
Molecular Investigations of Protein Aggregation in the Pathogenesis of Amyotrophic Lateral Sclerosis



School of Medicine and Surgery, University of Milano-Bicocca, 20900 Monza, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(1), 704; https://doi.org/10.3390/ijms24010704
Submission received: 23 November 2022
/
Revised: 20 December 2022
/
Accepted: 27 December 2022
/
Published: 31 December 2022
(This article belongs to the Special Issue Genetic and Molecular Susceptibility in Human Diseases)
Abstract
:Amyotrophic lateral sclerosis (ALS) is a devastating progressive neurodegenerative disorder characterized by selective loss of lower and upper motor neurons (MNs) in the brain and spinal cord, resulting in paralysis and eventually death due to respiratory insufficiency. Although the fundamental physiological mechanisms underlying ALS are not completely understood, the key neuropathological hallmarks of ALS pathology are the aggregation and accumulation of ubiquitinated protein inclusions within the cytoplasm of degenerating MNs. Herein, we discuss recent insights into the molecular mechanisms that lead to the accumulation of protein aggregates in ALS. This will contribute to a better understanding of the pathophysiology of the disease and may open novel avenues for the development of therapeutic strategies.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is an idiopathic disease characterized by progressive skeletal muscle paralysis caused by degeneration of the lower and/or upper motor neurons (MNs), resulting in severe disability [1]. ALS is a fatal disease that results in death caused by respiratory failure 3–5 years after diagnosis [2]. The rate of disease progression varies between individuals and can be affected by the site of onset, but it is usually rapid [3]. Comparative analyses of various European data confirm that ALS is estimated to have an annual incidence rate of 2–3/100,000 people, with a prevalence of 8–10/100,000 people in the West. This disease can affect people of any age, but onset under 20 years is extremely rare, and incidence rises to a peak between the ages of 65 and 75, after which it falls again. Moreover, Caucasians and males are slightly more likely to develop ALS than females, with a 1.2 times higher risk [3]. Unfortunately, available drugs cannot stop the disease but can only provide temporary relief and extend patients’ median survival by a few months [4].
Approximately 85–90% of ALS cases are sporadic (sALS), indicating a complex interaction between genetic and environmental risk factors. The remaining 10–15% of ALS cases are generally dominantly inherited and are thus classified as familial (fALS) [5]. Although over 30 ALS-related genes have been identified, the majority of fALS forms are caused by mutations in genes encoding superoxide dismutase 1 (SOD1), transactive response (TAR)-DNA binding protein 43 (TARDBP), fused in sarcoma (FUS), and chromosome 9 open reading frame 72 (C9ORF72) [6,7]. Proteins encoded by these genes are involved in a variety of biochemical processes and cellular pathways, such as autophagy, protein quality control, RNA metabolism, mitochondria, and ATP homeostasis. Interestingly, sALS and fALS are clinically indistinguishable [2].
The exact etiology of the disease is unknown; however, clinical evaluations have revealed the involvement of multiple cellular functions in patients’ MNs, abnormal increase in excitatory muscle tone, accumulation of misfolded protein, impaired axonal transport, and high calcium metabolism. Numerous studies have reported that the death of MNs is not due to a single event, but is caused by a combination of various mechanisms [8], including oxidative stress, mitochondrial dysfunction, cytotoxicity, the formation of protein aggregates, and changes in RNA metabolism [9,10,11]. The accumulation of insoluble proteins at the MN level is a key feature of ALS pathology. Most of these cytoplasmic inclusions are ubiquitinated [12] and contain primarily either TDP-43 [13,14], SOD1 [15], or FUS [16,17] proteins.
As these abnormal aggregates are toxic for cells and are responsible for neurodegeneration, this review discusses the molecular mechanisms underlying impaired protein homeostasis, also called proteostasis, in the pathogenesis of ALS.
2. Proteinopathies
Neurodegenerative diseases are usually age-related and characterized by a slow and progressive loss of different nerve functions. Among them, the most common are Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and ALS [18]. The majority of neurodegenerative diseases, with the exception of HD, can have a genetic or sporadic etiology, and familial forms typically manifest earlier with a more severe phenotype. In most cases the onset occurs in the fourth or fifth decade of life. Neurodegenerative disorders share common risk factors that include aging, oxidative stress, environmental stress, and protein malfunction. All of these affect cellular proteostasis, the process that maintains the proteome in the proper concentration (balancing protein production and degradation), in the correct folding (chaperones), and in the right place at the right time (trafficking). Two main cellular degradation systems prevent the formation of impaired proteins: the ubiquitin-proteasome system (UPS), which degrades functional as well as dysfunctional proteins, and the autophagy–lysosomal system (ALP) responsible for the degradation of whole organelles, large aggregates of proteins or macromolecules, and single proteins [19]. From a pathophysiological point of view, different pathologies are distinguished by the synthesis of aberrant proteins that can undergo conformational changes and acquire a toxic function or block their original biological activity [20]. Based on this feature, such diseases are classified as proteinopathies.
Although the proteins that play physiological roles in a healthy brain have monomeric structures, pathological conditions can cause them to undergo conformational changes that favor their association and result in the formation of oligomers, which can eventually aggregate into higher-order structures. These aggregates typically precipitate in different brain areas. Under these conditions, proteins might either acquire harmful qualities or cease their physiological function. Neurodegenerative disorders and proteinopathies are characterized by protein accumulation, both wild-type (wt) and mutant, with the altered conformation favoring aggregation [21]. These accumulations, which include TDP-43, FUS, SOD1, tau, and α-synuclein (α-syn), can develop intracellularly and in the surrounding area, as in the case of amyloid β (Aβ), and can determine the pathogenic forms of disease.
A single form of protein aggregation may cause certain proteinopathies, while others may be caused by multiple types. Proteinopathies are frequently mixed disorders, making their diagnosis and treatment challenging. Furthermore, they frequently share clinical features with other diseases. An example of this is the presence in AD brains of neurofibrillary tangles comprising tau aggregates and Aβ plaques, along with the accumulations of α-syn typical of Lewy body disease and PD. This characteristic is also found in ALS, which exhibits aggregates of proteins including TDP-43/FUS/SOD1, α-syn, tau, or Aβ [22,23,24].
3. Pathological Protein Aggregation Involved in ALS
As previously mentioned, protein aggregates are a pathological feature of several neurodegenerative diseases, including extracellular plaques of Aβ and intracellular neurofibrillary aggregates of tau protein in AD, or Lewy bodies in PD [26]. Pathological protein aggregates are also a feature of ALS, and occur in the form of ubiquitinated inclusions in neurons and glia (Figure 1).
Such inclusions contain different proteins, some of which may have an intrinsic tendency to aggregate following genetic mutations (SOD1, TDP-43, FUS), whereas others may simply be trapped in the aggregates [13,14,15,16,17,27]. In particular, cysteine-mediated aggregates of mutant SOD1 (mutSOD1) have been observed in ALS MNs, with the wt form of SOD1 also present, thus demonstrating the strong affinity and co-aggregation of the two forms of the protein [28]. It is widely believed that the toxic functions of mutSOD1 and other proteins typical of ALS are related to their tendency to aggregate. This hypothesis is supported by the presence of intracellular cytoplasmic inclusions, as well as mitochondrial inclusions rich in SOD1 identified in cell models and in animal spinal MNs, as well as in MNs of patients with ALS [27].
Numerous experimental theories have been proposed to explain how the aggregation of mutant proteins contributes to cellular toxicity in ALS patients. These suggestions have included the ability to sequester proteins necessary for the normal functioning of the MN [29], the ability to reduce the activity of the proteasome, which is essential for correct protein turnover, and the ability to inhibit the correct functioning of specific cellular organelles, such as mitochondria, by internal or external aggregation [30]. Cell degeneration depends on the sensitivity of MNs to the aggregation of proteins in the mutant form, supported by the observation thatTDP-43 and FUS also aggregate in patient tissues and ALS models in the same manner as SOD1 [31,32]. Indeed, anatomopathological observations of tissues derived from ALS patients show that these two proteins aggregate as cytoplasmic inclusions (positive for ubiquitin, but negative for SOD1) and that the mutations that affect them seem to increase the degree of aggregation [33]. Furthermore, it has been shown that the aggregation of FUS and TDP-43 is based on a conserved low-complexity domain (LCD), also called a prion-like domain (PrLD) [34], and that such aggregates can also sequester wt proteins and in their native form [10]. The LCD can mediate the liquid–liquid phase separation (LLPS) and therefore the formation of stress granules (SGs), which are cytoplasmic condensates lacking a membrane, composed primarily of RNA and RNA-binding proteins (RBPs), generated under unfavorable environmental conditions. SGs may serve to protect RNA from degradation by inhibiting the initiation of mRNA translation at the cellular level and starting the synthesis of cytoprotective proteins. They are intrinsically dynamic and dissolve quickly upon stress removal [35]. TDP-43 mislocalization and aggregation are observed in approximately 97% of all ALS cases, including all sALS cases, whereas SOD1 (2%) and FUS (1%) inclusions are associated with the remaining cases [36]. Not only are ubiquitin-immunoreactive inclusions most frequently reported in all forms of ALS, but the aggregates may also be reactive to p62, a protein that participates in sequestosome formation and autophagy [37].
