

Advances in Human Mitochondria-Based Therapies


Center of Experimental Orthopaedics, Saarland University Medical Center, Saarland University, Kirrbergerstr. Bldg 37, 66421 Homburg, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(1), 608; https://doi.org/10.3390/ijms24010608
Submission received: 26 November 2022
/
Revised: 19 December 2022
/
Accepted: 23 December 2022
/
Published: 29 December 2022
(This article belongs to the Special Issue Mitochondrial Function in Health and Disease 2022)
Abstract
:Mitochondria are the key biological generators of eukaryotic cells, controlling the energy supply while providing many important biosynthetic intermediates. Mitochondria act as a dynamic, functionally and structurally interconnected network hub closely integrated with other cellular compartments via biomembrane systems, transmitting biological information by shuttling between cells and tissues. Defects and dysregulation of mitochondrial functions are critically involved in pathological mechanisms contributing to aging, cancer, inflammation, neurodegenerative diseases, and other severe human diseases. Mediating and rejuvenating the mitochondria may therefore be of significant benefit to prevent, reverse, and even treat such pathological conditions in patients. The goal of this review is to present the most advanced strategies using mitochondria to manage such disorders and to further explore innovative approaches in the field of human mitochondria-based therapies.
1. Introduction
The symbiotic state of mitochondria and eukaryotic cells traces back to a ‘‘survival of the fittest’’ event that occurred about 1.5 billion years ago, when a prokaryotic cell engulfed an alpha-probacteria and retained it as a functional component, namely an organelle [1]. Mitochondria inherit the structural and genetic basis of their bacterial ancestors, consisting of two separate, functionally distinct outer mitochondrial membranes (OMMs) and inner mitochondrial membranes (IMMs) that enclose intermembranous spaces (IMs) and a stromal compartment possessing a partially independent circular genome—the mitochondrial DNA (mtDNA)—reduced during evolution by gene transfer to the nucleus [2,3,4]. Over a long period of natural adaptation and evolution, newly acquired intracellular symbionts produced new specialized aerobic respiration that efficiently extracts energy from glucose through the electron transport chain (ETC) and oxidative phosphorylation (OXPHOS) to produce adenosine triphosphate (ATP), the main ‘‘currency’’ that provides energy metabolism for living organisms [5]. This overwhelms the original energy operating rules of eukaryotic cells, becoming the main energy pathway, and is supported by the bacteriophage-related mitochondrial maintenance systems, supported by the observation that the closest relatives of many mtDNA modifying enzymes (such as mtDNA polymerases) are bacteriophage proteins [4].
The long-term intracellular symbiont state facilitates in-depth communication and compromise between mitochondria and eukaryotic cells that allows the intracellular grinding into a dynamic, interconnected network tightly bound to other cellular compartments. Mitochondria, as the central node in the operation of this system, are not only powerpacks of the cells but also perform a smooth shuttle with the nucleus, making the mitochondria–nucleus information delivery crucial for the maintenance of the intracellular metabolism in physiological conditions that may be affected during pathological situations including aging, oxidation, inflammation, immunological diseases, metabolic syndromes, obesity, cancer, and degenerative disorders. The mitochondrial retrograde signaling is an important way to affect the decision-making of the nucleus accomplished by retrograde signal-mediated protein activation of nuclear gene expression or by altering its epigenetics through DNA methylation and post-translational modification of histones [6,7]. The nucleus responds to signals from the mitochondria to assist cellular metabolic crises under the various pathological conditions listed above. In addition to having a strong network with the nucleus, mitochondria are equally tightly connected with other organelles and the membrane contact sites between them are critical for lipid and ion exchange, membrane dynamics, and signal transduction [8]. For example, mitochondria and lysosomes cooperate with each other to complete autophagy [9]. Mitochondrial binding to the endoplasmic reticulum (ER) via mitochondria-associated ER membranes (MAMs) [10] controls the transport of calcium, a regulator of the overall mitochondrial membrane potential (∆Ψm) and an important second messenger that transmits external or cytoplasmic signals to the mitochondria, and is affected in neurodegenerative diseases and during aging [11,12].
Mitochondria play an important role in the normal functioning of cells, either as an energy supply station or as a metabolic information hub. Mitochondrial dysfunction emerged to be implicated in a growing number of major diseases common in humans, such as aging, oxidative disorders, inflammatory diseases, mitochondrial diseases, and cancer (Figure 1).
To address this fundamental problem, a number of therapeutic strategies have been developed to target the mitochondria in these human pathologies and also in the field of regenerative medicine as presented thereafter.
2. Strategies Targeting Mitochondria in Aging
Aging is by far the greatest risk factor for a variety of common human diseases including cardiovascular disorders, arthritis, coronary heart disease, and neurodegenerative diseases. Mitochondria have long been implicated in aging, with a time-dependent accumulation of mtDNA variants and an imbalance of reactive oxygen species (ROS) [13]. Most of these aging-related disorders are first associated with shattered energy-demanding organs such as the myocardium, brain, and muscle, where energy is supplied by mitochondria performing the OXPHOS pathway to conduct electrons in the ETC for a conversion of electrical potential energy into bioenergy that supports cellular activities [14]. In senescent cells, electron transport in the ETC is blocked and leaks into the mitochondrial matrix [15]. This undesired electron robs the mitochondrial IM of protons, depleting the ΔΨm, which is extremely important for electron transport in the ETC and reacts with oxygen to generate ROS [16] causing irreversible damage to DNA (mtDNA and nuclear DNA), proteins, and biological membranes [17].
Due to their role as multifaceted regulators of aging and cellular senescence (Figure 2), mitochondria have therefore been targeted to generate anti-aging treatments by balancing mitochondrial metabolism and mitophagy (the “quality control” mechanism of mitochondria breaking down damaged mitochondria and removing dysfunctional and undesirable mitochondrial components and by-products), via maintaining mitochondrial calcium (mitoCa2+) homeostasis, and modulating mitochondrial dynamics [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39] (Table 1).
Mitochondrial metabolic alterations from OXPHOS to glycolysis are frequently observed in senescent cells [40,41], whereas defending the efficiency of OXPHOS from being impeded was shown to be beneficial for the lifespan of living organisms [42], suggesting that active repair and reversal of mitochondrial metabolism may be valuable to tackle aging. Specifically, Din et al. [18] demonstrated that overexpressing regulators of mitochondrial biogenesis (cellular Myelocytomatosis—c-myc, peroxisome proliferator activated receptor gamma coactivator 1 alpha—PGC-1α, proviral insertion in murine 1—Pim-1) in a mouse model of cardiomyocyte aging was capable of reversing cardiac aging by maintaining mitochondrial function. Other gene regulation targets may include pumilio2 (PUM2), 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), activating transcription factor 3 (ATF3), sirtuin 3 (SIRT3), sirtuin 4 (SIRT4), and nuclear enriched abundant transcript 1 (NEAT1) [22,23,24,25,26,27,28]. In addition, administering biomodulators may provide effective, alternative therapeutic approaches capable of modulating mitochondrial functions in response to aging such as coenzyme Q (CoQ), an important component in ETC receiving electrons from complex I and complex III [43]. CoQ-deficient ETCs frequently appear in senescent cells and CoQ supplementation may optimize mitochondrial functions by normalizing ∆Ψm and enhancing ATP synthesis in senescent cells [44]. Mitoquinone (MitoQ) or O-(3-piperidino-2-hydroxy-1-propyl)nicotinic amidoxime (BGP-15) have been proven to modulate the ∆Ψm and ROS contents to reverse aging-associated meiotic spindle defects in mice and humans [45]. Weimer et al. [32] also reported that D-glucosamine (GlcN) is able to increase mitochondrial respiration by promoting the dependence of energy metabolism on OXPHOS while impairing glycolysis to contribute to an extension of the lifespan in many biological species (nematodes, aged mice).
Calcium overload in mitochondria is an important sign of aging [46]. The ER is an important source of mitoCa2+ through MAMs which achieve spatial and functional coupling of mitochondria with the ER via the inositol 1,4,5-trisphosphate receptor (IP3R)-glucose-regulated protein 75 (Grp75)-voltage-dependent anion channel (VDAC) [47]. In senescent cells, IP3R and VDAC are overactivated, resulting in unconstrained calcium flux into mitochondria from the ER [48,49] where calcium concentrations are 5- to 10-fold higher than in the mitochondria [50]. The resulting calcium overload further triggers abnormal ∆Ψm and elevated ROS [51], thus accelerating the aging process in a “vicious circle”. Limiting calcium overload may therefore be another important approach to suppress aging. For instance, Wiel et al. [52] showed that inhibiting the IM mitoCa2+ uniporter holocomplex (MCU) membrane protein (the direct channel of calcium in the mitochondria) prevented a persistent accumulation of mitoCa2+ and that knocking down IP3R2 (the inositol 1,4,5-trisphosphate receptor type 2—ITPR2—and main channel of calcium exchange on MAMs) can reduce ROS levels and inhibit senescence. Other significant factors to counterbalance aging by inhibiting mitoCa2+ include the tumor suppressor candidate 2 (Tusc2/Fus1), ATF3, CDGSH iron sulfur domain 2 (Cisd2), isradipine, verapamil, and α-Klotho [24,53,54,55,56,57].
As dynamic cell organelles, the mitochondria perform a continuous cycle of fission and fusion referred to as mitochondrial dynamics. This process ensures proper mitochondrial functions in response to nutrient demands, signal transduction, and external stress [58]. Abnormal mitochondrial dynamics is compromised in aging and related disorders, as evidenced by the occurrence of an aberrant (giant) mitochondrial morphology [59] and maintaining or restoring the normal mitochondrial dynamics may allow researchers to tackle the aging process in affected cells. For instance, up-regulating the cytosolic dynamin-related protein 1 (DRP1—a central protein that coordinates mitochondrial fission by mediating membrane contraction and fission) [60] can prolong the lifespan of Drosophila by facilitating mitophagy and improving mitochondrial respiratory functions [38,61]. Other proteins that may be targeted include mitochondrial fission factor (MFF), fission protein-1 (FIS1), mitochondrial Rho 1 (MIRO1), mitochondrial dynamics protein-49 (MiD49), and mitochondrial dynamics protein-51 (MiD51), all involved in the mitochondrial dynamic networks and deviate from physiological expression during aging, by normalizing them in cells in order to restore natural mitochondrial functions, morphology, and lifespan in living organisms [62].
3. Strategies Targeting Mitochondria in Oxidative Disorders
Oxidative stress recapitulates a state of unbalanced antioxidant effects due to excessive ROS levels leading to oxidative damage in the body that affects various intracellular biomolecules such as DNA, proteins, and lipids in the course of aging and other human disorders [63]. As a major ROS producer, the mitochondria that play crucial roles in the cellular emergency responses (oxidative stress, physical stimulation, calcium overload) are the first systems attacked [64], resulting in loss of membrane elasticity, disruption of ∆Ψm, in mtDNA mutations, and reduced ATP synthesis efficiency leading to mitochondrial dysfunction [65]. Mitochondria-targeted antioxidant therapy to reduce excessive ROS levels may therefore be an effective means to diminish or even prevent oxidative damage in affected cells and organs. Such a strategy by ROS scavenging and by the pharmacological manipulation of mitochondrial biogenesis is based on the use of antioxidant moieties, of gene therapy, and of traditional Chinese medicine (TCM) monomers with antioxidant potency.
