
The Gut–Vascular Barrier as a New Protagonist in Intestinal and Extraintestinal Diseases
 , and
, and 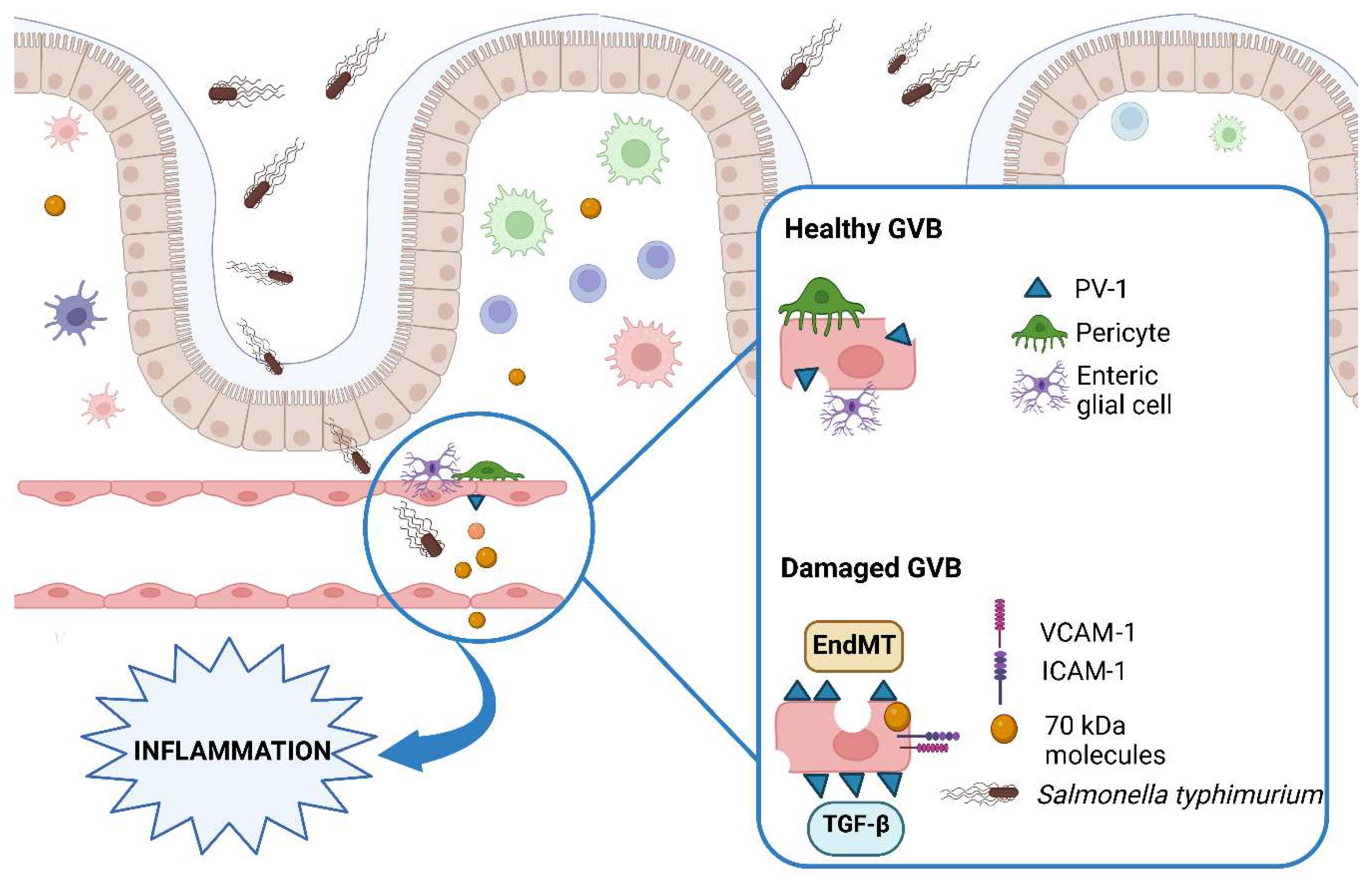{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Gut–Vascular Barrier
3. Gut–Vascular Barrier in Liver Disease
3.1. Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis
3.2. Alcoholic Liver Disease
3.3. Cirrhosis
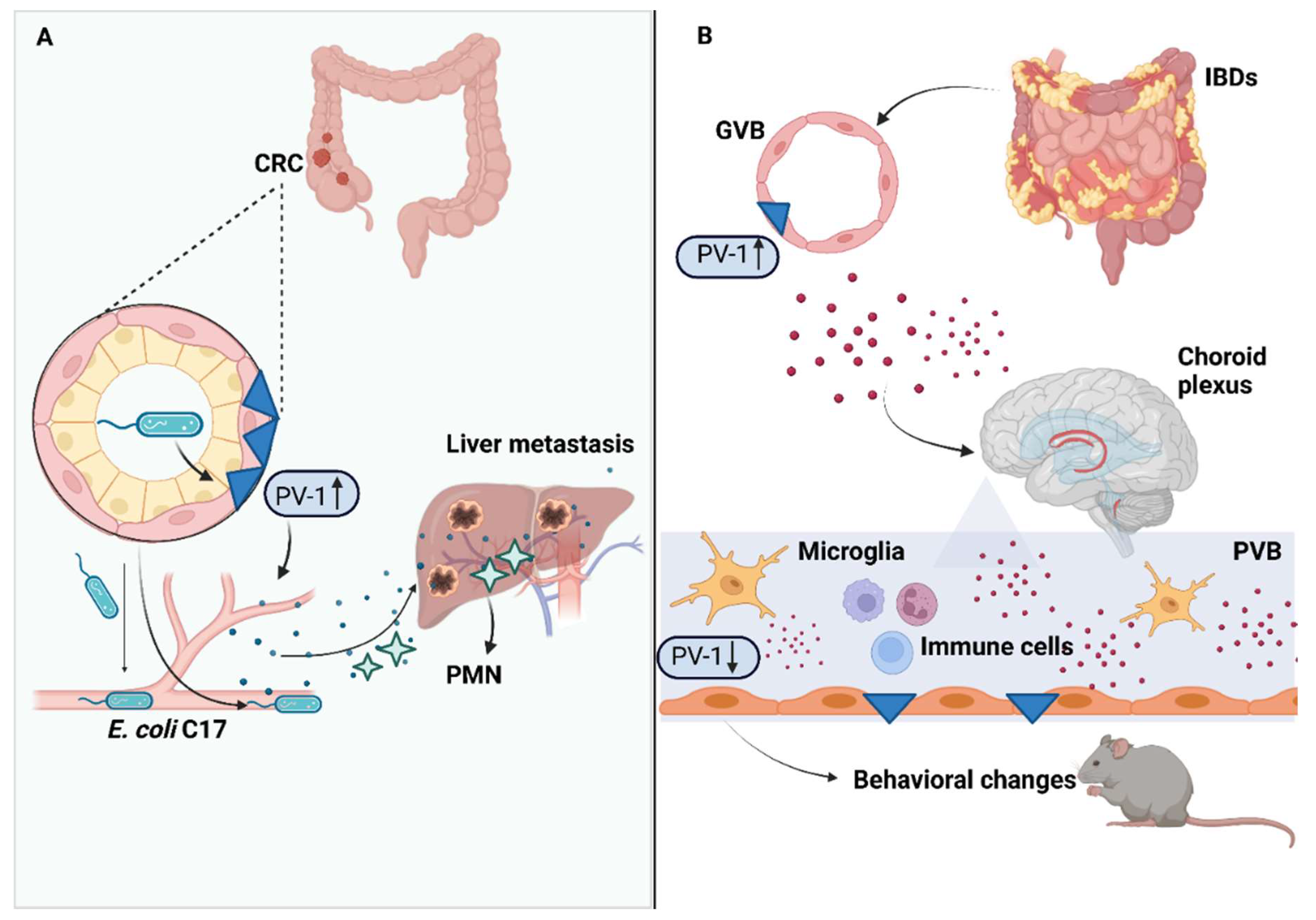
4. Gut–Vascular Barrier in Colorectal Cancer Progression
5. Gut–Vascular Barrier in Inflammatory Bowel Diseases
6. Gut–Vascular Barrier in Celiac Disease
7. Gut–Vascular Barrier in Spondyloarthritis
8. Gut–Vascular Barrier and The Gut–Brain Axis
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Kho, Z.Y.; Lal, S.K. The Human Gut Microbiome—A Potential Controller of Wellness and Disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.C.; Johansson, M.E. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Gut Microbes 2010, 1, 51–54. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, S.; Woodmansey, E.J.; Macfarlane, G.T. Colonization of Mucin by Human Intestinal Bacteria and Establishment of Biofilm Communities in a Two-Stage Continuous Culture System. Appl. Environ. Microbiol. 2005, 71, 7483–7492. [Google Scholar] [CrossRef] [Green Version]
- Birchenough, G.M.H.; Johansson, M.E.; Gustafsson, J.K.; Bergström, J.H.; Hansson, G.C. New developments in goblet cell mucus secretion and function. Mucosal Immunol. 2015, 8, 712–719. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2016, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Sturgeon, C.; Fasano, A. Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. Tissue Barriers 2016, 4, e1251384. [Google Scholar] [CrossRef] [Green Version]
- Di Tommaso, N.; Gasbarrini, A.; Ponziani, F.R. Intestinal Barrier in Human Health and Disease. Int. J. Environ. Res. Public Health 2021, 18, 12836. [Google Scholar] [CrossRef]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artis, D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 2008, 8, 411–420. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell Metab. 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Troy, E.B.; Kasper, D.L. Beneficial effects of Bacteroides fragilis polysaccharides on the immune system. Front. Biosci. (Landmark Ed.) 2010, 15, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, L.; Borrero, M.J.; Úbeda, M.; Conde, E.; Del Campo, R.; Rodríguez-Serrano, M.; Lario, M.; Sánchez-Díaz, A.M.; Pastor, O.; Díaz, D.; et al. Intestinal Immune Dysregulation Driven by Dysbiosis Promotes Barrier Disruption and Bacterial Translocation in Rats With Cirrhosis. Hepatology 2018, 70, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Balzan, S.; de Almeida Quadros, C.; De Cleva, R.; Zilberstein, B.; Cecconello, I. Bacterial translocation: Overview of mechanisms and clinical impact. J. Gastroenterol. Hepatol. 2007, 22, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Yadav, H. Bacterial Translocation from the Gut to the Distant Organs: An Overview. Ann. Nutr. Metab. 2017, 71 (Suppl. S1), 11–16. [Google Scholar] [CrossRef]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; Van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 571731. [Google Scholar] [CrossRef]
- Linares, R.; Francés, R.; Gutiérrez, A.; Juanola, O. Bacterial Translocation as Inflammatory Driver in Crohn’s Disease. Front. Cell Dev. Biol. 2021, 9, 703310. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Zocco, M.A.; Cerrito, L.; Gasbarrini, A.; Pompili, M. Bacterial translocation in patients with liver cirrhosis: Physiology, clinical consequences, and practical implications. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 641–656. [Google Scholar] [CrossRef]
- Kouzu, K.; Tsujimoto, H.; Kishi, Y.; Ueno, H.; Shinomiya, N. Bacterial Translocation in Gastrointestinal Cancers and Cancer Treatment. Biomedicines 2022, 10, 380. [Google Scholar] [CrossRef] [PubMed]
- Mowat, A.; Agace, W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef]
- Geboes, K.; Geboes, K.P.; Maleux, G. Vascular anatomy of the gastrointestinal tract. Best Pr. Res. Clin. Gastroenterol. 2001, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Brescia, P.; Rescigno, M. The gut vascular barrier: A new player in the gut–liver–brain axis. Trends Mol. Med. 2021, 27, 844–855. [Google Scholar] [CrossRef]
- Cong, X.; Kong, W. Endothelial tight junctions and their regulatory signaling pathways in vascular homeostasis and disease. Cell. Signal. 2019, 66, 109485. [Google Scholar] [CrossRef]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 794–809. [Google Scholar] [CrossRef] [Green Version]