To prevent the formation of protein aggregates, the cells are equipped with protein quality control systems, and the presence of chaperone proteins ensures that proteins in their native form fold correctly. These can intervene to avoid misfolding and therefore restore the proteins to the correct conformation shape: the UPS or the ALP. Dysfunction of these systems causes the formation of protein aggregates [38]. In this context, genetic screening has identified disease-associated mutations in many of the proteins identified within ALS inclusions [5]. These findings imply that aggregates are not merely a disease marker but are also strongly linked to the etiology and pathomechanisms of neurodegeneration.
3.1. SOD1
In 1993, Rosen et al. first described eleven disease-associated mutations in the SOD1 gene located on chromosome 21 [39], which encodes for the Cu/Zn superoxide dismutase, a cytoplasmic enzyme responsible for the catabolism of superoxide radicals to hydrogen peroxide and molecular oxygen [40]. SOD1 is ubiquitously expressed, highly conserved, and represents ~1% of all cytoplasmic proteins. It is a 32 kDa polypeptide of 153 amino acids, composed of a binding site with a zinc atom and another for a copper atom (Figure 2). This protein is predominantly localized in the cytoplasm of cells, although it is also apparently present in the intramembrane space of the mitochondria, nucleus, lysosomes, and peroxisomes [41]. Its role is to convert superoxide anions, which are toxic to the cell, into peroxides of hydrogen and oxygen [42]. SOD1 also exerts pro-oxidant activity including peroxidation with the production of hydroxyl radicals and nitration of tyrosines [43]. The ubiquitinated form of this protein is widely expressed and constitutes about 0.5–0.8% of soluble proteins in the human brain [44]. To date, over 200 mutations in SOD1 have been identified accounting for approximately 20% of fALS patients [45,46], characterized by inter-family and intra-family variability in phenotype with respect to severity of symptoms, age of onset, and disease duration [46]. The most common mutations are G93A, A4V, H46R, D90A, inherited as dominant traits, except for the latter that also shows a recessive inheritance pattern of transmission, but only in Scandinavian populations [32,47].
The two principal critical features of SOD1 mediated cytotoxicity are misfolding and protein aggregation. This means that disease onset is driven by mutant protein that is synthesized inside MNs. MutSOD1 protein interacts specifically with neurofilament light chain mRNA and the dynein–dynactin complex, thus inducing cytoskeletal defects or altering axonal transport [47]. Furthermore, it has an increased tendency to form aggregate-prone monomers, and the degree of instability correlates inversely with survival time, suggesting that increased propensity to aggregation may be the unifying common denominator for different SOD1 mutations [47]. Researchers identified misfolded SOD1 in MNs in a subset of patients with sALS without SOD1 mutations, thus suggesting a role for wt SOD1 in sALS, possibly after secondary (oxidative) modification [48,49]. Finally, the aggregation and spread of mutant SOD1 has been demonstrated in cultured cells [50], and its seeding ability via a prion-like mechanism illustrated using spinal cord homogenate [51].
There are currently no explanations for how mutSOD1 causes disease. Initially it was hypothesized that the mutations impair the enzymatic activity of the protein, causing increased levels of reactive oxygen species (ROS) with consequent oxidative stress and death of neuronal cells [52]. More recent studies have shown that mutant protein forms maintain their catalytic activity intact with no apparent causal relationship between residual enzyme activity, clinical progression, and disease phenotype [53].
The mutSOD1 protein accumulates in the oligomeric form and produces cytoplasmic aggregates, which can cause neuronal cell death by sequestering other cytoplasmic proteins required for neuronal survival, blocking the UPS with consequent loss of chaperone proteins, destruction of mitochondria, and the blockade of cytoskeletal or axonal transport [54].
3.2. TDP-43
TDP-43 was first isolated in 1995, when researchers observed its ability to bind the transactivation response region (TAR) of HIV DNA, hence the name TAR DNA binding protein [55]. It was subsequently found in the human brain and in several cell culture systems [56]. TDP-43 is a highly conserved protein across different species and shows ubiquitous expression in humans and rodents with a predominant localization in the nucleus. This protein consists of 414 amino acids and has a molecular weight of 43 kDa, encoded by the TARDBP gene located on chromosome 1, a member of a heterogeneous family of proteins that bind RNA, known as hnRNP (heterogeneous ribonucleoprotein) [57]. From a structural point of view, the protein contains an N-terminal region, a nuclear localization signal (NLS), two RNA recognition motifs (RRM1 and RRM2) which exhibit an export signal nuclear (NES), and a C-terminal region comprising a glycine-rich LCD which mediates the interaction with other proteins belonging to the hnRNP family (Figure 2) [57]. Mutations affecting the TDP-43 protein represent about 5% of sALS and about 3% of fALS cases [58] as well as patients affected by frontotemporal dementia (FTD) [13,14]. The majority of TARDBP mutations are clustered mainly within the glycine-rich C terminal, and have a crucial role in nucleocytoplasmic shuttling, aggregation propensity, and protein–protein interaction [59]. The cellular function of TDP-43 remains unknown, but different studies have shown that this protein plays a fundamental role in a number of biological activities, including regulation of gene transcription, control of splicing processes, and maintaining the stability of mRNA [60]. Protein levels are strictly controlled through a self-regulation system, with particular involvement of the C-terminal region, further supporting the suggestion that mutations in this domain can interfere with the homeostatic control process by altering the recruitment of complexes required for self-regulation [61].
In pathological conditions, this protein tends to form ubiquitinated inclusions in the central nervous system (CNS), in particular the hippocampus, neocortex, and spinal cord. A further feature that distinguishes it from the wt form is its localization: insoluble protein aggregates tend to form in the cytoplasm in the neurons of patients with either pathology, resulting in a decrease of TDP-43 protein in the nucleus, in contrast to healthy subjects where it is expressed in the nucleus [13]. In addition to ubiquitination, hyperphosphorylation of TDP-43 is important for protein aggregation in ALS pathogenesis. In particular, in samples from ALS patients TDP-43 appears to be hyperphosphorylated at the C-terminal level, thus favoring its aggregation [62]. According to the findings of Braak and colleagues, pTDP-43 is widespread throughout the CNS, particularly in the agranular neocortex, and in spinal and bulbar MNs, where the presence of pTDP-43 makes it impossible to distinguish between the two types of MNs [63]. Furthermore, hyperphosphorylation of TDP-43 has been clearly linked to cell death in areas of the CNS, in relation to disease progression [64]. Another study also demonstrated that pTDP-43 aggregates led to the death of dopaminergic neurons in the substantia nigra [65].
Regardless of the presence of genetic mutations, the aberrant localization of TDP-43 in the cytoplasm of neurons in ALS patients appears to be linked to a pathogenetic mechanism associated with a loss of function in nuclear protein responsible for the regulation of mRNA transcription and splicing processes [60]. The formation of cellular inclusions of TDP-43 induces toxicity in the cell, and in this case the protein acquires a toxic function (gain of function) [66,67,68].
Furthermore, the accumulation of TDP-43 within ubiquitinated inclusions (UBIs) can lead to the altered regulation of genes or factors involved in the degradation processes of intracellular proteins, thus contributing to TDP-43 proteinopathy.
TDP-43 levels are strictly regulated by an intrinsic autoregulatory pathway, as demonstrated by observations that inactivation of one copy of the gene does not reduce protein mRNA levels in mice. Autoregulation is believed to be mediated by TDP-43 dependent splicing of an intron in the 3′ UTR region of its own mRNA, and splicing of this gene leads to unstable RNA which undergoes decay [69]. Furthermore, overexpression of wt human TDP-43 in neurons results in neurodegeneration accompanied by decreased locomotor activity, motor impairment, shorter lifespan, and MN loss [70].
TDP-43 is primarily cleaved by caspase 3 into two extremely aggregation-prone fragments with molecular weights of 25 kDa and 35 kDa, namely TDP-25 and TDP-35, respectively [71]. These fragments derive from the entire wt TDP-43 protein chain, induce greater toxicity, and contribute to the loss of function of TDP-43. Therefore, they must be efficiently cleared from cells to prevent their aggregation and the sequestration of other important neuronal components. The clearance of aberrant or misfolded proteins is mediated by the protein quality control system (PQC) [72]. This system is composed of chaperone and co-chaperone proteins that recognize and bind damaged proteins and direct them towards degradation processes.
As mentioned previously, the LCD sequence of TDP-43 controls its ability to undergo LLPS [73], leading to the formation of insoluble aggregates [74]. In particular, when aggregate-prone TDP-43 is more concentrated, the critical concentration for LLPS is exceeded, making it difficult to maintain protein homeostasis by preventing aggregation [62]. Further evidence of the importance of proper interactions between TDP-43 and LLPS comes from a recent study by Gao et al. performed on LLPS-deficient TDP-43 mice, showing that low levels of TDP-43 and LLPS led to the alteration of neuronal cells [75].
3.3. FUS
The discovery of TDP-43 mutations in ALS rapidly led to the identification of mutations in another RNA binding protein, namely FUS [16,17], accounting for 4–5% of fALS and 1% of sALS forms associated with young age at onset and short survival time [5,76]. The FUS gene is located on chromosome 16 and encodes for a 526-amino acid protein of 75 kDa in weight, showing a structure similar to that of TDP-43 (Figure 2) [77,78]. Although FUS was initially identified as a component of a fusion oncogene resulting from a chromosomal translocation observed in liposarcomas [60], this protein also plays a role in RNA processes [79]. FUS is widely expressed in most human tissues [80] and is primarily localized at the nucleus, although it shuttles to the cytoplasm to mediate a wide range of cellular processes including DNA repair, genomic stability, transcriptional regulation, splicing, transport, and maturation of mRNAs [60,81]. Specifically in the CNS, FUS regulates mRNA transport towards the dendrites and supports synaptic plasticity upon activation of glutamate receptors [82].
