

Profound Modification of Fatty Acid Profile and Endocannabinoid-Related Mediators in PPARα Agonist Fenofibrate-Treated Mice

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
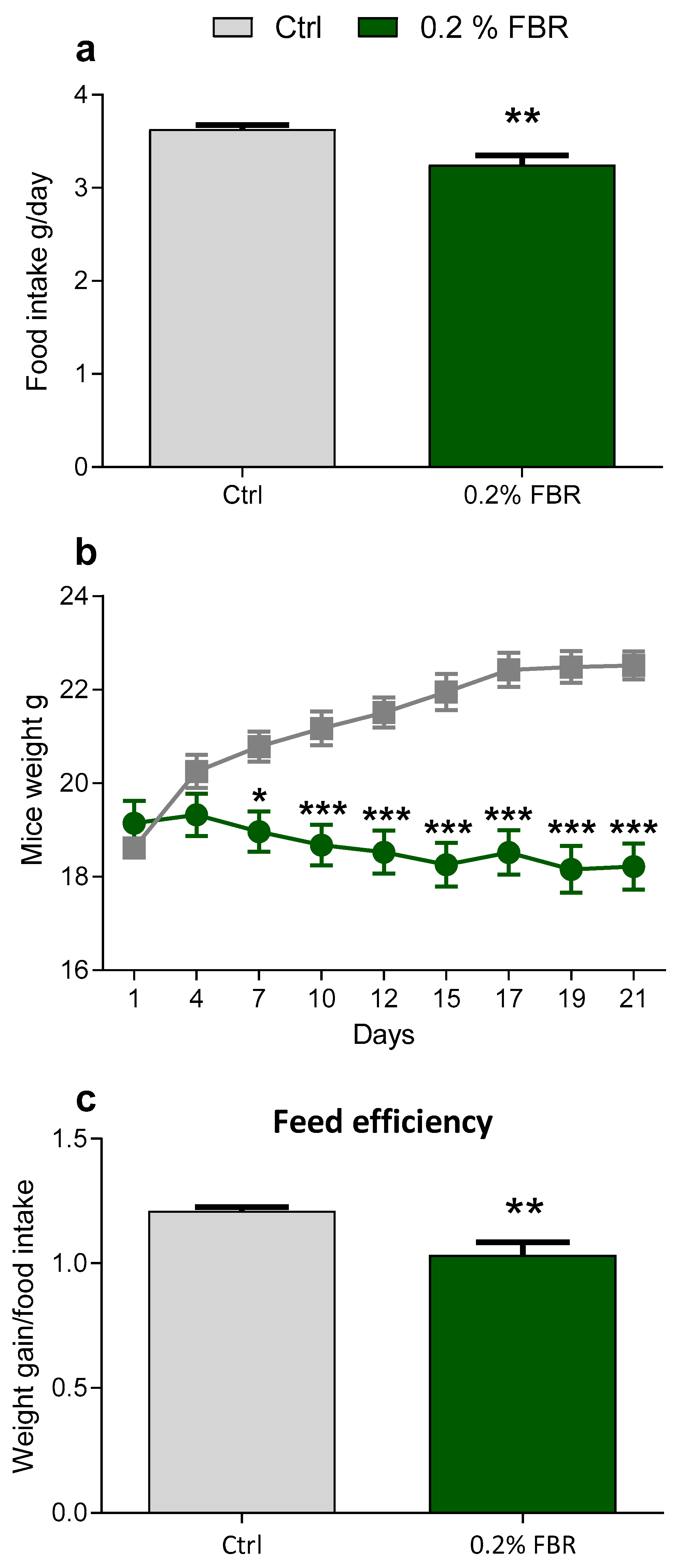
2.1. Body Weight, Food Intake and Growth
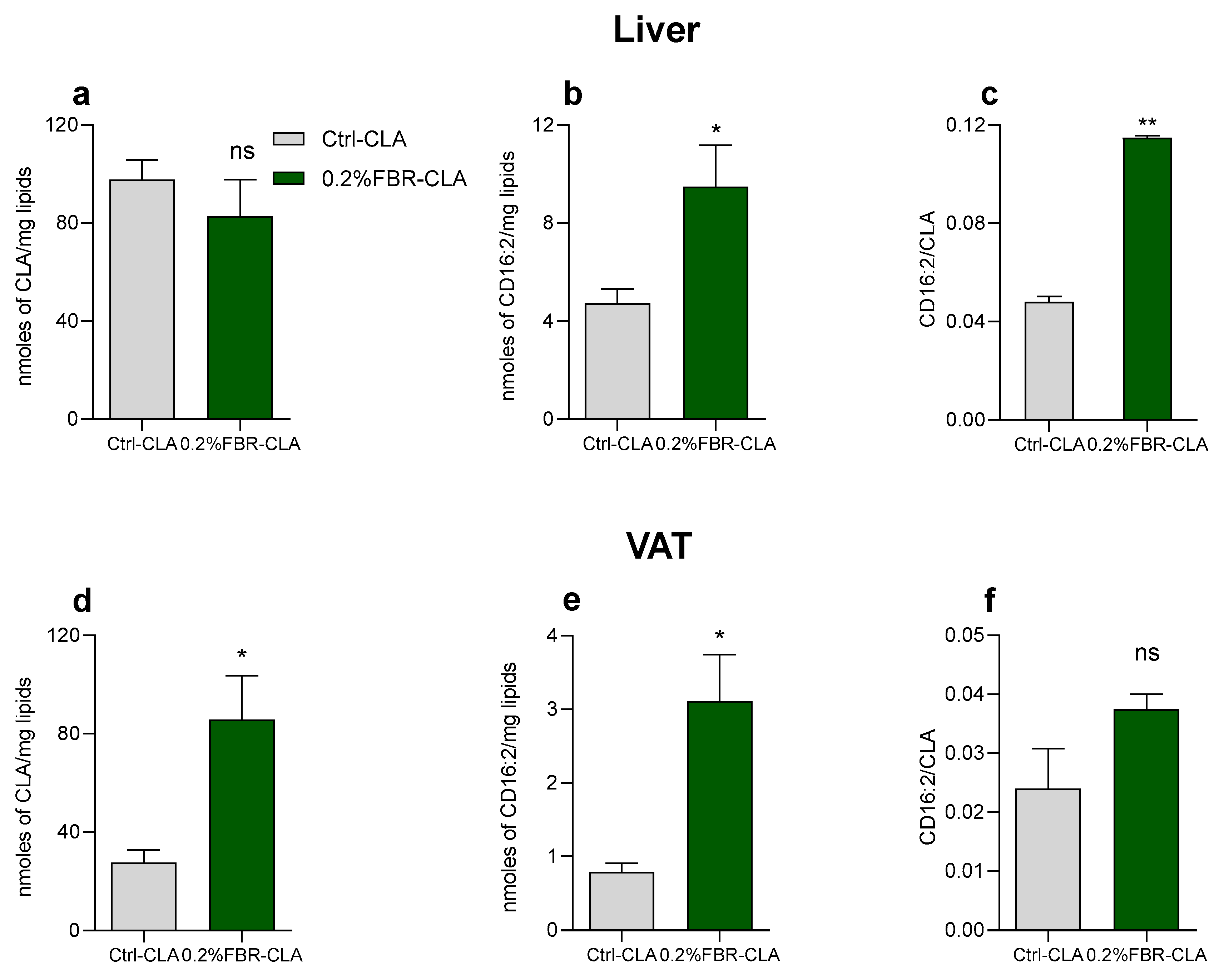
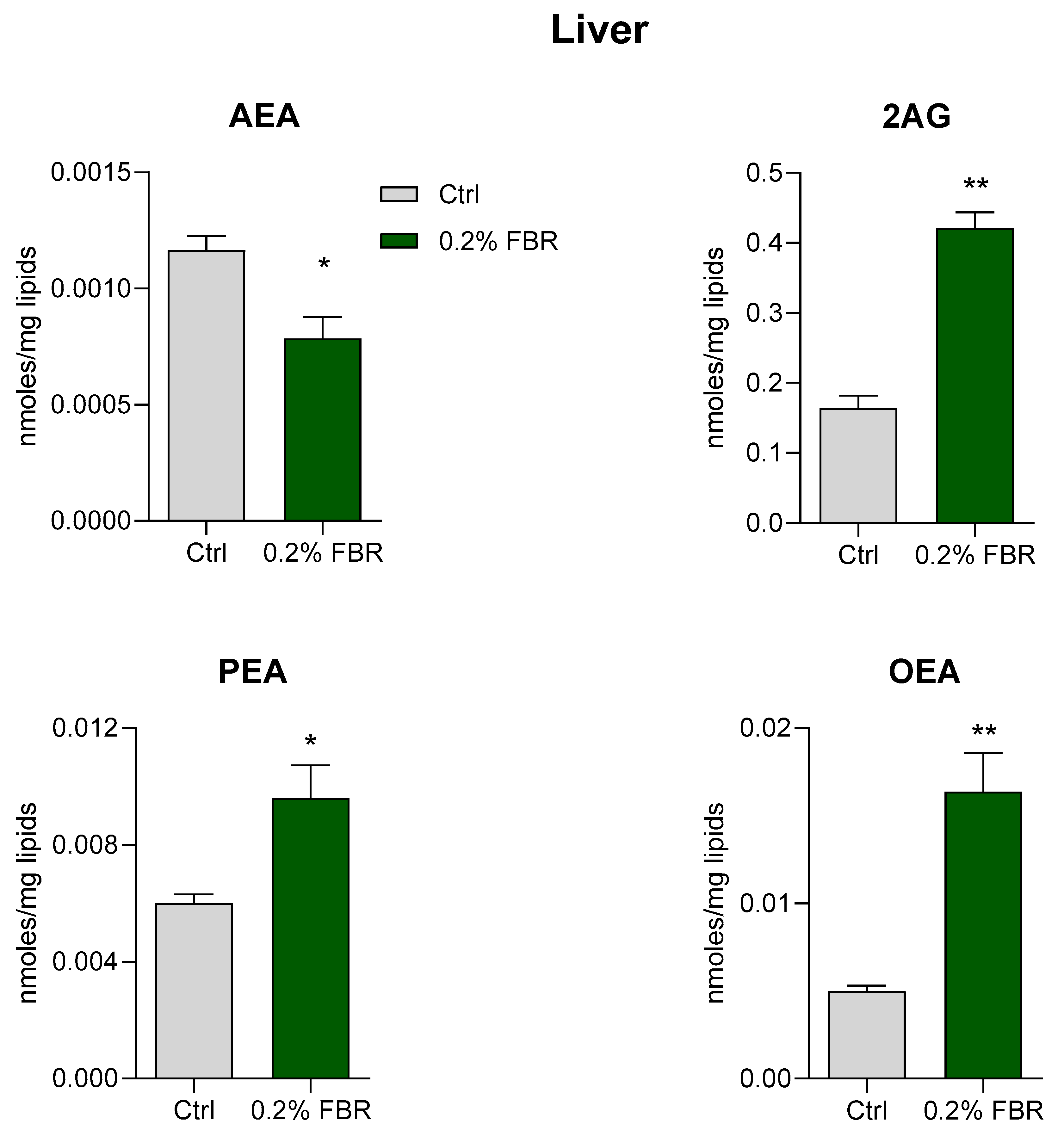
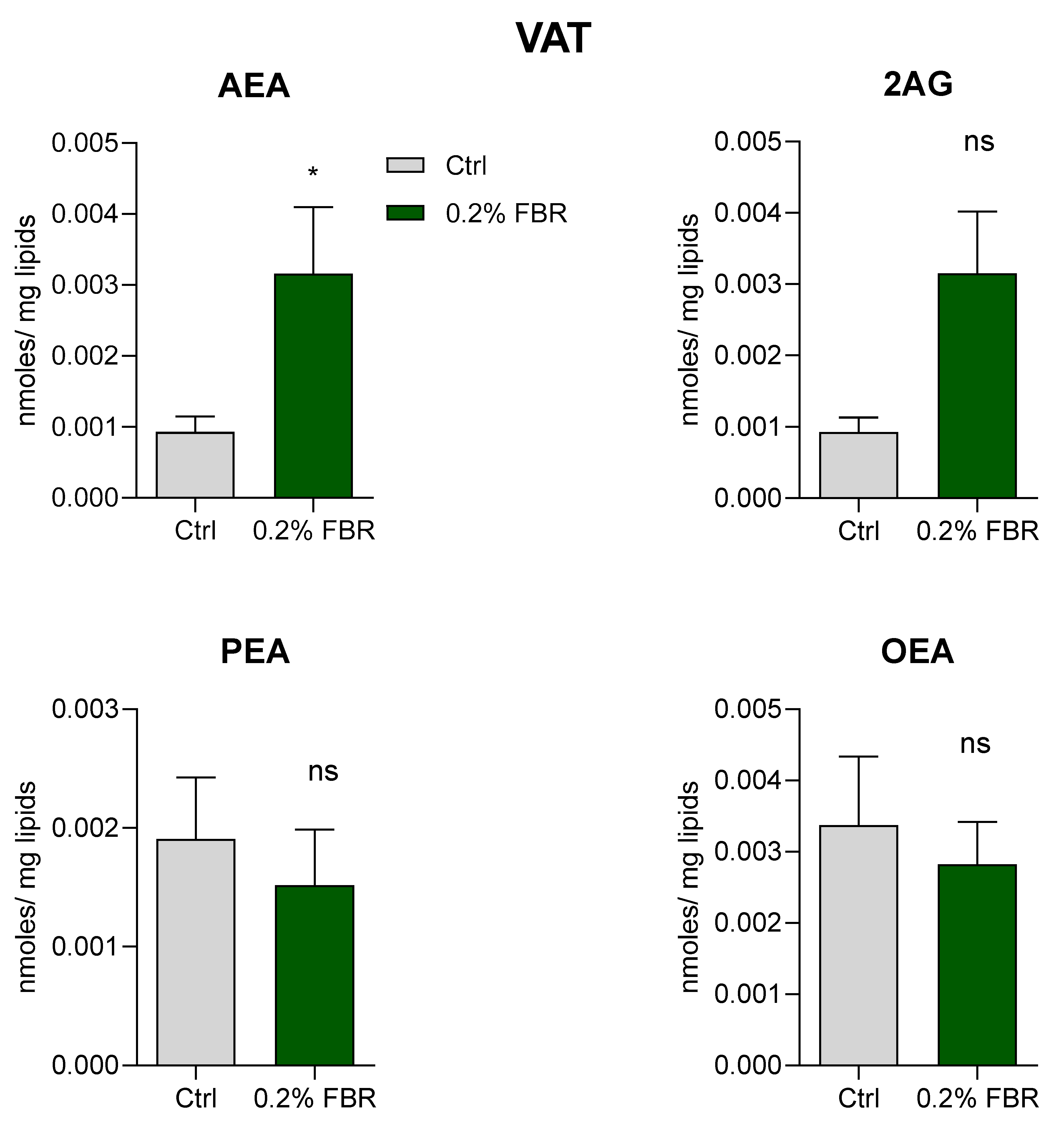
2.2. Tissue Fatty Acid Profile and eCB-Like Mediators
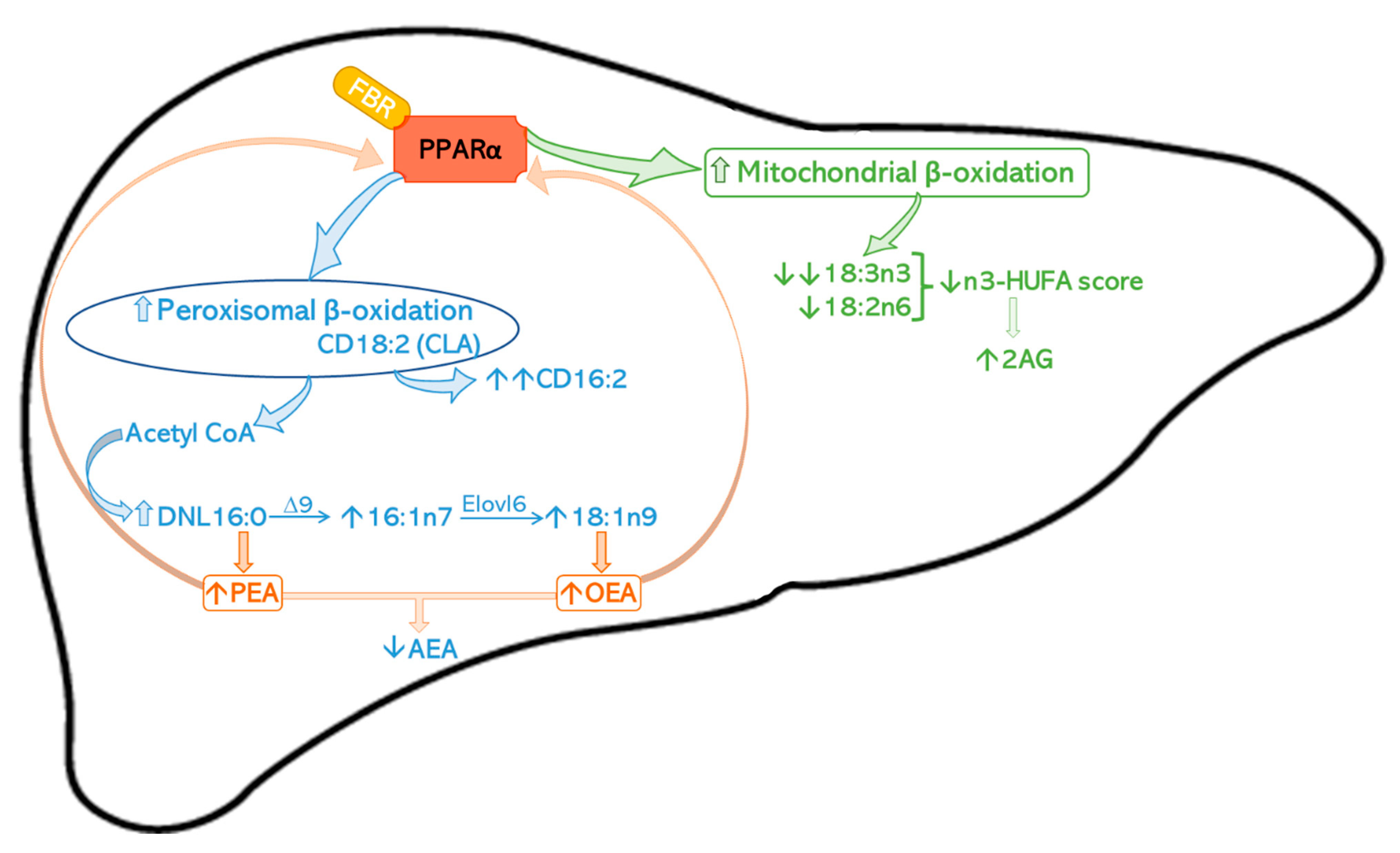
3. Discussion

4. Material and Methods
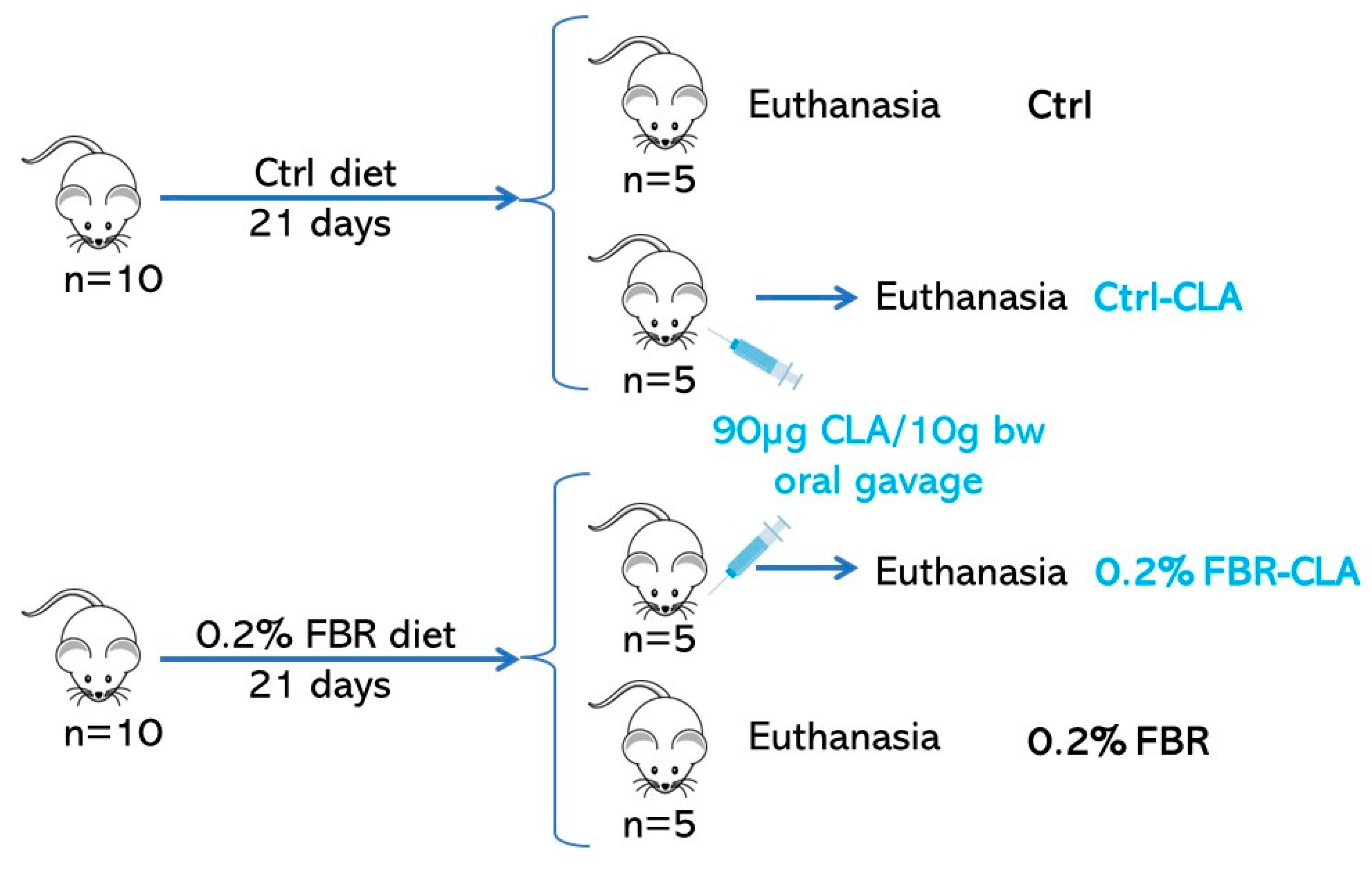
4.1. Animals and Diets
4.2. Lipid Analyses
4.2.1. Measurement of Fatty Acid Composition
4.2.2. Quantification of eCB and eCB-Like Molecules
4.3. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Tenenbaum, A.; Fisman, E.Z. Fibrates Are an Essential Part of Modern Anti-Dyslipidemic Arsenal: Spotlight on Atherogenic Dyslipidemia and Residual Risk Reduction. Cardiovasc. Diabetol. 2012, 11, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeage, K.; Keating, G.M. Fenofibrate: A Review of Its Use in Dyslipidaemia. Drugs 2011, 71, 1917–1946. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Park, J.-S. Exploring Fenofibrate Formulations for the Treatment of Lipid Disorders: Past, Present, and Future. Cardiometab. Syndr. J. 2022, 2, 77. [Google Scholar] [CrossRef]
- Liu, A.; Patterson, A.D.; Yang, Z.; Zhang, X.; Liu, W.; Qiu, F.; Sun, H.; Krausz, K.W.; Idle, J.R.; Gonzalez, F.J.; et al. Fenofibrate Metabolism in the Cynomolgus Monkey Using Ultraperformance Liquid Chromatography-Quadrupole Time-of-Flight Mass Spectrometry-Based Metabolomics. Drug Metab. Dispos. 2009, 37, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Todisco, S.; Santarsiero, A.; Convertini, P.; De Stefano, G.; Gilio, M.; Iacobazzi, V.; Infantino, V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology 2022, 11, 792. [Google Scholar] [CrossRef]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [Green Version]
- Patsouris, D.; Mandard, S.; Voshol, P.J.; Escher, P.; Tan, N.S.; Havekes, L.M.; Koenig, W.; März, W.; Tafuri, S.; Wahli, W.; et al. PPARα Governs Glycerol Metabolism. J. Clin. Investig. 2004, 114, 94–103. [Google Scholar] [CrossRef] [Green Version]
- Larsen, P.J.; Jensen, P.B.; Sørensen, R.V.; Larsen, L.K.; Vrang, N.; Wulff, E.M.; Wassermann, K. Differential Influences of Peroxisome Proliferator–Activated Receptorsγ and -α on Food Intake and Energy Homeostasis. Diabetes 2003, 52, 2249–2259. [Google Scholar] [CrossRef] [Green Version]
- Chinetti, G.; Fruchart, J.-C.; Staels, B. Peroxisome Proliferator-Activated Receptors (PPARs): Nuclear Receptors at the Crossroads between Lipid Metabolism and Inflammation. Inflamm. Res. 2000, 49, 497–505. [Google Scholar] [CrossRef]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; More, K.J.; Breitbart, R.E.; et al. Evidence That the Diabetes Gene Encodes the Leptin Receptor: Identification of a Mutation in the Leptin Receptor Gene in Db/Db Mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef]
