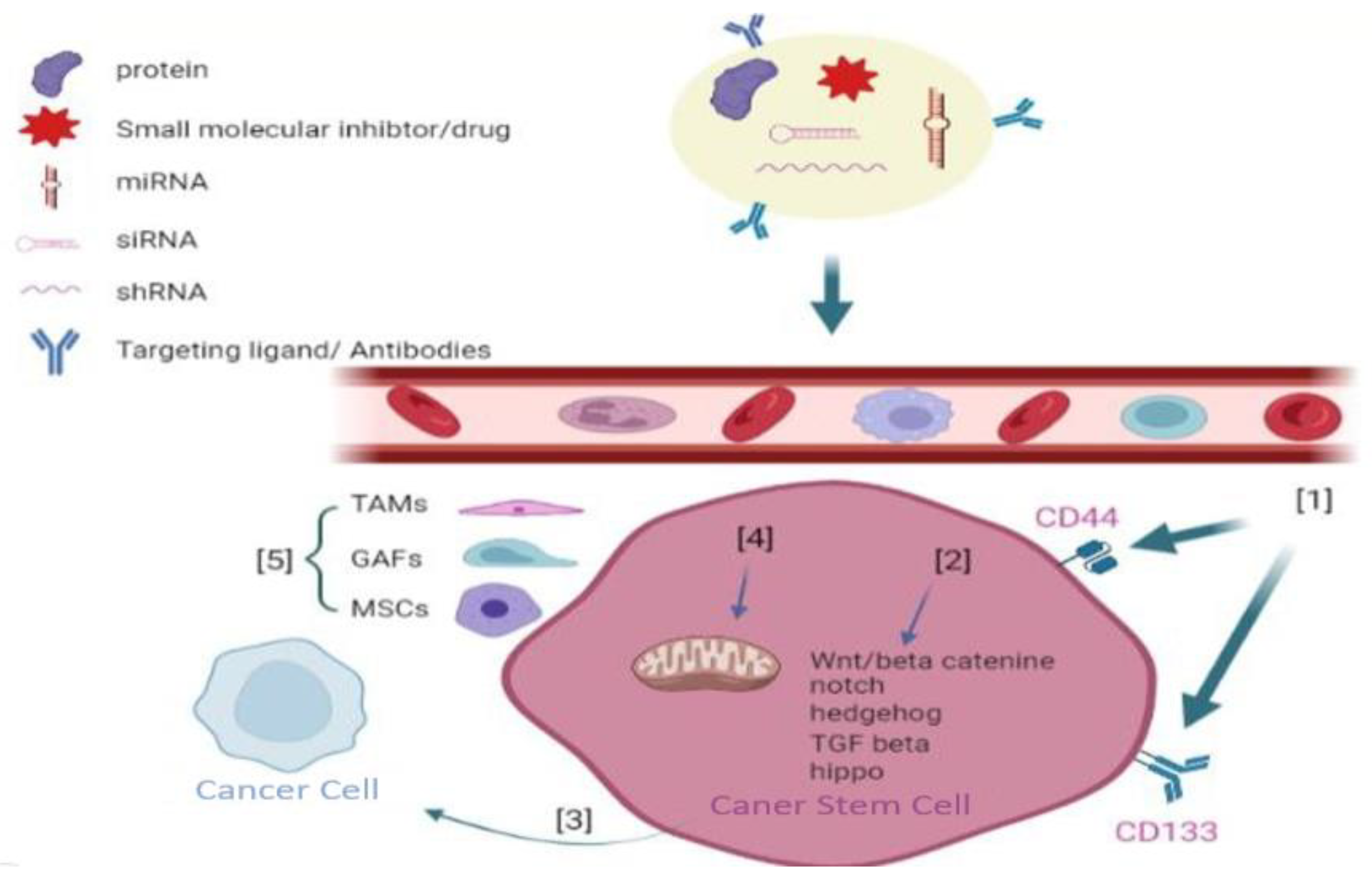
7.1. Targeting CSC Surface Markers
7.2. Inducing CSCs’ Differentiation
7.3. Targeting Metabolism in CSCs
7.4. Targeting the TME
7.5. Target Exosomes of CSCs
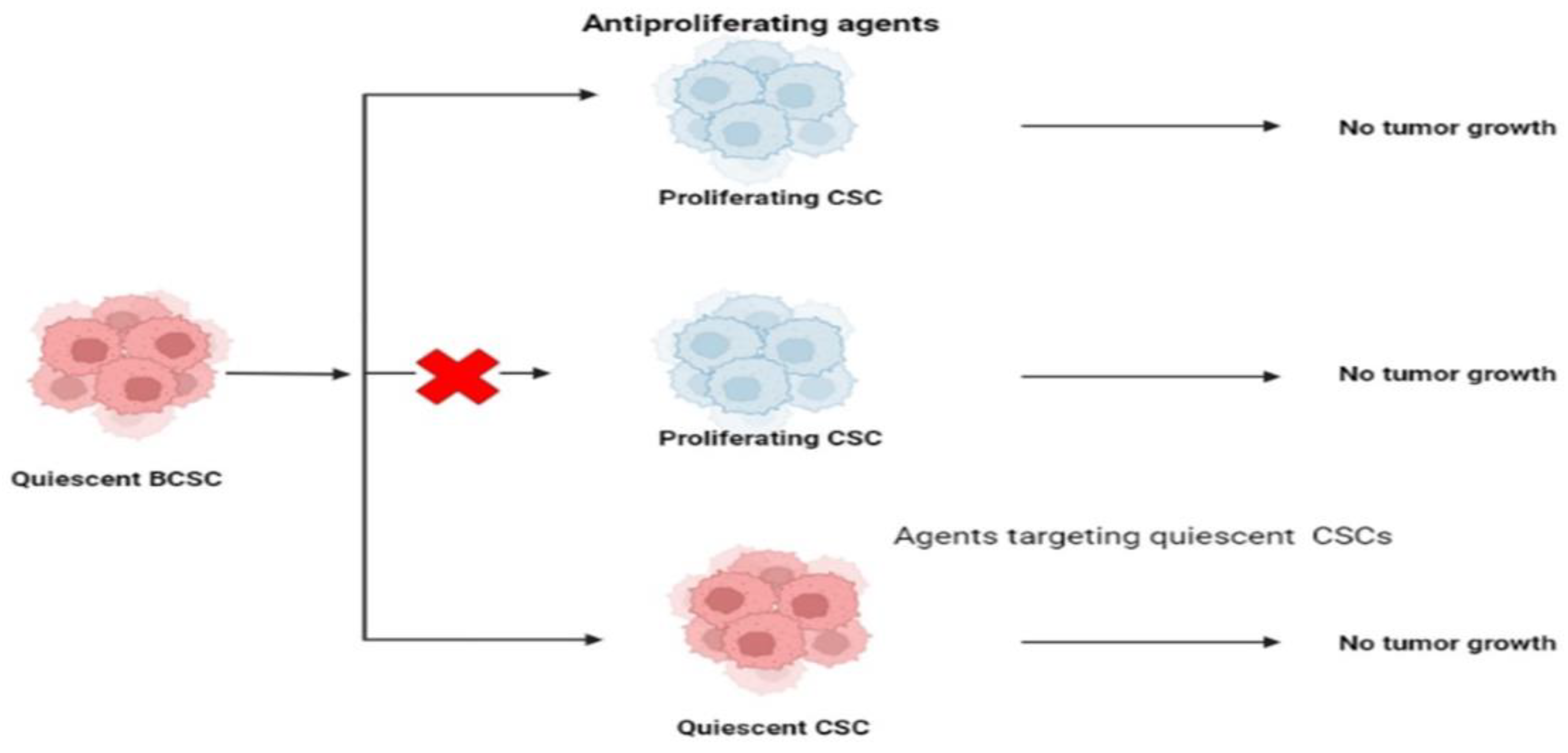
7.6. Targeting CSCs’ Quiescence
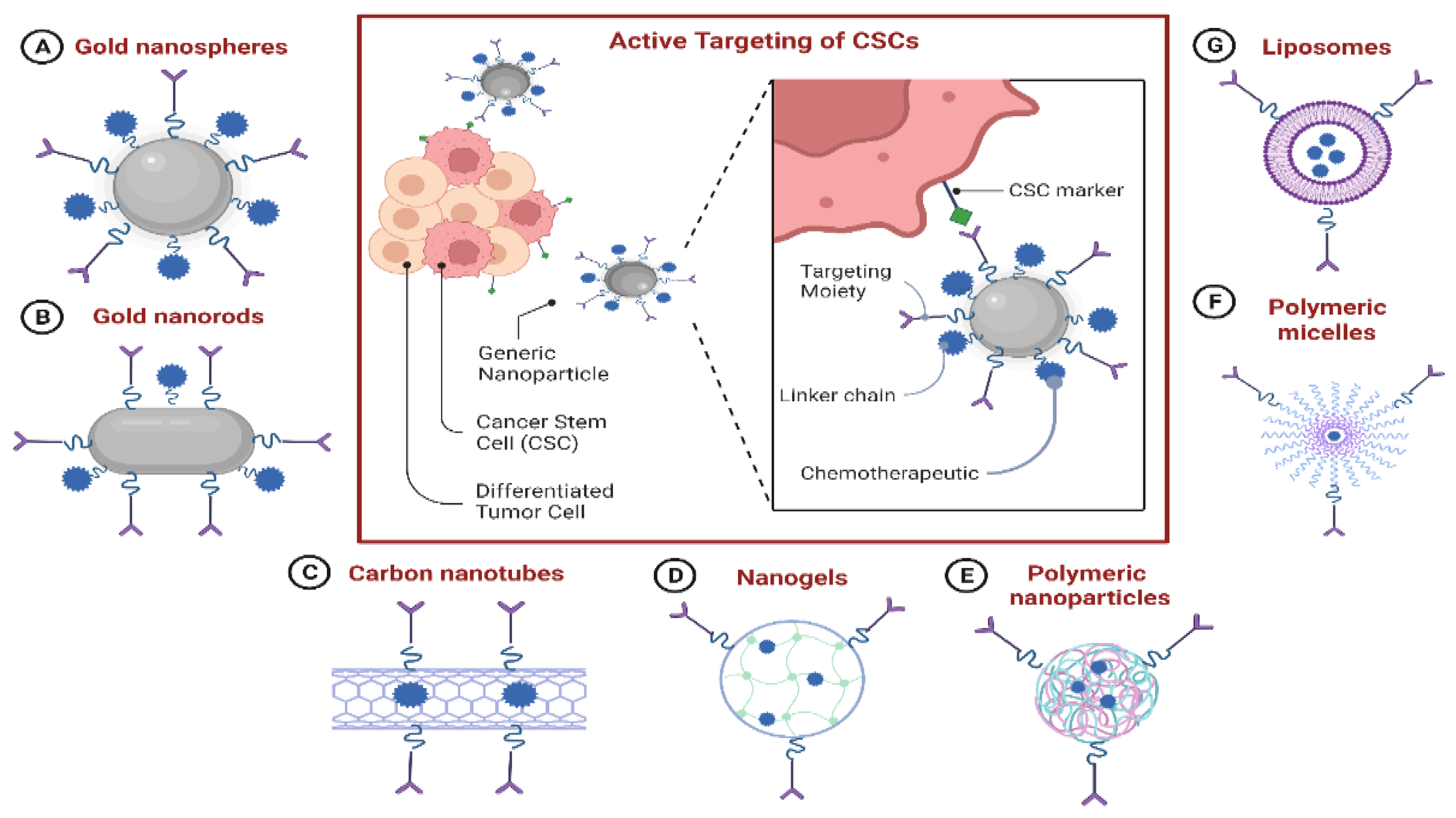
7.7. Nanoparticle-Based Drug-Delivery Systems (NDDSs) for Targeting CSCs
7.7.1. NDDS-Based Delivery of Chemotherapeutics to CSCs
7.7.2. NDDS-Based Delivery of Nucleic Acid Therapeutics to CSCs
7.7.3. Combinational Delivery of Chemotherapeutics and CSC-Specific Agents

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Application/Efficacy | Reference | |
|---|---|---|---|
| NDDS-Based Delivery of Chemotherapeutics to CSCs |
|
| [179] [145] |
| NDDS-Based Delivery of Nucleic Acid Therapeutics to CSCs |
|
| [173] [181] [181] [180] |
| Combinational Delivery of Chemotherapeutics and CSCs-Specific Agents |
|
| [178] [182] |
7.7.4. The Benefits and Drawbacks of Using Existing NDDSs against BCSCs
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Santosa, A.; Wall, S.; Fottrel, E.; Högberg, U.; Byass, P. The development and experience of epidemiological transition theory over four decades: A systematic review. Glob. Health Action 2014, 7 (Suppl. S1), 23574. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Qiu, H.; Cao, S.; Xu, R. Cancer incidence, mortality, and burden in China: A time-trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun. 2021, 41, 1037–1048. [Google Scholar] [CrossRef]
- Alonso, R.; Piñeros, M.; Laversanne, M.; Musetti, C.; Garau, M.; Barrios, E.; Bray, F. Lung cancer incidence trends in Uruguay 1990–2014: An age-period-cohort analysis. Cancer Epidemiol. 2018, 55, 17–22. [Google Scholar] [CrossRef]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Leon, G.; MacDonagh, L.; Finn, S.P.; Cuffe, S.; Barr, M.P. Cancer stem cells in drug resistant lung cancer: Targeting cell surface markers and signaling pathways. Pharmacol. Ther. 2016, 158, 71–90. [Google Scholar] [CrossRef]
- Afify, S.M.; Seno, M. Conversion of stem cells to cancer stem cells: Undercurrent of cancer initiation. Cancers 2019, 11, 345. [Google Scholar] [CrossRef] [Green Version]
- Bu, P.; Chen, K.Y.; Lipkin, S.M.; Shen, X. Asymmetric division: A marker for cancer stem cells? Oncotarget 2013, 4, 950. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Cabrera, M.C. Cancer stem cell plasticity and tumor hierarchy. World J. Stem. Cells 2015, 7, 27. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine 2016, 95, S2–S7. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Kang, Y. Cell fusion as a hidden force in tumor progression. Cancer Res. 2009, 69, 8536–8539. [Google Scholar] [CrossRef] [Green Version]
- Pawelek, J.M. Cancer cell fusion with migratory bone marrow-derived cells as an explanation for metastasis: New therapeutic paradigms. Future Oncol. 2008, 4, 449–452. [Google Scholar] [CrossRef]
- Dittmar, T.; Nagler, C.; Schwitalla, S.; Reith, G.; Niggemann, B.; Zänker, K.S. Recurrence cancer stem cells–Made by cell fusion? Med. Hypotheses 2009, 73, 542–547. [Google Scholar] [CrossRef]
- Bjerkvig, R.; Tysnes, B.B.; Aboody, K.S.; Najbauer, J.; Terzis, A.J.A. The origin of the cancer stem cell: Current controversies and new insights. Nat. Rev. Cancer 2005, 5, 899–904. [Google Scholar] [CrossRef]
- Johansson, C.B.; Youssef, S.; Koleckar, K.; Holbrook, C.; Doyonnas, R.; Corbel, S.Y.; Steinman, L.; Rossi, F.; Blau, H.M. Extensive fusion of haematopoietic cells with Purkinje neurons in response to chronic inflammation. Nat. Cell Biol. 2008, 10, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Houghton, J.; Stoicov, C.; Nomura, S.; Rogers, A.B.; Carlson, J.; Li, H.; Cai, X.; Fox, J.G.; Goldenring, J.R.; Wang, T.C. Gastric Cancer Originating from Bone Marrow-Derived Cells. Science 2004, 306, 1568–1571. [Google Scholar] [CrossRef]
- Ojha, R.; Bhattacharyya, S.; Singh, S.K. Autophagy in Cancer Stem Cells: A Potential Link Between Chemoresistance, Recurrence, and Metastasis. BioRes. Open Access 2015, 4, 97–108. [Google Scholar] [CrossRef]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [Green Version]
- Brown, Y.; Hua, S.; Tanwar, P.S. Extracellular matrix-mediated regulation of cancer stem cells and chemoresistance. Int. J. Biochem. Cell Biol. 2019, 109, 90–104. [Google Scholar] [CrossRef]
- McCarthy, N. Autophagy: Directed development. Nat. Rev. Cancer 2014, 14, 74–75. [Google Scholar] [CrossRef]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, D.; Liu, Y.; Su, Z.; Zhang, L.; Chen, F.; Zhou, Y.; Wu, Y.; Yu, M.; Zhang, Z.; et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133 + pancreatic cancer stem-like cells. Cancer Cell Int. 2013, 13, 119. [Google Scholar] [CrossRef] [Green Version]