As with TARDBP, the majority of pathogenic mutations in FUS map to the C terminal within the NLS region of the protein that regulates interaction with transportin-1 [83], thereby interfering with the nuclear–cytoplasmic balance of FUS [78,82]. Accordingly, this protein exhibits mainly nuclear localization under healthy conditions, but abnormal cytoplasmic aggregates have been found in the brains and spinal cords of ALS patients with FUS mutations [16,57,82]. Therefore, the toxic effects of FUS seem to be related to its aberrant cytoplasmic localization that possibly disrupts nucleocytoplasmic transport [84]. This evidence is supported by a study in Drosophila where deletion of the nuclear export signal reduced the toxicity of mutant FUS [85].
Furthermore, authors have reported that FUS can undergo phase transition by LCD sequence [86] and formation of GSs [80], similar to TDP-43 [75]. Study in vitro demonstrated that ALS mutations may accelerate the kinetics of phase separation and exacerbate the transition of FUS from liquid to solid phase [86]. Further studies confirmed that an aberrant phase transition is reflected in the molecular mechanism underpinning ALS pathogenesis [87,88,89,90].
Interestingly, neuropathological examination of tissues from patients harboring FUS mutations showed increased cytoplasmic FUS staining, FUS-immunoreactive dystrophic neurites, and cytoplasmic inclusions in lower MNs [16,17]. These mislocalized immunoreactive FUS inclusions were strikingly non-reactive for TDP-43, suggesting that neurodegenerative processes driven by FUS are independent from TDP-43 mislocalization.
4. Molecular Mechanisms Leading to Protein Aggregation in ALS
Protein folding is the process by which a protein develops a well-defined three-dimensional structure, i.e., the tertiary structure, the result of a series of polypeptide chain folding [91]. The misfolding of a protein is the basis of the phenomenon of protein aggregation. This occurs mainly due to hydrophobic forces that cause two proteins, similar or different, to interact and form oligomers or amorphous fibrils.
FUS and TDP-43 contain domains enriched for asparagine, glutamine, tyrosine, and glycine residues. Each can adopt one of two conformational states: an explained or unfolded state, and an aggregated state. Prion proteins in an aggregated state can sequester prion proteins in an unfolded state to adopt aggregation-prone conformation, and aggregation thus spreads. It has been shown that the aggregation of SOD1, FUS, and TDP-43 is based in regions similar to prion domains [51,92,93,94]. Many of the mutant proteins in ALS participate in the formation of RNA granules, which is normally a reversible process but under pathological conditions it is assumed that the formation of RNA SGs results in insoluble aggregates. Studies have verified the colocalization of positive inclusions of TDP-43 by observing markers of these SGs in MNs of sALS patients [95,96]. ALS-associated mutant proteins alter the formation of RNA SGs, interfering with local RNA translation and making proteins more easily aggregated. Defects in both the assembly and disassembly of SGs have been associated with neurodegenerative disorders [97]. The size of ALS-associated proteins and their interactors within cytoplasmic aggregates may result in loss of function. Meanwhile, the knockdown of FUS or TDP-43 results in the loss of nuclear foci known as GEM [98,99]. The majority of ALS-causing mutations harbor the LCD regions of SG-related RBPs, resulting in abnormal LLPS abilities that impair SG homeostasis and lead to irreversible and toxic aggregates [100]. In addition, reduced numbers of GEM have been observed in the spinal cords and fibroblasts of patients with ALS [99,101]. These findings indicate the ability of protein aggregates to sequester RNA and the resultant effects on cells.
Protein degradation occurs through the processes of UPS and autophagy, which are crucial for the removal of ubiquitinating proteins and are significantly implicated in the presence of protein aggregates. As mentioned above, chaperone molecules are involved in the folding of proteins and preventing protein aggregation in response to stress under physiological conditions, and also help in protein degradation in the proteasome and during the process of autophagy. Chaperones are overregulated in ALS and are present in MS aggregates [102], leading to increased solubility and reduced toxicity of FUS and TDP-43 [103,104]. Accordingly, the knockdown of molecular chaperones increases the accumulation of TDP-43 C-terminal fragments and increases the toxicity of TDP-43 overexpression [105], while proteasome inhibition was found to increase levels of endogenous TDP-43 and associated toxicity [106]. The formation of intracellular aggregates may depend on the accumulation of misfolded and generated proteins, or could be a direct consequence of the mutation or of oxidative stress. In both cases, malfunctioning of the misfolded protein response system (UPR) appears to play a key role [102].
The transport of misfolded proteins towards particular compartments, favoring their destruction, is a critical aspect of preventing protein accumulation and aggregation [107]. The mechanism behind the sequestration of ALS-related proteins (FUS, TDP-43, and SOD1) prevents interactions that could result in cytotoxic oligomers and cell damage [107,108]. The three types of cellular compartment include juxtanuclear quality control (JUNQ), insoluble protein depots (IPOD), and RNA-specific interaction compartments/inclusions (RISCI) [107,109]. Due to their colocalization with ubiquitin, JUNQs are dynamic and their ubiquitination process occurs more rapidly [107,109]. In contrast, IPOD inclusions are not dynamic and involve a longer and slower ubiquitination process [107]. Published research has reported that the sequestration of mutSOD1 proteins in JUNQ may explain why this protein becomes toxic. Indeed, it appears that mutSOD1 protein can impair the dynamic nature of the JUNQ compartment [108,110]. Furthermore, several studies have demonstrated that SOD1 aggregations might affect the UPS system and damage the cellular signaling that is essential for homeostasis [111,112]. When co-expressed, it appears that TDP-43 aggregates, which were initially believed to be located in IPOD, are localized differently within SOD1-positive inclusions (JUNQ) [109].
Numerous research efforts continue to be focused on the toxic effects of protein aggregates on ALS patients. Explanations have been proposed for how ALS protein inclusions result in extreme cytotoxicity. It has been revealed that in cultured cell models these inclusions have the ability to sequester other proteins necessary for their function, such as those involved in the proper development of the cytoskeleton, chromatin architecture, and RNA metabolism [113,114,115]. In an intriguing study, Woerner et al. found that protein accumulation in the cytoplasm inhibits nucleocytoplasmic transport, including mRNA transport, and as a result blocks the annexes of the cellular pathways [116].
Furthermore, significant mitochondrial degradation and dysfunction have frequently been linked to TDP-43, SOD1, and FUS. The apparent cause of this is the blockage of mitochondrial transport pores and the subsequent accumulation in the intermembrane space, thereby activating the mitochondrial UPR [117,118,119]. The sequestration of additional molecules and cellular elements necessary for cellular homeostasis may be another mechanism through which these aggregates instigate neuronal death. Together these phenomena result in loss of function in essential proteins [120]. In conclusion, several studies have demonstrated that protein aggregation causes complete disruption of primary protein-degradation pathways, from the UPS system to changes in the autophagy process [121,122]. However, future studies are necessary to investigate how these processes are affected in ALS, as they may represent potential therapeutic targets.
5. TDP-43, SOD1 and FUS Aggregates in Other Neurodegenerative Diseases
These altered proteins typically associated with ALS have also been found in other neurodegenerative disorders. In this section of our study we briefly review their involvement in frontotemporal lobar degeneration (FTLD), AD, PD, and HD (Figure 3).
In addition to ALS, FUS and TDP-43 are also associated with FTLD [123,124]. The TDP-43 aggregates in FTD are characterized by compact neuronal cytoplasmic inclusions (NCIs) and short dystrophic neurites (DNs) with neuronal intranuclear inclusions (NIIs), which are preferentially localized in the upper neocortical layers. These characteristics are particularly associated with the behavioral variant of FTD (bvFTD). Other types of TDP-43 aggregates are linked to FTD without the presence of MN disease (MND), and to semantic frontotemporal dementia (SD). These data provide evidence for the critical role of TDP-43 in FTLD pathology [124,125]. A subset of FTLD with ubiquitinated inclusions has been identified in the presence of FUS protein accumulations. A study published in 2009 showed that FUS aggregates formed only in the affected areas of the cortex in FTLD brains. It has also been demonstrated that FUS plays a significant role in neuronal homeostasis, and that the multifunctional interaction between FUS and SFPQ is a harmful factor in FTLD diseases [126,127].
Accumulations of these proteins have also been found in PD and AD. PD affects the dopaminergic neurons in the substantia nigra, and mutant TDP-43 has been found in PD patients [128]. Cytoplasmic aggregates were found in the CNS of PD patients, in particular the spinal cord and bulbar nuclei. TDP-43 aggregates in PD are reportedly associated with induced dopaminergic neuronal loss in PD patients with altered Parkin expression. In some physiological conditions, Parkin over-expression can alleviate neuronal death induced by TDP-43 [129,130].
