
GABA Release from Astrocytes in Health and Disease

Institute of Physiology, University Medical Center of the Johannes Gutenberg University, Duesbergweg 6, 55128 Mainz, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(24), 15859; https://doi.org/10.3390/ijms232415859
Submission received: 14 November 2022
/
Revised: 6 December 2022
/
Accepted: 10 December 2022
/
Published: 13 December 2022
(This article belongs to the Special Issue The Role of Glial Cells in Health and Disease)
Abstract
:Astrocytes are the most abundant glial cells in the central nervous system (CNS) mediating a variety of homeostatic functions, such as spatial K+ buffering or neurotransmitter reuptake. In addition, astrocytes are capable of releasing several biologically active substances, including glutamate and GABA. Astrocyte-mediated GABA release has been a matter of debate because the expression level of the main GABA synthesizing enzyme glutamate decarboxylase is quite low in astrocytes, suggesting that low intracellular GABA concentration ([GABA]i) might be insufficient to support a non-vesicular GABA release. However, recent studies demonstrated that, at least in some regions of the CNS, [GABA]i in astrocytes might reach several millimoles both under physiological and especially pathophysiological conditions, thereby enabling GABA release from astrocytes via GABA-permeable anion channels and/or via GABA transporters operating in reverse mode. In this review, we summarize experimental data supporting both forms of GABA release from astrocytes in health and disease, paying special attention to possible feedback mechanisms that might govern the fine-tuning of astrocytic GABA release and, in turn, the tonic GABAA receptor-mediated inhibition in the CNS.
1. Introduction
Astrocytes are the main glial cells in the central nervous system (CNS). Being non-excitable cells, astrocytes fulfill multiple functions, including extracellular ion homeostasis, neurotransmitter uptake, regulation of synapse number, or controlling local blood flow (for recent review, [1,2]). Astrocytes can sense neuronal activity via a variety of membrane-located ionotropic and metabotropic receptors, including the receptors for neurotransmitters glutamate, GABA or ATP, and thereby actively adapt their functions to the physiological requirements (for review, [3,4,5,6,7]). Receptor activation on astrocytes can induce Ca2+ and Na+ transients, which frequently propagate via the astrocytic syncytium in the form of waves [8,9].
In addition to their homeostatic uptake functions, astrocytes can release biologically active substances, the so-called gliotransmitters, including glutamate [10], D-serine [11], and ATP [12]. Astrocytes possess various mechanisms, which enable the release of substances into the extracellular space, including vesicular release ([10], for review [13]), release via purinergic P2X7 receptors [14] or “hemichannels” [15], via the reversal of the neurotransmitter transporter ([16], for review [17]) and through volume-sensitive anion channels [18]. Originally, all the above pathways have been described for glutamate, the main excitatory neurotransmitter in the brain. However, the results of recent studies demonstrate that GABA, the main inhibitory neurotransmitter in the CNS, can be also released from astrocytes. Similar to neuronal GABA transporters (GATs, for review [19]) astrocyte-located GATs can operate in the reverse mode, i.e., releasing GABA [20,21]. In addition, bestrophins 1 (Best1), originally characterized as a swelling- and Ca2+-activated Cl− channel [22], has been demonstrated to be permeable not only for glutamate but also for GABA [23]. Best1 channel was originally identified in the retinal pigment epithelium [24], and its mutations have been demonstrated to underlie several retinopathies (for review, [25]). The Best1-mediated GABA release can potentially affect the excitation-to-inhibition (E/I) balance in the brain. Thereby, mutations in this channel have been attributed to neurodevelopmental disorders, including autism spectrum disorder (ASD) and attention deficit/hyperactivity disorder (ADHS, [26]).
However, previous immunohistochemical data reported low levels of the GABA synthesizing enzyme glutamate decarboxylase (GAD) in astrocytes, suggesting that the intracellular GABA concentration ([GABA]i) is too low for non-vesicular release [27]. However, recent data demonstrate that astrocytes in various brain regions are not only capable of synthesizing sufficient amounts of GABA, partially via GAD independent pathways, but can also release GABA (see below). In addition, there is evidence that both processes are strongly modulated under pathological conditions [28,29].
In this review, we summarize the available results about GABA release from astrocytes in health and disease and discuss a possible involvement of several release mechanisms to tune the extracellular GABA concentration ([GABA]o) and in turn the tonic GABAA receptor (GABAAR)-mediated inhibition.
2. GABA Synthesis in Astrocytes
Up to recently, the GABA has been considered to be synthesized mostly by neurons. This conclusion was based on biochemical data, suggesting that the astrocyte expression of GAD, as the classical enzyme synthetizing GABA is too low to produce a physiologically relevant amount of GABA [27,28]. In line with this hypothesis, previous studies revealed that the GABA concentration in cultured astrocytes ranges from 0.2 to 0.9 mM [30]. Only after transfection of an astrocytic cell line (C8S) with isoform 67 of the of the glutamate decarboxylase (GAD67), these transfected astrocytes are capable of synthesizing and even releasing GABA via GATs [31]. However, in addition to the GAD-dependent pathway, GAD-independent pathways for the synthesis of GABA have been described (for recent reviews, [2,6]). Cultured astrocytes are able to synthesize GABA from the polyamine putrescine [32,33], thereby utilizing a GAD-independent pathway of GABA synthesis (Figure 1). Recent studies performed both in brain slices and in vivo confirmed the capability of astrocytes to synthesize GABA in both GAD-dependent and GAD-independent ways, and moreover, that the amount of synthesized GABA depends on physiological/pathophysiological conditions [23,33,34,35,36,37].