- Kantara, C.; O’Connell, M.; Sarkar, S.; Moya, S.; Ullrich, R.; Singh, P. Curcumin promotes autophagic survival of a subset of colon cancer stem cells, which are ablated by DCLK1-siRNA. Cancer Res. 2014, 74, 2487–2498. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.G.; Penfornis, P.; Oskowitz, A.Z.; Boonjindasup, A.G.; Cai, D.Z.; Dhule, S.S.; Rowan, B.G.; Kelekar, A.; Krause, D.S.; Pochampally, R.R. Activation of autophagy in mesenchymal stem cells provides tumor stromal support. Carcinogenesis 2011, 32, 964–972. [Google Scholar] [CrossRef]
- Yoo, Y.D.; Kwon, Y.T. Molecular mechanisms controlling asymmetric and symmetric self- renewal of CSCs. J. Anal. Sci. Technol. 2015, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, E.; Chen, T. A matter of life and death: Self-renewal in stem cells. EMBO Rep. 2013, 14, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.W.; Hirth, F. Genetic mechanisms regulating stem cell self-renewal and differentiation in the central nervous system of Drosophila. Cell Adhes. Migr. 2009, 3, 402–411. [Google Scholar] [CrossRef]
- Park, T.S.; Donnenberg, V.S.; Donnenberg, A.D.; Zambidis, E.T.; Zimmerlin, L. Dynamic Interactions Between CSCs And Their Stromal Partners. Curr. Pathobiol. Rep. 2014, 2, 41–52. [Google Scholar] [CrossRef] [Green Version]
- King, T.D.; Suto, M.J.; Li, Y. The Wnt/β-catenin signaling pathway: A potential therapeutic target in the treatment of triple negative breast cancer. J. Cell Biochem. 2012, 113, 13–18. [Google Scholar] [CrossRef]
- Han, H.B.; Gu, J.; Zuo, H.J.; Chen, Z.G.; Zhao, W.; Li, M.; Ji, D.B.; Lu, Y.Y.; Zhang, Z.Q. Let-7c functions as a metastasis suppressor by targeting MMP11 and PBX3 in colorectal cancer. J. Pathol. 2012, 226, 544–555. [Google Scholar] [CrossRef]
- Jawhari, A.U.; Buda, A.; Jenkins, M.; Shehzad, K.; Sarraf, C.; Noda, M.; Farthing, M.J.; Pignatelli, M.; Adams, J.C. Fascin, an actin-bundling protein, modulates colonic epithelial cell invasiveness and differentiation in vitro. Am. J. Pathol. 2003, 162, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhang, Y.; Leung, L.; Liu, L.; Yang, F.; Yao, X. Efficacy and safety of angiogenesis inhibitors in advanced gastric cancer: A systematic review and meta-analysis. J. Hematol. Oncol. 2016, 9, 111. [Google Scholar] [CrossRef] [Green Version]
- Goel, G.; Sun, W. Ramucirumab, another anti-angiogenic agent for metastatic colorectal cancer in second-line setting—its impact on clinical practice. J. Hematol. Oncol. 2015, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Trivanović, D.; Jauković, A.; Krstić, J.; Nikolić, S.; Okić Djordjević, I.; Kukolj, T.; Obradović, H.; Mojsilović, S.; Ilić, V.; Santibanez, J.F.; et al. Inflammatory cytokines prime adipose tissue mesenchymal stem cells to enhance malignancy of MCF-7 breast cancer cells via transforming growth factor-β1. IUBMB Life 2016, 68, 190–200. [Google Scholar] [CrossRef] [Green Version]
- Kasimir-Bauer, S.; Bittner, A.; König, L.; Reiter, K.; Keller, T.; Kimmig, R.; Hoffmann, O. Does primary neoadjuvant systemic therapy eradicate minimal residual disease? Analysis of disseminated and circulating tumor cells before and after therapy. Breast Cancer Res. 2016, 18, 20. [Google Scholar] [CrossRef]
- Mori, M.; Ito, F.; Shi, L.; Wang, Y.; Ishida, C.; Hattori, Y.; Niwa, M.; Hirayama, T.; Nagasawa, H.; Iwase, A.; et al. Ovarian endometriosis-associated stromal cells reveal persistently high affinity for iron. Redox Biol. 2015, 6, 578–586. [Google Scholar] [CrossRef] [Green Version]
- Kubota, Y. Tumor angiogenesis and anti-angiogenic therapy. Keio J. Med. 2012, 61, 47–56. [Google Scholar] [CrossRef]
- Kanwar, J.; Kamalapuram, S.; Krishnakumar, S.; Kanwar, R. Multimodal iron oxide (Fe3O4)-saturated lactoferrin nanocapsules as nanotheranostics for real-time imaging and breast cancer therapy of claudin-low, triple-negative (ER-/PR-/HER2-). Nanomedicine 2016, 11, 249–268. [Google Scholar] [CrossRef]
- Sharma, B.; Varney, M.; Saxena, S.; Wu, L.; Singh, R. Induction of CXCR2 ligands, stem cell-like phenotype, and metastasis in chemotherapy-resistant breast cancer cells. Cancer Lett. 2016, 372, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Liu, J.; Liu, H.; Liang, S.; Lin, M.; Gu, Y.; Liu, T.; Wang, D.; Ge, H.; Mo, S.L. The antitumor effect of tanshinone IIA on anti-proliferation and decreasing VEGF/VEGFR2 expression on the human non-small cell lung cancer A549 cell line. Acta Pharm. Sin. B 2015, 5, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, C.; Li, Y.; Zhu, Z. Involvement of breast CSCs in tumor angiogenesis. Oncol. Lett. 2017, 14, 8150–8155. [Google Scholar] [CrossRef]
- Ciccone, V.; Terzuoli, E.; Donnini, S.; Giachetti, A.; Morbidelli, L.; Ziche, M. Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1α/VEGF signalling in MCF-7 breast cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 311. [Google Scholar] [CrossRef]