- Borrelli, F.; Romano, B.; Petrosino, S.; Pagano, E.; Capasso, R.; Coppola, D.; Battista, G.; Orlando, P.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide, a Naturally Occurring Lipid, Is an Orally Effective Intestinal Anti-Inflammatory Agent: Palmitoylethanolamide and Colitis. Br. J. Pharmacol. 2015, 172, 142–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.D.; Karimian Azari, E.; Ayala, J.E. Oleoylethanolamide: A Fat Ally in the Fight against Obesity. Physiol. Behav. 2017, 176, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Campolongo, P.; Roozendaal, B.; Trezza, V.; Cuomo, V.; Astarita, G.; Fu, J.; McGaugh, J.L.; Piomelli, D. Fat-Induced Satiety Factor Oleoylethanolamide Enhances Memory Consolidation. Proc. Natl. Acad. Sci. USA 2009, 106, 8027–8031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahri-Joutey, M.; Andreoletti, P.; Surapureddi, S.; Nasser, B.; Cherkaoui-Malki, M.; Latruffe, N. Mechanisms Mediating the Regulation of Peroxisomal Fatty Acid Beta-Oxidation by PPARα. IJMS 2021, 22, 8969. [Google Scholar] [CrossRef]
- Lamas Bervejillo, M.; Ferreira, A.M. Understanding Peroxisome Proliferator-Activated Receptors: From the Structure to the Regulatory Actions on Metabolism. In Bioactive Lipids in Health and Disease; Trostchansky, A., Rubbo, H., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2019; Volume 1127, pp. 39–57. ISBN 978-3-030-11487-9. [Google Scholar]
- Murru, E.; Carta, G.; Cordeddu, L.; Melis, M.; Desogus, E.; Ansar, H.; Chilliard, Y.; Ferlay, A.; Stanton, C.; Coakley, M.; et al. Dietary Conjugated Linoleic Acid-Enriched Cheeses Influence the Levels of Circulating n-3 Highly Unsaturated Fatty Acids in Humans. IJMS 2018, 19, 1730. [Google Scholar] [CrossRef] [Green Version]
- Mollica, M.P.; Trinchese, G.; Cavaliere, G.; De Filippo, C.; Cocca, E.; Gaita, M.; Della-Gatta, A.; Marano, A.; Mazzarella, G.; Bergamo, P. C9,T11-Conjugated Linoleic Acid Ameliorates Steatosis by Modulating Mitochondrial Uncoupling and Nrf2 Pathway. J. Lipid Res. 2014, 55, 837–849. [Google Scholar] [CrossRef] [Green Version]
- Oosterveer, M.H.; Grefhorst, A.; van Dijk, T.H.; Havinga, R.; Staels, B.; Kuipers, F.; Groen, A.K.; Reijngoud, D.-J. Fenofibrate Simultaneously Induces Hepatic Fatty Acid Oxidation, Synthesis, and Elongation in Mice. J. Biol. Chem. 2009, 284, 34036–34044. [Google Scholar] [CrossRef] [Green Version]
- Hebbachi, A.M.; Knight, B.L.; Wiggins, D.; Patel, D.D.; Gibbons, G.F. Peroxisome Proliferator-Activated Receptor α Deficiency Abolishes the Response of Lipogenic Gene Expression to Re-Feeding. J. Biol. Chem. 2008, 283, 4866–4876. [Google Scholar] [CrossRef] [Green Version]
- Matias, I. Effect of Polyunsaturated Fatty Acids on Endocannabinoid and N-Acyl-Ethanolamine Levels in Mouse Adipocytes. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2008, 1781, 52–60. [Google Scholar] [CrossRef]