Immunohistochemical approaches revealed that both Bergmann glial cells and lamellar astrocytes in the cerebellum contain a detectable amount of GABA under physiological conditions. Using an electrophysiological approach, the minimal [GABA]i required to support the measured level of tonic GABAAR-mediated inhibition is estimated to be about 3 mM [23]. GABA synthesis in Bergmann glial cells is driven by monoamine oxidase B (MAO-B) from the polyamine putrescine ([23], Figure 1). Using immunogold labeling technique, the GABA concentration in Bergmann glial cells is estimated to be 5–10 mM [36,38]. In Alzheimer’s disease mouse models, MAO-B-mediated GABA synthesis in reactive astrocytes in the hippocampus is potentiated by about five times as compared to control animals [35,37]. Additionally, it has been demonstrated that thalamic astrocytes synthesize GABA via diamine oxidase (DAO) and aldehyde dehydrogenase 1a1 (Ald1a) ([34], Figure 1). In this case, intra-astrocytic GABA concentration has been estimated to be between 4 and 14 mM [34]. Intriguingly, in hippocampal astrocytes from human Alzheimer patients and from 5xFAD mice, an animal model of Alzheimer disease [39], elevated levels of GAD67 have been reported [28]. It was demonstrated that under these conditions, astrocytes are not only GABA synthesizing cells, but can release GABA, supporting tonic inhibition of granule cells in the dental gyrus [28].
The above-mentioned data demonstrates that the [GABA]i of astrocytes is region dependent and may be as high, at about 10 mM. The extracellular GABA concentration ([GABA]o) is region dependent as well and amounts to 0.25 µM in the cortex [40], 0.8 µM in the hippocampus [41] and 3.5 µM in thalamus [34]. Such a strong transmembrane GABA gradient might enable GABA release, both via GABA-permeable channels, following the chemical gradient, or via secondary active GABA transporters, using the electrochemical driving force.
3. GABA Release from Astrocytes in Health
3.1. Volume-Regulated Anionic Channels—Bestrophin1
As astrocytes express aquaporins [42], their membrane is permeable for water. Osmotic gradients between the extracellular and intracellular solutions drive water fluxes across the cellular membrane, influencing the cell volume in this way. An orchestrated functioning of ionic channels and transporters is required to keep the cell volume unchanged or allow controlled cell swelling or shrinkage (for recent reviews, [43,44]). Several volume-gated ion channels contribute to the volume control of astrocytes. Volume-regulated anionic channels are outwardly rectifying anion channels activated by cell swelling, encoded by the SWELL1 gene, and permeable for larger anionic or uncharged molecules such as glutamate or GABA [45].
Bestrophins are a family of swelling- and Ca2+-activated Cl− channels [46]. Four bestrophins (Best1–4) have been identified [47]. Bestrophins were identified in the human genome in relation with the disease Best vitelliform macular dystrophy (BVMD, [48]). More than 200 mutations in Best1 gene are known to be linked with different retinopathies [25,49]. The Best1 channel is predominantly expressed in the retinal pigment epithelium and is localized to the basolateral plasma membrane [24]. Best1 is characterized as a protein with multiple functions, including a Ca2+-activated anion channel, a Ca2+ channel modulator and a volume-sensitive anion channel (for review, [47,50]). In addition to Cl− ions, all bestrophins are permeable for HCO3− [51] and for relatively large anions, such as aspartate and glutamate (Figure 2A, for recent review; [52,53]). These properties of Best1-channel lead to the hypothesis that distorted regulation of intracellular Ca2+ signaling [54,55] and/or pH homeostasis [51] may underlie the Best1 mutation-related retinopathies (for recent review, [25]). Moreover, diabetic retinopathy is accompanied by a reduction in GABA release in the retina, leading to hyperexcitation (for review, [56]), but the role of possible Best1-mediated GABA release in the retina needs further investigation.
In situ hybridization and immunohistochemical data demonstrated a strong Best1 expression in the hippocampus, thalamus and cerebellum, both in neurons and astrocytes [57]. The functional expression of Best1 has been confirmed by electrophysiological experiments in cultured mouse hippocampal astrocytes [57]. In addition, the observation that the application of the hypo-osmic solution stimulated the non-synaptic, astrocyte-mediated release of both GABA and glutamate in the mouse hippocampus, suggests a possible involvement of volume-regulated channels permeable for both glutamate and GABA [58]. Recent studies reported that Best1 is also permeable for GABA, with a GABA permeability of 0.27 as compared to Cl− [59,60], which is lower than that for glutamate (0.47–0.67). However, the GABA permeability of Best1 was estimated electrophysiologically, despite the fact that GABA is a zwitterionic amino acid and mostly uncharged at physiological pH. Thus, it can be assumed that the GABA permeability of Best1 may be much higher. This question definitely needs further investigations.