- Stan, R.-V.; Kubitza, M.; Palade, G.E. PV-1 is a component of the fenestral and stomatal diaphragms in fenestrated endothelia. Proc. Natl. Acad. Sci. USA 1999, 96, 13203–13207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, D.; Stan, R.V. Morphological Heterogeneity of Endothelium. Semin. Thromb. Hemost. 2010, 36, 236–245. [Google Scholar] [CrossRef]
- Stan, R.V.; Tse, D.; Deharvengt, S.J.; Smits, N.C.; Xu, Y.; Luciano, M.R.; McGarry, C.L.; Buitendijk, M.; Nemani, K.V.; Elgueta, R.; et al. The Diaphragms of Fenestrated Endothelia: Gatekeepers of Vascular Permeability and Blood Composition. Dev. Cell 2012, 23, 1203–1218. [Google Scholar] [CrossRef] [Green Version]
- Elkadri, A.; Thoeni, C.; Deharvengt, S.J.; Murchie, R.; Guo, C.; Stavropoulos, J.D.; Marshall, C.R.; Wales, P.; Bandsma, R.H.; Cutz, E.; et al. Mutations in Plasmalemma Vesicle Associated Protein Result in Sieving Protein-Losing Enteropathy Characterized by Hypoproteinemia, Hypoalbuminemia, and Hypertriglyceridemia. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 381–394.e7. [Google Scholar] [CrossRef]
- Broekaert, I.J.; Becker, K.; Gottschalk, I.; Körber, F.; Dötsch, J.; Thiele, H.; Altmüller, J.; Nürnberg, P.; Hünseler, C.; Cirak, S. Mutations in plasmalemma vesicle-associated protein cause severe syndromic protein-losing enteropathy. J. Med. Genet. 2018, 55, 637–640. [Google Scholar] [CrossRef]
- Shue, E.H.; Carson-Walter, E.B.; Liu, Y.; Winans, B.N.; Ali, Z.S.; Chen, J.; Walter, K.A. Plasmalemmal Vesicle Associated Protein-1 (PV-1) is a marker of blood-brain barrier disruption in rodent models. BMC Neurosci. 2008, 9, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidemann, J.; Domschke, W.; Kucharzik, T.; Maaser, C. Intestinal Microvascular Endothelium and Innate Immunity in Inflammatory Bowel Disease: A Second Line of Defense? Infect. Immun. 2006, 74, 5425–5432. [Google Scholar] [CrossRef] [Green Version]
- Cromer, W.E.; Mathis, J.M.; Granger, D.N.; Chaitanya, G.V.; Alexander, J.S. Role of the endothelium in inflammatory bowel diseases. World J. Gastroenterol. 2011, 17, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.C.; Banks, R.E.; Haidar, A.; Gearing, A.J.; Hemingway, I.K.; Ibbotson, S.H.; Dixon, M.F.; Axon, A.T. Adhesion molecules in inflammatory bowel disease. Gut 1995, 36, 724–730. [Google Scholar] [CrossRef] [Green Version]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Spadoni, I.; Pietrelli, A.; Pesole, G.; Rescigno, M. Gene expression profile of endothelial cells during perturbation of the gut vascular barrier. Gut Microbes 2016, 7, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M.; et al. Wnt/β-catenin signaling controls development of the blood–brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spadoni, I.; Fornasa, G.; Rescigno, M. Organ-specific protection mediated by cooperation between vascular and epithelial barriers. Nat. Rev. Immunol. 2017, 17, 761–773. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef]
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.-S.; Kim, J. Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front. Immunol. 2018, 9, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.-J.; Dijke, P.T. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol. 2020, 8, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, L.; Du, X.; Li, W.; Mao, Y.; Sun, L.; Li, X. EndMT: A promising and controversial field. Eur. J. Cell Biol. 2018, 97, 493–500. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Schwartz, M.A.; Simons, M. Endothelial-to-Mesenchymal Transition, Vascular Inflammation, and Atherosclerosis. Front. Cardiovasc. Med. 2020, 7, 53. [Google Scholar] [CrossRef]
- Vergara, D.; Simeone, P.; Damato, M.; Maffia, M.; Lanuti, P.; Trerotola, M. The Cancer Microbiota: EMT and Inflammation as Shared Molecular Mechanisms Associated with Plasticity and Progression. J. Oncol. 2019, 2019, 1253727. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Hancock, B.M.; Bermudez, A.; Del Cid, N.; Reyes, E.; van Sorge, N.M.; Lauth, X.; Smurthwaite, C.A.; Hilton, B.J.; Stotland, A.; et al. Bacterial induction of Snail1 contributes to blood-brain barrier disruption. J. Clin. Investig. 2015, 125, 2473–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtani, N.; Kawada, N. Role of the Gut-Liver Axis in Liver Inflammation, Fibrosis, and Cancer: A Special Focus on the Gut Microbiota Relationship. Hepatol. Commun. 2019, 3, 456–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [Green Version]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Plaza-Díaz, J.; Solís-Urra, P.; Rodríguez-Rodríguez, F.; Olivares-Arancibia, J.; Navarro-Oliveros, M.; Abadía-Molina, F.; Álvarez-Mercado, A. The Gut Barrier, Intestinal Microbiota, and Liver Disease: Molecular Mechanisms and Strategies to Manage. Int. J. Mol. Sci. 2020, 21, 8351. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.-Y.; Zhai, Z.-Z.; Li, Z.-F.; Wang, L. High fat diet-triggered non-alcoholic fatty liver disease: A review of proposed mechanisms. Chem. Interact. 2020, 330, 109199. [Google Scholar] [CrossRef] [PubMed]