- Murru, E.; Lopes, P.A.; Carta, G.; Manca, C.; Abolghasemi, A.; Guil-Guerrero, J.L.; Prates, J.A.M.; Banni, S. Different Dietary N-3 Polyunsaturated Fatty Acid Formulations Distinctively Modify Tissue Fatty Acid and N-Acylethanolamine Profiles. Nutrients 2021, 13, 625. [Google Scholar] [CrossRef]
- Artmann, A.; Petersen, G.; Hellgren, L.I.; Boberg, J.; Skonberg, C.; Nellemann, C.; Hansen, S.H.; Hansen, H.S. Influence of Dietary Fatty Acids on Endocannabinoid and N-Acylethanolamine Levels in Rat Brain, Liver and Small Intestine. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2008, 1781, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D. A Fatty Gut Feeling. Trends Endocrinol. Metab. 2013, 24, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Schoonjans, K.; Peinado-Onsurbe, J.; Lefebvre, A.M.; Heyman, R.A.; Briggs, M.; Deeb, S.; Staels, B.; Auwerx, J. PPARalpha and PPARgamma Activators Direct a Distinct Tissue-Specific Transcriptional Response via a PPRE in the Lipoprotein Lipase Gene. EMBO J. 1996, 15, 5336–5348. [Google Scholar] [CrossRef]
- Auboeuf, D.; Rieusset, J.; Fajas, L.; Vallier, P.; Frering, V.; Riou, J.P.; Staels, B.; Auwerx, J.; Laville, M.; Vidal, H. Tissue Distribution and Quantification of the Expression of MRNAs of Peroxisome Proliferator–Activated Receptors and Liver X Receptor-α in Humans: No Alteration in Adipose Tissue of Obese and NIDDM Patients. Diabetes 1997, 46, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Jow, L.; Croston, G.E.; Paterniti, J.R. Identification, Characterization, and Tissue Distribution of Human Peroxisome Proliferator-Activated Receptor (PPAR) Isoforms PPARγ2 versus PPARγ1 and Activation with Retinoid X Receptor Agonists and Antagonists. J. Biol. Chem. 1997, 272, 8071–8076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.-K.; Han, Y.; Kim, M.S.; Seo, E.; Kang, S.; Park, S.-Y.; Koh, H.; Kim, D.K.; Lee, H.-J. Reduction of Food Intake by Fenofibrate Is Associated with Cholecystokinin Release in Long-Evans Tokushima Rats. Korean J. Physiol. Pharmacol. 2012, 16, 181. [Google Scholar] [CrossRef] [Green Version]
- Mancini, F.P.; Lanni, A.; Sabatino, L.; Moreno, M.; Giannino, A.; Contaldo, F.; Colantuoni, V.; Goglia, F. Fenofibrate Prevents and Reduces Body Weight Gain and Adiposity in Diet-Induced Obese Rats. FEBS Lett. 2001, 491, 154–158. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.S.; Pineau, T.; Drago, J.; Lee, E.J.; Owens, J.W.; Kroetz, D.L.; Fernandez-Salguero, P.M.; Westphal, H.; Gonzalez, F.J. Targeted Disruption of the Alpha Isoform of the Peroxisome Proliferator-Activated Receptor Gene in Mice Results in Abolishment of the Pleiotropic Effects of Peroxisome Proliferators. Mol. Cell. Biol. 1995, 15, 3012–3022. [Google Scholar] [CrossRef] [Green Version]
- Costet, P.; Legendre, C.; Moré, J.; Edgar, A.; Galtier, P.; Pineau, T. Peroxisome Proliferator-Activated Receptor α-Isoform Deficiency Leads to Progressive Dyslipidemia with Sexually Dimorphic Obesity and Steatosis. J. Biol. Chem. 1998, 273, 29577–29585. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandusse, S.; Denis, S.; Mooijer, P.A.W.; Zhang, Z.; Reddy, J.K.; Spector, A.A.; Wanders, R.J.A. Identification of the Peroxisomal β-Oxidation Enzymes Involved in the Biosynthesis of Docosahexaenoic Acid. J. Lipid Res. 2001, 42, 1987–1995. [Google Scholar] [CrossRef]