Lee at al. [23] performed the first detailed study that confirmed the Best1-mediated GABA release from astrocytes in the cerebellum. Best1 is a Ca2+-activated anionic channel and its half-maximal activation was determined to be at [Ca2+]i of 150 nM. This concentration is quite close to the resting [Ca2+]i of astrocytes, suggesting the possibility of tonic GABA release under resting conditions. Pharmacological blockade or genetic silencing of Best1 in cerebellar astrocytes reduces the GABAA receptor (GABAAR)-mediated tonic current in cerebellar granule cells by about 75%. These results indicate that Bergmann glia cells and lamellar astrocytes in the mouse cerebellum tonically release GABA via Best1 channels under resting physiological conditions [23]. Subsequent studies by Yoon et al. revealed that Best1 channels are strongly expressed in several thalamic nuclei [61]. Thalamo-cortical projection neurons in the ventrobasal (VB) nucleus of thalamus exhibit sustained tonic GABAAR-mediated currents, which remain unchanged after blocking phasic GABA release, indicating a non-vesicular source of extracellular GABA [34]. As [GABA]o in thalamus amounts to 3.5 µM [34], it seems unlikely (see Equation (1) and Figure 2) that the reverse mode of these GABA transporters mediates sufficient GABA release. However, the tonic GABAAR-mediated current is reduced to about 30% by the pharmacological blockage of Best1 or selective genetic ablation of Best1 in thalamic astrocytes [34]. Chelating of intracellular Ca2+ with BAPTA significantly reduces the tonic GABAAR-mediated currents in thalamic neurons, while they are potentiated by an elevation of [Ca2+]i in astrocytes, confirming the involvement of the Ca2+-modulated Best1 channel. Thus, similar to cerebellar astrocytes, [GABA]i is quite high in thalamic astrocytes and Best1 channels the mediate constitutive [Ca2+]i dependent release GABA under resting physiological conditions [34].
In addition to the cerebellum and thalamus, the measurable expression of Best1 channels has been observed in hippocampal astrocytes [57,61]. However, in the hippocampus, the main source of the extracellular GABA is the phasic GABAergic transmission mediated by interneurons [62], and the pyramidal cell do not demonstrate any tonic GABAergic inhibition unless GAT1 is blocked [63]. As only about 20% of hippocampal astrocytes are GABA positive, the GABA release from astrocytes under physiological conditions is not detectable despite the strong Best1 expression in hippocampal astrocytes [61]. Moreover, astrocytic Best1 channels are located in the astrocytic distal processes and positioned closely to excitatory synapses, indicating that probably glutamate, rather than GABA, is released via Best1 channels in the hippocampus under physiological conditions [61,64].
The hypothesis that the spatial distribution of Best1 channels in the astrocytic membrane may depend on [GABA]i has been addressed in a recent study [65]. In control mice, astrocytes in the hippocampal CA1 region containing a low amount of GABA and Best1 channels are mostly located in the proximity of glutamatergic synapses. In the APP/PS1 mouse model of Alzheimer’s disease, the MAO-B activity is potentiated in hippocampal astrocytes, resulting in an elevated [GABA]i [35]. Under these conditions, the spatial Best1 distribution is shifted closer to GABAergic synapses, while in the hippocampus of MAO-B-KO mice (lower [GABA]i), the Best1 channels were located closer to glutamatergic synapses [65].
In summary, in brain regions where resting GABA concentration in astrocytes is high, namely in the cerebellum and thalamus, the tonic Best1-mediated GABA release takes place under resting physiological conditions. The amount of GABA released is modulated by intracellular Ca2+ activity and the transmembrane GABA gradient. However, the Best1 permeability for GABA, and so the selectivity for GABA as compared to glutamate, remains to be further investigated. Low intracellular GABA concentration probably results in the Best1 channel relocation closer to glutamatergic synaptic contacts and switches the released gliotransmitter to glutamate.
3.2. GAT-Mediated GABA Release
In the classical view, GATs are supposed to remove GABA from the extracellular space. GAT-mediated uptake of one GABA molecule is accompanied with the cotransport of one Cl− and two Na+ ions [66]. Because under physiological pH GABA is predominantly a zwitterionic molecule, the transport process is electrogenic.
As all three molecules/ions, namely GABA, Na+ and Cl−, are transported independently from each other, the GAT reversal potential can be estimated using the following thermodynamic equation [17]:
Importantly, the GAT-mediated transport process can be switched into the opposite direction, the reverse mode, thereby releasing GABA. GAT reversal occurs if the membrane potential is positive to the GAT reversal potential [17], which can be caused by a membrane depolarization or if the intracellular Na+ concentration ([Na+]i) increases high enough (Figure 2B). Indeed, the depolarization-induced GAT-mediated GABA release was reported from retinal horizontal cells, growth cones of neurons and various neurons in cultures or acute brain slices [19,67,68,69]. In addition, tonic GABAAR-mediated inhibition was reported to be mediated by the GAT3-mediated GABA release in the acute slice preparation of the neocortex [20]. In the marginal zone of the early postnatal cortex, the GAT3-mediated GABA release was demonstrated to result in a presynaptic GABABR-mediated suppression of GABAergic transmission [70]. Interestingly, GAT1 is reported to be expressed both in neurons and astrocytes [71], while GAT3 is predominantly located to astroglial processes [72], suggesting that GAT-3, in particular, contributes to the tonic GABA release from astrocytes in the neocortex. In the hippocampus, however, astrocytes are capable of releasing GABA only after a strong elevation of [Na+]i, as a result of facilitating the EAAT-mediated glutamate uptake, occurring, for example, during epileptic-like activity [21,73].