- Do, M.H.; Lee, E.; Oh, M.-J.; Kim, Y.; Park, H.-Y. High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change. Nutrients 2018, 10, 761. [Google Scholar] [CrossRef] [Green Version]
- Seki, K.; Kitade, M.; Nishimura, N.; Kaji, K.; Asada, K.; Namisaki, T.; Moriya, K.; Kawaratani, H.; Okura, Y.; Takaya, H.; et al. Oral administration of fructose exacerbates liver fibrosis and hepatocarcinogenesis via increased intestinal permeability in a rat steatohepatitis model. Oncotarget 2018, 9, 28638–28651. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, K.; Kanmura, S.; Morinaga, Y.; Tanaka, A.; Makino, T.; Fujita, T.; Arima, S.; Sasaki, F.; Nasu, Y.; Tanoue, S.; et al. A high-fructose diet induces epithelial barrier dysfunction and exacerbates the severity of dextran sulfate sodium-induced colitis. Int. J. Mol. Med. 2018, 43, 1487–1496. [Google Scholar] [CrossRef]
- Cho, Y.E.; Kim, D.K.; Seo, W.; Gao, B.; Yoo, S.; Song, B.J. Fructose Promotes Leaky Gut, Endotoxemia, and Liver Fibrosis Through Ethanol-Inducible Cytochrome P450-2E1–Mediated Oxidative and Nitrative Stress. Hepatology 2019, 73, 2180–2195. [Google Scholar] [CrossRef]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A High-Fat Diet Is Associated With Endotoxemia That Originates From the Gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Jin, R.; Willment, A.; Patel, S.S.; Sun, X.; Song, M.; Mannery, Y.O.; Kosters, A.; McClain, C.J.; Vos, M.B. Fructose Induced Endotoxemia in Pediatric Nonalcoholic Fatty Liver Disease. Int. J. Hepatol. 2014, 2014, 560620. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-A.; Gu, W.; Lee, I.-A.; Joh, E.-H.; Kim, D.-H. High Fat Diet-Induced Gut Microbiota Exacerbates Inflammation and Obesity in Mice via the TLR4 Signaling Pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet-Induced Obesity and Diabetes in Mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2012, 62, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Rabot, S.; Membrez, M.; Bruneau, A.; Gérard, P.; Harach, T.; Moser, M.; Raymond, F.; Mansourian, R.; Chou, C.J. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010, 24, 4948–4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front. Immunol. 2021, 11, 594150. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Sterbini, F.P.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated With Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.K.; Bhattacharya, D.; Borgerding, J.N.; Fiel, M.I.; Faith, J.J.; Friedman, S.L. Modeling dysbiosis of human NASH in mice: Loss of gut microbiome diversity and overgrowth of Erysipelotrichales. PLoS ONE 2021, 16, e0244763. [Google Scholar] [CrossRef]
- Neag, M.A.; Catinean, A.; Muntean, D.M.; Pop, M.R.; Bocsan, C.I.; Botan, E.C.; Buzoianu, A.D. Probiotic Bacillus Spores Protect Against Acetaminophen Induced Acute Liver Injury in Rats. Nutrients 2020, 12, 632. [Google Scholar] [CrossRef]
- Cheng, C.; Tan, J.; Qian, W.; Zhang, L.; Hou, X. Gut inflammation exacerbates hepatic injury in the high-fat diet induced NAFLD mouse: Attention to the gut-vascular barrier dysfunction. Life Sci. 2018, 209, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houttu, V.; Csader, S.; Nieuwdorp, M.; Holleboom, A.G.; Schwab, U. Dietary Interventions in Patients With Non-alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Front. Nutr. 2021, 8, 437. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Yan, J.; Wu, L.; Wu, J.; Chen, Z.; Jiang, J.; Chen, Z.; He, B. Probiotics Alleviated Nonalcoholic Fatty Liver Disease in High-Fat Diet-Fed Rats via Gut Microbiota/FXR/FGF15 Signaling Pathway. J. Immunol. Res. 2021, 2021, 2264737. [Google Scholar] [CrossRef]
- Panzitt, K.; Zollner, G.; Marschall, H.-U.; Wagner, M. Recent advances on FXR-targeting therapeutics. Mol. Cell. Endocrinol. 2022, 552, 111678. [Google Scholar] [CrossRef]
- Massey, V.L.; Arteel, G.E. Acute Alcohol-Induced Liver Injury. Front. Physiol. 2012, 3, 193. [Google Scholar] [CrossRef] [Green Version]
- Farooq, M.O.; Bataller, R. Pathogenesis and Management of Alcoholic Liver Disease. Dig. Dis. 2016, 34, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Ferrier, L.; Bérard, F.; Debrauwer, L.; Chabo, C.; Langella, P.; Buéno, L.; Fioramonti, J. Impairment of the Intestinal Barrier by Ethanol Involves Enteric Microflora and Mast Cell Activation in Rodents. Am. J. Pathol. 2006, 168, 1148–1154. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zhong, W. Targeting the gut barrier for the treatment of alcoholic liver disease. Liver Res. 2017, 1, 197–207. [Google Scholar] [CrossRef]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Stärkel, P.; Windey, K.; Tremaroli, V.; Bäckhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef]