- Burdge, G.C. Metabolism of α-Linolenic Acid in Humans. Prostaglandins Leukot. Essent. Fat. Acids 2006, 75, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Carta, G.; Murru, E.; Manca, C.; Serra, A.; Mele, M.; Banni, S. Natural CLA-Enriched Lamb Meat Fat Modifies Tissue Fatty Acid Profile and Increases n-3 HUFA Score in Obese Zucker Rats. Biomolecules 2019, 9, 751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piras, A.; Carta, G.; Murru, E.; Lopes, P.A.; Martins, S.V.; Prates, J.A.M.; Banni, S. Effects of Dietary CLA on N-3 HUFA Score and N-Acylethanolamides Biosynthesis in the Liver of Obese Zucker Rats. Prostaglandins Leukot. Essent. Fat. Acids 2015, 98, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Pintus, S.; Murru, E.; Carta, G.; Cordeddu, L.; Batetta, B.; Accossu, S.; Pistis, D.; Uda, S.; Elena Ghiani, M.; Mele, M.; et al. Sheep Cheese Naturally Enriched in α-Linolenic, Conjugated Linoleic and Vaccenic Acids Improves the Lipid Profile and Reduces Anandamide in the Plasma of Hypercholesterolaemic Subjects. Br. J. Nutr. 2013, 109, 1453–1462. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Hudgins, L.C.; Hirsch, J.; Shike, M. Adipose-Tissue Fatty Acid Composition in Recipients of Long-Term Total Parenteral Nutrition (TPN). Am. J. Clin. Nutr. 1991, 53, 1487–1492. [Google Scholar] [CrossRef] [Green Version]
- Banni, S.; Petroni, A.; Blasevich, M.; Carta, G.; Angioni, E.; Murru, E.; Day, B.W.; Melis, M.P.; Spada, S.; Ip, C. Detection of Conjugated C16 PUFAs in Rat Tissues as Possible Partial Beta-Oxidation Products of Naturally Occurring Conjugated Linoleic Acid and Its Metabolites. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2004, 1682, 120–127. [Google Scholar] [CrossRef]
- Heller, F.; Harvengt, C. Effects of Clofibrate, Bezafibrate, Fenofibrate and Probucol on Plasma Lipolytic Enzymes in Normolipaemic Subjects. Eur. J. Clin. Pharmacol. 1983, 25, 57–63. [Google Scholar] [CrossRef]
- Iannotti, F.A.; Vitale, R.M. The Endocannabinoid System and PPARs: Focus on Their Signalling Crosstalk, Action and Transcriptional Regulation. Cells 2021, 10, 586. [Google Scholar] [CrossRef]
- Melis, M.; Carta, G.; Pistis, M.; Banni, S. Physiological Role of Peroxisome Proliferator-Activated Receptors Type Alpha on Dopamine Systems. CNSNDDT 2013, 12, 70–77. [Google Scholar] [CrossRef]
- Priestley, R.S.; Nickolls, S.A.; Alexander, S.P.H.; Kendall, D.A. A Potential Role for Cannabinoid Receptors in the Therapeutic Action of Fenofibrate. FASEB J. 2015, 29, 1446–1455. [Google Scholar] [CrossRef]
- Stasiulewicz, A.; Znajdek, K.; Grudzień, M.; Pawiński, T.; Sulkowska, J.I. A Guide to Targeting the Endocannabinoid System in Drug Design. IJMS 2020, 21, 2778. [Google Scholar] [CrossRef] [PubMed]