- Maiti, A.; Qi, Q.; Peng, X.; Yan, L.; Takabe, K.; Hait, N. Class I histone deacetylase inhibitor suppresses vasculogenic mimicry by enhancing the expression of tumor suppressor and anti-angiogenesis genes in aggressive human TNBC cells. Int. J. Oncol. 2019, 55, 116–130. [Google Scholar] [CrossRef] [Green Version]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Ramadan, F.; Fahs, A.; Ghayad, S.E.; Saab, R. Signaling pathways in Rhabdomyosarcoma invasion and metastasis. Cancer Metastasis Rev. 2020, 39, 287–301. [Google Scholar] [CrossRef]
- Kim, S.Y.; Hong, S.H.; Basse, P.H.; Wu, C.; Bartlett, D.L.; Kwon, Y.T.; Lee, Y.J. Cancer Stem Cells Protect Non-Stem Cells From Anoikis: Bystander Effects. J. Cell Biochem. 2016, 117, 2289–2301. [Google Scholar] [CrossRef]
- Kim, Y.; Koo, K.; Sung, J.; Yun, U.; Kim, H. Anoikis Resistance: An Essential Prerequisite for Tumor Metastasis. Int. J. Cell Biol. 2012, 2012, 306879. [Google Scholar] [CrossRef]
- Aramini, B.; Masciale, V.; Grisendi, G.; Bertolini, F.; Maur, M.; Guaitoli, G.; Chrystel, I.; Morandi, U.; Stella, F.; Dominici, M.; et al. Dissecting Tumor Growth: The Role of Cancer Stem Cells in Drug Resistance and Recurrence. Cancers 2022, 14, 976. [Google Scholar] [CrossRef]
- Zhu, P.; Fan, Z. Cancer stem cells and tumorigenesis. Biophys. Rep. 2018, 4, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Ablett, M.; O’Brien, C.; Sims, A.; Farnie, G.; Clarke, R. A differential role for CXCR4 in the regulation of normal versus malignant breast stem cell activity. Oncotarget 2013, 5, 599–612. [Google Scholar] [CrossRef] [Green Version]
- Lamb, R.; Ablett, M.; Spence, K.; Landberg, G.; Sims, A.; Clarke, R. Wnt Pathway Activity in Breast Cancer Sub-Types and Stem-Like Cells. PLoS ONE 2013, 8, e67811. [Google Scholar] [CrossRef] [Green Version]
- Baskar, R.; Dai, J.; Wenlong, N.; Yeo, R.; Yeoh, K. Biological response of cancer cells to radiation treatment. Front. Mol. Biosci. 2014, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Balaji, K.; Subramanian, B.; Yadav, P.; Anu Radha, C.; Ramasubramanian, V. Radiation therapy for breast cancer: Literature review. Med. Dosim. 2016, 41, 253–257. [Google Scholar] [CrossRef]
- Wang, Q. DNA damage responses in CSCs: Implications for cancer therapeutic strategies. World J. Biol. Chem. 2015, 6, 57. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in CSCs. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.M.; Li, J.J. NF-kappa B-mediated adaptive resistance to ionizing radiation. Free. Radic. Biol. Med. 2008, 44, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.; Liu, Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2010, 21, 103–115. [Google Scholar] [CrossRef]
- Duru, N.; Fan, M.; Candas, D.; Menaa, C.; Liu, H.C.; Nantajit, D.; Wen, Y.; Xiao, K.; Eldridge, A.; Chromy, B.A.; et al. HER2-Associated Radioresistance of Breast CSCs Isolated from HER2-Negative Breast Cancer Cells. Clin. Cancer Res. 2012, 18, 6634–6647. [Google Scholar] [CrossRef] [Green Version]
- Kamble, D.; Mahajan, M.; Dhat, R.; Sitasawad, S. Keap1-Nrf2 Pathway Regulates ALDH and Contributes to Radioresistance in Breast CSCs. Cells 2021, 10, 83. [Google Scholar] [CrossRef]
- Arnold, K.; Opdenaker, L.; Flynn, N.; Appeah, D.; Sims-Mourtada, J. Radiation induces an inflammatory response that results in STAT3-dependent changes in cellular plasticity and radioresistance of breast cancer stem-like cells. Int. J. Radiat. Biol. 2020, 96, 434–447. [Google Scholar] [CrossRef]
- Hittelman, W.N.; Liao, Y.; Wang, L.; Milas, L. Are CSCs radioresistant? Future Oncol. 2010, 6, 1563–1576. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.M.; Zhang, J.G.; Zhang, X.; Li, Q. Targeting CSCs for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target Ther. 2021, 6, 62. [Google Scholar] [CrossRef]
- Bao, B.; Ahmad, A.; Azmi, A.S.; Ali, S.; Sarkar, F.H. Overview of CSCs (CSCs) and mechanisms of their regulation: Implications for cancer therapy. Curr. Protoc Pharmacol. 2013, 61, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Powell, K.; Li, L. Breast CSCs: Biomarkers, Identification and Isolation Methods, Regulating Mechanisms, Cellular Origin, and Beyond. Cancers 2020, 12, 3765. [Google Scholar] [CrossRef]
- Feng, Z.M.; Qiu, J.; Chen, X.W.; Liao, R.X.; Liao, X.Y.; Zhang, L.P.; Chen, X.; Li, Y.; Chen, Z.T.; Sun, J.G. Essential role of miR-200c in regulating self-renewal of breast CSCs and their counterparts of mammary epithelium. BMC Cancer 2015, 15, 645. [Google Scholar] [CrossRef] [Green Version]
- Akbarzadeh, M.; Maroufi, N.F.; Tazehkand, A.P.; Akbarzadeh, M.; Bastani, S.; Safdari, R.; Farzane, A.; Fattahi, A.; Nejabati, H.R.; Nouri, M.; et al. Current approaches in identification and isolation of CSCs. J. Cell. Physiol. 2019, 234, 14759–14772. [Google Scholar] [CrossRef]
- Keysar, S.B.; Jimeno, A. More than markers: Biological significance of cancer stem cell- defining molecules. Mol. Cancer Ther. 2010, 9, 2450–2457. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Dick, J.E. Acute myeloid leukemia stem cells. Ann. N. York Acad. Sci. 2005, 1044, 1–5. [Google Scholar] [CrossRef]