GAT reversal potential in astrocytes can be estimated from Equation (1) and values available in the literature ([Na+]o = 140 mM, [Cl−]i = 135 mM, [Na+]i = 15 mM [74,75,76,77], [Cl−]i = 30–40 mM [78,79]. The extracellular GABA concentration ([GABA]o) strongly depends on the brain region. In the early postnatal cerebral cortex, [GABA]o has been reported to be 0.25 µM [40], in the adult hippocampus ~0.8 µM [41] and in the thalamus ~3.5 µM [34]. Using these numbers, one can estimate the [GABA]i required to set the GAT reversal potential close to the resting potential of astrocytes (ca. −80 mV), obtaining a [GABA]i of ~1 mM in the cerebral cortex, ~4 mM in the hippocampus, and ~10 mM in the thalamus (Figure 2C). These estimates demonstrate that GAT reversal under resting conditions is more probable in the cerebral cortex [20,76]. Moreover, the relative position of the reversal potential, and thus the direction and strength of the astrocytic tonic GABA release can be modulated by [Na+]i transients, occurring for example via the activation of EAATs (Figure 3A [80]).
In the hippocampus, the main source of extracellular GABA is supposed to have a neuronal origin, and GAT1 operating in the uptake mode is the dominant GAT isoform [41,82]. However, further studies revealed that astrocytic GAT3 contributes to setting the GABA levels in the extracellular space [83]. However, under physiological conditions, the astrocytic [GABA]i appears to be relatively low, and the GAT3 operate in the uptake mode, reducing extracellular [GABA]o. This suggests that in the hippocampus astrocytic, [GABA]i is similar to that in the cerebral cortex, i.e., about 1 mM [73], and [GABA]o amounts to 0.8 µM [41,73]; an elevation of [Na+]i to about 25–30 mM is required to result in GAT reversal (Figure 2D). Because EAATs cotransport three Na+ ions with one glutamate molecule, EAAT-mediated glutamate uptake strongly affects [Na+]i [76]. Consequently, elevated neuronal activity, for example epileptic-like activity, could potentially reverse GAT3. Indeed, the activation of EAATs with the EAAT substrates glutamate or aspartate leads to the GABA release from astrocytes [21]. Interestingly, the GAT1-mediated GABA uptake does not reverse during epileptic-like activity [84]. As GAT1 in the hippocampus is located predominantly to neurons, this result can be explained by a lower resting [Na+]i, of about 8 mM in neurons [85]. Thus, because of the relatively high [Na+]i in glial cells, the astrocytic GAT3 may fulfill a specific protecting role under pathophysiological conditions. Indeed, using a low [Mg2+]o in vitro model of epilepsy, a GAT3-mediated GABA release has been demonstrated to reduce the duration of seizure-like events. In addition, the blockade of GAT3-mediated GABA release in vivo reduces the power of the gamma-range oscillation, indicating that the GAT3-mediated GABA release is functional under physiological conditions [73].
Similar to the hippocampus, GAT1 is the dominant GABA transporter in the striatum, a brain region in which extracellular GABA tunes the activity of output neurons [86] and dopamine release [87]. Inhibition of GAT1-mediated GABA uptake discloses an additional GAT3-mediated removal of GABA in the striatum [88]. The physiological role of GAT3-mediated release from astrocytes was investigated in the recent work of Yu at al. [81]. This group reduced intracellular Ca2+ activity selectively in striatal astrocytes by viral overexpression of the plasma membrane Ca2+ ATPase type 2 (PLMCA2). Interestingly, the dumping of astrocytic [Ca2+]i transients affected mouse behavior, as evident by potentiated self-grooming. Electrical activity of medium spiny neurons (MSNs), striatal output neurons, was reduced, although neither glutamatergic nor GABAergic phasic synaptic transmission has been modified. The observed reduction in MSN activity was mediated by tonic GABAergic inhibition via extrasynaptic GABAA receptors. Unexpectedly, the blockade of GAT3, but not of GAT1, rescued the strength of tonic GABAAR-mediated inhibition and alleviated the self-grooming behavior. The further experiments demonstrated that the reduction in Ca2+ activity reduced the number of GAT3 inserted in the plasma membrane, while the mere GAT3 expression was unaffected [81]. Thus Ca2+-dependent GAT3 trafficking determines the extracellular GABA concentration. This work demonstrates that astrocytes may control tonic GABAAR-mediated inhibition not necessarily via GABA release but also via the tunable GABA uptake.