- Lowe, P.P.; Gyongyosi, B.; Satishchandran, A.; Iracheta-Vellve, A.; Ambade, A.; Kodys, K.; Catalano, D.; Ward, D.V.; Szabo, G. Alcohol-related changes in the intestinal microbiome influence neutrophil infiltration, inflammation and steatosis in early alcoholic hepatitis in mice. PLoS ONE 2017, 12, e0174544, Erratum in PLoS ONE 2017, 12, e0179070. [Google Scholar] [CrossRef] [Green Version]
- Addolorato, G.; Ponziani, F.R.; Dionisi, T.; Mosoni, C.; Vassallo, G.A.; Sestito, L.; Petito, V.; Picca, A.; Marzetti, E.; Tarli, C.; et al. Gut microbiota compositional and functional fingerprint in patients with alcohol use disorder and alcohol-associated liver disease. Liver Int. 2020, 40, 878–888. [Google Scholar] [CrossRef]
- Yang, A.-M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841. [Google Scholar] [CrossRef] [Green Version]
- Fairfield, B.; Schnabl, B. Gut dysbiosis as a driver in alcohol-induced liver injury. JHEP Rep. 2020, 3, 100220. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Duan, Y.; Lang, S.; Jiang, L.; Wang, Y.; Llorente, C.; Liu, J.; Mogavero, S.; Bosques-Padilla, F.; Abraldes, J.G.; et al. The Candida albicans exotoxin candidalysin promotes alcohol-associated liver disease. J. Hepatol. 2020, 72, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.F.; Matschinsky, F.M. Ethanol metabolism: The good, the bad, and the ugly. Med. Hypotheses 2020, 140, 109638. [Google Scholar] [CrossRef]
- Chen, P.; Torralba, M.; Tan, J.; Embree, M.; Zengler, K.; Stärkel, P.; van Pijkeren, J.-P.; DePew, J.; Loomba, R.; Ho, S.B.; et al. Supplementation of Saturated Long-Chain Fatty Acids Maintains Intestinal Eubiosis and Reduces Ethanol-induced Liver Injury in Mice. Gastroenterology 2015, 148, 203–214.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, C.; Zaramela, L.S.; Gao, B.; Embree, M.; Tarasova, J.; Parker, S.J.; Wang, Y.; Chu, H.; Chen, P.; Lee, K.-C.; et al. Acetate reprograms gut microbiota during alcohol consumption. Nat. Commun. 2022, 13, 4630. [Google Scholar] [CrossRef]
- Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009, 50, 638–644. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547. [Google Scholar] [CrossRef]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Stärkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2010, 53, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Fouts, D.E.; Stärkel, P.; Hartmann, P.; Chen, P.; Llorente, C.; DePew, J.; Moncera, K.; Ho, S.B.; Brenner, D.A.; et al. Intestinal REG3 Lectins Protect against Alcoholic Steatohepatitis by Reducing Mucosa-Associated Microbiota and Preventing Bacterial Translocation. Cell Host Microbe 2016, 19, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrikx, T.; Duan, Y.; Wang, Y.; Oh, J.-H.; Alexander, L.M.; Huang, W.; Stärkel, P.; Ho, S.B.; Gao, B.; Fiehn, O.; et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut 2018, 68, 1504–1515. [Google Scholar] [CrossRef]
- Keir, M.E.; Yi, T.; Lu, T.T.; Ghilardi, N. The role of IL-22 in intestinal health and disease. J. Exp. Med. 2020, 217, e20192195. [Google Scholar] [CrossRef]
- Maccioni, L.; Gao, B.; Leclercq, S.; Pirlot, B.; Horsmans, Y.; De Timary, P.; Leclercq, I.; Fouts, D.; Schnabl, B.; Stärkel, P. Intestinal permeability, microbial translocation, changes in duodenal and fecal microbiota, and their associations with alcoholic liver disease progression in humans. Gut Microbes 2020, 12, 1782157. [Google Scholar] [CrossRef] [PubMed]
- Grander, C.; Grabherr, F.; Spadoni, I.; Enrich, B.; Oberhuber, G.; Rescigno, M.; Tilg, H. The role of gut vascular barrier in experimental alcoholic liver disease and A. muciniphila supplementation. Gut Microbes 2020, 12, 1851986. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Martin-Mateos, R.M.; Shah, V.H. Gut–liver axis, cirrhosis and portal hypertension: The chicken and the egg. Hepatol. Int. 2017, 12 (Suppl. S1), 24–33. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Albillos, A.; Lario, M.; Álvarez-Mon, M. Cirrhosis-associated immune dysfunction: Distinctive features and clinical relevance. J. Hepatol. 2014, 61, 1385–1396. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.S. Altered Microbiota in Cirrhosis and Its Relationship to the Development of Infection. Clin. Liver Dis. 2019, 14, 107–111. [Google Scholar] [CrossRef]