In recent years, TDP-43 accumulation has been reported in several AD cases [131,132,133]. Studies have shown that these cytoplasmic aggregates have significative negative effects in patients, including brain atrophy and memory loss [134,135], and these effects are due to direct interaction between TDP-43 and Aβ or tau [133,136]. Furthermore, it has been suggested that TDP-43 accumulation may aggravate AD pathology. Moreover, various studies of these two diseases have suggested a role for SOD1 protein in neuronal death in the substantia nigra of patients with AD or PD. As occurs in ALS, the protein aggregation of SOD1 along with the alteration of proteins typically involved in AD and PD causes increased damage following oxidative stress, in turn leading to neuronal death [137,138]. In PD, SOD1 aggregation is localized only in the regions with neuronal loss. A recent study reported clear evidence of the role of this protein in other degenerative neurological pathologies [137], while previous studies revealed that SOD1 promotes the formation in AD brains of protein aggregates associated with Aβ plaques and neurofibrillary tangles [139].
The final neurodegenerative disease briefly covered by this review is HD, a disorder caused by huntingtin (HTT) inclusions in the CNS (Table 1). TDP-43 accumulation has been found in this disease, but the question whether this protein co-localizes with HTT to form inclusions remains controversial [140]. Sampedro and colleagues recently demonstrated that alterations of TDP-43 in the plasma of HD patients were correlated with typical features of this pathology and played a role in the severity of typical HD symptoms [141]. A very recent study revealed that cells responded in different manners to various types of aggregate: the presence of HTT-polyQ aggregation induced a proteotoxic stress response, while aggregation of mutant FUS led to malfunctioning proteostasis in the HEK293T human cell line and primary neuronal cells [142]. In these contexts, the same authors further considered the possible role of molecular chaperones, which are important regulators of protein folding and pathological aggregation. They found that cells obtain protection from mutant FUS aggregation by a complex of full-length (FL) DNAJB14 and DNAJB12 interacting with HSP70 [142]. However, DNAJB12-FL exacerbated HTT-polyQ aggregation, suggesting differential roles for DNAJ isoforms in the regulation of different aggregated proteins [142].
6. Misfolding Proteins Typical of Other Neurodegenerative Diseases in ALS
In addition to recognized ALS hallmark proteins, recent research has uncovered novel proteins that appear to be involved in the disease. In the CNS cells of ALS patients, aggregations of additional proteins such as α-syn, tau, or Aβ have been identified, suggesting their involvement and complex interplay in the pathophysiology of ALS [143].
Recent research by Calvo et al. has shown that ALS exhibits characteristics resembling those of other pathologies, particularly synucleinopathies [144]. Several studies have reported the presence of α-syn aggregates in the spinal cords and glial cells of ALS patients, and these apparently play an important role in neuronal degeneration and related onset of symptoms typically associated with PD [145,146]. Additionally, evidence of interaction between a-syn aggregates and SOD1 has been found in numerous investigations, specifically in mutant transgenic mice hSOD1G93A and in the brain tissue of ALS patients [24,147,148].
Recent medical research has shown that tau may serve as a biomarker for ALS diagnosis. In fact, the study of cerebrospinal fluid (CSF) from ALS patients revealed significantly altered levels of tau and associated protein accumulations [149,150]. Furthermore, a fascinating study published in 2016 identified that neurotoxic tau fragments were detected in brain and spinal cord samples from sALS patients, but not in healthy individuals [151]. A polyclonal antibody that recognizes pThr 175 tau was used in the analysis of tau-immunoreactive inclusions in ALS patients, revealing that widespread alteration of tau is also associated with an increase in TDP-43 immunoreactivity [23].
The literature also provides evidence of a potential role for Aβ accumulation during the process of neuronal neurodegeneration occurring in ALS [152]. In fact, it has been discovered that Aβ accumulates in the anterior horn of the spinal cord at the MN level in patients with ALS. Furthermore, the aberrant accumulation of Aβ42 in spinal cord MNs of ALS patients is linked to oxidative stress-induced cytotoxicity and may contribute to neurodegeneration [153]. Clinical observations resulting from the analysis of Aβ levels in CSF suggested that the tau protein may be a potential biomarker for the diagnosis of ALS [154].
7. Conclusions
To date, it is not yet completely understood whether and how these protein aggregates cause cell death in ALS, although studies on TDP-43, SOD1 and FUS aggregates have been illuminating. There are a number of possible mechanisms for how protein aggregation in ALS might result in cell toxicity, and there is strong evidence linking many aggregating proteins, including SOD1, TDP-43, and FUS, to mitochondrial dysfunction and indeed complete mitochondrial degeneration [117,155]. Although the exact mechanism is yet to be elucidated, the dysfunction may be caused by the blockage of mitochondrial transport pores, accumulation inside the intermembrane space, and/or activation of the mitochondrial unfolded protein response. It is also recognized that protein aggregation interferes with the proteostasis network, affecting the key protein degradation pathways of autophagy and the UPS [121]. The sequestration of other vital cellular molecules into protein aggregates is likewise detrimental to cell health by inducing loss of function in essential proteins [120]. It has been shown to affect the expression of amyloidogenic sequesters of other proteins involved in crucial biochemical pathways such as proteostasis, cytoskeletal maintenance, chromatin organization, and RNA metabolism [114], some of which are disrupted in ALS [5,113]. Finally, evidence also suggests that protein aggregation in the cytoplasm prevents mRNA transport as well as other types of nucleocytoplasmic transport, compromising global RNA metabolism. Given the variety of downstream consequences that protein aggregation has on cellular health, it remains difficult to identify specific interactions that would be viable therapeutic targets, aside from the main aggregating proteins themselves [116].
The identification of biomarkers and therapies for early diagnosis and effective treatment of ALS will be aided in future by a better understanding of the precise role of protein aggregation and changes of proteostasis within the pathological mechanisms of the disease.
Author Contributions