- Pham, P.V.; Phan, N.L.; Nguyen, N.T.; Truong, N.H.; Duong, T.T.; Le, D.V.; Truong, K.D.; Phan, N.K. Differentiation of breast CSCs by knockdown of CD44: Promising differentiation therapy. J. Transl. Med. 2011, 9, 209. [Google Scholar] [CrossRef] [Green Version]
- Vikram, R.; Chou, W.C.; Hung, S.C.; Shen, C.Y. Tumorigenic and Metastatic Role of CD44-/low/CD24-/low Cells in Luminal Breast Cancer. Cancers 2020, 12, 1239. [Google Scholar] [CrossRef]
- Meyer, M.J.; Fleming, J.M.; Lin, A.F.; Hussnain, S.A.; Ginsburg, E.; Vonderhaar, B.K. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor- negative breast cancer. Cancer Res. 2010, 70, 4624–4633. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; Li, H.; Wang, H.; Shi, X.; Fan, Y.; Ding, X.; Lin, C.; Zhan, Q.; Qian, H.; Xu, B. Enriched CD44(+)/CD24(-) population drives the aggressive phenotypes presented in triple-negative breast cancer (TNBC). Cancer Lett. 2014, 353, 153–159. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.; El-Rayes, B. Identifying and targeting CSCs in the treatment of gastric cancer. Cancer 2017, 123, 1303–1312. [Google Scholar] [CrossRef] [Green Version]
- Moltzahn, F.; Thalmann, G.N. CSCs in prostate cancer. Transl. Androl. Urol. 2013, 2, 242–253. [Google Scholar] [CrossRef]
- Huang, S.D.; Yuan, Y.; Tang, H.; Liu, X.H.; Fu, C.G.; Cheng, H.Z.; Bi, J.W.; Yu, Y.W.; Gong, D.J.; Zhang, W.; et al. Tumor cells positive and negative for the common cancer stem cell markers are capable of initiating tumor growth and generating both progenies. PLoS ONE 2013, 8, e54579. [Google Scholar] [CrossRef] [Green Version]
- Fojo, T.; Coley, H.M. The role of efflux pumps in drug-resistant metastatic breast cancer: New insights and treatment strategies. Clin. Breast Cancer 2007, 7, 749–756. [Google Scholar] [CrossRef]
- Zinzi, L.; Contino, M.; Cantore, M.; Capparelli, E.; Leopoldo, M.; Colabufo, N.A. ABC transporters in CSCs membranes as a novel target for treating tumor relapse. Front. Pharmacol. 2014, 5, 163. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Birnbaum, D. Breast CSCs: Tools and models to rely on. BMC Cancer 2009, 9, 202. [Google Scholar] [CrossRef]
- Yeo, S.K.; Guan, J.L. Breast Cancer: Multiple Subtypes within a Tumor? Trends Cancer 2017, 3, 753–760. [Google Scholar] [CrossRef]
- Goodell, M.A. Stem cell identification and sorting using the Hoechst 33342 side population (SP). Curr. Protoc Cytom. 2005, 34, 9–18. [Google Scholar] [CrossRef]
- Kim, M.C.; D’Costa, S.; Suter, S.; Kim, Y. Evaluation of a side population of canine lymphoma cells using Hoechst 33342 dye. J. Vet. Sci. 2013, 14, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Zhao, Y.; Ouyang, T.; Zhao, T.; Zhang, S.; Chen, J.; Yu, J.; Lei, T. Label-retaining assay enriches tumor-initiating cells in glioblastoma spheres cultivated in serum-free medium. Oncol Lett. 2016, 12, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Boutonnat, J.; Faussat, A.M.; Marie, J.P.; Bignon, J.; Wdzieczak-Bakala, J.; Barbier, M.; Thierry, J.; Ronot, X.; Colle, P.E. Usefulness of PKH fluorescent labelling to study leukemic cell proliferation with various cytostatic drugs or acetyl tetrapeptide--AcSDKP. BMC Cancer 2005, 5, 120. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Ma, H.; Zhang, Y.; Li, B.; Zhou, M.; Wang, X.; Zhao, Y. Application of technique of labeling BMSCs with PKH26 to tissue engineered bone construction. Chin. J. Reparative Reconstr. Surg. 2009, 23, 1246–1249. [Google Scholar]
- Velasco-Velázquez, M.A.; Homsi, N.; De La Fuente, M.; Pestell, R.G. Breast cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 573–577. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [Green Version]
- Hart, L.S.; Dolloff, N.G.; Dicker, D.T.; Koumenis, C.; Christensen, J.G.; Grimberg, A.; El-Deiry, W.S. Human colon CSCs are enriched by insulin-like growth factor-1 and are sensitive to figitumumab. Cell Cycle 2011, 10, 2331–2338. [Google Scholar] [CrossRef]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Poturnajova, M.; Kozovska, Z.; Matuskova, M. Aldehyde dehydrogenase 1A1 and 1A3 isoforms - mechanism of activation and regulation in cancer. Cell Signal. 2021, 87, 110120. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Delort, L.; Bougaret, L.; Cholet, J.; Vermerie, M.; Billard, H.; Decombat, C.; Bourgne, C.; Berger, M.; Dumontet, C.; Caldefie-Chezet, F. Hormonal Therapy Resistance and Breast Cancer: Involvement of Adipocytes and Leptin. Nutrients 2019, 11, 2839. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Cruzado, L.; Tornin, J.; Santos, L.; Rodriguez, A.; García-Castro, J.; Morís, F.; Rodriguez, R. Aldh1 Expression and Activity Increase During Tumor Evolution in Sarcoma Cancer Stem Cell Populations. Sci. Rep. 2016, 6, 27878. [Google Scholar] [CrossRef] [Green Version]
- Greve, B.; Kelsch, R.; Spaniol, K.; Eich, H.T.; Götte, M. Flow cytometry in cancer stem cell analysis and separation. Cytom. A 2012, 81, 284–293. [Google Scholar] [CrossRef]