In summary, tonic GAT3-mediated GABA release from astrocytes occurs in the cerebral cortex probably because of low [GABA]o. In case of higher extracellular GABA concentration, such as in the hippocampus, additional elevation of [Na+]i is required to change the direction of GAT3-mediated GABA transport. Furthermore, [Ca2+]i influences the number of plasma membrane located in GAT3 and in turn the strength of the GAT3-mediated release/uptake.
4. GABA Release from Astrocytes in Disease
4.1. Alzheimer’s Disease
Alzheimer´s disease (AD) is a neurodegenerative disorder with an increasing prevalence in aging populations. Several lines of evidence suggest that amyloid β (Aβ) peptide and tau protein aggregations trigger a cascade of pathological changes, including the weakening of synaptic transmission and synapse loss (for review, [89]). Astrocytes as a part of the tripartite synapse are involved in the development of AD pathology (for recent review, [90]).
Elevated tonic GABAAR-mediated inhibition in the dental gyrus in the hippocampus has been reported in the 5xFAD mouse model of AD [28]. Immunochemical data demonstrate increased GABA and glutamate levels in GFAP-positive reactive astrocytes. The elevated GABA concentration in astrocytes is accompanied by an increased level of GAD67. These changes are observed in 6- to 8-months old animals after the appearance of Aβ plaques, but not at younger ages (3- to 4 months). A similar elevation of GABA level in reactive astrocytes was detected in human AD patient brains [28]. In addition, immunostaining experiments revealed an increased expression of GAT3 both in mouse reactive astrocytes and human AD brain samples, suggesting that GAT3-mediated GABA release underlies the elevated tonic inhibition in these animals [28]. Indeed, the selective GAT3 blockade significantly reduces the tonic GABA current in granule cells and also rescues the synaptic plasticity and memory deficit in this mouse model [28]. In contrast, the blockade of GAT1 enhances tonic GABAAR-mediated currents, confirming that GAT1 in the AD hippocampus still operates in uptake mode [83]. Thus, in the hippocampus of the 5xFAD mouse model of AD, both GABA synthesis and release via GAT3 are specifically facilitated in reactive astrocytes, but not in neurons [28]. Similar results were obtained in another model of AD, the APPNL−F/NL−F knock-in mouse model [91]. Moreover, in this case, both GABA content and GAD67 expression are significantly increased in reactive astrocytes, leading to the GAT3-mediated GABA release and increased tonic inhibition of principal neurons in the CA1 and DG regions of the hippocampus [91]. Unfortunately, the expression of Best1 channel has not been inspected in these publications.
GABA release from reactive astrocytes has also been reported in the hippocampus of APP/PS1 mice, another mouse model of AD [35]. Extracellular GABA, but not glutamate levels are significantly increased in the APP/PS1 mice as compared with WT mice, leading to elevated tonic GABAAR-mediated currents. Immunostainings with antibodies against GABA demonstrated that the GABA content, as estimated from the intensity of immunoreactivity, in reactive astrocytes is comparable with that in GABAergic interneurons. In contrast to the results from the 5xFAD AD mouse model [28], the GAD activity is not potentiated in these APP/PS1 mice. Pharmacological tools revealed that the increased level of GABA in astrocytes results from the elevated MAO-B activity, confirming that GABA is synthesized from putrescine (Figure 1) in this case. Moreover, in contrast to the 5xFAD AD mouse [28], GAT1 and GAT3 expressions are not significantly altered in the APP/PS1 mice. The GABA release from reactive astrocytes has been demonstrated to occur via the Best1 channels [35]. Moreover, the Best1 membrane distribution is strongly changed, demonstrating in reactive astrocytes of APP/PS1 mice a major localization in soma and proximal dendrites, instead of the distal microdomain-like distribution observed in WT animals [35]. The GABA release from astrocytes in the hippocampus of APP/PS1 mice has been demonstrated to activate GABAB receptors at GABAergic interneurons, leading to the disinhibition of granule cells [92]. Interestingly, Jo at al. [35] reported that the Best1 silencing by means of shRNA blocks GABA release in the 5xFAD AD mouse model as well, which also suggests the Best1-mediated GABA release in this AD mouse model. These results contradict the GAT3-mediated release reported by Wu et al. and Aldabbagh et al. [28,91]. Unfortunately, this contradiction was not discussed in this publication. As the GABA elevation in reactive astrocytes and accumulation of Aβ plagues demonstrate a bell-shaped relationship [93], one may suggest that both the level of astrocytic GABA and active GABA release mechanism depend on the disease stage, similar to the data reported for epilepsy (see below, [94]).