E.D. and C.V. carried out the literature review, conceptualization, and writing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the University of Milano-Bicocca, grant number 2020-ATE-0024.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Figures were created with BioRender.com (accessed on 19 December 2022).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kruger, D. Amyotrophic lateral sclerosis. JAAPA 2012, 25, 53–54. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couratier, P.; Corcia, P.; Lautrette, G.; Nicol, M.; Preux, P.M.; Marin, B. Epidemiology of amyotrophic lateral sclerosis: A review of literature. Rev. Neurol. 2016, 172, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Shen, D.; Gao, Y.; Zhou, Q.; Ni, Y.; Meng, H.; Shi, H.; Le, W.; Chen, S. A perspective on therapies for amyotrophic lateral sclerosis: Can disease progression be curbed? Transl. Neurodegener. 2021, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.; Weishaupt, J.H. Update on amyotrophic lateral sclerosis genetics. Curr. Opin. Neurol. 2019, 32, 735–739. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 262. [Google Scholar] [CrossRef] [Green Version]
- Blokhuis, A.M.; Groen, E.J.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef]
- Ramesh, N.; Pandey, U.B. Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand. Front. Mol. Neurosci. 2017, 10, 263. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D.; Ohama, E.; Reaume, A.G.; Scott, R.W.; Cleveland, D.W. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Höhn, A.; Tramutola, A.; Cascella, R. Proteostasis Failure in Neurodegenerative Diseases: Focus on Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 5497046. [Google Scholar] [CrossRef] [Green Version]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Takalo, M.; Salminen, A.; Soininen, H.; Hiltunen, M.; Haapasalo, A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2013, 2, 1–14. [Google Scholar] [PubMed]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Strong, M.J. Widespread neuronal and glial hyperphosphorylated tau deposition in ALS with cognitive impairment. Amyotroph. Lateral Scler. 2012, 13, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Oguchi, K.; Koshihara, H.; Hineno, A.; Nakamura, A.; Ohara, S. α-Synuclein coaggregation in familial amyotrophic lateral sclerosis with SOD1 gene mutation. Hum. Pathol. 2013, 44, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [Green Version]
- Lansbury, P.T.; Lashuel, H.A. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 2006, 443, 774–779. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, M.; Ferri, A.; Carrì, M.T. Amyotrophic lateral sclerosis: From current developments in the laboratory to clinical implications. Antioxid. Redox Signal. 2008, 10, 405–443. [Google Scholar] [CrossRef]
- Guareschi, S.; Cova, E.; Cereda, C.; Ceroni, M.; Donetti, E.; Bosco, D.A.; Trotti, D.; Pasinelli, P. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. USA 2012, 109, 5074–5079. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.J.; Meyerowitz, J.; James, J.L.; Liddell, J.R.; Crouch, P.J.; Kanninen, K.M.; White, A.R. Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochem. Int. 2012, 60, 415–424. [Google Scholar] [CrossRef]
- Trist, B.G.; Hilton, J.B.; Hare, D.J.; Crouch, P.J.; Double, K.L. Superoxide Dismutase 1 in Health and Disease: How a Frontline Antioxidant Becomes Neurotoxic. Angew. Chem. Int. Ed. 2021, 60, 9215–9246. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef] [PubMed]
- King, O.D.; Gitler, A.D.; Shorter, J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012, 1462, 61–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asadi, M.R.; Sadat Moslehian, M.; Sabaie, H.; Jalaiei, A.; Ghafouri-Fard, S.; Taheri, M.; Rezazadeh, M. Stress Granules and Neurodegenerative Disorders: A Scoping Review. Front. Aging Neurosci. 2021, 13, 650740. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Attarwala, I.Y.; Xie, X.Q. SQSTM1/p62: A Potential Target for Neurodegenerative Disease. ACS Chem. Neurosci. 2019, 10, 2094–2114. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Buettner, G.R. Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anticancer Agents Med. Chem. 2011, 11, 341–346. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Pardo, C.A.; Xu, Z.; Borchelt, D.R.; Price, D.L.; Sisodia, S.S.; Cleveland, D.W. Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc. Natl. Acad. Sci. USA 1995, 92, 954–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, O.; Shatunov, A.; Jones, A.R.; Andersen, P.M.; Powell, J.F.; Al-Chalabi, A. Development of a Smartphone App for a Genetics Website: The Amyotrophic Lateral Sclerosis Online Genetics Database (ALSoD). JMIR Mhealth Uhealth 2013, 1, e18. [Google Scholar] [CrossRef] [PubMed]
- Berdyński, M.; Miszta, P.; Safranow, K.; Andersen, P.M.; Morita, M.; Filipek, S.; Żekanowski, C.; Kuźma-Kozakiewicz, M. SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci. Rep. 2022, 12, 103. [Google Scholar] [CrossRef]
- Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 2006, 6, 37–46. [Google Scholar] [CrossRef]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef]
- Bosco, D.A.; Morfini, G.; Karabacak, N.M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; Fontaine, B.A.; Lemay, N.; McKenna-Yasek, D.; et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010, 13, 1396–1403. [Google Scholar] [CrossRef] [Green Version]
- Grad, L.I.; Guest, W.C.; Yanai, A.; Pokrishevsky, E.; O’Neill, M.A.; Gibbs, E.; Semenchenko, V.; Yousefi, M.; Wishart, D.S.; Plotkin, S.S.; et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16398–16403. [Google Scholar] [CrossRef]
- Chia, R.; Tattum, M.H.; Jones, S.; Collinge, J.; Fisher, E.M.; Jackson, G.S. Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS ONE 2010, 5, e10627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radunović, A.; Delves, H.T.; Robberecht, W.; Tilkin, P.; Enayat, Z.E.; Shaw, C.E.; Stević, Z.; Apostolski, S.; Powell, J.F.; Leigh, P.N. Copper and zinc levels in familial amyotrophic lateral sclerosis patients with CuZnSOD gene mutations. Ann. Neurol. 1997, 42, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Ticozzi, N.; Tiloca, C.; Morelli, C.; Colombrita, C.; Poletti, B.; Doretti, A.; Maderna, L.; Messina, S.; Ratti, A.; Silani, V. Genetics of familial Amyotrophic lateral sclerosis. Arch. Ital. Biol. 2011, 149, 65–82. [Google Scholar] [CrossRef]
- Ou, S.H.; Wu, F.; Harrich, D.; García-Martínez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, I.; Arai, T.; Hasegawa, M. The basis of clinicopathological heterogeneity in TDP-43 proteinopathy. Acta Neuropathol. 2019, 138, 751–770. [Google Scholar] [CrossRef] [Green Version]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef]
- Andersen, P.M.; Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef]
- Khosravi, B.; Hartmann, H.; May, S.; Möhl, C.; Ederle, H.; Michaelsen, M.; Schludi, M.H.; Dormann, D.; Edbauer, D. Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum. Mol. Genet. 2017, 26, 790–800. [Google Scholar] [CrossRef] [Green Version]