- Croker, A.K.; Goodale, D.; Chu, J.; Postenka, C.; Hedley, B.D.; Hess, D.A.; Allan, A.L. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell Mol. Med. 2009, 13, 2236–2252. [Google Scholar] [CrossRef] [Green Version]
- Saeg, F.; Anbalagan, M. Breast CSCs and the challenges of eradication: A review of novel therapies. Stem Cell Investig. 2018, 5, 39. [Google Scholar] [CrossRef]
- Muraro, M.G.; Mele, V.; Däster, S.; Han, J.; Heberer, M.; Cesare Spagnoli, G.; Iezzi, G. CD133+, CD166+CD44+, and CD24+CD44+ phenotypes fail to reliably identify cell populations with cancer stem cell functional features in established human colorectal cancer cell lines. Stem Cells Transl Med. 2012, 1, 592–603. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Cheaito, K.; Chalhoub, R.M.; Hadadeh, O.; Monzer, A.; Ballout, F.; El-Hajj, A.; Mukherji, D.; Liu, Y.N.; Daoud, G.; et al. Sphere-Formation Assay: Three- Dimensional in vitro Culturing of Prostate Cancer Stem/Progenitor Sphere-Forming Cells. Front. Oncol. 2018, 8, 347. [Google Scholar] [CrossRef]
- Fu, J.J.; Zhou, Y.; Shi, X.X.; Kang, Y.J.; Lu, Z.S.; Li, Y.; Li, C.M.; Yu, L. Spontaneous formation of tumor spheroid on a hydrophilic filter paper for cancer stem cell enrichment. Colloids Surf. B Biointerfaces 2019, 174, 426–434. [Google Scholar] [CrossRef]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [Green Version]
- Nandi, A.; Chakrabarti, R. Assessment of Breast Cancer Stem Cell Activity Using a Spheroid Formation Assay. Methods Mol. Biol. 2022, 2429, 485–500. [Google Scholar] [CrossRef]
- Apostolou, P.; Toloudi, M.; Chatziioannou, M.; Ioannou, E.; Papasotiriou, I. CSCs stemness transcription factors expression correlates with breast cancer disease stage. Curr. Stem. Cell Res. Ther. 2012, 7, 415–419. [Google Scholar] [CrossRef]
- Yilmazer, A. Evaluation of cancer stemness in breast cancer and glioblastoma spheroids in vitro. 3 Biotech 2018, 8, 390. [Google Scholar] [CrossRef]
- Zhou, W.; Lv, R.; Qi, W.; Wu, D.; Xu, Y.; Liu, W.; Mou, Y.; Wang, L. Snail contributes to the maintenance of stem cell-like phenotype cells in human pancreatic cancer. PLoS ONE 2014, 9, e87409. [Google Scholar] [CrossRef]
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95 (Suppl. S1), S8–S19. [Google Scholar] [CrossRef]
- Vesuna, F.; Lisok, A.; Kimble, B.; Raman, V. Twist modulates breast CSCs by transcriptional regulation of CD24 expression. Neoplasia 2009, 11, 1318–1328. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Peng, Y.; Liu, Y.; Xin, H.; Zhan, X.; Tan, W. Twist1 and Snail link Hedgehog signaling to tumor-initiating cell-like properties and acquired chemoresistance independently of ABC transporters. Stem Cells 2015, 33, 1063–1074. [Google Scholar] [CrossRef]
- Duchartre, Y.; Kim, Y.M.; Kahn, M. The Wnt signaling pathway in cancer. Crit. Rev. Oncol. Hematol. 2016, 99, 141–149. [Google Scholar] [CrossRef]
- Kim, Y.M.; Kahn, M. The role of the Wnt signaling pathway in CSCs: Prospects for drug development. Res. Rep. Biochem. 2014, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. Canonical and non-canonical WNT signaling in CSCs and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M.; Katoh, M. WNT signaling pathway and stem cell signaling network. Clin. Cancer Res. 2007, 13, 4042–4045. [Google Scholar] [CrossRef] [Green Version]
- Pećina-Slaus, N. Wnt signal transduction pathway and apoptosis: A review. Cancer Cell Int. 2010, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Butti, R.; Gunasekaran, V.P.; Kumar, T.V.S.; Banerjee, P.; Kundu, G.C. Breast cancer stem cells: Biology and therapeutic implications. Int. J. Biochem. Cell Biol. 2019, 107, 38–52. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of CSCs. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef]
- Bhateja, P.; Cherian, M.; Majumder, S.; Ramaswamy, B. The Hedgehog Signaling Pathway: A Viable Target in Breast Cancer? Cancers 2019, 11, 1126. [Google Scholar] [CrossRef] [Green Version]
- Signorelli, D.; Proto, C.; Botta, L.; Trama, A.; Tiseo, M.; Pasello, G.; Lo Russo, G.; Fabbri, A.; Imbimbo, M.; Busico, A.; et al. SMO mutations confer poor prognosis in malignant pleural mesothelioma. Transl. Lung Cancer Res. 2020, 9, 1940–1951. [Google Scholar] [CrossRef]
- Pasca di Magliano, M.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef]