4.2. Huntington’s Disease
Huntington´s disease (HD) is a neurodegenerative disorder triggered by poly-glutamine expansion in the huntington protein [95]. Previous studies reported decreased levels of EAATs, the astrocytic glutamate transporters [96,97,98], and suggested elevated extracellular concentration of glutamate [99] and excitotoxic cell death as a possible cause of HD symptoms´ manifestation. Parallel to glutamatergic system malfunctions, GABAergic signaling, including tonic GABAAR-mediated inhibition, is demonstrated to be distorted as well (for recent review, [100]). In the mouse striatum tonic, the GABAAR-mediated currents in striatal output neurons (SONs) under resting conditions were detected from P19 on. The blocking of GAT1, but not GAT3, discloses tonic currents at younger ages and potentiated them at older ones, indicating that GAT1 is the dominant GABA uptake mechanism in the striatum and operates in the uptake mode [86]. When GAT1 is pharmacologically blocked, the GAT3 also operates in the uptake mode [88]. However, as GAT1 is the main GABA uptake mechanism, its blockade would result in an elevation of [GABA]o, potentially affecting the direction of the GAT3-mediated GABA transport and thereby obscuring a putative release mode under these experimental conditions (Figure 2C).
Further experiments revealed that in WT animals about half of the striatal extracellular GABA is released synaptically, while the remaining half represents the GABA released via GAT3 from astrocytes [101]. In two mouse models of HD, namely Z-Q175-KI and R2/6, however, both the GAT3-mediated GABA release and tonic GABAAR-mediated currents in SONs have been strongly reduced. The increase in [Na+]i by the D-aspartate-triggered stimulation of EAAT switched the GAT3 in the release mode, similar to that in WT animals. Thus, it was suggested that in the healthy striatum, the EAAT-mediated glutamate uptake keeps GAT3 in the reverse mode by means of elevated [Na+]i (Figure 2D); meanwhile, in HD astrocytes, the reduced glutamate uptake and lower [Na+]i fail to reverse GAT3, and it operates in the uptake mode, leading to the hyperexcitability of SONs [101].
Using the R2/6 mouse model of HD, Jiang et al. [102] reported the reduced spontaneous, but potentiated evoked Ca2+ signaling in striatal astrocytes. The reduced expression of EAAT2 and the K+ channel Kir4.1 has been observed in this model, leading to the elevated extracellular glutamate levels and activation of metabotropic GluRs. Interestingly, these R2/6 mice demonstrate increased self-grooming behavior [103], comparable to the behavior observed after attenuating the tonic GABAAR-mediated inhibition in the striatum (by the enhanced Ca2+-dependent GAT3-mediated astrocytic GABA uptake upon PLMCA2 overexpression) [81]. Ca2+ signaling in striatal astrocytes was reduced in this HD mouse model [103], leading to reduced extracellular GABA and reduced tonic GABAAR-mediated inhibition. Thus, it is possible that the facilitated GAT3-mediated GABA uptake underlies the observed changes, similar to the results obtained in the WT mice [81].
The source of astrocytic GABA has been investigated by Yoon et al. [38]. They show that astrocytes in the striatum are capable of synthesizing GABA from putrescine via MAO-B pathway (Figure 1). In line with Wojtowicz et al. [101], astrocytes release GABA under resting physiological conditions. Although the Best1-mediated GABA release from astrocytes has been suggested by Yoon et al., the authors did not provide any data that would allow one to distinguish between GAT3- and Best1-mediated release mechanisms [38].
In summary, the experimental data demonstrate that the GABA release from striatal astrocytes is reduced in HD. However, the question whether the observed reduction results from (1) reduced [Na+]i and switch of GAT3 in uptake mode; (2) reduced number of membrane-located GAT3; or (3) reduced [Ca2+]i with decreased Ca2+-dependent Best1 conductance, remains elusive.
4.3. Autism Spectrum Disorder (ASD)
ASD is a neurodevelopmental disorder characterized by repetitive behaviors, communication deficits and disturbed sensory perception. The etiology of ASD remains elusive. The main working hypothesis is that the excitation/inhibition (E/I) imbalance leads to the manifestation of ASD symptoms (for recent review, [104,105]). An elevated E/I ratio might result from changes in glutamatergic and/or GABAergic signaling. Indeed, the selective stimulation of excitatory neurons in the mouse medial prefrontal cortex in vivo is sufficient to induce social behavior deficits, while the selective activation of inhibitory interneurons rescues the observed impairments [106]. In addition to the potentiated glutamatergic synaptic inputs, elevated levels of glutamate in the extracellular space may also lead to excitotoxicity in ASD [107]. Extracellular glutamate concentration is set by the activity of EAATs. Astrocyte-located EAAT2 plays a critical role in controlling extracellular glutamate level and its genetic silencing results in epileptic seizures and premature death [108]. The conditional deletion of EAAT2 in astrocytes reduced its expression in the cortex and striatum by 60–80% [109]. Such a reduction in astrocytic EAAT2 failed to affect either the kinetics of cortico-striatal EPSCs or the paired-pulse ratio of EPSCs, but significantly attenuated the reduction in the EPSC amplitude during repetitive stimulation. These results indicate that the reduced uptake of glutamate by astrocytes potentiates the glutamatergic drive, i.e., shifts the E/I balance to excitation, leading in turn to self-grooming (ASD-like) behavior [109].