- Law, W.J.; Cann, K.L.; Hicks, G.G. TLS, EWS and TAF15: A model for transcriptional integration of gene expression. Brief. Funct. Genom. 2006, 5, 8–14. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruijs da Silva, L.A.; Simonetti, F.; Hutten, S.; Riemenschneider, H.; Sternburg, E.L.; Pietrek, L.M.; Gebel, J.; Dötsch, V.; Edbauer, D.; Hummer, G.; et al. Disease-linked TDP-43 hyperphosphorylation suppresses TDP-43 condensation and aggregation. EMBO J. 2022, 41, e108443. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis—A model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef] [Green Version]
- Brettschneider, J.; Arai, K.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Lee, E.B.; Kuwabara, S.; Shibuya, K.; Irwin, D.J.; Fang, L.; et al. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 2014, 128, 423–437. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.; Rademakers, R. The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 2008, 21, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Vanden Broeck, L.; Callaerts, P.; Dermaut, B. TDP-43-mediated neurodegeneration: Towards a loss-of-function hypothesis? Trends Mol. Med. 2014, 20, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Ederle, H.; Dormann, D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017, 591, 1489–1507. [Google Scholar] [CrossRef] [Green Version]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ray, P.; Rao, E.J.; Shi, C.; Guo, W.; Chen, X.; Woodruff, E.A., 3rd; Fushimi, K.; Wu, J.Y. A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2010, 107, 3169–3174. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Xu, Y.F.; Dickey, C.A.; Buratti, E.; Baralle, F.; Bailey, R.; Pickering-Brown, S.; Dickson, D.; Petrucelli, L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J. Neurosci. 2007, 27, 10530–10534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crippa, V.; Cicardi, M.E.; Ramesh, N.; Seguin, S.J.; Ganassi, M.; Bigi, I.; Diacci, C.; Zelotti, E.; Baratashvili, M.; Gregory, J.M.; et al. The chaperone HSPB8 reduces the accumulation of truncated TDP-43 species in cells and protects against TDP-43-mediated toxicity. Hum. Mol. Genet. 2016, 25, 3908–3924. [Google Scholar] [CrossRef] [PubMed]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Ren, X.; Dunn, J.R.; Peters, A.; Miyagi, M.; Fujioka, H.; Zhao, F.; Askwith, C.; Liang, J.; et al. Translational regulation in the brain by TDP-43 phase separation. J. Cell Biol. 2021, 220, e202101019. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Lin, H.X.; Liu, G.L.; Tao, Q.Q.; Ni, W.; Xiao, B.G.; Wu, Z.Y. The investigation of genetic and clinical features in Chinese patients with juvenile amyotrophic lateral sclerosis. Clin. Genet. 2017, 92, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Lanson, N.A., Jr.; Pandey, U.B. FUS-related proteinopathies: Lessons from animal models. Brain Res. 2012, 1462, 44–60. [Google Scholar] [CrossRef]
- Zhao, M.; Kim, J.R.; van Bruggen, R.; Park, J. RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Mol. Cells 2018, 41, 818–829. [Google Scholar] [CrossRef]
- Andersson, M.K.; Ståhlberg, A.; Arvidsson, Y.; Olofsson, A.; Semb, H.; Stenman, G.; Nilsson, O.; Aman, P. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 2008, 9, 37. [Google Scholar] [CrossRef]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, S.; Cleveland, D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chook, Y.M.; Süel, K.E. Nuclear import by karyopherin-βs: Recognition and inhibition. Biochim. Biophys. Acta 2011, 1813, 1593–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanson, N.A., Jr.; Maltare, A.; King, H.; Smith, R.; Kim, J.H.; Taylor, J.P.; Lloyd, T.E.; Pandey, U.B. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet. 2011, 20, 2510–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 2018, 173, 706–719.e713. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Gui, X.; Zhou, H.; Gu, J.; Li, Y.; Liu, X.; Zhao, M.; Li, D.; Li, X.; Liu, C. Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation. Nat. Struct. Mol. Biol. 2018, 25, 341–346. [Google Scholar] [CrossRef]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; McKnight, S.L.; Tycko, R. Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell 2017, 171, 615–627.e616. [Google Scholar] [CrossRef] [Green Version]
- Yoshizawa, T.; Ali, R.; Jiou, J.; Fung, H.Y.J.; Burke, K.A.; Kim, S.J.; Lin, Y.; Peeples, W.B.; Saltzberg, D.; Soniat, M.; et al. Nuclear Import Receptor Inhibits Phase Separation of FUS through Binding to Multiple Sites. Cell 2018, 173, 693–705.e622. [Google Scholar] [CrossRef]
- Khan, P.; Parkash, A.; Islam, A.; Ahmad, F.; Hassan, M.I. Molecular basis of the structural stability of hemochromatosis factor E: A combined molecular dynamic simulation and GdmCl-induced denaturation study. Biopolymers 2016, 105, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, B.; Faure, A.J.; Seuma, M.; Schmiedel, J.M.; Tartaglia, G.G.; Lehner, B. The mutational landscape of a prion-like domain. Nat. Commun. 2019, 10, 4162. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udan-Johns, M.; Bengoechea, R.; Bell, S.; Shao, J.; Diamond, M.I.; True, H.L.; Weihl, C.C.; Baloh, R.H. Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Hum. Mol. Genet. 2014, 23, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentmann, E.; Neumann, M.; Tahirovic, S.; Rodde, R.; Dormann, D.; Haass, C. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2012, 287, 23079–23094. [Google Scholar] [CrossRef] [Green Version]
- Krause, L.J.; Herrera, M.G.; Winklhofer, K.F. The Role of Ubiquitin in Regulating Stress Granule Dynamics. Front. Physiol. 2022, 13, 910759. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef]
- Jiang, L.; Ngo, S.T. Altered TDP-43 Structure and Function: Key Insights into Aberrant RNA, Mitochondrial, and Cellular and Systemic Metabolism in Amyotrophic Lateral Sclerosis. Metabolites 2022, 12, 709. [Google Scholar] [CrossRef]
- Tsuiji, H.; Iguchi, Y.; Furuya, A.; Kataoka, A.; Hatsuta, H.; Atsuta, N.; Tanaka, F.; Hashizume, Y.; Akatsu, H.; Murayama, S.; et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol. Med. 2013, 5, 221–234. [Google Scholar] [CrossRef]
- Baradaran-Heravi, Y.; Van Broeckhoven, C.; van der Zee, J. Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol. Dis. 2020, 134, 104639. [Google Scholar] [CrossRef]
- Yamazaki, T.; Chen, S.; Yu, Y.; Yan, B.; Haertlein, T.C.; Carrasco, M.A.; Tapia, J.C.; Zhai, B.; Das, R.; Lalancette-Hebert, M.; et al. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2012, 2, 799–806. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.P.; Smith, E.F.; Shaw, P.J.; De Vos, K.J. Protein Homeostasis in Amyotrophic Lateral Sclerosis: Therapeutic Opportunities? Front. Mol. Neurosci. 2017, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Miguel, L.; Avequin, T.; Delarue, M.; Feuillette, S.; Frébourg, T.; Campion, D.; Lecourtois, M. Accumulation of insoluble forms of FUS protein correlates with toxicity in Drosophila. Neurobiol. Aging 2012, 33, e1001–e1015. [Google Scholar] [CrossRef]