Since mitochondrial oxidative damage involves lipid peroxidation and as the binding of alkyl polypropylenes to the IMMs strongly exacerbates its damage [66], initial research first focused on the use of antioxidants that are effective against lipid peroxidation. Ubiquinone [67], tocopherol [68], lipoic acid [69], the peroxidase mimetic ebselen (MitoPrx) [70], and their derivatives have been extensively reported for their effectiveness against mitochondrial oxidative damage. MitoQ that is reduced by the complex II to an active ubiquinone antioxidant in the respiratory chain has been also particularly validated by driving the ∆Ψm and lipid peroxidation in numerous clinical trials [71]. In addition, the superoxide dismutase mimetic M40403 (MitoSOD) (resist O2•−), MitoPrx (resist H2O2), TEMPOL (MitoTEMPOL) (resist OH•), vitamin E (MitoE) (resist OH•), and lipoic acid (MitoLip) (resist O2•−, H2O2 and OH•) are additional antioxidants recognized for their ROS scavenging effects [72]. To increase mitochondrial targeting, lipophilic cations such as triphenylphosphonium (TPP) that can bind to antioxidants and allow their passage through the mitochondrial membrane [73,74,75], were introduced in a follow-up attempt, greatly (100-fold) enhancing mitochondrial antioxidant uptake [76].
Regarding potential gene therapy interventions, several antioxidant genes were described to regulate the mitochondrial metabolism and to block oxidative stress by regulating OXPHOS metabolites. Among them, the mitochondrial signal transducer and activator of transcription 3 (STAT3) that manipulates the complex Ⅰdehydrogenase activity through a retrograde nicotinamide adenine dinucleotide (NAD+) signal may improve cellular antioxidant activities [77]. In addition to targeting OXPHOS metabolites, other gene regulation strategies may further suppress oxidative stress. For instance, the use of the hypoxia-inducible factor alpha (HIF-1α), a master regulator of tissue responses to oxidative pathological stimuli, may modulate damaging ROS levels that impair cardiac function in myocardial fibrosis after myocardial infarction [78]. Suppressive effects of HIF-α on mitochondrial oxidative stress were also documented for the treatment of liver fibrosis [79], obesity [80,81], and insulin resistance [82]. In addition, overexpression of the neurofibromatosis-1 (NF1) was shown to advantageously increase resistance to oxidative and mitochondrial respiration while reducing the levels of ROS production by 60% in Drosophila melanogaster through adenylyl cyclase (AC)/cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) signaling [83].
The application of TCM has been performed for more than 2000 years and in recent years, various TCM monomers have been reported in pharmacological research as effective antioxidants to treat a number of human diseases. The herbal salvia miltiorrhiza has been widely used for cardiovascular diseases as it can balance the production of ROS in cardiomyocytes [84,85] and reduce the oxidative damage of cardiac ischemia-reperfusion [86,87]. The anti-inflammatory and anti-tumoral Atractylodes lactone extracted from the rhizome of Atractylodes macrocephala Koidz can counteract the oxidative stress associated with chronic kidney disease, reducing muscle wasting via inhibition of the phosphoinositide 3-kinase (PI3K)/protein kinase B (PKB or AKT)/mammalian target of rapamycin (mTOR) pathway [88]. Other beneficial TCM compounds may include curcumin, an inhibitor of oxidative stress and mitochondrial dysfunction in astrocytes [89], cuscuta pedicellata extract that improves oxidative stress caused by high-quality diet through gene protection [90], and artemisinin protecting glutamate-mediated neuronal oxidative apoptosis by manipulating the ∆Ψm and reducing ROS levels [91].
4. Strategies Targeting Mitochondria in Inflammatory Diseases
Growing evidence supports the contribution of mitochondrial dysregulation to an inflammatory phenotype in numerous diseases such as rheumatoid arthritis [92], multiple sclerosis [93], thyroiditis [94], and type 1 diabetes [95]. With aging or upon tissue damage, the mitochondria release undesired tricarboxylic acid metabolites and damaged mitochondrial components (mtDNA, cardiolipin, N-formyl peptides) recognized as damage-associated molecular patterns (DAMPs) that act as danger signals to trigger the immune system via pattern recognition receptors (PRRs) [96]. For instance, circulating mtDNA gradually increases after 50 years of age [97] correlating with an enhanced production of pro-inflammatory cytokines such as in cultured monocytes [97] and in elderly individuals [97]. Succinate, a tricarboxylic acid metabolite, induces HIF-1-mediated interleukin 1 beta (IL-1β) production [98] and its accumulation promotes reverse electron transport from complex Ⅱ to complex Ⅰ, a pattern resulting in dramatic elevation of ROS levels [99]. Other mitochondrial metabolites with immunostimulatory effects include citrate, acetate, acetyl-CoA, and itaconate contributing to inflammation and immune responses [100]. When stimulated by pathogen-associated molecular patterns (PAMPs) and DAMPs, MAMs are also involved in sensitive molecular signals by providing sites for the activation of the inflammasome as large protein complexes controlling the activation of the proteolytic caspase 1 enzyme. In turn, this regulates the maturation of IL-1β and IL-18 and cell death (pyroptosis via the formation of gasdermin D-mediated lytic pores in the plasma membrane), for instance the nucleotide oligomerization domain (NOD)-like receptor protein 3 (NLRP3) inflammasome [101]. Owing to the importance of mitochondria in the development of inflammation, several mitochondria-targeted strategies have been explored as anti-inflammatory therapeutic options focusing on mitophagy, ROS control, and on repressing the inflammasome.
The removal of dysfunctional and undesirable mitochondrial components such as mtDNA and ROS by mitophagy contributes to inflammation as DAMPs that are sensed by inflammatory signals [96]. Interestingly, there is evidence that mitophagy combined with the mitochondrial unfolded protein response (UPRmt) can significantly mitigate LPS-mediated inflammatory myocardial injury [102] and that the nuclear factor kappa B (NF-κB) and downstream p62 that recognize damaged mitochondria may activate mitophagy, contributing to homeostasis and tissue repair [103]. The protection of mitophagy against chronic inflammation has also been demonstrated in other disorders including colitis [104], diabetes [105], and Alzheimer’s disease [106].
Another promising therapeutic strategy for inflammation is to control the production of ROS, such as using the various approaches and compounds cited (MitoQ, BGP-15, MitoSOD, MitoPrx, MitoTEMPOL, MitoE, MitoLip, HIF-1α, NF1, TCM, etc.) [45,52,72,78,83,84,85,86,87,91,107].
Restricting the inflammasome via mitochondrial regulation may further be beneficial to control inflammatory pathological phenotypes [108] such as during pathogen infection (human immunodeficiency virus—HIV) [109], sepsis [110], colitis [111], particulate matter (PM) 2.5-mediated pulmonary pyroptosis [112], and more recently during the COVID-19 pandemic with its associated severe acute respiratory syndrome (SARS) due to the SARS coronavirus (SARS-CoV) caused by an excessive inflammatory storm led by NLRP3 [113] such as using the MitoTEMPOL antioxidant that can regulate formation of the NLRP3 inflammasome and reduce SARS-CoV viroporin 3a protein-induced IL-1β secretion for instance [114].
5. Strategies Targeting Mitochondria in Mitochondrial Diseases
Mitochondrial diseases represent a group of maternally inherited metabolic disorders caused by mutations in mtDNA that lead to mitochondrial dysfunction, mostly affecting OXPHOS and the levels of ATP synthesis [115] together with a loss of enzymatic intermediates, the accumulation of toxic metabolites, and the disruption of the Krebs and folate cycles [116]. Current therapeutic strategies for mitochondrial diseases are divided into small molecule therapy and mtDNA gene editing.
Supplementation with small molecules is currently the most commonly employed treatment modality for mitochondrial diseases as a means to remove toxic compounds, replenish defective enzymes or their analogs, or balance cofactors since toxic metabolites often trigger the clinical phenotype of mitochondrial diseases. For instance, accumulation of hydrogen sulfide in patients with ethylmalonic encephalopathy (a disease caused by mutations in the ethylmalonic encephalopathy protein 1 (ETHE1) gene coding for a sulfur dioxygenase) may be reduced by the combined application of N-acetylcysteine and metronidazole [117,118,119]. In addition, erythrocyte encapsulation of thymidine kinase phosphorylase was reported to improve mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) syndrome in patients, a mitochondrial disease caused by thymidine phosphorylasethymine phosphorylase (TYMP) mutations leading to thymidine phosphorylase deficiency [120,121,122]. A similar approach was applied to primary CoQ deficiency disorders such as encephalonephropathies with nephrotic syndrome, childhood-onset mitochondrial diseases, and isolated cerebellar ataxia using CoQ supplementation [123,124]. Complementary therapy of enzyme cofactors (non-protein small molecules, metal hydrate ions) has also been successfully used such as by supplementation of vitamin B12, a cofactor for flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), to treat mitochondrial diseases associated with FMN and FAD deficiencies (complex Ⅰ deficiency due to acyl-CoA dehydrogenase protein 9—ACAD9—mutations, severe X-linked mitochondrial encephalomyopathy due to mutations in the apoptosis-inducing factor mitochondrion-associated 1—AIFM1—precursor with NAD-dependent NADH oxidase activity) [125,126,127,128].
The mtDNA gene editing technology may also offer powerful options to treat mitochondrial diseases [129] based on antigenomic mtDNA therapy or on the use of restriction endonucleases [130,131,132], zinc-finger nucleases (ZFNs) [133,134], transcription activator-like effectors nucleases (TALENs) [135,136,137], and the clustered regularly interspaced short palindromic repeats/CRISPR-associated 9 (CRISPR/Cas9) [138,139,140] to correct mtDNA variants [141,142,143,144,145], especially with the emergence of double-stranded DNA deaminase (DddA)-derived cytosine base editors (DdCBEs) that can support the editing of C•G to T•A in mutated mtDNA sequences [142,143,144,145]. Still, single base editing is challenging due to genome complexity and probably insufficient to fully reverse a diseased phenotype while it may lead to off-target modifications [146], all impacting the clinical translation potential of mtDNA gene editing.
6. Strategies Targeting Mitochondria in Cancer
Mitochondrial damage in tumor cells is associated with an abnormally weakened OXPHOS, supported by the observation that these cells perform the glycolytic pathway to catabolize glucose in lactate to generate ATP even in the presence of sufficient oxygen (Warburg effect) (Figure 3) [147,148] while maintaining high ROS levels [149] that activate molecular signaling pathways promoting tumor cell proliferation [63].
Regulating mitochondrial energy metabolism may therefore be a powerful strategy for tumor therapy and several approaches have been attempted based on the use of specific pathway inhibitors or on the targeting of mitochondrial material metabolism and of mitophagy, including in conjunction with nanodelivery technologies to improve efficacy and reduce adverse reactions [150].
Different inhibitors of critical mitochondrial energy metabolic processes have been applied such as KUNB31 and geldanamycin, two inhibitors of the heat shock protein 90 (HSP90) that block ATP binding and hydrolysis in non-small cell lung cancer, multiple myeloma, ovarian cancer, melanoma, and renal cell carcinoma [151,152] or metformin, an ETC blocker of the complex I that can reduce the production of ATP in breast cancer, non-small cell lung cancer, renal cell carcinoma, melanoma, and colon cancer [153,154].