- Tsiaoussis, G.I.; Assimakopoulos, S.F.; Tsamandas, A.C.; Triantos, C.K.; Thomopoulos, K.C. Intestinal barrier dysfunction in cirrhosis: Current concepts in pathophysiology and clinical implications. World J. Hepatol. 2015, 7, 2058–2068. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Gustot, T.; Mookerjee, R.P.; Garcia-Pagan, J.C.; Fallon, M.B.; Shah, V.H.; Moreau, R.; Jalan, R. Inflammation and portal hypertension—The undiscovered country. J. Hepatol. 2014, 61, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Sorribas, M.; Jakob, M.O.; Yilmaz, B.; Li, H.; Stutz, D.; Noser, Y.; de Gottardi, A.; Moghadamrad, S.; Hassan, M.; Albillos, A.; et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J. Hepatol. 2019, 71, 1126–1140. [Google Scholar] [CrossRef] [PubMed]
- Vlahcevic, Z.; Buhac, I.; Farrar, J.; Bell, C.C.; Swell, L. Bile acid metabolism in patients with cirrhosis. I. Kinetic aspects of cholic acid metabolism. Gastroenterology 1971, 60, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Alves, J.M.; Hylemon, P.B.; Bajaj, J.S. Cirrhosis, bile acids and gut microbiota: Unraveling a complex relationship. Gut Microbes 2013, 4, 382–387. [Google Scholar] [CrossRef] [Green Version]
- Schwabl, P.; Hambruch, E.; Seeland, B.A.; Hayden, H.; Wagner, M.; Garnys, L.; Strobel, B.; Schubert, T.-L.; Riedl, F.; Mitteregger, D.; et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J. Hepatol. 2017, 66, 724–733. [Google Scholar] [CrossRef] [Green Version]
- Klindt, C.; Reich, M.; Hellwig, B.; Stindt, J.; Rahnenführer, J.; Hengstler, J.G.; Köhrer, K.; Schoonjans, K.; Häussinger, D.; Keitel, V. The G Protein-Coupled Bile Acid Receptor TGR5 (Gpbar1) Modulates Endothelin-1 Signaling in Liver. Cells 2019, 8, 1467. [Google Scholar] [CrossRef] [Green Version]
- Bertocchi, A.; Carloni, S.; Ravenda, P.S.; Bertalot, G.; Spadoni, I.; Cascio, A.L.; Gandini, S.; Lizier, M.; Braga, D.; Asnicar, F.; et al. Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell 2021, 39, 708–724.e11. [Google Scholar] [CrossRef]
- Wang, H.; Pan, J.; Barsky, L.; Jacob, J.C.; Zheng, Y.; Gao, C.; Wang, S.; Zhu, W.; Sun, H.; Lu, L.; et al. Characteristics of pre-metastatic niche: The landscape of molecular and cellular pathways. Mol. Biomed. 2021, 2, 3. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Zhang, C.; Zhou, C.; Wu, X.-J.; Yang, M.; Yang, Z.-H.; Xiong, H.-Z.; Zhou, C.-P.; Lu, Y.-X.; Li, Y.; Li, X.-N. Human CD133-positive hematopoietic progenitor cells initiate growth and metastasis of colorectal cancer cells. Carcinogenesis 2014, 35, 2771–2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, M.; Huang, K.; Liu, Y.; Yang, Y.; Tang, H.; Liu, X.; Wang, C.; Chen, H.; Xiong, Y.; Zhang, J.; et al. Modulation of intestinal microbiota by glycyrrhizic acid prevents high-fat diet-enhanced pre-metastatic niche formation and metastasis. Mucosal Immunol. 2019, 12, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, X.; Li, X.; Fan, Y.; Li, G.; Guo, C.; Zhu, F.; Zhang, L.; Shi, Y. CD68+HLA-DR+ M1-like macrophages promote motility of HCC cells via NF-κB/FAK pathway. Cancer Lett. 2014, 345, 91–99. [Google Scholar] [CrossRef]
- Jedinak, A.; Dudhgaonkar, S.; Sliva, D. Activated macrophages induce metastatic behavior of colon cancer cells. Immunobiology 2010, 215, 242–249. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Chemopreventive Effects of Licorice and Its Components. Curr. Pharmacol. Rep. 2015, 1, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-J.; Choi, J.-S.; Kim, K.-W.; Jeong, J.-W. The Anti-Angiogenic Activities of Glycyrrhizic Acid in Tumor Progression. Phytother. Res. 2012, 27, 841–846. [Google Scholar] [CrossRef]
- Zhang, S.; Dogan, B.; Guo, C.; Herlekar, D.; Stewart, K.; Scherl, E.J.; Simpson, K.W. Short Chain Fatty Acids Modulate the Growth and Virulence of Pathosymbiont Escherichia coli and Host Response. Antibiotics 2020, 9, 462. [Google Scholar] [CrossRef]
- Dalmasso, G.; Cougnoux, A.; Delmas, J.; Darfeuille-Michaud, A.; Bonnet, R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes 2014, 5, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Wassenaar, T.M. E. coli and colorectal cancer: A complex relationship that deserves a critical mindset. Crit. Rev. Microbiol. 2018, 44, 619–632. [Google Scholar] [CrossRef]
- McDowell, R.; Perrott, S.; Murchie, P.; Cardwell, C.; Hughes, C.; Samuel, L. Oral antibiotic use and early-onset colorectal cancer: Findings from a case-control study using a national clinical database. Br. J. Cancer 2021, 126, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Ponder, A.; Long, M.D. A clinical review of recent findings in the epidemiology of inflammatory bowel disease. Clin. Epidemiol. 2013, 5, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Kwon, J.E.; Cho, M.-L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhoury, M.; Negrulj, R.; Mooranian, A.; Al-Salami, H. Inflammatory bowel disease: Clinical aspects and treatments. J. Inflamm. Res. 2014, 7, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Deban, L.; Correale, C.; Vetrano, S.; Malesci, A.; Danese, S. Multiple Pathogenic Roles of Microvasculature in Inflammatory Bowel Disease: A Jack of All Trades. Am. J. Pathol. 2008, 172, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Hatoum, O.A.; Miura, H.; Binion, D.G. The vascular contribution in the pathogenesis of inflammatory bowel disease. Am. J. Physiol. Circ. Physiol. 2003, 285, H1791–H1796. [Google Scholar] [CrossRef] [Green Version]