- Crippa, V.; Galbiati, M.; Boncoraglio, A.; Rusmini, P.; Onesto, E.; Giorgetti, E.; Cristofani, R.; Zito, A.; Poletti, A. Motoneuronal and muscle-selective removal of ALS-related misfolded proteins. Biochem. Soc. Trans. 2013, 41, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, H.; Guo, Q.; Bader, J.; Frottin, F.; Farny, D.; Kleinberger, G.; Haass, C.; Mann, M.; Hartl, F.U.; Baumeister, W.; et al. Gel-like inclusions of C-terminal fragments of TDP-43 sequester stalled proteasomes in neurons. EMBO Rep. 2022, 23, e53890. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Coyne, A.N.; Pei, F.; Vaughan, S.; Chaung, M.; Zarnescu, D.C.; Buchan, J.R. Endocytosis regulates TDP-43 toxicity and turnover. Nat. Commun. 2017, 8, 2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Villanueva, J.F.; Díaz-Molina, R.; García-González, V. Protein Folding and Mechanisms of Proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef] [Green Version]
- Farrawell, N.E.; Lambert-Smith, I.A.; Warraich, S.T.; Blair, I.P.; Saunders, D.N.; Hatters, D.M.; Yerbury, J.J. Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci. Rep. 2015, 5, 13416. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, S.J.; Lyakhovetsky, R.; Werdiger, A.C.; Gitler, A.D.; Soen, Y.; Kaganovich, D. Compartmentalization of superoxide dismutase 1 (SOD1G93A) aggregates determines their toxicity. Proc. Natl. Acad. Sci. USA 2012, 109, 15811–15816. [Google Scholar] [CrossRef]
- Basso, M.; Massignan, T.; Samengo, G.; Cheroni, C.; De Biasi, S.; Salmona, M.; Bendotti, C.; Bonetto, V. Insoluble mutant SOD1 is partly oligoubiquitinated in amyotrophic lateral sclerosis mice. J. Biol. Chem. 2006, 281, 33325–33335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrawell, N.E.; Lambert-Smith, I.; Mitchell, K.; McKenna, J.; McAlary, L.; Ciryam, P.; Vine, K.L.; Saunders, D.N.; Yerbury, J.J. SOD1(A4V) aggregation alters ubiquitin homeostasis in a cell model of ALS. J. Cell Sci. 2018, 131, jcs209122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciryam, P.; Lambert-Smith, I.A.; Bean, D.M.; Freer, R.; Cid, F.; Tartaglia, G.G.; Saunders, D.N.; Wilson, M.R.; Oliver, S.G.; Morimoto, R.I.; et al. Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E3935–E3943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olzscha, H.; Schermann, S.M.; Woerner, A.C.; Pinkert, S.; Hecht, M.H.; Tartaglia, G.G.; Vendruscolo, M.; Hayer-Hartl, M.; Hartl, F.U.; Vabulas, R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 2011, 144, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, Z.L.; Brito, R.M.M. Structure and Aggregation Mechanisms in Amyloids. Molecules 2020, 25, 1195. [Google Scholar] [CrossRef] [Green Version]
- Woerner, A.C.; Frottin, F.; Hornburg, D.; Feng, L.R.; Meissner, F.; Patra, M.; Tatzelt, J.; Mann, M.; Winklhofer, K.F.; Hartl, F.U.; et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 2016, 351, 173–176. [Google Scholar] [CrossRef]
- Nakaya, T.; Maragkakis, M. Amyotrophic Lateral Sclerosis associated FUS mutation shortens mitochondria and induces neurotoxicity. Sci. Rep. 2018, 8, 15575. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [Green Version]
- Méndez-López, I.; Sancho-Bielsa, F.J.; Engel, T.; García, A.G.; Padín, J.F. Progressive Mitochondrial SOD1(G93A) Accumulation Causes Severe Structural, Metabolic and Functional Aberrations through OPA1 Down-Regulation in a Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 8194. [Google Scholar] [CrossRef]
- Yang, H.; Hu, H.Y. Sequestration of cellular interacting partners by protein aggregates: Implication in a loss-of-function pathology. FEBS J. 2016, 283, 3705–3717. [Google Scholar] [CrossRef]
- Medinas, D.B.; Valenzuela, V.; Hetz, C. Proteostasis disturbance in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017, 26, R91–R104. [Google Scholar] [CrossRef]
- Jones, C.L.; Tepe, J.J. Proteasome Activation to Combat Proteotoxicity. Molecules 2019, 24, 2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, D.G.; Neumann, M.; Kusaka, H.; Yokota, O.; Ishihara, K.; Terada, S.; Kuroda, S.; Mackenzie, I.R. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009, 118, 617–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.R.; Neumann, M. Subcortical TDP-43 pathology patterns validate cortical FTLD-TDP subtypes and demonstrate unique aspects of C9orf72 mutation cases. Acta Neuropathol. 2020, 139, 83–98. [Google Scholar] [CrossRef]
- Tan, R.H.; Shepherd, C.E.; Kril, J.J.; McCann, H.; McGeachie, A.; McGinley, C.; Affleck, A.; Halliday, G.M. Classification of FTLD-TDP cases into pathological subtypes using antibodies against phosphorylated and non-phosphorylated TDP43. Acta Neuropathol. Commun. 2013, 1, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishigaki, S.; Fujioka, Y.; Okada, Y.; Riku, Y.; Udagawa, T.; Honda, D.; Yokoi, S.; Endo, K.; Ikenaka, K.; Takagi, S.; et al. Altered Tau Isoform Ratio Caused by Loss of FUS and SFPQ Function Leads to FTLD-like Phenotypes. Cell Rep. 2017, 18, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, S.; Riku, Y.; Fujioka, Y.; Endo, K.; Iwade, N.; Kawai, K.; Ishibashi, M.; Yokoi, S.; Katsuno, M.; Watanabe, H.; et al. Aberrant interaction between FUS and SFPQ in neurons in a wide range of FTLD spectrum diseases. Brain 2020, 143, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Tiloca, C.; Goldwurm, S.; Calcagno, N.; Verde, F.; Peverelli, S.; Calini, D.; Zecchinelli, A.L.; Sangalli, D.; Ratti, A.; Pezzoli, G.; et al. TARDBP mutations in a cohort of Italian patients with Parkinson’s disease and atypical parkinsonisms. Front. Aging Neurosci. 2022, 14, 1020948. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Duan, Y.; Qin, C.; Li, J.C.; Duan, G.; Deng, X.; Ni, J.; Cao, X.; Xiang, K.; Tian, K.; et al. Distinct multilevel misregulations of Parkin and PINK1 revealed in cell and animal models of TDP-43 proteinopathy. Cell Death Dis. 2018, 9, 953. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, R.; Beck, G.; Yonenobu, Y.; Inoue, K.; Mitsutake, A.; Ishiura, H.; Hasegawa, M.; Murayama, S.; Mochizuki, H. TDP-43 Proteinopathy Presenting with Typical Symptoms of Parkinson’s Disease. Mov. Disord. 2022, 37, 1561–1563. [Google Scholar] [CrossRef]
- Robinson, J.L.; Lee, E.B.; Xie, S.X.; Rennert, L.; Suh, E.; Bredenberg, C.; Caswell, C.; Van Deerlin, V.M.; Yan, N.; Yousef, A.; et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018, 141, 2181–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.S.; Yu, L.; White, C.C.; Chibnik, L.B.; Chhatwal, J.P.; Sperling, R.A.; Bennett, D.A.; Schneider, J.A.; De Jager, P.L. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE ε4 haplotype status: A community-based cohort study. Lancet Neurol. 2018, 17, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.H.; Tu, L.H.; Chang, T.Y.; Ganesan, K.; Chang, W.W.; Chang, P.S.; Fang, Y.S.; Lin, Y.T.; Jin, L.W.; Chen, Y.R. TDP-43 interacts with amyloid-β, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease. Nat. Commun. 2020, 11, 5950. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Dickson, D.W.; Tosakulwong, N.; Weigand, S.D.; Murray, M.E.; Petrucelli, L.; Liesinger, A.M.; Senjem, M.L.; Spychalla, A.J.; Knopman, D.S.; et al. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer’s disease: A longitudinal retrospective study. Lancet Neurol. 2017, 16, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Matej, R.; Tesar, A.; Rusina, R. Alzheimer’s disease and other neurodegenerative dementias in comorbidity: A clinical and neuropathological overview. Clin. Biochem. 2019, 73, 26–31. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Cascio, F.L.; Sengupta, U.; Garcia, S.; Bhatt, N.; Ellsworth, A.; Heidelman, E.A.; Johnson, O.D.; Doskocil, S.; et al. TDP-43 and Tau Oligomers in Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiol. Dis. 2020, 146, 105130. [Google Scholar] [CrossRef]