In addition, as tumor cells need a sufficient material on a regular basis to maintain their rapid proliferation and metabolism, targeting mitochondrial material metabolism presents another attractive treatment modality against cancer. Tricarboxylic acid (TCA) cycle intermediates provide raw materials for the biosynthesis of macromolecules [155] and glutamine is the major carbon source that replenishes TCA cycle intermediates while maintaining their use in biosynthesis in many tumor cells. Glutamine is converted to glutamate by the glutaminase (GLS) for further catabolism to produce beneficial α-ketoglutarate for the cellular material cycle [156]. Blocking this metabolic pathway is therefore an attractive approach in cancer therapy and indeed, multiple specific GLS inhibitors have been developed including the compound 968 and bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES), reducing glutamine catabolism and modulating the growth of glutamine-dependent tumors [157,158].
Mitophagy is also involved in the rapid metabolism of tumor cells as another part of the mitochondrial “quality control” process by generating TCA cycle intermediates for tumor cell metabolism [159]. Interestingly, knocking-out the essential autophagy related 7 (ATG7) gene for mitophagy allows increased tolerance to starvation and prolonged lifespan of mice with lung cancer [160]. Chloroquine, another well-known inhibitor of autophagy, and its derivative hydroxychloroquine have also been reported for their excellent tumor suppressive effects [161,162,163]. In addition, given that tumor cells are in a delicate oxidative balance adapted to different ROS levels than normal cells, disrupting this balance with high selectivity has been shown as a potential therapeutic approach for targeting tumors [74,164,165].
7. Strategies Targeting Mitochondria for Regenerative Medicine
The repair of damaged tissues is generally supported by the differentiation of progenitor (stem) cells, a critical process associated with pivotal biological activities (protein synthesis, genome replication, carbohydrate preparation) that necessitate large amounts of energy from the mitochondria. However, the contribution of mitochondria to stem cell differentiation is not limited to providing energy but also probably associated with mitochondrial biogenesis and dynamics [166,167]. This is in light of evidence showing that mitochondria are present at higher mass in differentiating cells, with a more elongated morphology, larger surface area, and lower membrane potential compared with their situation in pre-differentiated cells [168,169,170]. Regulating mitochondrial biological behavior may therefore be of strong value to promote stem cell differentiation for tissue repair such as by regulating mitochondrial dynamics (fission, fusion), mitochondrial respiration, and mitoCa2+ uptake.
Changes in mitochondrial morphology controlled by mitochondrial dynamics are closely related to the modulation of mitochondrial functions. Owing to the central role of the mitochondria in cellular processes, mitochondrial dynamics is extremely important in the cell cycle at the level of cell proliferation, differentiation, and aging and regulating mitochondrial fission. Fusion is thus an important way to influence the fate of stem cells [171]. Several studies, for instance, reported the significant impact of the dynamin-related protein 1 (Drp1), a molecule directing mitochondrial fission, on the differentiation of eukaryotic cells both in vitro and in vivo. Specifically, ablation of Drp1 in the mouse brain was shown to cause cerebellar hypoplasia in association with the presence of few giant mitochondria in Purkinje cells (instead of normally many short tubular mitochondria) [171] while neural cell-specific Drp1(−/−) mice were reported to develop brain hypoplasia with reduced neurite numbers and abnormal synapse formation, dying shortly after birth [172]. Accordingly, treatment with Drp1 inhibitors such as the mitochondrial division inhibitor 1 (mdivi-1, an inhibitor of the guanosine triphosphate hydrolase (GTPase) activity of Drp1) that prevents myotube formation and impairs the myogenic differentiation of myoblasts [173] or with the mitochondrial fission 1 protein (FIS1) involved in myeloid differentiation [174] may provide strong tools to address these critical problems. On the other side, there is evidence that mitofusin 2 (Mfn2), a receptor located in OMMs and contributing to mitochondrial fusion, plays a key role in mammalian stem cell differentiation and that overexpression of Mfn2 in human-induced pluripotent stem cells (hIPSCs) promotes their differentiation and maturation in neurons [175].
Mitochondrial respiration, in particular the intermediate ROS product, is also important for stem cell differentiation. Interestingly, while ROS is considered detrimental at unrestricted levels, closely regulated ROS levels may have a beneficial impact on cellular functions such as cell proliferation and differentiation [176]. For instance, appropriate ROS levels are capable of enhancing the adipogenic differentiation of mesenchymal stem cells (MSCs) by controlling specific signaling cascades (c-jun NH2-terminal kinase—JNK, p38 mitogen-activated protein kinase—p38 MAPK, extracellular regulated kinase—ERK, phosphatidylinositol 3-kinase/protein kinase B—PI3K/Akt—pathways) in these cells [176]. In contrast, inhibition of mitochondrial respiration via hypoxia and mitochondrial ETC inhibitors was shown to significantly suppress the adipogenic differentiation of MSCs [177]. Other studies demonstrated that the overexpression of superoxide dismutase 2 (SOD2), a mitochondrial antioxidant metalloenzyme, can significantly improve bone differentiation and bone formation in mice with osteogenic differentiation defects by assisting SIRT3 and regulating the mitochondrial oxidative transition and respiratory activities [178]. Finally, as an important intermediate of OXPHOS, providing the main carbon source for macromolecular synthesis and displaying antioxidant properties, glutathione was reported to increase the differentiation capacity of MSCs for bone regeneration [179,180].
Apart from its effects on mitochondrial metabolism, an imbalance in mitoCa2+ may also affect the regenerative capacity of cells and tissues [49,181,182]. This concept is supported by the observation that increasing mitoCa2+ via inhibition of the mitochondrial calcium uptake 1 (MICU1, the “gatekeeper” of mitoCa2+ uptake in the MCU) attenuates the regenerative capacity of liver cells [183]. In contrast, overexpression of MICU1 in a Streptococcus pneumoniae-mediated lung injury model can significantly reduce mitoCa2+ uptake and induce AT2 cells to alveolar cell differentiation as a beneficial process to promote alveolar repair [184], while MICU1 can increase α-ketoglutarate by reducing mitoCa2+ uptake and influence myofibroblast differentiation [185].
8. Conclusions and Perspectives
Mitochondria are complex organelles that control multiple molecular signals and cellular activities via the formation of interactive networks with other organelles and the nucleus to coordinate cellular behavior and to defend cells against external stress factors. Mitochondria play a central role in this signaling network that can receive instructions from the nucleus to focus on cellular tasks such as ATP synthesis. In addition, they can provide feedback information to the nucleus through retrograde signals to initiate a balance mechanism. Mitochondria also cooperate with the ER and lysosomes to complete various cellular functions such as calcium ion conduction, biomembrane flow, and autophagy.
However, when this balance is altered, the functions of the mitochondria become compromised such as during human aging, oxidative disorders, inflammatory and mitochondrial diseases, cancer, and degenerative pathologies as reviewed herein, and the resulting mitochondrial dysfunction will turn this fine signaling network into a vicious circle. Therefore, targeting the mitochondria as candidates for therapy may be a potent strategy to control and manage such human disorders.
As further reported herein, the feasibility of such a therapeutic paradigm has been explored in human medicine and due to an increased knowledge of mitochondria, a number of mitochondria-based therapeutic approaches have been developed using drug therapy, gene regulation, gene therapy, and genetic engineering procedures to tackle mitochondrial dysfunction and manage human mitochondria-related diseases. However, a number of challenges and issues still remain to be critically addressed prior to clinical translation. While the use of cost-effective drugs and compounds may be readily applicable in a clinical situation considering the high numbers of patients affected by these various pathologies, the effects may not be sufficiently robust for an effective, long-lasting treatment without the manifestation of undesirable adverse effects in generally irreversible human pathologies [186,187,188]. On the other hand, strategies that aim at durably or even permanently modulating genetic phenotypes involved in mitochondrial dysfunction are more challenging and invasive than the use of recombinant agents since they are based on the arduous production of complex, mitochondria-specific gene transfer vectors in a consistent, scalable, and globally affordable manner, requiring considerable amounts of research (academic and industrial) funding and being constrained by regulatory agencies [189,190,191,192,193] especially when involving genome editing procedures [129,193]. Alternatively, based on the observation that mitochondrial transfer naturally occurs between cells through tunneling nanotubes (TNTs) and extracellular vesicles (EVs) [194,195,196,197], somatic mitochondrial transfer, and to a broader extent mitochondrial transplantation, emerged as possible exogenous replacement therapies to compensate for the functional deficit of damaged mitochondria in somatic human adult cells such as in disorders of the brain (Alzheimer’s and Parkinson’s diseases, cerebral ischemia), heart (myocardial infarction, cardiomyopathies), lungs (acute distress syndrome, acute lung injury), liver, kidneys (diabetic nephropathy), and musculoskeletal system (osteoarthritis, skeletal muscle atrophy, tendinopathies, spinal cord injury) [196,197]. Yet, such options are still in their infancy, facing a number of critical hurdles and challenges that need to be carefully addressed for the purpose of human disease control, including (1) ethical concerns, (2) the maintenance of mitochondrial integrity at isolation and upon storage prior to transplantation, (3) the choice of methodology for mitochondria delivery into the recipient as it may interfere with the efficiency of transplantation and lead to dissemination to non-target cells and tissues, (4) the viability and integrity of the transplanted mitochondria upon exposure to the extracellular environment (Ca2+ levels, temperature), (5) the compatibility of the transplanted mitochondria with the host nuclear DNA (and with the mtDNA) that may affect its functionality, expression, and retention, and (6) the cell source, dose, time point(s) and cycles of mitochondria administration to avoid a potential lack of sustained therapeutic effects [196,197].
In conclusion and as described in this review, despite the availability of advanced, promising strategies targeting the mitochondria to manage several prevalent, serious human disorders, a number of significant challenges remain to be carefully addressed for a future safe, effective, and practicable application of any of these approaches in clinical settings.