- Murru, E.; Carta, G.; Manca, C.; Sogos, V.; Pistis, M.; Melis, M.; Banni, S. Conjugated Linoleic Acid and Brain Metabolism: A Possible Anti-Neuroinflammatory Role Mediated by PPARα Activation. Front. Pharmacol. 2021, 11, 587140. [Google Scholar] [CrossRef] [PubMed]
- Lands, W.E.M. Stories about Acyl Chains. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2000, 1483, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Maccarrone, M. Endocannabinoid Signaling and Its Regulation by Nutrients: Endocannabinoid Signaling and Nutrients. BioFactors 2014, 40, 373–380. [Google Scholar] [CrossRef]
- Di Marzo, V.; Griinari, M.; Carta, G.; Murru, E.; Ligresti, A.; Cordeddu, L.; Giordano, E.; Bisogno, T.; Collu, M.; Batetta, B.; et al. Dietary Krill Oil Increases Docosahexaenoic Acid and Reduces 2-Arachidonoylglycerol but Not N-Acylethanolamine Levels in the Brain of Obese Zucker Rats. Int. Dairy J. 2010, 20, 231–235. [Google Scholar] [CrossRef]
- Piscitelli, F.; Carta, G.; Bisogno, T.; Murru, E.; Cordeddu, L.; Berge, K.; Tandy, S.; Cohn, J.S.; Griinari, M.; Banni, S.; et al. Effect of Dietary Krill Oil Supplementation on the Endocannabinoidome of Metabolically Relevant Tissues from High-Fat-Fed Mice. Nutr. Metab. 2011, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Batetta, B.; Griinari, M.; Carta, G.; Murru, E.; Ligresti, A.; Cordeddu, L.; Giordano, E.; Sanna, F.; Bisogno, T.; Uda, S.; et al. Endocannabinoids May Mediate the Ability of (n-3) Fatty Acids to Reduce Ectopic Fat and Inflammatory Mediators in Obese Zucker Rats. J. Nutr. 2009, 139, 1495–1501. [Google Scholar] [CrossRef] [Green Version]
- Banni, S.; Carta, G.; Murru, E.; Cordeddu, L.; Giordano, E.; Sirigu, A.; Berge, K.; Vik, H.; Maki, K.C.; Di Marzo, V.; et al. Krill Oil Significantly Decreases 2-Arachidonoylglycerol Plasma Levels in Obese Subjects. Nutr. Metab. 2011, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Veilleux, A.; Di Marzo, V.; Silvestri, C. The Expanded Endocannabinoid System/Endocannabinoidome as a Potential Target for Treating Diabetes Mellitus. Curr. Diab. Rep. 2019, 19, 117. [Google Scholar] [CrossRef]
- Gatta-Cherifi, B.; Cota, D. New Insights on the Role of the Endocannabinoid System in the Regulation of Energy Balance. Int. J. Obes. 2016, 40, 210–219. [Google Scholar] [CrossRef]
- Muccioli, G.G.; Naslain, D.; Bäckhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The Endocannabinoid System Links Gut Microbiota to Adipogenesis. Mol. Syst. Biol. 2010, 6, 392. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.N.; Kantae, V.; Maarse, B.C.E.; van den Berg, S.M.; van Eenige, R.; Nahon, K.J.; Reifel-Miller, A.; Coskun, T.; de Winther, M.P.J.; Lutgens, E.; et al. High Fat Diet Increases Circulating Endocannabinoids Accompanied by Increased Synthesis Enzymes in Adipose Tissue. Front. Physiol. 2019, 9, 1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Oveisi, F.; Gaetani, S.; Lin, E.; Piomelli, D. Oleoylethanolamide, an Endogenous PPAR-α Agonist, Lowers Body Weight and Hyperlipidemia in Obese Rats. Neuropharmacology 2005, 48, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Diep, T.A.; Madsen, A.N.; Holst, B.; Kristiansen, M.M.; Wellner, N.; Hansen, S.H.; Hansen, H.S. Dietary Fat Decreases Intestinal Levels of the Anorectic Lipids through a Fat Sensor. FASEB J. 2011, 25, 765–774. [Google Scholar] [CrossRef]