- Candi, E.; Amelio, I.; Agostini, M.; Melino, G. MicroRNAs and p63 in epithelial stemness. Cell Death Differ. 2015, 22, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Hou, Y.; Yang, G.; Zhang, H.; Tu, G.; Du, Y.E.; Wen, S.; Xu, L.; Tang, X.; Tang, S.; et al. LncRNA-Hh Strengthen CSCs Generation in Twist- Positive Breast Cancer via Activation of Hedgehog Signaling Pathway. Stem. Cells 2016, 34, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Janghorban, M.; Xin, L.; Rosen, J.M.; Zhang, X.H. Notch Signaling as a Regulator of the Tumor Immune Response: To Target or Not To Target? Front. Immunol. 2018, 9, 1649. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, V.; Nataraj, R.; Thangaraj, G.S.; Karthikeyan, M.; Gnanasekaran, A.; Kaginelli, S.B.; Kuppanna, G.; Kallappa, C.G.; Basalingappa, K.M. Targeting Notch signalling pathway of CSCs. Stem Cell Investig. 2018, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Espinoza, I.; Pochampally, R.; Xing, F.; Watabe, K.; Miele, L. Notch signaling: Targeting CSCs and epithelial-to-mesenchymal transition. Onco Targets Ther. 2013, 6, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Avila, J.L.; Kissil, J.L. Notch signaling in pancreatic cancer: Oncogene or tumor suppressor? Trends Mol. Med. 2013, 19, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Duan, G.; Walther, D. The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput. Biol. 2015, 11, e1004049. [Google Scholar] [CrossRef]
- Xiao, W.; Gao, Z.; Duan, Y.; Yuan, W.; Ke, Y. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 41. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Yu, A.; Deng, L.; Zhang, H. Eradicating the Roots: Advanced Therapeutic Approaches Targeting Breast CSCs. Curr. Pharm. Des. 2020, 26, 2009–2021. [Google Scholar] [CrossRef]
- García-Heredia, J.M.; Otero-Albiol, D.; Pérez, M.; Pérez-Castejón, E.; Muñoz-Galván, S.; Carnero, A. Breast tumor cells promotes the horizontal propagation of EMT, stemness, and metastasis by transferring the MAP17 protein between subsets of neoplastic cells. Oncogenesis 2020, 9, 96. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Woosley, A.N.; Dalton, A.C.; Hussey, G.S.; Howley, B.V.; Mohanty, B.K.; Grelet, S.; Dincman, T.; Bloos, S.; Olsen, S.K.; Howe, P.H. TGFβ promotes breast cancer stem cell self-renewal through an ILEI/LIFR signaling axis. Oncogene 2019, 38, 3794–3811. [Google Scholar] [CrossRef]
- Elia, I.; Rossi, M.; Stegen, S.; Broekaert, D.; Doglioni, G.; van Gorsel, M.; Boon, R.; Escalona- Noguero, C.; Torrekens, S.; Verfaillie, C.; et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 2019, 568, 117–121. [Google Scholar] [CrossRef]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast CSCs. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef] [Green Version]
- Kai, M.; Kanaya, N.; Wu, S.V.; Mendez, C.; Nguyen, D.; Luu, T.; Chen, S. Targeting breast CSCs in triple-negative breast cancer using a combination of LBH589 and salinomycin. Breast. Cancer Res. Treat. 2015, 151, 281–294. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, I.G.; Choi, B.H.; Ku, S.K.; Kwak, M.K. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: Implications for cancer stem cell resistance. Redox Biol. 2018, 17, 246–258. [Google Scholar] [CrossRef]
- Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in CSCs: A Promising Biomarker and Therapeutic Target. Stem Cells Transl. Med. 2015, 4, 1033–1043. [Google Scholar] [CrossRef]
- Marangoni, E.; Lecomte, N.; Durand, L.; de Pinieux, G.; Decaudin, D.; Chomienne, C.; Smadja- Joffe, F.; Poupon, M.F. CD44 targeting reduces tumour growth and prevents post- chemotherapy relapse of human breast cancers xenografts. Br. J. Cancer 2009, 100, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Brugnoli, F.; Grassilli, S.; Al-Qassab, Y.; Capitani, S.; Bertagnolo, V. CD133 in Breast Cancer Cells: More than a Stem Cell Marker. J. Oncol. 2019, 2019, 7512632. [Google Scholar] [CrossRef] [Green Version]
- Ohlfest, J.R.; Zellmer, D.M.; Panyam, J.; Swaminathan, S.K.; Oh, S.; Waldron, N.N.; Toma, S.; Vallera, D.A. Immunotoxin targeting CD133(+) breast carcinoma cells. Drug. Deliv. Transl. Res. 2013, 3, 195–204. [Google Scholar] [CrossRef]
- Naujokat, C. Monoclonal antibodies against human CSCs. Immunotherapy 2014, 6, 290–308. [Google Scholar] [CrossRef]
- Kubo, M.; Umebayashi, M.; Kurata, K.; Mori, H.; Kai, M.; Onishi, H.; Katano, M.; Nakamura, M.; Morisaki, T. Catumaxomab with Activated T-cells Efficiently Lyses Chemoresistant EpCAM-positive Triple-negative Breast Cancer Cell Lines. Anticancer Res. 2018, 38, 4273–4279. [Google Scholar] [CrossRef]
- Sun, R.; Liu, Y.; Li, S.Y.; Shen, S.; Du, X.J.; Xu, C.F.; Cao, Z.T.; Bao, Y.; Zhu, Y.H.; Li, Y.P.; et al. Corrigendum to “Co-delivery of all-trans-retinoic acid and doxorubicin for cancer therapy with synergistic inhibition of CSCs”. Biomaterials 2020, 263, 120373. [Google Scholar] [CrossRef]
- Snyder, V.; Reed-Newman, T.C.; Arnold, L.; Thomas, S.M.; Anant, S. Cancer Stem Cell Metabolism and Potential Therapeutic Targets. Front. Oncol. 2018, 8, 203. [Google Scholar] [CrossRef] [Green Version]