Author Contributions
Conceptualization, G.Z. and M.C.; validation, G.Z., J.K.V., H.M. and M.C.; resources, M.C.; data curation, G.Z. and M.C.; writing—original draft preparation, G.Z. and M.C.; writing—review and editing, G.Z., J.K.V., H.M. and M.C.; supervision, M.C.; funding acquisition, H.M. and M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the World Arthrosis Foundation (M.C., H.M.) and by the National Natural Science Foundation of China (NSFC) (grant 82002355 to G.Z.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Gabaldon, T.; Huynen, M.A. Shaping the mitochondrial proteome. Biochim. Biophys. Acta 2004, 1659, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecrenier, N.; Van Der Bruggen, P.; Foury, F. Mitochondrial DNA polymerases from yeast to man: A new family of polymerases. Gene 1997, 185, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; Savoia, A.; Forti, F.; D’Apolito, M.F.; Centra, M.; Rocchi, M.; Zeviani, M. Identification of the gene encoding the human mitochondrial RNA polymerase (h-mtRPOL) by cyberscreening of the Expressed Sequence Tags database. Hum. Mol. Genet. 1997, 6, 615–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Butow, R.A.; Avadhani, N.G. Mitochondrial signaling: The retrograde response. Mol. Cell 2004, 14, 1–15. [Google Scholar] [CrossRef]
- Desai, R.; East, D.A.; Hardy, L.; Faccenda, D.; Rigon, M.; Crosby, J.; Alvarez, M.S.; Singh, A.; Mainenti, M.; Hussey, L.K.; et al. Mitochondria form contact sites with the nucleus to couple prosurvival retrograde response. Sci. Adv. 2020, 6, eabc9955–eabc9969. [Google Scholar] [CrossRef]
- Murley, A.; Nunnari, J. The emerging network of mitochondria-organelle contacts. Mol. Cell 2016, 61, 648–653. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.M.; Jung, Y.K. A molecular approach to mitophagy and mitochondrial dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar]
- Barazzuol, L.; Giamogante, F.; Cali, T. Mitochondria associated membranes (MAMs): Architecture and physiopathological role. Cell Calcium 2021, 94, 102343. [Google Scholar] [CrossRef]
- Bao, F.X.; Shi, H.Y.; Long, Q.; Yang, L.; Wu, Y.; Ying, Z.F.; Qin, D.J.; Zhang, J.; Guo, Y.P.; Li, H.M.; et al. Mitochondrial membrane potential-dependent endoplasmic reticulum fragmentation is an important step in neuritic degeneration. CNS Neurosci. Ther. 2016, 22, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549–aaf5557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatefi, Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu. Rev. Biochem. 1985, 54, 1015–1069. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, F.T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [Green Version]
- Din, S.; Konstandin, M.H.; Johnson, B.; Emathinger, J.; Völkers, M.; Toko, H.; Collins, B.; Ormachea, L.; Samse, K.; Kubli, D.A.; et al. Metabolic dysfunction consistent with premature aging results from deletion of Pim kinases. Circ. Res. 2014, 115, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.; Oh, S.S.; Weaver, D.; Lewandowska, A.; Maxfield, D.; Schuler, M.H.; Smith, N.K.; Macfarlane, J.; Saunders, G.; Palmer, C.A.; et al. Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc. Natl. Acad. Sci. USA 2014, 111, E3631–E3640. [Google Scholar] [CrossRef] [Green Version]
- Wenz, T. Mitochondria and PGC-1alpha in aging and age-associated diseases. J. Aging Res. 2011, 2011, 810619–810631. [Google Scholar] [CrossRef] [Green Version]
- Muraski, J.A.; Rota, M.; Misao, Y.; Fransioli, J.; Cottage, C.; Gude, N.; Esposito, G.; Delucchi, F.; Arcarese, M.; Alvarez, R.; et al. Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat. Med. 2007, 13, 1467–1475. [Google Scholar] [CrossRef]
- D’Amico, D.; Mottis, A.; Potenza, F.; Sorrentino, V.; Li, H.; Romani, M.; Lemos, V.; Schoonjans, K.; Zamboni, N.; Knott, G.; et al. The RNA-Binding protein PUM2 impairs mitochondrial dynamics and mitophagy during aging. Mol. Cell 2019, 73, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmistrova, O.; Olias-Arjona, A.; Lapresa, R.; Jimenez-Blasco, D.; Eremeeva, T.; Shishov, D.; Romanov, S.; Zakurdaeva, K.; Almeida, A.; Fedichev, P.O.; et al. Targeting PFKFB3 alleviates cerebral ischemia-reperfusion injury in mice. Sci. Rep. 2019, 9, 11670–11682. [Google Scholar] [CrossRef] [Green Version]
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; St Croix, C.; Sembrat, J.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17, e12720–e12732. [Google Scholar] [CrossRef]
- Ling, W.; Krager, K.; Richardson, K.K.; Warren, A.D.; Ponte, F.; Aykin-Burns, N.; Manolagas, S.C.; Almeida, M.; Kim, H.N. Mitochondrial Sirt3 contributes to the bone loss caused by aging or estrogen deficiency. JCI Insight 2021, 6, e146728–e146745. [Google Scholar] [CrossRef]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2010, 2, 914–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, A.; Anand, R.; Altinoluk-Hambüchen, S.; Ezzahoini, H.; Stefanski, A.; Iram, A.; Bergmann, L.; Urbach, J.; Böhler, P.; Hänsel, J.; et al. SIRT4 interacts with OPA1 and regulates mitochondrial quality control and mitophagy. Aging 2017, 9, 2163–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Zhang, C.F.; Guo, J.L.; Su, J.L.; Guo, Z.K.; Li, H.Y. Involvement of NEAT1/PINK1-mediated mitophagy in chronic obstructive pulmonary disease induced by cigarette smoke or PM2.5. Ann. Transl. Med. 2022, 10, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Genova, M.L.; Castelluccio, C.; Fato, R.; Parenti Castelli, G.; Merlo Pich, M.; Formiggini, G.; Bovina, C.; Marchetti, M.; Lenaz, G. Major changes in complex I activity in mitochondria from aged rats may not be detected by direct assay of NADH:coenzyme Q reductase. Biochem. J. 1995, 311, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Gioscia-Ryan, R.A.; LaRocca, T.J.; Sindler, A.L.; Zigler, M.C.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 2014, 592, 2549–2561. [Google Scholar] [CrossRef]
- Kozma, M.; Bombicz, M.; Varga, B.; Priksz, D.; Gesztelyi, R.; Tarjanyi, V.; Kiss, R.; Szekeres, R.; Takacs, B.; Menes, A.; et al. Cardioprotective role of BGP-15 in ageing zucker diabetic fatty rat (ZDF) model: Extended mitochondrial longevity. Pharmaceutics 2022, 14, 226. [Google Scholar] [CrossRef] [PubMed]
- Weimer, S.; Priebs, J.; Kuhlow, D.; Groth, M.; Priebe, S.; Mansfeld, J.; Merry, T.L.; Dubuis, S.; Laube, B.; Pfeiffer, A.F.; et al. D-glucosamine supplementation extends life span of nematodes and of ageing mice. Nat. Commun. 2014, 5, 3563–3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, D.V.; Vindrieux, D.; Goehrig, D.; Jaber, S.; Collin, G.; Griveau, A.; Wiel, C.; Bendridi, N.; Djebali, S.; Farfariello, V.; et al. Calcium channel ITPR2 and mitochondria-ER contacts promote cellular senescence and aging. Nat. Commun. 2021, 12, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Kao, C.H.; Chen, Y.T.; Wang, C.H.; Wu, C.Y.; Tsai, C.Y.; Liu, F.C.; Yang, C.W.; Wei, Y.H.; Hsu, M.T.; et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009, 23, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Guzman, J.N.; Ilijic, E.; Yang, B.; Sanchez-Padilla, J.; Wokosin, D.; Galtieri, D.; Kondapalli, J.; Schumacker, P.T.; Surmeier, D.J. Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J. Clin. Investig. 2018, 128, 2266–2280. [Google Scholar] [CrossRef] [Green Version]
- Mansell, E.; Sigurdsson, V.; Deltcheva, E.; Brown, J.; James, C.; Miharada, K.; Soneji, S.; Larsson, J.; Enver, T. Mitochondrial potentiation ameliorates age-related heterogeneity in hematopoietic stem cell function. Cell Stem. Cell 2021, 28, 241–256. [Google Scholar] [CrossRef]
- Sahu, A.; Mamiya, H.; Shinde, S.N.; Cheikhi, A.; Winter, L.L.; Vo, N.V.; Stolz, D.; Roginskaya, V.; Tang, W.Y.; St Croix, C.; et al. Age-related declines in alpha-Klotho drive progenitor cell mitochondrial dysfunction and impaired muscle regeneration. Nat. Commun. 2018, 9, 4859–4872. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Oliveira, M.P.; Khamoui, A.V.; Aparicio, R.; Rera, M.; Rossiter, H.B.; Walker, D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017, 8, 448–461. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.T.; Chen, P.L.; Su, M.P.; Li, J.C.; Chang, Y.W.; Liu, R.W.; Juan, H.F.; Yang, J.M.; Chan, S.P.; Tsai, Y.C.; et al. Loss of Fis1 impairs proteostasis during skeletal muscle aging in Drosophila. Aging Cell 2021, 20, e13379–e13397. [Google Scholar] [CrossRef]