- Rodríguez de Fonseca, F.; Navarro, M.; Gómez, R.; Escuredo, L.; Nava, F.; Fu, J.; Murillo-Rodríguez, E.; Giuffrida, A.; LoVerme, J.; Gaetani, S.; et al. An Anorexic Lipid Mediator Regulated by Feeding. Nature 2001, 414, 209–212. [Google Scholar] [CrossRef] [Green Version]
- Thabuis, C.; Destaillats, F.; Landrier, J.-F.; Tissot-Favre, D.; Martin, J.-C. Analysis of Gene Expression Pattern Reveals Potential Targets of Dietary Oleoylethanolamide in Reducing Body Fat Gain in C3H Mice. J. Nutr. Biochem. 2010, 21, 922–928. [Google Scholar] [CrossRef]
- Blüher, M.; Engeli, S.; Klöting, N.; Berndt, J.; Fasshauer, M.; Bátkai, S.; Pacher, P.; Schön, M.R.; Jordan, J.; Stumvoll, M. Dysregulation of the Peripheral and Adipose Tissue Endocannabinoid System in Human Abdominal Obesity. Diabetes 2006, 55, 3053–3060. [Google Scholar] [CrossRef] [Green Version]
- Côté, M.; Matias, I.; Lemieux, I.; Petrosino, S.; Alméras, N.; Després, J.-P.; Di Marzo, V. Circulating Endocannabinoid Levels, Abdominal Adiposity and Related Cardiometabolic Risk Factors in Obese Men. Int. J. Obes. 2007, 31, 692–699. [Google Scholar] [CrossRef] [Green Version]
- Parlar, A.; Arslan, S.; Doğan, M.; Ayşenur Çam, S.; Yalçin, A.; Elibol, E.; Kaya Özer, M.; Üçkardeş, F.; Kara, H. The Exogenous Administration of CB2 Specific Agonist, GW405833, Inhibits Inflammation by Reducing Cytokine Production and Oxidative Stress. Exp. Ther. Med. 2018, 16, 4900–4908. [Google Scholar] [CrossRef] [Green Version]
- Belfort, R.; Berria, R.; Cornell, J.; Cusi, K. Fenofibrate Reduces Systemic Inflammation Markers Independent of Its Effects on Lipid and Glucose Metabolism in Patients with the Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2010, 95, 829–836. [Google Scholar] [CrossRef]
- Burdge, G.C.; Wootton, S.A. Conversion of α-Linolenic Acid to Palmitic, Palmitoleic, Stearic and Oleic Acids in Men and Women. Prostaglandins Leukot. Essent. Fat. Acids 2003, 69, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.J. Incorporation of Radioactive Polyunsaturated Fatty Acids into Liver and Brain of Developing Rat. Lipids 1975, 10, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Botolin, D.; Christian, B.; Busik, J.; Xu, J.; Jump, D.B. Tissue-Specific, Nutritional, and Developmental Regulation of Rat Fatty Acid Elongases. J. Lipid Res. 2005, 46, 706–715. [Google Scholar] [CrossRef] [Green Version]