- Pindiprolu, S.K.S.S.; Krishnamurthy, P.T.; Chintamaneni, P.K.; Karri, V.V.S.R. Nanocarrier based approaches for targeting breast CSCs. Artif. Cells Nanomed. Biotechnol. 2018, 46, 885–898. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, H.A.; Iliopoulos, D.; Struhl, K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc. Natl. Acad. Sci. USA 2013, 110, 972–977. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; Monville, F.; Wicinski, J.; Cabaud, O.; Cervera, N.; Josselin, E.; Finetti, P.; Guille, A.; Larderet, G.; Viens, P.; et al. Mevalonate metabolism regulates Basal breast CSCs and is a potential therapeutic target. Stem Cells 2012, 30, 1327–1337. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liao, D.; Chen, C.; Liu, Y.; Chuang, T.H.; Xiang, R.; Markowitz, D.; Reisfeld, R.A.; Luo, Y. Tumor-associated macrophages regulate murine breast CSCs through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells 2013, 31, 248–258. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Gong, H.; Luo, S.; Cui, Y. The Role of Exosomes and Their Applications in Cancer. Int. J. Mol. Sci. 2021, 22, 12204. [Google Scholar] [CrossRef]
- Aiello, N.; Kang, Y. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.S.; Ma, S.; Dou, H.; Liu, F.; Zhang, S.Y.; Jiang, C.; Xiao, M.; Huang, Y.X. Breast cancer-derived exosomes regulate cell invasion and metastasis in breast cancer via miR-146a to activate cancer associated fibroblasts in tumor microenvironment. Exp. Cell Res. 2020, 391, 111983. [Google Scholar] [CrossRef]
- Hayday, A. γδ T Cell Update: Adaptate Orchestrators of Immune Surveillance. J. Immunol. 2019, 203, 311–320. [Google Scholar] [CrossRef]
- Yin, Z.; Yu, M.; Ma, T.; Zhang, C.; Huang, S.; Karimzadeh, M.R. Mechanisms underlying low-clinical responses to PD-1/PD-L1 blocking antibodies in immunotherapy of cancer: A key role of exosomal PD-L1. J. Immunother. Cancer 2021, 9, e001698. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; Van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Guo, B.; Bellingham, S.; Hill, A. The Neutral Sphingomyelinase Pathway Regulates Packaging of the Prion Protein into Exosomes. J. Biol. Chem. 2015, 290, 3455–3467. [Google Scholar] [CrossRef] [Green Version]
- Gernapudi, R.; Yao, Y.; Zhang, Y.; Wolfson, B.; Roy, S.; Duru, N.; Eades, G.; Yang, P.; Zhou, Q. Targeting exosomes from preadipocytes inhibits preadipocyte to cancer stem cell signaling in early-stage breast cancer. Breast Cancer Res. Treat. 2015, 150, 685–695. [Google Scholar] [CrossRef] [Green Version]
- Martynoga, B.; Mateo, J.L.; Zhou, B.; Andersen, J.; Achimastou, A.; Urbán, N.; van den Berg, D.; Georgopoulou, D.; Hadjur, S.; Wittbrodt, J.; et al. Epigenomic enhancer annotation reveals a key role for NFIX in neural stem cell quiescence. Genes Dev. 2013, 27, 1769–1786. [Google Scholar] [CrossRef] [Green Version]
- Paik, J.H.; Ding, Z.; Narurkar, R.; Ramkissoon, S.; Muller, F.; Kamoun, W.S.; Chae, S.S.; Zheng, H.; Ying, H.; Mahoney, J.; et al. FoxOs Cooperatively Regulate Diverse Pathways Governing Neural Stem Cell Homeostasis. Cell Stem Cell 2009, 5, 540–553. [Google Scholar] [CrossRef]
- Pece, S.; Tosoni, D.; Confalonieri, S.; Mazzarol, G.; Vecchi, M.; Ronzoni, S.; Bernard, L.; Viale, G.; Pelicci, P.G.; Di Fiore, P.P. Biological and Molecular Heterogeneity of Breast Cancers Correlates with Their Cancer Stem Cell Content. Cell 2010, 140, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Lorenzana, D.; Avilés-Vazquez, S.; Sandoval Esquivel, M.A.; Alvarado-Moreno, A.; Ortiz-Navarrete, V.; Torres-Martínez, H.; Ayala-Sánchez, M.; Mayani, H.; Chavez-Gonzalez, A. CDKIs p18INK4c and p57Kip2 are involved in quiescence of CML leukemic stem cells after treatment with TKI. Cell Cycle 2016, 15, 1276–1287. [Google Scholar] [CrossRef] [Green Version]
- Takeishi, S.; Matsumoto, A.; Onoyama, I.; Naka, K.; Hirao, A.; Nakayama, K. Ablation of Fbxw7 Eliminates Leukemia-Initiating Cells by Preventing Quiescence. Cancer Cell 2013, 23, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Shi, G.; Zhang, X.; Dong, W.; Zhang, L. Transgenic expression of BRCA1 disturbs hematopoietic stem and progenitor cells quiescence and function. Exp. Cell Res. 2013, 319, 2739–2746. [Google Scholar] [CrossRef]
- Kim, H.; Lin, Q.; Yun, Z. BRCA1 regulates the cancer stem cell fate of breast cancer cells in the context of hypoxia and histone deacetylase inhibitors. Sci. Rep. 2019, 9, 9702. [Google Scholar] [CrossRef] [Green Version]
- Kakarala, M.; Brenner, D.E.; Korkaya, H.; Cheng, C.; Tazi, K.; Ginestier, C.; Liu, S.; Dontu, G.; Wicha, M.S. Targeting breast stem cells with the cancer preventive compounds curcumin and piperine. Breast Cancer Res. Treat. 2010, 122, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Benes, P.; Knopfova, L.; Trcka, F.; Nemajerova, A.; Pinheiro, D.; Soucek, K.; Fojta, M.; Smarda, J. Inhibition of topoisomerase IIα: Novel function of wedelolactone. Cancer Lett. 2011, 303, 29–38. [Google Scholar] [CrossRef]
- Gülçür, E.; Thaqi, M.; Khaja, F.; Kuzmis, A.; Önyüksel, H. Curcumin in VIP-targeted sterically stabilized phospholipid nanomicelles: A novel therapeutic approach for breast cancer and breast CSCs. Drug. Deliv. Transl. Res. 2013, 3, 562–574. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Shin, D.H.; Kim, J.S. Anticancer Effect of Metformin in Herceptin-Conjugated Liposome for Breast Cancer. Pharmaceutics 2019, 12, 11. [Google Scholar] [CrossRef]