- Kang, H.T.; Park, J.T.; Choi, K.; Kim, Y.; Choi, H.J.C.; Jung, C.W.; Lee, Y.S.; Park, S.C. Chemical screening identifies ATM as a target for alleviating senescence. Nat. Chem. Biol. 2017, 13, 616–623. [Google Scholar] [CrossRef]
- Kang, H.T.; Park, J.T.; Choi, K.; Choi, H.J.C.; Jung, C.W.; Kim, G.R.; Lee, Y.S.; Park, S.C. Chemical screening identifies ROCK as a target for recovering mitochondrial function in Hutchinson-Gilford progeria syndrome. Aging Cell 2017, 16, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.H.; Bandy, B.; Tahara, E.B.; Kowaltowski, A.J. Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 49883–49888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcazar-Fabra, M.; Navas, P.; Brea-Calvo, G. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta 2016, 1857, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Sawashita, J.; Kubo, H.; Nishio, S.Y.; Hashimoto, S.; Suzuki, N.; Yoshimura, H.; Tsuruoka, M.; Wang, Y.; Liu, Y.; et al. Ubiquinol-10 supplementation activates mitochondria functions to decelerate senescence in senescence-accelerated mice. Antioxid. Redox Signal 2014, 20, 2606–2620. [Google Scholar] [CrossRef] [PubMed]
- Al-Zubaidi, U.; Adhikari, D.; Cinar, O.; Zhang, Q.H.; Yuen, W.S.; Murphy, M.P.; Rombauts, L.; Robker, R.L.; Carroll, J. Mitochondria-targeted therapeutics, MitoQ and BGP-15, reverse aging-associated meiotic spindle defects in mouse and human oocytes. Hum. Reprod. 2021, 36, 771–784. [Google Scholar] [CrossRef]
- Gao, P.; Jiang, Y.; Wu, H.; Sun, F.; Li, Y.; He, H.; Wang, B.; Lu, Z.; Hu, Y.; Wei, X.; et al. Inhibition of mitochondrial calcium overload by SIRT3 prevents obesity- or age-related whitening of brown adipose tissue. Diabetes 2020, 69, 165–180. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Monaco, G.; Missiaen, L.; De Smedt, H.; Parys, J.B.; Bultynck, G. IP(3) receptors, mitochondria, and Ca signaling: Implications for aging. J. Aging Res. 2011, 2011, 920178–920198. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Wiel, C.; Lallet-Daher, H.; Gitenay, D.; Gras, B.; Le Calvé, B.; Augert, A.; Ferrand, M.; Prevarskaya, N.; Simonnet, H.; Vindrieux, D.; et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 2014, 5, 3792–3801. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Chou, T.Y.; Lin, I.H.; Chen, C.G.; Kao, C.H.; Huang, G.J.; Chen, L.K.; Wang, P.N.; Lin, C.P.; Tsai, T.F. Upregulation of Cisd2 attenuates Alzheimer’s-related neuronal loss in mice. J. Pathol. 2020, 250, 299–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.F.; Kao, C.H.; Kirby, R.; Tsai, T.F. Cisd2 mediates mitochondrial integrity and life span in mammals. Autophagy 2009, 5, 1043–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Li, X.; Jiang, G.; Wang, X.; Chang, H.C.; Hsu, W.H.; Li, Q. Isradipine prevents rotenone-induced intracellular calcium rise that accelerates senescence in human neuroblastoma SH-SY5Y cells. Neuroscience 2013, 246, 243–253. [Google Scholar] [CrossRef]
- Liu, W.; Lin, H.; Mao, Z.; Zhang, L.; Bao, K.; Jiang, B.; Xia, C.; Li, W.; Hu, Z.; Li, J. Verapamil extends lifespan in Caenorhabditis elegans by inhibiting calcineurin activity and promoting autophagy. Aging 2020, 12, 5300–5317. [Google Scholar] [CrossRef]
- Delcroix, V.; Mauduit, O.; Tessier, N.; Montillaud, A.; Lesluyes, T.; Ducret, T.; Chibon, F.; Van Coppenolle, F.; Ducreux, S.; Vacher, P. The role of the anti-aging protein Klotho in IGF-1 signaling and reticular calcium leak: Impact on the chemosensitivity of dedifferentiated liposarcomas. Cancers 2018, 10, 439. [Google Scholar] [CrossRef] [Green Version]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Wilson, P.D.; Franks, L.M. The effect of age on mitochondrial ultrastructure. Gerontologia 1975, 21, 81–94. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Smith, H.J.; Yao, P.; Mair, W.B. Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 2019, 20, e48395–e48417. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Radical medicine: Treating ageing to cure disease. Nat. Rev. Mol. Cell Biol. 2005, 6, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid. Med. Cell Longev. 2019, 2019, 5080843–5080856. [Google Scholar] [CrossRef] [Green Version]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.; Yin, J.J.; Yu, L.L. Phenolic acid, tocopherol and carotenoid compositions, and antioxidant functions of hard red winter wheat bran. J. Agric. Food Chem. 2005, 53, 3916–3922. [Google Scholar] [CrossRef]
- Brown, S.E.; Ross, M.F.; Sanjuan-Pla, A.; Manas, A.R.; Smith, R.A.; Murphy, M.P. Targeting lipoic acid to mitochondria: Synthesis and characterization of a triphenylphosphonium-conjugated alpha-lipoyl derivative. Free Radic. Biol. Med. 2007, 42, 1766–1780. [Google Scholar] [CrossRef]
- Filipovska, A.; Kelso, G.F.; Brown, S.E.; Beer, S.M.; Smith, R.A.; Murphy, M.P. Synthesis and characterization of a triphenylphosphonium-conjugated peroxidase mimetic. Insights into the interaction of ebselen with mitochondria. J. Biol. Chem. 2005, 280, 24113–24126. [Google Scholar] [CrossRef] [Green Version]
- James, A.M.; Sharpley, M.S.; Manas, A.R.; Frerman, F.E.; Hirst, J.; Smith, R.A.; Murphy, M.P. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 2007, 282, 14708–14718. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Adlam, V.J.; Blaikie, F.H.; Manas, A.R.; Porteous, C.M.; James, A.M.; Ross, M.F.; Logan, A.; Cochemé, H.M.; Trnka, J.; et al. Mitochondria-targeted antioxidants in the treatment of disease. Ann. N. Y. Acad. Sci. 2008, 1147, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 1997, 15, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Smith, R.R. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef]
- Liberman, E.A.; Topaly, V.P.; Tsofina, L.M.; Jasaitis, A.A.; Skulachev, V.P. Mechanism of coupling of oxidative phosphorylation and the membrane potential of mitochondria. Nature 1969, 222, 1076–1078. [Google Scholar] [CrossRef]
- Jauslin, M.L.; Meier, T.; Smith, R.A.; Murphy, M.P. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003, 17, 1972–1974. [Google Scholar] [CrossRef]
- Lahiri, T.; Brambilla, L.; Andrade, J.; Askenazi, M.; Ueberheide, B.; Levy, D.E. Mitochondrial STAT3 regulates antioxidant gene expression through complex I-derived NAD in triple negative breast cancer. Mol. Oncol. 2021, 15, 1432–1439. [Google Scholar] [CrossRef]
- Janbandhu, V.; Tallapragada, V.; Patrick, R.; Li, Y.; Abeygunawardena, D.; Humphreys, D.T.; Martin, E.M.M.A.; Ward, A.O.; Contreras, O.; Farbehi, N.; et al. Hif-1a suppresses ROS-induced proliferation of cardiac fibroblasts following myocardial infarction. Cell Stem Cell 2022, 29, 281–297. [Google Scholar] [CrossRef]
- Li, H.S.; Zhou, Y.N.; Li, L.; Li, S.F.; Long, D.; Chen, X.L.; Zhang, J.B.; Feng, L.; Li, Y.P. HIF-1alpha protects against oxidative stress by directly targeting mitochondria. Redox. Biol. 2019, 25, 101109–101123. [Google Scholar] [CrossRef]
- Gaspar, J.M.; Velloso, L.A. Hypoxia inducible factor as a central regulator of metabolism—Implications for the development of obesity. Front. Neurosci. 2018, 12, 813–827. [Google Scholar] [CrossRef]
- Lee, K.Y.; Gesta, S.; Boucher, J.; Wang, X.L.; Kahn, C.R. The differential role of Hif1beta/Arnt and the hypoxic response in adipose function, fibrosis, and inflammation. Cell Metab. 2011, 14, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, J.W.; Osborne, O.; Oh, D.Y.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.J.; Schriner, S.E.; McCleary, D.; Day, B.J.; Wallace, D.C. Life extension through neurofibromin mitochondrial regulation and antioxidant therapy for neurofibromatosis-1 in Drosophila melanogaster. Nat. Genet. 2007, 39, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Fei, A.H.; Cao, Q.; Chen, S.Y.; Wang, H.R.; Wang, F.L.; Pan, S.M.; Lin, Z.F. Salvianolate inhibits reactive oxygen species production in H2O2-treated mouse cardiomyocytes in vitro via the TGFbeta pathway. Acta Pharmacol. Sin. 2013, 34, 496–500. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Fu, L.; Nile, S.H.; Zhang, J.; Kai, G. Salvia miltiorrhiza in treating cardiovascular diseases: A review on its pharmacological and clinical applications. Front. Pharmacol. 2019, 10, 753–767. [Google Scholar] [CrossRef]