- Langer, V.; Vivi, E.; Regensburger, D.; Winkler, T.H.; Waldner, M.J.; Rath, T.; Schmid, B.; Skottke, L.; Lee, S.; Jeon, N.L.; et al. IFN-γ drives inflammatory bowel disease pathogenesis through VE-cadherin–directed vascular barrier disruption. J. Clin. Investig. 2019, 129, 4691–4707. [Google Scholar] [CrossRef] [Green Version]
- Oshimaa, T.; Larouxa, F.S.; Coe, L.L.; Morise, Z.; Kawachia, S.; Bauera, P.; Grisham, M.B.; Specian, R.D.; Cartera, P.; Jenningsb, S.; et al. Interferon-γ and Interleukin-10 Reciprocally Regulate Endothelial Junction Integrity and Barrier Function. Microvasc. Res. 2001, 61, 130–143, Erratum in Microvasc. Res. 2001, 61, 291. [Google Scholar] [CrossRef]
- Catinean, A.; Neag, M.A.; Krishnan, K.; Muntean, D.M.; Bocsan, C.I.; Pop, R.M.; Mitre, A.O.; Melincovici, C.S.; Buzoianu, A.D. Probiotic Bacillus Spores Together with Amino Acids and Immunoglobulins Exert Protective Effects on a Rat Model of Ulcerative Colitis. Nutrients 2020, 12, 3607. [Google Scholar] [CrossRef]
- Alkim, C.; Alkim, H.; Koksal, A.R.; Boga, S.; Sen, I. Angiogenesis in Inflammatory Bowel Disease. Int. J. Inflamm. 2015, 2015, 970890. [Google Scholar] [CrossRef]
- Danese, S.; Sans, M.; De La Motte, C.; Graziani, C.; West, G.; Phillips, M.H.; Pola, R.; Rutella, S.; Willis, J.; Gasbarrini, A.; et al. Angiogenesis as a Novel Component of Inflammatory Bowel Disease Pathogenesis. Gastroenterology 2006, 130, 2060–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickland, L.A.; Jubb, A.M.; Hongo, J.-A.; Zhong, F.; Burwick, J.; Fu, L.; Frantz, G.D.; Koeppen, H. Plasmalemmal vesicle-associated protein (PLVAP) is expressed by tumour endothelium and is upregulated by vascular endothelial growth factor-A (VEGF). J. Pathol. 2005, 206, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Bertocchi, A.; Mancinelli, S.; Bellini, M.; Erreni, M.; Borreca, A.; Braga, D.; Giugliano, S.; Mozzarelli, A.M.; Manganaro, D.; et al. Identification of a choroid plexus vascular barrier closing during intestinal inflammation. Science 2021, 374, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Hanley, N.; Campbell, M. Claudin-5: Gatekeeper of neurological function. Fluids Barriers CNS 2019, 16, 3. [Google Scholar] [CrossRef] [Green Version]
- Zanini, B.; Baschè, R.; Ferraresi, A.; Pigozzi, M.G.; Ricci, C.; Lanzarotto, F.; Villanacci, V.; Lanzini, A. Factors That Contribute to Hypertransaminasemia in Patients With Celiac Disease or Functional Gastrointestinal Syndromes. Clin. Gastroenterol. Hepatol. 2013, 12, 804–810.e2. [Google Scholar] [CrossRef]
- Volta, U. Pathogenesis and Clinical Significance of Liver Injury in Celiac Disease. Clin. Rev. Allergy Immunol. 2008, 36, 62–70. [Google Scholar] [CrossRef]
- Villavicencio, K.J.; Wu, G.Y. Celiac Disease and Elevated Liver Enzymes: A Review. J. Clin. Transl. Hepatol. 2021, 9, 116–124. [Google Scholar] [CrossRef]
- De Leo, L.; Naviglio, S.; Vatta, S.; Benelli, E.; Stera, G.; Santon, D.; Ziberna, F.; Taddio, A.; Martelossi, S.; Giudici, F.; et al. Circulating PV-1 as a marker of celiac disease-associated liver injury. Biomark. Med. 2020, 14, 1675–1681. [Google Scholar] [CrossRef]
- Auvinen, K.; Lokka, E.; Mokkala, E.; Jäppinen, N.; Tyystjärvi, S.; Saine, H.; Peurla, M.; Shetty, S.; Elima, K.; Rantakari, P.; et al. Fenestral diaphragms and PLVAP associations in liver sinusoidal endothelial cells are developmentally regulated. Sci. Rep. 2019, 9, 15698. [Google Scholar] [CrossRef] [Green Version]
- Wendling, D. The gut in spondyloarthritis. Jt. Bone Spine 2016, 83, 401–405. [Google Scholar] [CrossRef]
- Costello, M.-E.; Ciccia, F.; Willner, D.; Warrington, N.; Robinson, P.C.; Gardiner, B.; Marshall, M.; Kenna, T.J.; Triolo, G.; Brown, M.A. Brief Report: Intestinal Dysbiosis in Ankylosing Spondylitis. Arthritis Rheumatol. 2015, 67, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Alessandro, R.; Luchetti, M.M.; Milling, S.; Saieva, L.; Cypers, H.; Stampone, T.; Di Benedetto, P.; et al. Dysbiosis and zonulin upregulation alter gut epithelial and vascular barriers in patients with ankylosing spondylitis. Ann. Rheum. Dis. 2017, 76, 1123–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appleton, J. The Gut-Brain Axis: Influence of Microbiota on Mood and Mental Health. Integr. Med. 2018, 17, 28–32. [Google Scholar]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar]