Similar to the excitatory glutamate action, the reduction in the extracellular GABA level may shift the E/I ratio towards excitation. Diminishing the Ca2+ signaling by the overexpression of Ca2+ extruding ATPase in hippocampal astrocytes results in repetitive behavior, typical for ASD. Further analysis revealed no change in EAAT2 expression and a strong overexpression of GAT3, the astrocytic GABA transporter [81]. As GAT3 in the hippocampus under control conditions operates in the uptake mode [83], the GAT3 over-activity reduces the extracellular GABA level and shifts the E/I balance towards excitation. As astrocytic EAAT activity may reverse the Na+/Ca2+ exchanger via the [Na+]i increase [110], intra-astrocytic sodium and calcium signaling may set the EAAT/GAT expression balance and E/I ratio. Interestingly, the E/I imbalance resulting from a distorted balance of astrocytic GAT3 and EAAT2 seems to also underlie the obsessive compulsive disorders (for recent review, [111]).
4.4. Epilepsy
Epilepsy is a disorder characterized by excessive network activity. The shift of excitation-to-inhibition (E/I) balance towards excitation is generally suggested to underlie the occurrence of spontaneous network discharges, which manifest as seizures (for review [112,113]). The pharmacological enhancement of GABAergic (inhibitory) transmission may be beneficial to suppress hyperexcitability and prevent epileptic seizures [114,115,116]. In this context, the GABA release from astrocytes and resulting tonic GABAAR-mediated inhibition might be a potent tool to rescue the distorted E/I balance [117].
As GABA concentration in hippocampal astrocytes under physiological conditions is relatively low, GATs operate in uptake mode under the control condition [84]. However, the EAAT blockade in vitro and in vivo results in an elevation in the extracellular GABA concentration [21,73]. The observed GABA release is mediated by astrocytic GAT3 [21], leading to the conclusion that the EAAT-induced [Na+]i elevation can switch the GAT3 into reverse mode (Figure 2D). Such increased [Na+]i may occur under epileptic conditions. Indeed, Heja et al. [21,73] have demonstrated that hippocampal astrocytes are capable of both GABA synthesis from putrescine (Figure 1) and EAAT-dependent GAT3-mediated GABA release. In the low-Mg2+ model of epilepsy, the GAT3-mediated GABA release significantly shortens seizure-like events, confirming the inhibitory role of GABA release from astrocytes [21,73]. Similar results were obtained in vivo in a rat model of absence epilepsy, the WAG/Rij model [118,119]. Moreover, these in vivo studies also revealed that the astrocytic polyamine metabolism may modulate the neuronal activity in several ways, including the GAT3-mediated GABA release [118,119].
The strong reduction in the GABAergic interneuron number has been demonstrated in the human and different mouse model of temporal lobe epilepsy [29,120,121,122]. GABAergic interneurons represent the main source of extracellular GABA in the healthy hippocampus [62], and thus the decreased interneuron number should in turn weaken the phasic GABA release. However, unexpectedly, the tonic GABAAR-mediated inhibition was elevated already at day 5 after epilepsy induction in the intracortical kainate mouse model [29]. The [GABA]i in hippocampal astrocytes increased from day 5 on and became more pronounced at day 14 and 28. The facilitated GABA synthesis in reactive astrocytes was demonstrated to be mediated by both MAO-B and GAD [29]. Interestingly, the potentiation of MAO-B and GAD activity in astrocytes has been observed also contralaterally; however, without any change in the strength of tonic inhibition on this side. In contrast to the results obtained in the WAG/Rij rat model [118,119], GAT3 did not mediate the facilitated GABA release from the astrocytes in this model of epilepsy [29].
While the mechanism of GABA release was not analyzed by Müller et al. [29], a recent study by Pandit et al. addressed the mechanism of GABA release from astrocytes in the mouse hippocampus in the kainic acid (KA) and pilocarpine (PI) models of epilepsy [94]. In both models on the third day after KA or PI injection, the hippocampal astrocytes demonstrate an increased expression of GFAP, a sign of reactive astrocytes, and an elevated [GABA]i. Interestingly, [GABA]i returns to control level at day 7 after KA/PI injection [94], in contrast to the elevated astrocytic [GABA]i at day 14 reported in [29]. Similar to [GABA]i, Best1 expression is strongly increased at day 3 and returns almost to the control level thereafter [94]. Phasic GABAergic synaptic transmission is not affected, while the tonic GABAAR-mediated currents in CA1 pyramidal neurons are strongly increased. The pharmacological blockade of Best1 channels reduces the strength of the tonic GABAAR-mediated inhibition to about 30%. Moreover, the selective expression of Best1 channels in hippocampal astrocytes in Best1-KO mice essentially suppresses seizure susceptibility, indicating an essential role of the Best1-mediated GABA release under epileptic conditions [94]. GAT1 was demonstrated to operate in the uptake mode. Unfortunately, nipecotic acid, a non-selective GAT antagonist, was used to exclude the GAT3-mediated GABA release. As the GAT1 blockade would definitely increase [GABA]o and potentially switch GAT3 in the uptake mode (Figure 2C), a possible role of the GAT3-mediated release for the increased tonic GABAAR-mediated inhibition requires further investigations.
In summary, the epileptic activity stimulates (1) GABA synthesis in hippocampal astrocytes and (2) results in astrocytic GABA release. The question of whether the GABA release is driven by the EAAT-mediated [Na+]i increase and GAT3 reversal or whether it occurs via the Best1-mediated GABA release requires further experiments.