- Puligheddu, M.; Melis, M.; Pillolla, G.; Milioli, G.; Parrino, L.; Terzano, G.M.; Aroni, S.; Sagheddu, C.; Marrosu, F.; Pistis, M.; et al. Rationale for an Adjunctive Therapy with Fenofibrate in Pharmacoresistant Nocturnal Frontal Lobe Epilepsy. Epilepsia 2017, 58, 1762–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puligheddu, M.; Pillolla, G.; Melis, M.; Lecca, S.; Marrosu, F.; De Montis, M.G.; Scheggi, S.; Carta, G.; Murru, E.; Aroni, S.; et al. PPAR-Alpha Agonists as Novel Antiepileptic Drugs: Preclinical Findings. PLoS ONE 2013, 8, e64541. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Bi, L.; Hulke, M.; Li, T. Fish Oil and Fenofibrate Prevented Phosphorylation-Dependent Hepatic Sortilin 1 Degradation in Western Diet-Fed Mice. J. Biol. Chem. 2014, 289, 22437–22449. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, T.; Kamei, Y.; Kato, H.; Sugita, S.; Takeya, M.; Suganami, T.; Ogawa, Y. Effect of Peroxisome Proliferator-Activated Receptor-α Ligands in the Interaction Between Adipocytes and Macrophages in Obese Adipose Tissue. Obesity 2008, 16, 1199–1207. [Google Scholar] [CrossRef]
- Caillaud, M.; Patel, N.H.; Toma, W.; White, A.; Thompson, D.; Mann, J.; Tran, T.H.; Roberts, J.L.; Poklis, J.L.; Bigbee, J.W.; et al. A Fenofibrate Diet Prevents Paclitaxel-Induced Peripheral Neuropathy in Mice. Cancers 2020, 13, 69. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Chiang, S.P.; Gessert, C.F.; Lowry, O.H. Colorimetric Determination of Extracted Lipids. An Adaptation for Microgram Amounts of Lipids Obtained from Cerumen. Curr. List Med. Lit. Res. Rep. 1957, 33, 56–113. [Google Scholar]
- Banni, S.; Carta, G.; Contini, M.S.; Angioni, E.; Deiana, M.; Dessì, M.A.; Melis, M.P.; Corongiu, F.P. Characterization of Conjugated Diene Fatty Acids in Milk, Dairy Products, and Lamb Tissues. J. Nutr. Biochem. 1996, 7, 150–155. [Google Scholar] [CrossRef]
- Melis, M.P.; Angioni, E.; Carta, G.; Murru, E.; Scanu, P.; Spada, S.; Banni, S. Characterization of Conjugated Linoleic Acid and Its Metabolites by RP-HPLC with Diode Array Detector. Eur. J. Lipid Sci. Technol. 2001, 103, 617–621. [Google Scholar] [CrossRef]
- Richardson, D.; Ortori, C.A.; Chapman, V.; Kendall, D.A.; Barrett, D.A. Quantitative Profiling of Endocannabinoids and Related Compounds in Rat Brain Using Liquid Chromatography–Tandem Electrospray Ionization Mass Spectrometry. Anal. Biochem. 2007, 360, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.D. The Percentage of N-3 Highly Unsaturated Fatty Acids in Total HUFA as a Biomarker for Omega-3 Fatty Acid Status in Tissues. Lipids 2008, 43, 45–53. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ctrl | 0.2% FBR | ||||||
|---|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | ||||
| 12:0 | 3.96 | ± | 0.42 | 0.75 | ± | 0.03 | ** |
| 14:0 | 28.73 | ± | 2.67 | 10.87 | ± | 0.46 | ** |
| 16:0 | 1368.80 | ± | 117.21 | 1117.72 | ± | 15.18 | ns |
| 18:0 | 353.80 | ± | 45.13 | 249.95 | ± | 10.58 | ns |
| 20:0 | 5.94 | ± | 0.96 | 1.79 | ± | 0.34 | * |
| 16:1n7 | 108.82 | ± | 12.44 | 159.18 | ± | 9.57 | * |
| 18:1n9 | 818.39 | ± | 49.72 | 1248.86 | ± | 21.48 | ** |
| 20:3n9 | 3.63 | ± | 0.09 | 19.99 | ± | 0.80 | ** |
| 18:3n3 | 39.19 | ± | 3.72 | 5.39 | ± | 0.33 | ** |
| 20:5n3 | 6.46 | ± | 0.46 | 6.24 | ± | 0.75 | ns |
| 22:5n3 | 7.49 | ± | 1.59 | 14.06 | ± | 1.65 | * |