- Cryan, J.F.; O’Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The gut microbiome in neurological disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef]
- Liang, S.; Wu, X.; Hu, X.; Wang, T.; Jin, F. Recognizing Depression from the Microbiota–Gut–Brain Axis. Int. J. Mol. Sci. 2018, 19, 1592. [Google Scholar] [CrossRef] [Green Version]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Nemani, K.; Ghomi, R.H.; McCormick, B.; Fan, X. Schizophrenia and the gut–brain axis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 56, 155–160. [Google Scholar] [CrossRef]
- Kong, G.; Ellul, S.; Narayana, V.K.; Kanojia, K.; Ha, H.T.T.; Li, S.; Renoir, T.; Cao, K.-A.L.; Hannan, A.J. An integrated metagenomics and metabolomics approach implicates the microbiota-gut-brain axis in the pathogenesis of Huntington’s disease. Neurobiol. Dis. 2020, 148, 105199. [Google Scholar] [CrossRef]
- Bisgaard, T.H.; Allin, K.H.; Keefer, L.; Ananthakrishnan, A.N.; Jess, T. Depression and anxiety in inflammatory bowel disease: Epidemiology, mechanisms and treatment. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Craig, C.F.; Filippone, R.T.; Stavely, R.; Bornstein, J.C.; Apostolopoulos, V.; Nurgali, K. Neuroinflammation as an etiological trigger for depression comorbid with inflammatory bowel disease. J. Neuroinflamm. 2022, 19, 4. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. The liver–brain axis in liver failure: Neuroinflammation and encephalopathy. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 522–528. [Google Scholar] [CrossRef]
- Butterworth, R.F. Hepatic encephalopathy: A central neuroinflammatory disorder? Hepatology 2011, 53, 1372–1376. [Google Scholar] [CrossRef]
- D’Mello, C.; Le, T.; Swain, M.G. Cerebral Microglia Recruit Monocytes into the Brain in Response to Tumor Necrosis Factor Signaling during Peripheral Organ Inflammation. J. Neurosci. 2009, 29, 2089–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.; D’Mello, C.; Le, T.; Urbanski, S.; Swain, M.G. Regulatory T cells suppress sickness behaviour development without altering liver injury in cholestatic mice. J. Hepatol. 2012, 56, 626–631. [Google Scholar] [CrossRef]
- McMillin, M.; Grant, S.; Frampton, G.; Petrescu, A.D.; Williams, E.; Jefferson, B.; Thomas, A.; Brahmaroutu, A.; DeMorrow, S. Elevated circulating TGFβ1 during acute liver failure activates TGFβR2 on cortical neurons and exacerbates neuroinflammation and hepatic encephalopathy in mice. J. Neuroinflamm. 2019, 16, 69. [Google Scholar] [CrossRef]
- McMillin, M.A.; Frampton, G.A.; Seiwell, A.P.; Patel, N.S.; Jacobs, A.N.; DeMorrow, S. TGFβ1 exacerbates blood–brain barrier permeability in a mouse model of hepatic encephalopathy via upregulation of MMP9 and downregulation of claudin-5. Lab. Investig. 2015, 95, 903–913. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Jiang, Y.; Xu, K.; Cui, M.; Ye, W.; Zhao, G.; Jin, L.; Chen, X. The progress of gut microbiome research related to brain disorders. J. Neuroinflamm. 2020, 17, 25. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.J.; Betrapally, N.; Ghosh, S.A.; Sartor, R.B.; Hylemon, P.B.P.B.; Gillevet, P.M.P.M.; Sanyal, A.J.A.J.; Heuman, D.M.D.M.; Carl, D.; Zhou, H.; et al. Gut microbiota drive the development of neuroinflammatory response in cirrhosis in mice. Hepatology 2016, 64, 1232–1248. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Kang, J.D.; Sartor, R.B.; Sikaroodi, M.; Fagan, A.; Gavis, E.A.; Zhou, H.; Hylemon, P.B.; Herzog, J.W.; Li, X.; et al. Neuroinflammation in Murine Cirrhosis Is Dependent on the Gut Microbiome and Is Attenuated by Fecal Transplant. Hepatology 2019, 71, 611–626. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, J.; Chen, Y. Regulation of Neurotransmitters by the Gut Microbiota and Effects on Cognition in Neurological Disorders. Nutrients 2021, 13, 2099. [Google Scholar] [CrossRef] [PubMed]
- Huo, R.; Zeng, B.; Zeng, L.; Cheng, K.; Li, B.; Luo, Y.; Wang, H.; Zhou, C.; Fang, L.; Li, W.; et al. Microbiota Modulate Anxiety-Like Behavior and Endocrine Abnormalities in Hypothalamic-Pituitary-Adrenal Axis. Front. Cell. Infect. Microbiol. 2017, 7, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota Modulate Behavioral and Physiological Abnormalities Associated with Neurodevelopmental Disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-K.; Shin, C. The Microbiota-Gut-Brain Axis in Neuropsychiatric Disorders: Pathophysiological Mechanisms and Novel Treatments. Curr. Neuropharmacol. 2018, 16, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Fung, T.C. The microbiota-immune axis as a central mediator of gut-brain communication. Neurobiol. Dis. 2019, 136, 104714. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Ghersi-Egea, J.