- Yin, Y.; Guan, Y.; Duan, J.; Wei, G.; Zhu, Y.; Quan, W.; Guo, C.; Zhou, D.; Wang, Y.; Xi, M.; et al. Cardioprotective effect of Danshensu against myocardial ischemia/reperfusion injury and inhibits apoptosis of H9c2 cardiomyocytes via Akt and ERK1/2 phosphorylation. Eur. J. Pharmacol. 2013, 699, 219–226. [Google Scholar] [CrossRef]
- Zhao, B.L.; Jiang, W.; Zhao, Y.; Hou, J.W.; Xin, W.J. Scavenging effects of salvia miltiorrhiza on free radicals and its protection for myocardial mitochondrial membranes from ischemia-reperfusion injury. Biochem. Mol. Biol. Int. 1996, 38, 1171–1182. [Google Scholar]
- Wang, M.; Hu, R.; Wang, Y.; Liu, L.; You, H.; Zhang, J.; Wu, X.; Pei, T.; Wang, F.; Lu, L.; et al. Atractylenolide III attenuates muscle wasting in chronic kidney disease via the oxidative stress-mediated PI3K/AKT/mTOR pathway. Oxid. Med. Cell Longev. 2019, 2019, 1875471–1875487. [Google Scholar] [CrossRef] [Green Version]
- Daverey, A.; Agrawal, S.K. Curcumin alleviates oxidative stress and mitochondrial dysfunction in astrocytes. Neuroscience 2016, 333, 92–103. [Google Scholar] [CrossRef]
- Mehanna, E.T.; El-Sayed, N.M.; Ibrahim, A.K.; Ahmed, S.A.; Abo-Elmatty, D.M. Isolated compounds from Cuscuta pedicellata ameliorate oxidative stress and upregulate expression of some energy regulatory genes in high fat diet induced obesity in rats. Biomed. Pharmacother. 2018, 108, 1253–1258. [Google Scholar] [CrossRef]
- Lin, S.P.; Li, W.; Winters, A.; Liu, R.; Yang, S.H. Artemisinin prevents glutamate-induced neuronal cell death via Akt pathway activation. Front. Cell Neurosci. 2018, 12, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, U.; Canavan, M.; Biniecka, M.; Veale, D.J. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Voet, S.; Prinz, M.; van Loo, G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol. Med. 2019, 25, 112–123. [Google Scholar] [CrossRef]
- Burek, C.L.; Rose, N.R. Autoimmune thyroiditis and ROS. Autoimmun. Rev. 2008, 7, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Vig, S.; Lambooij, J.M.; Zaldumbide, A.; Guigas, B. Endoplasmic reticulum-mitochondria crosstalk and beta-cell destruction in type 1 diabetes. Front. Immunol. 2021, 12, 669492–669501. [Google Scholar] [CrossRef]
- Dela Cruz, C.S.; Kang, M.J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016, 167, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Wang, Y.; Jasper, H.; Toan, S.; Muid, D.; Chang, X.; Zhou, H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox. Biol. 2021, 45, 102049–102058. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2020, 16, 3–17. [Google Scholar] [CrossRef] [PubMed]
- de Maranon, A.M.; Díaz-Pozo, P.; Canet, F.; Díaz-Morales, N.; Abad-Jiménez, Z.; López-Domènech, S.; Vezza, T.; Apostolova, N.; Morillas, C.; Rocha, M.; et al. Metformin modulates mitochondrial function and mitophagy in peripheral blood mononuclear cells from type 2 diabetic patients. Redox Biol. 2022, 53, 102342–102348. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- To, E.E.; Erlich, J.R.; Liong, F.; Luong, R.; Liong, S.; Esaq, F.; Oseghale, O.; Anthony, D.; McQualter, J.; Bozinovski, S.; et al. Mitochondrial reactive oxygen species contribute to pathological inflammation during Influenza A virus infection in mice. Antioxid. Redox Signal. 2020, 32, 929–942. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36–57. [Google Scholar] [CrossRef]
- Hernandez, J.C.; Latz, E.; Urcuqui-Inchima, S. HIV-1 induces the first signal to activate the NLRP3 inflammasome in monocyte-derived macrophages. Intervirology 2014, 57, 36–42. [Google Scholar] [CrossRef]
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and septic shock. Lancet 2018, 392, 75–87. [Google Scholar] [CrossRef]
- Chen, C.; Liu, X.; Gong, L.; Zhu, T.; Zhou, W.; Kong, L.; Luo, J. Identification of Tubocapsanolide A as a novel NLRP3 inhibitor for potential treatment of colitis. Biochem. Pharmacol. 2021, 190, 114645. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Shi, S.; Wang, Y.; Wang, X.; Shen, Z.; Wang, M.; Pei, C.; Wu, Y.; He, Y.; Wang, Z. Astragaloside IV alleviates PM2.5-caused lung toxicity by inhibiting inflammasome-mediated pyroptosis via NLRP3/caspase-1 axis inhibition in mice. Biomed. Pharmacother. 2022, 150, 112978–112988. [Google Scholar] [CrossRef] [PubMed]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 inflammasome in severe COVID-19. Front. Immunol. 2020, 11, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 2019, 10, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial diseases: Hope for the future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; Zeviani, M. Altered sulfide (H(2)S) metabolism in ethylmalonic encephalopathy. Cold Spring Harb. Perspect. Biol. 2013, 5, a011437–a011448. [Google Scholar] [CrossRef]
- Viscomi, C.; Burlina, A.B.; Dweikat, I.; Savoiardo, M.; Lamperti, C.; Hildebrandt, T.; Tiranti, V.; Zeviani, M. Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat. Med. 2010, 16, 869–871. [Google Scholar] [CrossRef]
- Tiranti, V.; D’Adamo, P.; Briem, E.; Ferrari, G.; Mineri, R.; Lamantea, E.; Mandel, H.; Balestri, P.; Garcia-Silva, M.T.; Vollmer, B.; et al. Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am. J. Hum. Genet. 2004, 74, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Nishino, I.; Spinazzola, A.; Hirano, M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 1999, 283, 689–692. [Google Scholar] [CrossRef]
- Garone, C.; Tadesse, S.; Hirano, M. Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain 2011, 134, 3326–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bax, B.E.; Bain, M.D.; Scarpelli, M.; Filosto, M.; Tonin, P.; Moran, N. Clinical and biochemical improvements in a patient with MNGIE following enzyme replacement. Neurology 2013, 81, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Emmanuele, V.; Hirano, M. Clinical presentations of coenzyme q10 deficiency syndrome. Mol. Syndromol. 2014, 5, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Emmanuele, V.; López, L.C.; Berardo, A.; Naini, A.; Tadesse, S.; Wen, B.; D’Agostino, E.; Solomon, M.; DiMauro, S.; Quinzii, C.; et al. Heterogeneity of coenzyme Q10 deficiency: Patient study and literature review. Arch. Neurol. 2012, 69, 978–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giancaspero, T.A.; Colella, M.; Brizio, C.; Difonzo, G.; Fiorino, G.M.; Leone, P.; Brandsch, R.; Bonomi, F.; Iametti, S.; Barile, M. Remaining challenges in cellular flavin cofactor homeostasis and flavoprotein biogenesis. Front. Chem. 2015, 3, 30–43. [Google Scholar] [CrossRef] [Green Version]
- Haack, T.B.; Danhauser, K.; Haberberger, B.; Hoser, J.; Strecker, V.; Boehm, D.; Uziel, G.; Lamantea, E.; Invernizzi, F.; Poulton, J.; et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat. Genet. 2010, 42, 1131–1134. [Google Scholar] [CrossRef]
- Ghezzi, D.; Sevrioukova, I.; Invernizzi, F.; Lamperti, C.; Mora, M.; D’Adamo, P.; Novara, F.; Zuffardi, O.; Uziel, G.; Zeviani, M. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am. J. Hum. Genet. 2010, 86, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Nouws, J.; Wibrand, F.; van den Brand, M.; Venselaar, H.; Duno, M.; Lund, A.M.; Trautner, S.; Nijtmans, L.; Ostergard, E. A patient with complex I deficiency caused by a novel ACAD9 mutation not responding to riboflavin treatment. JIMD Rep. 2014, 12, 37–45. [Google Scholar]
- Zhong, G.; Madry, H.; Cucchiarini, M. Mitochondrial genome editing to treat human osteoarthritis—A narrative review. Int. J. Mol. Sci. 2022, 23, 1467. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Taylor, R.W.; Diekert, K.; Lill, R.; Turnbull, D.M.; Lightowlers, R.N. Peptide nucleic acid delivery to human mitochondria. Gene Ther. 1999, 6, 1919–1928. [Google Scholar] [CrossRef] [Green Version]