| 22:6n3 | 191.61 | ± | 11.24 | 142.41 | ± | 11.06 | * |
| 18:2n6 | 1136.45 | ± | 42.77 | 568.52 | ± | 12.42 | ** |
| 18:3n6 | 40.39 | ± | 3.08 | 13.04 | ± | 0.60 | ** |
| 20:2n6 | 20.33 | ± | 1.52 | 18.78 | ± | 2.50 | ns |
| 20:3n6 | 25.63 | ± | 1.00 | 187.26 | ± | 3.69 | ** |
| 20:4n6 | 357.35 | ± | 25.03 | 445.65 | ± | 21.28 | * |
| 22:4n6 | 11.39 | ± | 0.58 | 12.74 | ± | 0.27 | ns |
| 22:5n6 | 15.05 | ± | 1.09 | 11.58 | ± | 0.84 | ns |
| SFA 1 | 1778.71 | ± | 160.81 | 1392.31 | ± | 23.85 | * |
| MUFA 1 | 927.21 | ± | 60.42 | 1408.03 | ± | 21.77 | ** |
| n3-PUFA 1 | 244.76 | ± | 11.41 | 168.10 | ± | 9.47 | ** |
| n6-PUFA 1 | 1606.59 | ± | 61.37 | 1257.58 | ± | 35.19 | ** |
| n3/n6 PUFA 1 | 0.152 | ± | 0.002 | 0.133 | ± | 0.005 | * |
| 16:1n7/16:0 | 0.080 | ± | 0.008 | 0.143 | ± | 0.010 | ** |
| 18:1n9/18:0 | 2.492 | ± | 0.406 | 5.032 | ± | 0.233 | ** |
| 20:3n9/18:1n9 | 0.005 | ± | 0.000 | 0.016 | ± | 0.001 | ** |
| mg lipids /g tissue | 61.84 | ± | 6.44 | 45.52 | ± | 1.25 | ** |
| Ctrl | 0.2% FBR | ||||||
|---|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | ||||
| 16:1n7 | 133.49 | ± | 8.40 | 305.95 | ± | 30.32 | ** |
| 18:1n9 | 1155.96 | ± | 29.38 | 1163.60 | ± | 45.49 | ns |
| 18:3n3 | 42.34 | ± | 5.66 | 25.47 | ± | 1.77 | ** |
| 22:6n3 | 4.64 | ± | 1.25 | 5.87 | ± | 1.83 | ns |
| 18:2n6 | 1055.44 | ± | 33.16 | 640.98 | ± | 30.62 | ** |
| 18:3n6 | 5.04 | ± | 0.74 | 4.83 | ± | 0.81 | ns |
| 20:3n6 | 16.09 | ± | 3.38 | 12.97 | ± | 1.74 | ns |
| 20:4n6 | 18.63 | ± | 4.97 | 16.08 | ± | 1.93 | ns |
| MUFA 1 | 1289.45 | ± | 30.22 | 1469.55 | ± | 69.17 | ns |
| n3-PUFA 1 | 46.97 | ± | 5.88 | 30.16 | ± | 2.57 | ** |
| n6-PUFA 1 | 1095.20 | ± | 36.79 | 673.89 | ± | 31.22 | ** |
| n3/n6 PUFA 1 | 0.043 | ± | 0.006 | 0.045 | ± | 0.004 | ns |
| mg lipids /g tissue | 477.49 | ± | 53.25 | 362.70 | ± | 60.37 | ns |
| FA | g/100 g |
|---|---|
| 16:0 | 0.5 |
| 18:0 | 0.1 |
| 18:1n9 | 0.7 |
| 18:2n6 | 2.0 |
| 18:3n3 | 0.1 |
| SFA 1 | 0.6 |
| MUFA 1 | 0.7 |
| PUFA 1 | 2.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murru, E.; Muntoni, A.L.; Manca, C.; Aroni, S.; Pistis, M.; Banni, S.; Carta, G. Profound Modification of Fatty Acid Profile and Endocannabinoid-Related Mediators in PPARα Agonist Fenofibrate-Treated Mice. Int. J. Mol. Sci. 2023, 24, 709. https://doi.org/10.3390/ijms24010709
Murru E, Muntoni AL, Manca C, Aroni S, Pistis M, Banni S, Carta G. Profound Modification of Fatty Acid Profile and Endocannabinoid-Related Mediators in PPARα Agonist Fenofibrate-Treated Mice. International Journal of Molecular Sciences. 2023; 24(1):709. https://doi.org/10.3390/ijms24010709
Chicago/Turabian StyleMurru, Elisabetta, Anna Lisa Muntoni, Claudia Manca, Sonia Aroni, Marco Pistis, Sebastiano Banni, and Gianfranca Carta. 2023. "Profound Modification of Fatty Acid Profile and Endocannabinoid-Related Mediators in PPARα Agonist Fenofibrate-Treated Mice" International Journal of Molecular Sciences 24, no. 1: 709. https://doi.org/10.3390/ijms24010709