- Trist, B.G.; Davies, K.M.; Cottam, V.; Genoud, S.; Ortega, R.; Roudeau, S.; Carmona, A.; De Silva, K.; Wasinger, V.; Lewis, S.J.G.; et al. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain. Acta Neuropathol. 2017, 134, 113–127. [Google Scholar] [CrossRef]
- Abe, H.; Nakanishi, H. Effect of pH on the aggregate formation of a non-amyloid component (1–13). J. Pept. Sci. 2003, 9, 177–186. [Google Scholar] [CrossRef]
- Choi, J.; Rees, H.D.; Weintraub, S.T.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative modifications and aggregation of Cu,Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J. Biol. Chem. 2005, 280, 11648–11655. [Google Scholar] [CrossRef] [Green Version]
- St-Amour, I.; Turgeon, A.; Goupil, C.; Planel, E.; Hébert, S.S. Co-occurrence of mixed proteinopathies in late-stage Huntington’s disease. Acta Neuropathol. 2018, 135, 249–265. [Google Scholar] [CrossRef]
- Sampedro, F.; Martínez-Horta, S.; Pérez-Pérez, J.; Pérez-González, R.; Horta-Barba, A.; Campolongo, A.; Izquierdo, C.; Aracil-Bolaños, I.; Rivas, E.; Puig-Davi, A.; et al. Plasma TDP-43 Reflects Cortical Neurodegeneration and Correlates with Neuropsychiatric Symptoms in Huntington’s Disease. Clin. Neuroradiol. 2022, 32, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Rozales, K.; Younis, A.; Saida, N.; Meller, A.; Goldman, H.; Kellerman, L.; Heinrich, R.; Berlin, S.; Shalgi, R. Differential roles for DNAJ isoforms in HTT-polyQ and FUS aggregation modulation revealed by chaperone screens. Nat. Commun. 2022, 13, 516. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Kayed, R. Amyloid β, Tau, and α-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases. Prog. Neurobiol. 2022, 214, 102270. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.; Chiò, A.; Pagani, M.; Cammarosano, S.; Dematteis, F.; Moglia, C.; Solero, L.; Manera, U.; Martone, T.; Brunetti, M.; et al. Parkinsonian traits in amyotrophic lateral sclerosis (ALS): A prospective population-based study. J. Neurol. 2019, 266, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.J.; Bird, T.D.; Leverenz, J.B. α-synuclein in motor neuron disease: An immunohistologic study. Acta Neuropathol. 2004, 107, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Mezey, E.; Dehejia, A.; Harta, G.; Papp, M.I.; Polymeropoulos, M.H.; Brownstein, M.J. Alpha synuclein in neurodegenerative disorders: Murderer or accomplice? Nat. Med. 1998, 4, 755–757. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.H.; Joo, K.M.; Kim, M.J.; Cha, C.I. Immunohistochemical study on the distribution of alpha-synuclein in the central nervous system of transgenic mice expressing a human Cu/Zn superoxide dismutase mutation. Neurosci. Lett. 2003, 342, 151–154. [Google Scholar] [CrossRef]
- Koch, Y.; Helferich, A.M.; Steinacker, P.; Oeckl, P.; Walther, P.; Weishaupt, J.H.; Danzer, K.M.; Otto, M. Aggregated α-Synuclein Increases SOD1 Oligomerization in a Mouse Model of Amyotrophic Lateral Sclerosis. Am. J. Pathol. 2016, 186, 2152–2161. [Google Scholar] [CrossRef] [Green Version]
- Grossman, M.; Elman, L.; McCluskey, L.; McMillan, C.T.; Boller, A.; Powers, J.; Rascovsky, K.; Hu, W.; Shaw, L.; Irwin, D.J.; et al. Phosphorylated tau as a candidate biomarker for amyotrophic lateral sclerosis. JAMA Neurol. 2014, 71, 442–448. [Google Scholar] [CrossRef] [Green Version]
- Agnello, L.; Colletti, T.; Lo Sasso, B.; Vidali, M.; Spataro, R.; Gambino, C.M.; Giglio, R.V.; Piccoli, T.; Bivona, G.; La Bella, V.; et al. Tau protein as a diagnostic and prognostic biomarker in amyotrophic lateral sclerosis. Eur. J. Neurol. 2021, 28, 1868–1875. [Google Scholar] [CrossRef]
- Vintilescu, C.R.; Afreen, S.; Rubino, A.E.; Ferreira, A. The Neurotoxic TAU(45-230) Fragment Accumulates in Upper and Lower Motor Neurons in Amyotrophic Lateral Sclerosis Subjects. Mol. Med. 2016, 22, 477–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, S.; Iwata, M. Immunoreactivity of β-amyloid precursor protein in amyotrophic lateral sclerosis. Acta Neuropathol. 1999, 97, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Calingasan, N.Y.; Chen, J.; Kiaei, M.; Beal, M.F. β-amyloid 42 accumulation in the lumbar spinal cord motor neurons of amyotrophic lateral sclerosis patients. Neurobiol. Dis. 2005, 19, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Colletti, T.; Agnello, L.; Spataro, R.; Guccione, L.; Notaro, A.; Lo Sasso, B.; Blandino, V.; Graziano, F.; Gambino, C.M.; Giglio, R.V.; et al. Prognostic Role of CSF β-amyloid 1-42/1-40 Ratio in Patients Affected by Amyotrophic Lateral Sclerosis. Brain Sci. 2021, 11, 302. [Google Scholar] [CrossRef] [PubMed]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef]
Figure 1.
The pathways involved in protein aggregation in neuronal cells, leading to neuronal death typically found in neurodegenerative diseases.
Figure 1.
The pathways involved in protein aggregation in neuronal cells, leading to neuronal death typically found in neurodegenerative diseases.

Figure 2.
Structures and functional domains of SOD1, TDP-43, and FUS proteins.

Figure 3.
The protein aggregates typically associated with ALS are linked to protein inclusions in other neurodegenerative diseases, also promoting neuronal death in these pathologies.
Figure 3.
The protein aggregates typically associated with ALS are linked to protein inclusions in other neurodegenerative diseases, also promoting neuronal death in these pathologies.


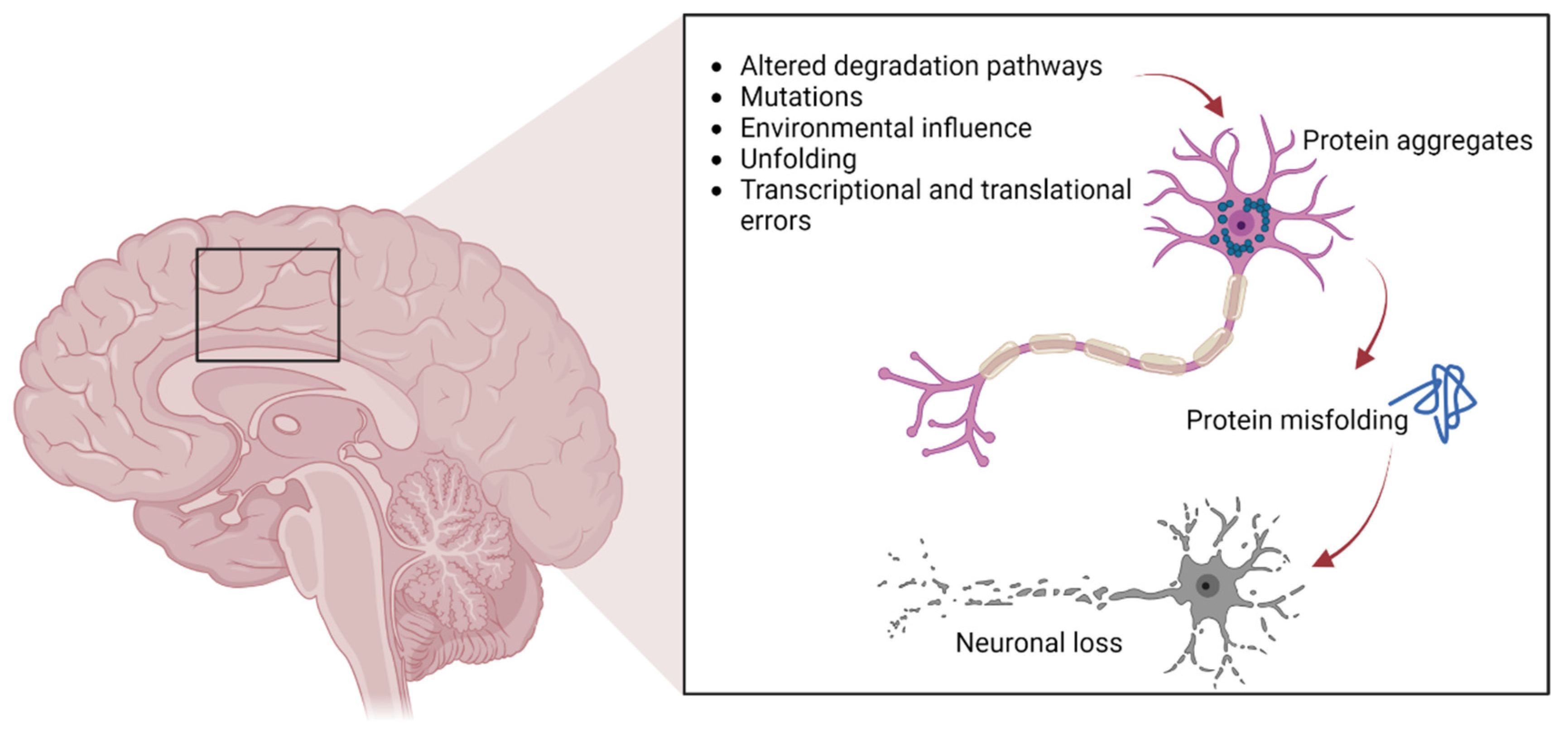{kind=link}
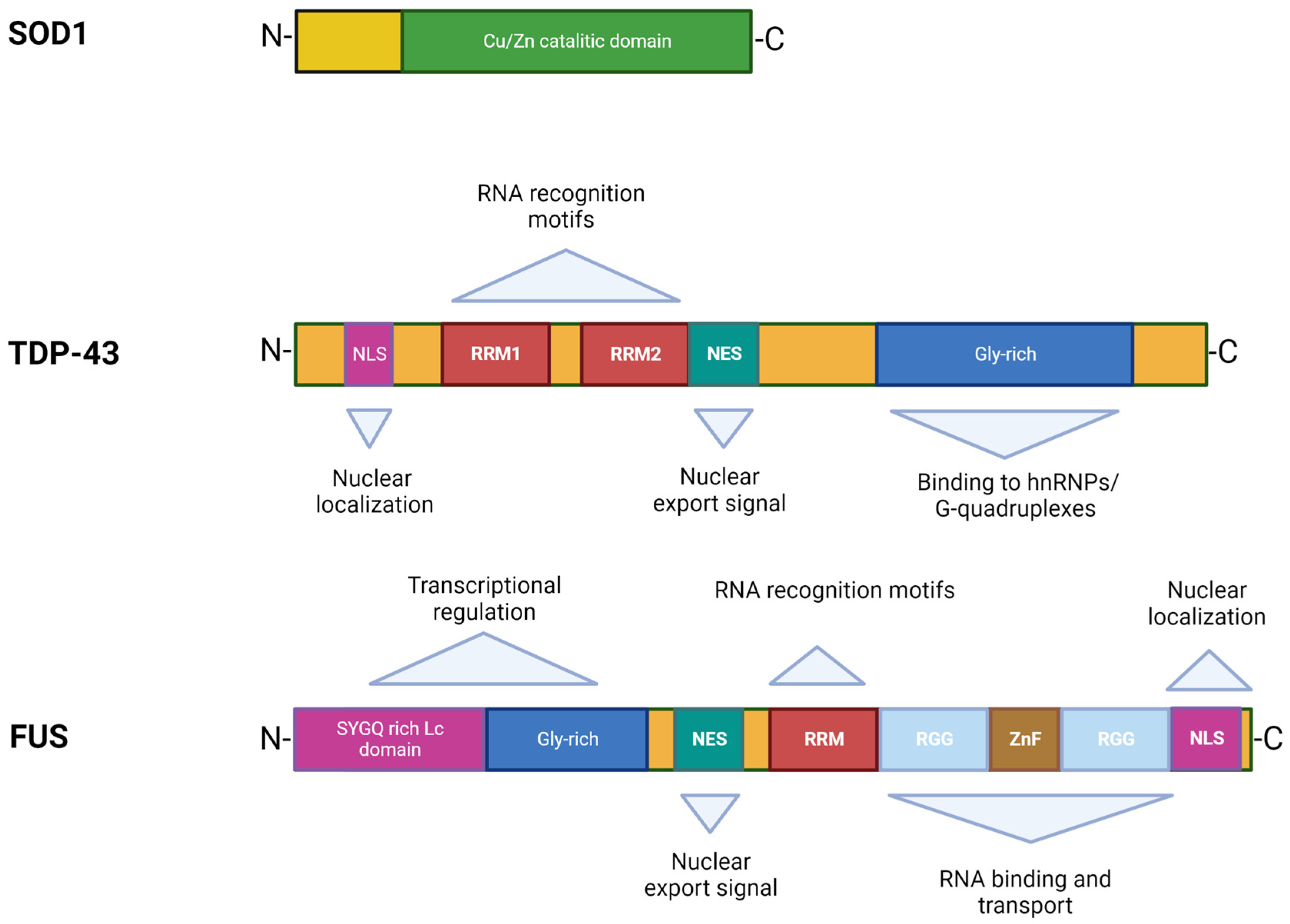{kind=link}
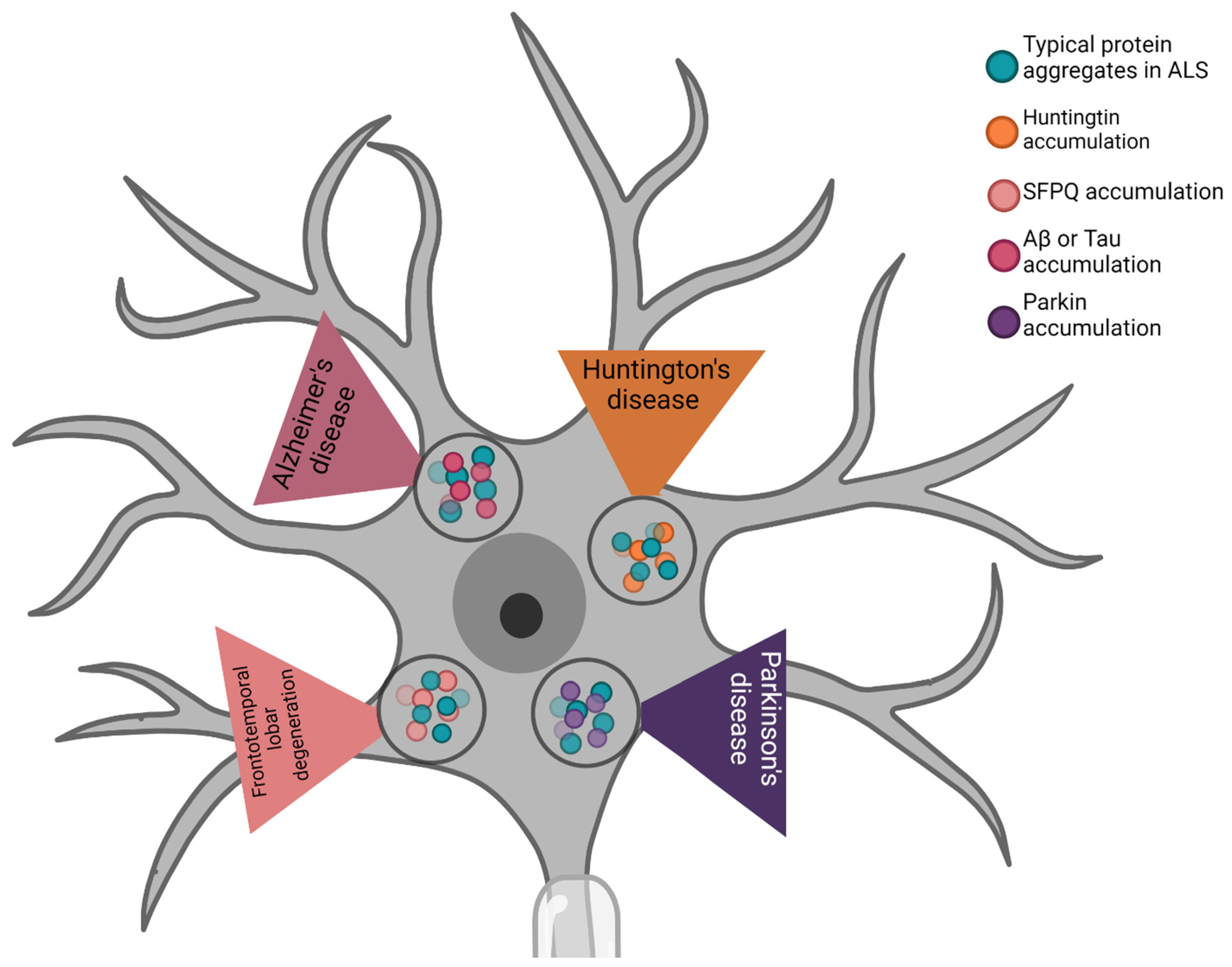{kind=link}
Table 1.
ALS proteins also found in other neurological diseases.
| Neurodegenerative Disease | ALS Proteins | Specific Protein |
|---|---|---|
| Alzheimer’s disease | TDP-43 | Aβ |
| SOD1 | Tau | |
| Frontotemporal lobar degeneration | FUS | SFPQ |
| TDP-43 | ||
| Huntington’s disease | FUS | HTT |
| TDP-43 | ||
| Parkinson’s disease | TDP-43 | Parkin |
| SOD1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Duranti, E.; Villa, C. Molecular Investigations of Protein Aggregation in the Pathogenesis of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2023, 24, 704. https://doi.org/10.3390/ijms24010704
AMA Style
Duranti E, Villa C. Molecular Investigations of Protein Aggregation in the Pathogenesis of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2023; 24(1):704. https://doi.org/10.3390/ijms24010704
Chicago/Turabian StyleDuranti, Elisa, and Chiara Villa. 2023. "Molecular Investigations of Protein Aggregation in the Pathogenesis of Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 24, no. 1: 704. https://doi.org/10.3390/ijms24010704
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.