- Muratovska, A.; Lightowlers, R.N.; Taylor, R.W.; Turnbull, D.M.; Smith, R.A.; Wilce, J.A.; Martin, S.W.; Murphy, M.P. Targeting peptide nucleic acid (PNA) oligomers to mitochondria within cells by conjugation to lipophilic cations: Implications for mitochondrial DNA replication, expression and disease. Nucleic Acids Res. 2001, 29, 1852–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonin, Y.; Heckel, A.M.; Vysokikh, M.; Dovydenko, I.; Meschaninova, M.; Rötig, A.; Munnich, A.; Venyaminova, A.; Tarassov, I.; Entelis, N. Modeling of antigenomic therapy of mitochondrial diseases by mitochondrially addressed RNA targeting a pathogenic point mutation in mitochondrial DNA. J. Biol. Chem. 2014, 289, 13323–13334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammage, P.A.; Viscomi, C.; Simard, M.L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Gaude, E.; Van Haute, L.; Rebelo-Guiomar, P.; Jackson, C.B.; Rorbach, J.; Pekalski, M.L.; Robinson, A.J.; Charpentier, M.; Concordet, J.P.; et al. Near-complete elimination of mutant mtDNA by iterative or dynamic dose-controlled treatment with mtZFNs. Nucleic Acids Res. 2016, 44, 7804–7816. [Google Scholar] [CrossRef] [Green Version]
- Bacman, S.R.; Williams, S.L.; Pinto, M.; Peralta, S.; Moraes, C.T. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013, 19, 1111–1113. [Google Scholar] [CrossRef]
- Hashimoto, M.; Bacman, S.R.; Peralta, S.; Falk, M.J.; Chomyn, A.; Chan, D.C.; Williams, S.L.; Moraes, C.T. MitoTALEN: A general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol. Ther. 2015, 23, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wu, H.; Kang, X.; Liang, Y.; Lan, T.; Li, T.; Tan, T.; Peng, J.; Zhang, Q.; An, G.; et al. Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein Cell 2018, 9, 283–297. [Google Scholar] [CrossRef] [Green Version]
- Jo, A.; Ham, S.; Lee, G.H.; Lee, Y.I.; Kim, S.; Lee, Y.S.; Shin, J.H.; Lee, Y. Efficient mitochondrial genome editing by CRISPR/Cas9. Biomed. Res. Int. 2015, 2015, 305716–305726. [Google Scholar] [CrossRef] [Green Version]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial genome engineering: The revolution may not be CRISPR-ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Schoeb, T.R.; Bajpai, P.; Slominski, A.; Singh, K.K. Reversing wrinkled skin and hair loss in mice by restoring mitochondrial function. Cell Death Dis. 2018, 9, 735–748. [Google Scholar] [CrossRef] [Green Version]
- Nakazato, I.; Okuno, M.; Zhou, C.; Itoh, T.; Tsutsumi, N.; Takenaka, M.; Arimura, S.I. Targeted base editing in the mitochondrial genome of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2022, 119, e2121177119–e2121177127. [Google Scholar] [CrossRef] [PubMed]
- Silva-Pinheiro, P.; Nash, P.A.; Van Haute, L.; Mutti, C.D.; Turner, K.; Minczuk, M. In vivo mitochondrial base editing via adeno-associated viral delivery to mouse post-mitotic tissue. Nat. Commun. 2022, 13, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, S.; Baek, G.; Kim, A.; Kang, B.C.; Seo, H.; Kim, J.S. Mitochondrial DNA editing in mice with DddA-TALE fusion deaminases. Nat. Commun. 2021, 12, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.I.; Lee, S.; Mok, Y.G.; Lim, K.; Lee, J.; Lee, J.M.; Chung, E.; Kim, J.S. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell 2022, 185, 1764–1776. [Google Scholar] [CrossRef]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Meng, H.; Liu, L.; Zhao, H.; Rao, X.; Yan, Y.; Wu, H.; Liu, M.; He, A.; Yi, C. Mitochondrial base editor induces substantial nuclear off-target mutations. Nature 2022, 606, 804–811. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Cross, C.E.; Halliwell, B.; Borish, E.T.; Pryor, W.A.; Ames, B.N.; Saul, R.L.; McCord, J.M.; Harman, D. Oxygen radicals and human disease. Ann. Intern. Med. 1987, 107, 526–545. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef]
- Mishra, S.J.; Khandelwal, A.; Banerjee, M.; Balch, M.; Peng, S.; Davis, R.E.; Merfeld, T.; Munthali, V.; Deng, J.; Matts, R.L.; et al. Selective inhibition of the Hsp90alpha isoform. Angew Chem. Int. Ed. Engl. 2021, 60, 10547–11051. [Google Scholar] [CrossRef] [PubMed]
- Jurczyszyn, A.; Zebzda, A.; Czepiel, J.; Perucki, W.; Bazan-Socha, S.; Cibor, D.; Owczarek, D.; Majka, M. Geldanamycin and its derivatives inhibit the growth of myeloma cells and reduce the expression of the MET receptor. J. Cancer 2014, 5, 480–490. [Google Scholar] [CrossRef] [PubMed]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in cancer prevention and therapy. Ann. Transl. Med. 2014, 2, 57–69. [Google Scholar] [PubMed]
- Lukey, M.J.; Wilson, K.F.; Cerione, R.A. Therapeutic strategies impacting cancer cell glutamine metabolism. Future Med. Chem. 2013, 5, 1685–1700. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.A.; Chen, C.L.; Huang, Y.H.; Evans, E.E.; Cheng, C.C.; Chuang, Y.J.; Zhang, C.; Le, A. Inhibition of glutaminolysis in combination with other therapies to improve cancer treatment. Curr. Opin. Chem. Biol. 2021, 62, 64–81. [Google Scholar] [CrossRef]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [Green Version]
- Briceno, E.; Reyes, S.; Sotelo, J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg. Focus 2003, 14, e3–e8. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelicano, H.; Feng, L.; Zhou, Y.; Carew, J.S.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Inhibition of mitochondrial respiration: A novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J. Biol. Chem. 2003, 278, 37832–37839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Gao, Z.; Chen, Y.; Guan, M.X. The role of mitochondria in osteogenic, adipogenic and chondrogenic differentiation of mesenchymal stem cells. Protein Cell 2017, 8, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarty, R.P.; Chandel, N.S. Mitochondria as signaling organelles control mammalian stem cell fate. Cell Stem Cell 2021, 28, 394–408. [Google Scholar] [CrossRef]
- Hofmann, A.D.; Beyer, M.; Krause-Buchholz, U.; Wobus, M.; Bornhäuser, M.; Rödel, G. OXPHOS supercomplexes as a hallmark of the mitochondrial phenotype of adipogenic differentiated human MSCs. PLoS ONE 2012, 7, e35160–e35168. [Google Scholar] [CrossRef] [Green Version]
- Quinn, K.P.; Sridharan, G.V.; Hayden, R.S.; Kaplan, D.L.; Lee, K.; Georgakoudi, I. Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Sci. Rep. 2013, 3, 3432–3441. [Google Scholar] [CrossRef] [Green Version]
- Forni, M.F.; Peloggia, J.; Trudeau, K.; Shirihai, O.; Kowaltowski, A.J. Murine mesenchymal stem cell commitment to differentiation is regulated by mitochondrial dynamics. Stem Cells 2016, 34, 743–755. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Kim, B.; Kim, J.S.; Yoon, Y.; Santiago, M.C.; Brown, M.D.; Park, J.Y. Inhibition of Drp1-dependent mitochondrial division impairs myogenic differentiation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R927–R938. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell 2018, 23, 86–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, D.; Yan, S.; Yu, Q.; Chen, D.; Yan, S.S. Mfn2 is required for mitochondrial development and synapse formation in human induced pluripotent stem cells/hiPSC derived cortical neurons. Sci. Rep. 2016, 6, 31462–31474. [Google Scholar] [CrossRef] [Green Version]
- Atashi, F.; Modarressi, A.; Pepper, M.S. The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: A review. Stem Cells Dev. 2015, 24, 1150–1163. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Marsboom, G.; Toth, P.T.; Rehman, J. Mitochondrial respiration regulates adipogenic differentiation of human mesenchymal stem cells. PLoS ONE 2013, 8, e77077–e77088. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Feng, Z.; Wang, X.; Zeng, M.; Liu, J.; Han, S.; Xu, J.; Chen, L.; Cao, K.; Long, J.; et al. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. 2018, 25, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Shaban, S.; El-Husseny, M.W.A.; Abushouk, A.I.; Salem, A.M.A.; Mamdouh, M.; Abdel-Daim, M.M. Effects of antioxidant supplements on the survival and differentiation of stem cells. Oxid Med. Cell Longev. 2017, 2017, 5032102–5032118. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Yang, Y.; Chen, Q.; Xie, L. Glutamine metabolism is essential for stemness of bone marrow mesenchymal stem cells and bone homeostasis. Stem Cells Int. 2019, 2019, 8928934–8928947. [Google Scholar] [CrossRef] [Green Version]