-F.; Strazielle, N.; Catala, M.; Silva-Vargas, V.; Doetsch, F.; Engelhardt, B. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol. 2018, 135, 337–361. [Google Scholar] [CrossRef] [Green Version]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158, Erratum in Sci. Transl. Med. 2014, 6, 266er7. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Wang, Y.; Yang, G.; Zhang, Q.; Meng, L.; Xin, Y.; Jiang, X. The role of short-chain fatty acids in intestinal barrier function, inflammation, oxidative stress, and colonic carcinogenesis. Pharmacol. Res. 2021, 165, 105420. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, Y.; Wang, P.; Huang, Y.; Wang, F. Short-Chain Fatty Acids Manifest Stimulative and Protective Effects on Intestinal Barrier Function Through the Inhibition of NLRP3 Inflammasome and Autophagy. Cell. Physiol. Biochem. 2018, 49, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Van Esch, B.C.A.M.; Henricks, P.A.J.; Garssen, J.; Folkerts, G. Time and Concentration Dependent Effects of Short Chain Fatty Acids on Lipopolysaccharide- or Tumor Necrosis Factor α-Induced Endothelial Activation. Front. Pharmacol. 2018, 9, 233. [Google Scholar] [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Bosma, E.K.; Van Noorden, C.J.F.; Schlingemann, R.O.; Klaassen, I. The role of plasmalemma vesicle-associated protein in pathological breakdown of blood–brain and blood–retinal barriers: Potential novel therapeutic target for cerebral edema and diabetic macular edema. Fluids Barriers CNS 2018, 15, 24. [Google Scholar] [CrossRef] [Green Version]
- Keuschnigg, J.; Henttinen, T.; Auvinen, K.; Karikoski, M.; Salmi, M.; Jalkanen, S. The prototype endothelial marker PAL-E is a leukocyte trafficking molecule. Blood 2009, 114, 478–484. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Xu, H.; Lehtinen, M.K. Macrophages on the margin: Choroid plexus immune responses. Trends Neurosci. 2021, 44, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.; Sousa, J.C.; Correia-Neves, M.; Oliveira, P.; Sousa, N.; Palha, J.A. The choroid plexus response to peripheral inflammatory stimulus. Neuroscience 2006, 144, 424–430. [Google Scholar] [CrossRef]
- Carloni, S.; Rescigno, M. Unveiling the gut-brain axis: Structural and functional analogies between the gut and the choroid plexus vascular and immune barriers. Semin. Immunopathol. 2022, 44, 869–882. [Google Scholar] [CrossRef]
- Hopkins, C.W.P.; Powell, N.; Norton, C.; Dumbrill, J.L.; Hayee, B.; Moulton, C.D. Cognitive Impairment in Adult Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. J. Acad. Consult.-Liaison Psychiatry 2021, 62, 387–403. [Google Scholar] [CrossRef]
- Van Langenberg, D.R.; Yelland, G.W.; Robinson, S.R.; Gibson, P.R. Cognitive impairment in Crohn’s disease is associated with systemic inflammation, symptom burden and sleep disturbance. United Eur. Gastroenterol. J. 2017, 5, 579–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajadinejad, M.S.; Asgari, K.; Molavi, H.; Kalantari, M.; Adibi, P. Psychological Issues in Inflammatory Bowel Disease: An Overview. Gastroenterol. Res. Pr. 2012, 2012, 106502. [Google Scholar] [CrossRef]
- Vicentini, F.A.; Szamosi, J.C.; Rossi, L.; Griffin, L.; Nieves, K.; Bihan, D.; Lewis, I.A.; Pittman, Q.J.; Swain, M.G.; Surette, M.G.; et al. Colitis-associated microbiota drives changes in behaviour in male mice in the absence of inflammation. Brain, Behav. Immun. 2022, 102, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Matisz, C.E.; Vicentini, F.A.; Hirota, S.A.; Sharkey, K.A.; Gruber, A.J. Behavioral adaptations in a relapsing mouse model of colitis. Physiol. Behav. 2020, 216, 112802. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C.A.; Steffen, J.; Morton, L.; Arumugam, S.; Liesenfeld, O.; Deli, M.A.; Kröger, A.; Schüler, T.; Dunay, I.R. Immune response and pathogen invasion at the choroid plexus in the onset of cerebral toxoplasmosis. J. Neuroinflamm. 2022, 19, 17. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Tommaso, N.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R. The Gut–Vascular Barrier as a New Protagonist in Intestinal and Extraintestinal Diseases. Int. J. Mol. Sci. 2023, 24, 1470. https://doi.org/10.3390/ijms24021470
Di Tommaso N, Santopaolo F, Gasbarrini A, Ponziani FR. The Gut–Vascular Barrier as a New Protagonist in Intestinal and Extraintestinal Diseases. International Journal of Molecular Sciences. 2023; 24(2):1470. https://doi.org/10.3390/ijms24021470
Chicago/Turabian StyleDi Tommaso, Natalia, Francesco Santopaolo, Antonio Gasbarrini, and Francesca Romana Ponziani. 2023. "The Gut–Vascular Barrier as a New Protagonist in Intestinal and Extraintestinal Diseases" International Journal of Molecular Sciences 24, no. 2: 1470. https://doi.org/10.3390/ijms24021470