- Rafael, D.; Gener, P.; Andrade, F.; Seras-Franzoso, J.; Montero, S.; Fernández, Y.; Hidalgo, M.; Arango, D.; Sayós, J.; Florindo, H.F.; et al. AKT2 siRNA delivery with amphiphilic-based polymeric micelles show efficacy against CSCs. Drug Deliv. 2018, 25, 961–972. [Google Scholar] [CrossRef]
- Kendellen, M.F.; Bradford, J.W.; Lawrence, C.L.; Clark, K.S.; Baldwin, A.S. Canonical and non- canonical NF-κB signaling promotes breast cancer tumor-initiating cells. Oncogene 2014, 33, 1297–1305. [Google Scholar] [CrossRef] [Green Version]
- Rao, D.D.; Vorhies, J.S.; Senzer, N.; Nemunaitis, J. siRNA vs. shRNA: Similarities and differences. Adv. Drug. Deliv. Rev. 2009, 61, 746–759. [Google Scholar] [CrossRef]
- Ke, X.; Yang, C.; Cheng, W.; Yang, Y.Y. Delivery of NF-κB shRNA using carbamate- mannose modified PEI for eliminating CSCs. Nanomedicine 2018, 14, 405–414. [Google Scholar] [CrossRef]
- Lin, X.; Chen, W.; Wei, F.; Zhou, B.P.; Hung, M.C.; Xie, X. Nanoparticle Delivery of miR-34a Eradicates Long-term-cultured Breast Cancer Stem Cells via Targeting C22ORF28 Directly. Theranostics 2017, 7, 4805–4824. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Liu, Y.; Li, S.; Rohrs, J.; Zhang, R.; Zhang, X.; Wang, P. Co-Eradication of Breast Cancer Cells and CSCs by Cross-Linked Multilamellar Liposomes Enhances Tumor Treatment. Mol. Pharm. 2015, 12, 2811–2822. [Google Scholar] [CrossRef]
- Zhang, J.; Kinoh, H.; Hespel, L.; Liu, X.; Quader, S.; Martin, J.; Chida, T.; Cabral, H.; Kataoka, K. Effective treatment of drug resistant recurrent breast tumors harboring cancer stem- like cells by staurosporine/epirubicin co-loaded polymeric micelles. J. Control Release 2017, 264, 127–135. [Google Scholar] [CrossRef]
- Shen, S.; Xia, J.X.; Wang, J. Nanomedicine-mediated cancer stem cell therapy. Biomaterials 2016, 74, 1–18. [Google Scholar] [CrossRef]
- Yang, Z.; Sun, N.; Cheng, R.; Zhao, C.; Liu, Z.; Li, X.; Liu, J.; Tian, Z. pH multistage responsive micellar system with charge-switch and PEG layer detachment for co-delivery of paclitaxel and curcumin to synergistically eliminate breast cancer stem cells. Biomaterials 2017, 147, 53–67. [Google Scholar] [CrossRef]
- Park, E.Y.; Chang, E.; Lee, E.J.; Lee, H.W.; Kang, H.G.; Chun, K.H.; Woo, Y.M.; Kong, H.K.; Ko, J.Y.; Suzuki, H.; et al. Targeting of miR34a-NOTCH1 axis reduced breast cancer stemness and chemoresistance. Cancer Res. 2014, 74, 7573–7582. [Google Scholar] [CrossRef]
- Zhao, Y.; Alakhova, D.Y.; Kabanov, A.V. Can nanomedicines kill cancer stem cells? Adv. Drug Deliv. Rev. 2013, 65, 1763–1783. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Li, W.; Guo, Y.; Feng, S.S. Nanomedicine strategies for sustained, controlled and targeted treatment of cancer stem cells. Nanomedicine 2016, 11, 3261–3282. [Google Scholar] [CrossRef]
- Lang, T.; Liu, Y.; Zheng, Z.; Ran, W.; Zhai, Y.; Yin, Q.; Zhang, P.; Li, Y. Cocktail Strategy Based on Spatio-Temporally Controlled Nano Device Improves Therapy of Breast Cancer. Adv. Mater. 2019, 31, e1903844. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Lane, L.A.; Nie, S. Stimuli-responsive nanoparticles for targeting the tumor microenvironment. J. Control. Release Off. J. Control. Release Soc. 2015, 219, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, H.; Chen, Y.; Jin, Q.; Ren, K.; Ji, J. Mixed-charge nanoparticles for long circulation, low reticuloendothelial system clearance, and high tumor accumulation. Adv. Healthc. Mater. 2014, 3, 1439–1447. [Google Scholar] [CrossRef]







| Origin | Characteristics | Isolation Techniques | Signaling Pathways Governing CSC Behavior | Novel Therapeutic Approaches for Targeting CSCs | |
|---|---|---|---|---|---|
| Cancer Stem Cells |
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eid, R.A.; Alaa Edeen, M.; Shedid, E.M.; Kamal, A.S.S.; Warda, M.M.; Mamdouh, F.; Khedr, S.A.; Soltan, M.A.; Jeon, H.W.; Zaki, M.S.A.; et al. Targeting Cancer Stem Cells as the Key Driver of Carcinogenesis and Therapeutic Resistance. Int. J. Mol. Sci. 2023, 24, 1786. https://doi.org/10.3390/ijms24021786
Eid RA, Alaa Edeen M, Shedid EM, Kamal ASS, Warda MM, Mamdouh F, Khedr SA, Soltan MA, Jeon HW, Zaki MSA, et al. Targeting Cancer Stem Cells as the Key Driver of Carcinogenesis and Therapeutic Resistance. International Journal of Molecular Sciences. 2023; 24(2):1786. https://doi.org/10.3390/ijms24021786
Chicago/Turabian StyleEid, Refaat A., Muhammad Alaa Edeen, Eslam M. Shedid, Al Shaimaa S. Kamal, Mona M. Warda, Farag Mamdouh, Sohila A. Khedr, Mohamed A. Soltan, Hee Won Jeon, Mohamed Samir A. Zaki, and et al. 2023. "Targeting Cancer Stem Cells as the Key Driver of Carcinogenesis and Therapeutic Resistance" International Journal of Molecular Sciences 24, no. 2: 1786. https://doi.org/10.3390/ijms24021786