- Szabadkai, G.; Duchen, M.R. Mitochondria: The hub of cellular Ca2+ signaling. Physiology 2008, 23, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Antony, A.N.; Paillard, M.; Moffat, C.; Juskeviciute, E.; Correnti, J.; Bolon, B.; Rubin, E.; Csordás, G.; Seifert, E.L.; Hoek, J.B.; et al. MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nat. Commun. 2016, 7, 10955–10964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Zhang, X.; LaCanna, R.; Tomar, D.; Elrod, J.W.; Tian, Y. MICU1-dependent mitochondrial calcium uptake regulates lung alveolar type 2 cell plasticity and lung regeneration. JCI Insight 2022, 7, e154447–e154462. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.A.; Gibb, A.A.; Arif, E.; Kolmetzky, D.W.; Tomar, D.; Luongo, T.S.; Jadiya, P.; Murray, E.K.; Lorkiewicz, P.K.; Hajnóczky, G.; et al. Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat. Commun. 2019, 10, 4509–4525. [Google Scholar] [CrossRef] [Green Version]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef]
- Nicolson, G.L.; Conklin, K.A. Molecular replacement for cancer metabolic and mitochondrial dysfunction, fatigue and the adverse effects of cancer therapy. Cancer Genom. Proteom. 2006, 3, 159–168. [Google Scholar]
- Viscomi, C.; Zeviani, M. Strategies for fighting mitochondrial diseases. J. Intern. Med 2020, 287, 665–684. [Google Scholar] [CrossRef]
- Evans, C.H.; Ghivizzani, S.C.; Robbins, P.D. Orthopedic gene therapy—Lost in translation? J. Cell Physiol. 2012, 227, 416–420. [Google Scholar] [CrossRef] [Green Version]
- Pitceathly, R.D.S.; Keshavan, N.; Rahman, J.; Rahman, S. Moving towards clinical trials for mitochondrial diseases. J. Inherit. Metab. Dis. 2021, 44, 22–41. [Google Scholar] [CrossRef]
- Olry de Labry-Lima, A.; Ponce-Polo, A.; García-Mochón, L.; Ortega-Ortega, M.; Pérez-Troncoso, D.; Epstein, D. Challenges for economic evaluations of advanced therapy medicinal products: A systematic review. Value Health 2022. [Google Scholar] [CrossRef] [PubMed]
- Beetler, D.J.; Di Florio, D.N.; Law, E.W.; Groen, C.M.; Windebank, A.J.; Peterson, Q.P.; Fairweather, D. The evolving regulatory landscape in regenerative medicine. Mol. Aspects Med. 2022, 88, 101138. [Google Scholar] [CrossRef] [PubMed]
- Falabella, M.; Minczuk, M.; Hanna, M.G.; Viscomi, C.; Pitceathly, R.D.S. Gene therapy for primary mitochondrial diseases: Experimental advances and clinical challenges. Nat. Rev. Neurol. 2022, 18, 689–698. [Google Scholar] [CrossRef]
- Vignais, M.L.; Caicedo, A.; Brondello, J.M.; Jorgensen, C. Cell connections by tunneling nanotubes: Effects of mitochondrial trafficking on target cell metabolism, homeostasis, and response to therapy. Stem Cells Int. 2017, 2017, 6917941–6917955. [Google Scholar] [CrossRef]
- Abraham, A.; Krasnodembskaya, A. Mesenchymal stem cell-derived extracellular vesicles for the treatment of acute respiratory distress syndrome. Stem Cells Transl. Med. 2020, 9, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Park, A.; Oh, M.; Lee, S.J.; Oh, K.J.; Lee, E.W.; Lee, S.C.; Bae, K.H.; Han, B.S.; Kim, W.K. Mitochondrial transplantation as a novel therapeutic strategy for mitochondrial diseases. Int. J. Mol. Sci. 2021, 22, 4793. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Y.; Qi, Z.; Cao, L.; Ding, S. Mitochondrial transfer/transplantation: An emerging therapeutic approach for multiple diseases. Cell Biosci. 2021, 12, 66–96. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Central roles of mitochondria in human diseases. Abbreviations: CoA, coenzyme A; NADH, nicotinamide adenine dinucleotide; ATP, adenosine triphosphate; ADP, adenosine diphosphate; FADH2, flavine adenine dinucleotide; TCA, tricarboxylic acid; MCU, mitochondrial calcium uniporter; VDAC1, voltage-dependent anion channel 1; IP3R, inositol 1,4,5-trisphosphate receptor; RyR, ryanodine receptor.
Figure 1.
Central roles of mitochondria in human diseases. Abbreviations: CoA, coenzyme A; NADH, nicotinamide adenine dinucleotide; ATP, adenosine triphosphate; ADP, adenosine diphosphate; FADH2, flavine adenine dinucleotide; TCA, tricarboxylic acid; MCU, mitochondrial calcium uniporter; VDAC1, voltage-dependent anion channel 1; IP3R, inositol 1,4,5-trisphosphate receptor; RyR, ryanodine receptor.

Figure 2.
Mitochondrial biology in age-related disorders. Abbreviations: MAMs, mitochondria-associated endoplasmic reticulum membranes; MCU, mitochondrial calcium uniporter; VDAC1, voltage-dependent anion channel 1; IP3R, inositol 1,4,5-trisphosphate receptor; RyR, ryanodine receptor; OXPHOS, oxidative phosphorylation; ETC, electron transport chain; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis.
Figure 2.
Mitochondrial biology in age-related disorders. Abbreviations: MAMs, mitochondria-associated endoplasmic reticulum membranes; MCU, mitochondrial calcium uniporter; VDAC1, voltage-dependent anion channel 1; IP3R, inositol 1,4,5-trisphosphate receptor; RyR, ryanodine receptor; OXPHOS, oxidative phosphorylation; ETC, electron transport chain; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis.

Figure 3.
The Warburg effect in cancer.


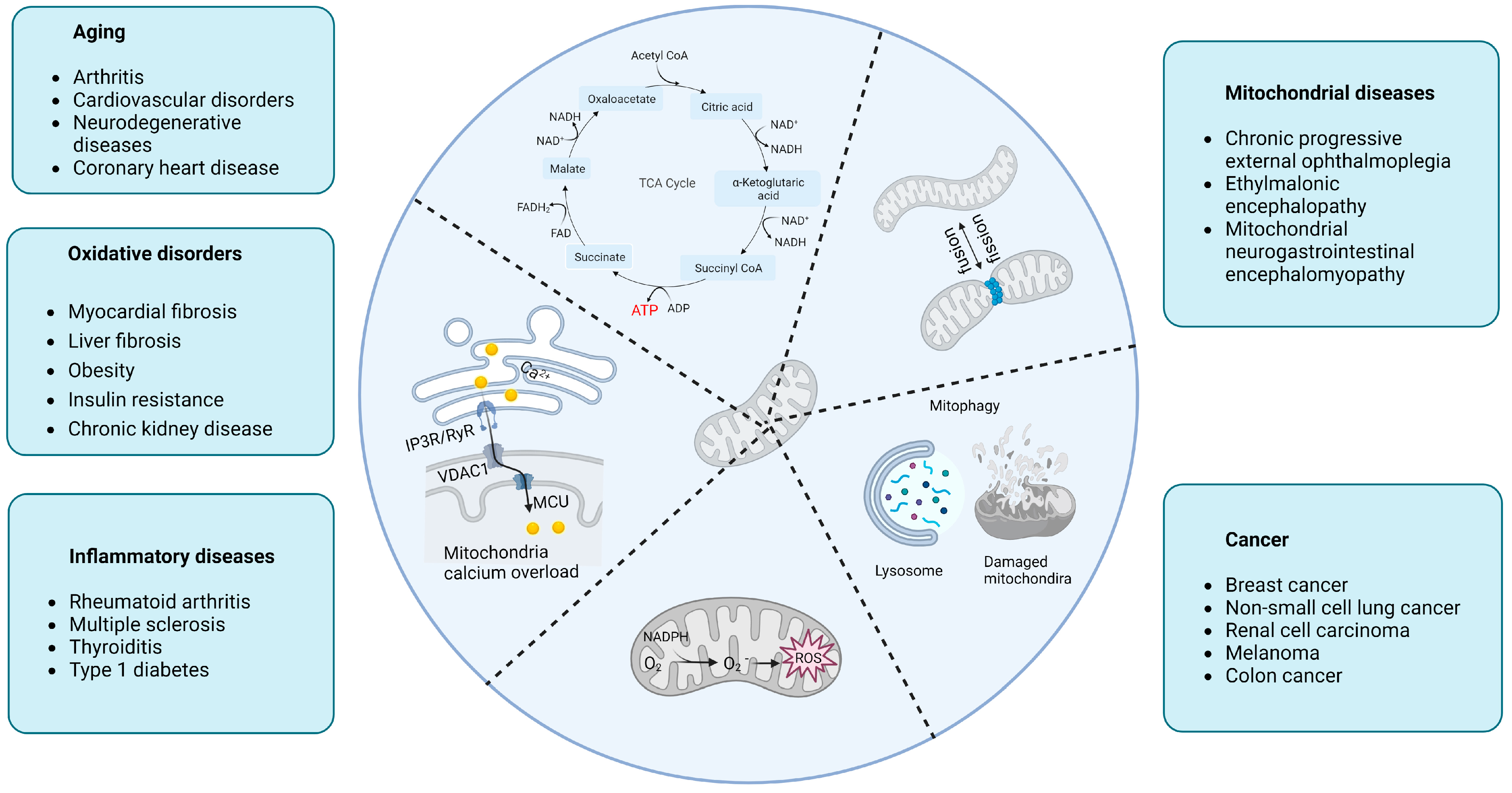{kind=link}
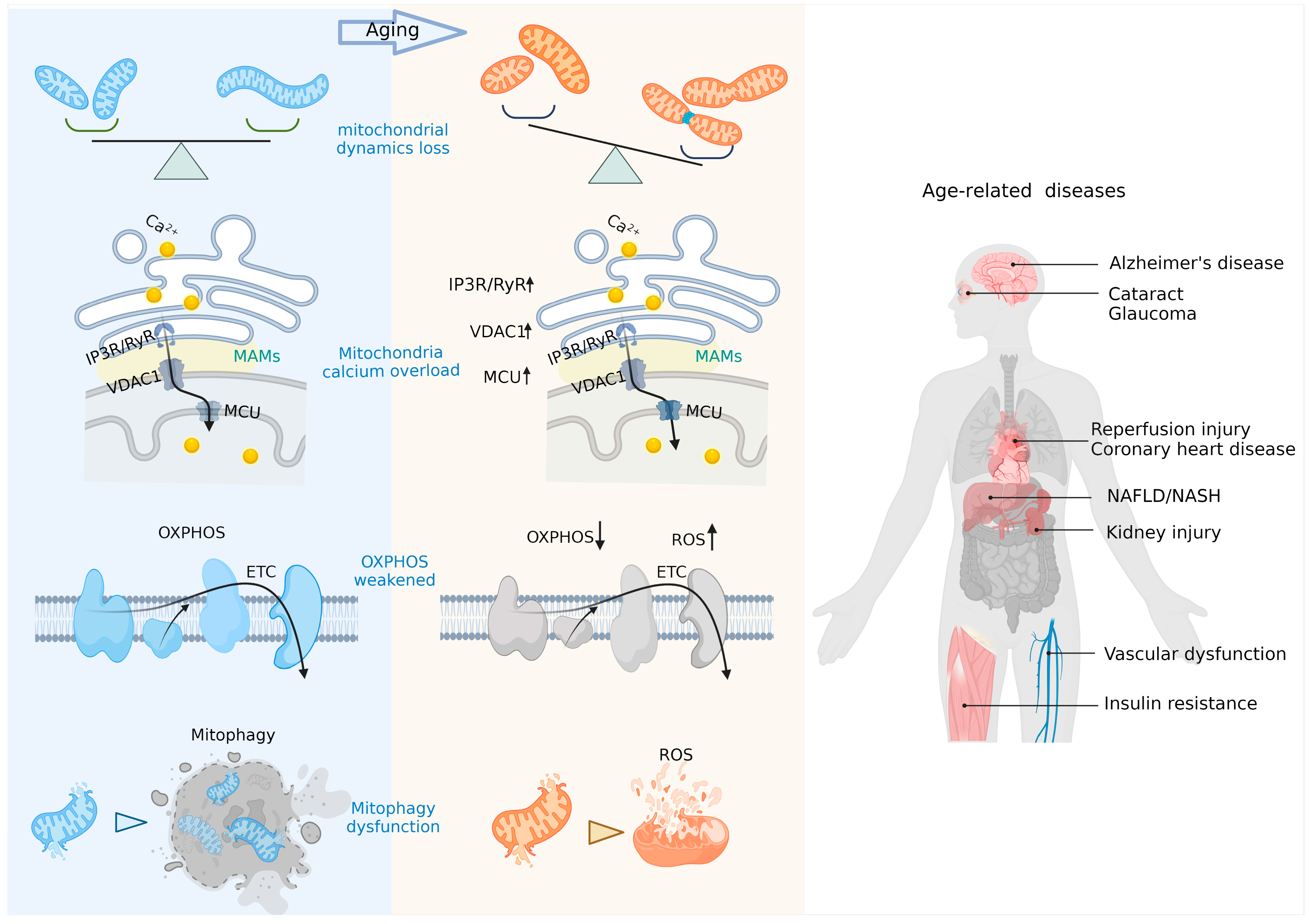{kind=link}
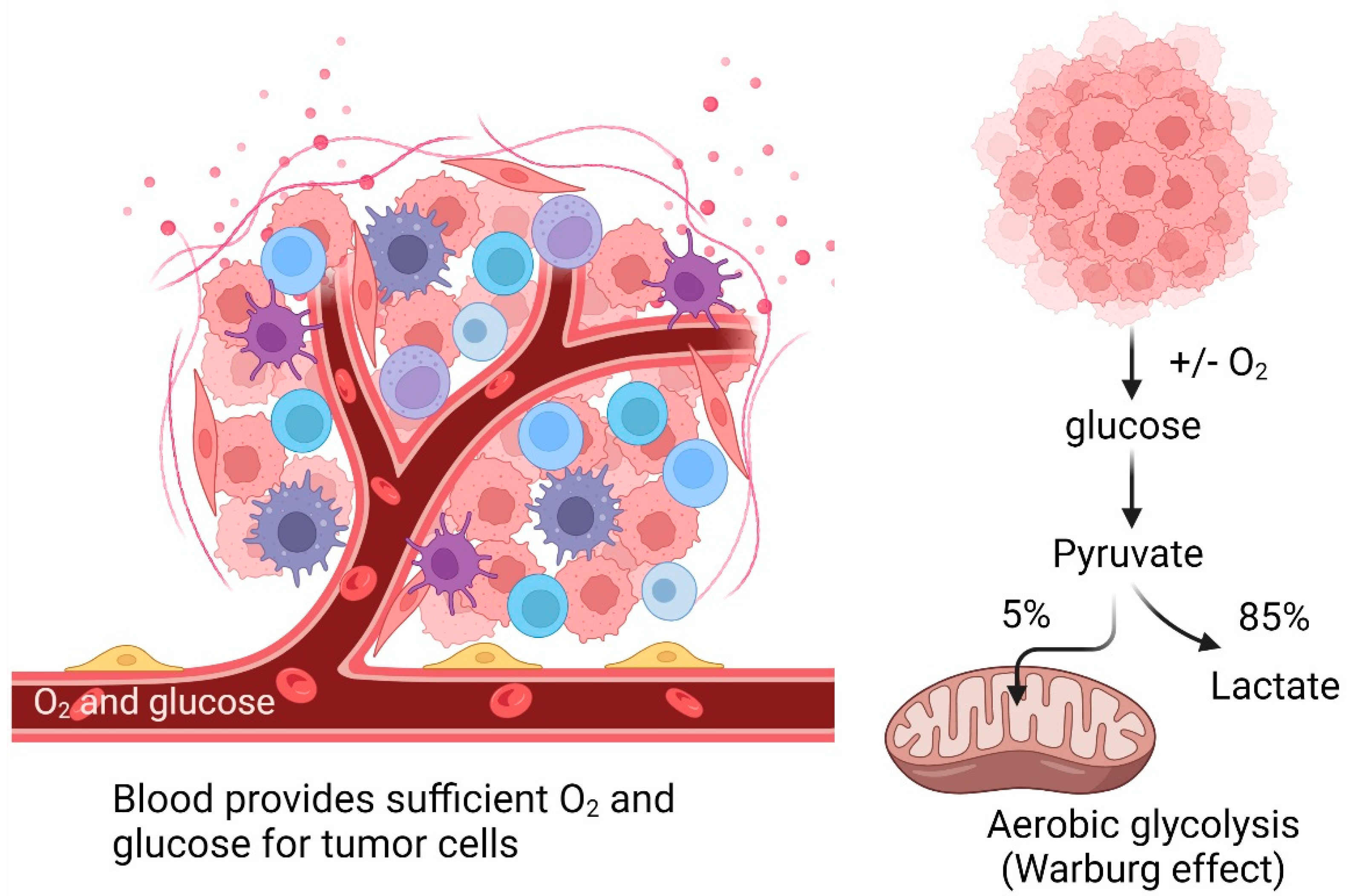{kind=link}
Table 1.
Factors involved in age-related human mitochondrial diseases.
| Factors | Models | Mechanisms | Refs. |
|---|---|---|---|
| c-myc | aging cardiomyocytes | mitochondrial metabolism | [18] |
| PGC-1α | age-related pathologies (muscle, heart, liver, brain) | mitochondrial metabolism | [19,20] |
| Pim-1 | myocardial infarction | calcium homeostasis, mitochondrial function | [18,21] |
| PUM2 | aged nematodes, aged mouse muscle cells | mitochondrial dynamics, mitophagy | [22] |
| PFKFB3 | cerebral ischemia-reperfusion injury | mitochondrial energy metabolism | [23] |
| ATF3 | idiopathic pulmonary fibrosis | mitochondrial homeostasis | [24] |
| SIRT3 | osteoporosis, cardiac hypertrophy | mitochondrial permeability, mitophagy | [25,26] |
| SIRT4 | ionizing radiation aging | mitochondrial dynamics, mitophagy | [27] |
| NEAT1 | chronic obstructive pulmonary disease | mitophagy | [28] |
| CoQ | heart and liver of aged mice | ETC | [29] |
| MitoQ | age-related endothelial dysfunction | oxidative damage to mitochondria | [30] |
| BGP-15 | type 2 diabetes mellitus-associated cardiac dysfunction | ETC | [31] |
| GlcN | extends lifespan in nematodes and mice | mitochondrial metabolism | [32] |
| IP3R2 | age-related liver fibrosis | MAMs | [33] |
| CISD2 | premature aging | autophagy | [34] |
| isradipine | Parkinson’s disease | calcium uptake | [35] |
| verapamil | age-related hematopoietic dysfunction | age-related hematopoietic stem cell dysfunction | [36] |
| α-Klotho | regeneration of aging muscles | maintenance of mtDNA integrity, mitochondrial function | [37] |
| DRP1 | extending the lifespan of Drosophila melanogaster | mitochondrial fission | [38] |
| FIS1 | skeletal muscle aging | mitochondrial morphology | [39] |
| MIRO1 | neuron disease | mitochondrial dynamics | [19] |
Abbreviations: c-myc, cellular Myelocytomatosis; PGC-1α, peroxisome proliferator activated receptor gamma coactivator 1 alpha; Pim-1, proviral insertion in murine 1; PUM-2, pumilio2; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; ATF3, activating transcription factor 3; SIRT3, sirtuin 3; SIRT4, sirtuin 4; NEAT1, nuclear enriched abundant transcript 1; CoQ, coenzyme Q; MitoQ, mitoquinone; BGP-15, O-(3-piperidino-2-hydroxy-1-propyl)nicotinic amidoxime; GlcN, D-glucosamine; IP3R2, inositol 1,4,5-triphosphate receptor 2; CISD2, CDGSH iron sulfur domain 2; DRP1, cytosolic dynamin-related protein 1; FIS1, fission protein-1; MIRO1, mitochondrial Rho 1; ETC, electron transport chain; MAMs, mitochondria-associated endoplasmic reticulum membranes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhong, G.; Venkatesan, J.K.; Madry, H.; Cucchiarini, M. Advances in Human Mitochondria-Based Therapies. Int. J. Mol. Sci. 2023, 24, 608. https://doi.org/10.3390/ijms24010608
AMA Style
Zhong G, Venkatesan JK, Madry H, Cucchiarini M. Advances in Human Mitochondria-Based Therapies. International Journal of Molecular Sciences. 2023; 24(1):608. https://doi.org/10.3390/ijms24010608
Chicago/Turabian StyleZhong, Gang, Jagadeesh K. Venkatesan, Henning Madry, and Magali Cucchiarini. 2023. "Advances in Human Mitochondria-Based Therapies" International Journal of Molecular Sciences 24, no. 1: 608. https://doi.org/10.3390/ijms24010608
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.