5. Summary: Is GABA Release from Astrocytes Mediated by Best1- or GAT3 or Both?
Theoretical assumptions indicate that, depending on the region and functional state, both the channel- and/or transporter-mediated GABA release is possible, given an astrocytic [GABA]I in the range of 0.5–4 mM and [GABA]o in the range of 0.5–1 µM. For both channel- and/or transporter-mediated GABA releases, experimental evidence has been provided, as summarized in the previous paragraphs and in Table 1. However, to be functionally relevant, the astrocytic GABA release has to be controlled by physiological demands. The physiologically required amount of extracellular GABA has to be adjusted to the detected/measured levels of neuronal activity and, accordingly, astrocytic GABA release/uptake has to be adapted. Below, we suggest some possible theoretical feedback mechanisms.
5.1. GAT3-Mediated Release
The GAT3-mediated GABA release is by definition equipped with a negative feedback mechanism. If the GAT3 operates in the release mode, the elevation of [GABA]o shifts its reversal towards more positive values, thus favoring the uptake mode (Figure 2C). In addition to the transmembrane GABA gradient, [Na+]i, and, to a lesser extent, [Cl−]i, determines the reversal potential and, in turn, the distinctive extracellular GABA level. The local astrocytic [Na+]i depends on the activity of several channels and secondary active transporters, and therefore reflects the neuronal activity levels (for reviews, [123,124,125]). Na+-coupled mechanisms, especially EAATs [21], may shift the GAT reversal potential and in turn [GABA]o (Figure 3A). Similar to this process, the EAAT activity may reverse the Na+/Ca2+ exchanger via the [Na+]i increase [126]. The resulting intracellular Ca2+ transients may not only trigger the release of other gliotransmitters, they influence GAT3 trafficking, modulating the number of membrane-located GAT3 and in turn the amount of GABA released or taken up [81]. Thus, the strength of the GAT3-mediated GABA release depends not only on the transmembrane GABA gradient but also, via changes in intracellular ionic concentrations on the neuronal activity levels, enabling a (patho-)physiologic condition dependent on the fine-tuning of the GABA release.
5.2. Best1-Mediated Release
Best1 is a Ca2+-activated anionic channel. This enables the possibility that local [Ca2+]i levels determine both the threshold and rate of tonic GABA release via this pathway. Interestingly, Bergmann glia cells in the cerebellum demonstrate variable resting [Ca2+]i, from about 50 to 250 nM, and the amplitude of somatic ATP-induced Ca2+ responses is negatively correlated to the resting somatic [Ca2+]i [127]. Similar results were obtained in hippocampal and cortical astrocytes in vitro and in vivo using two-photon microscopy [128]. Moreover, even within a single astrocyte, [Ca2+]i is spatially heterogeneous, ranging from 40 to 120 nM, and local [Ca2+]i controls the amplitudes of sensory-induced Ca2+ transients in vivo [128]. Because a half-maximal activation of Best1 channels occurs at a [Ca2+]i of about 150 nM [23], the reported levels of [Ca2+]i ranging from about 50 to 250 nM are within the dynamic range of Best1, thus supporting the activity-dependent modulation of the Best1-mediated GABA release. However, mechanisms underlying spatially restricted [Ca2+]i levels and thus the focal control of the Best1-mediated GABA release needs further investigations.
If the Best1-mediated release is tonic, such as what is reported in the cerebellum and thalamus [23,34], then all GATs probably operate in the uptake mode, thereby setting [GABA]o. The reversal of GAT directionality in Bergmann glial cells was reported to occur when [GABA]i reaches about 10 mM [129]. Using sniffer HEK293T cells expressing high-affinity GABAC receptors, a detectable Best1-mediated GABA release was already observed at the [GABA]i of 3 mM [23], suggesting that the Best1-mediated release probably dominates in the cerebellum. In turn, the elevated [GABA]o will shift the GAT reversal potential to more positive values. Thus, the Best1-mediated release and GAT3-mediated uptake operate in concert to determine [GABA]o (Figure 3B). Interesting, the pharmacologic or genetic blockade of Best1 did not completely eliminate the tonic GABAAR-mediated current but reduced it to about 30%, both in the cerebellum and thalamus [23,34]. Definitely, one cannot exclude the possibility that another channel, for example Best2 channel [47], contributes to GABA release, but an alternative explanation may be that a Best1 blockade results in lower [GABA]o, which in turn switches GAT3 into the release mode (Figure 3B). The observation that the non-selective GAT blockade with nipecotic acid induces larger tonic currents in the WT thalamus as compared to Best1-KO mice [34] indirectly supports this hypothesis.
5.3. Best1-/GAT3-Mediated Release
Interestingly, even if GAT3 operates in the uptake mode, it can indirectly suppress glutamatergic activity. In hippocampal slices, the GAT3-mediated GABA uptake leads to an elevation of [Na+]i, which in turn switches the Na+-Ca2+ exchange (NCE) into the reverse mode. The NCE-mediated Ca2+ influx can stimulate the ATP release from astrocytes, leading to an adenosine-mediated inhibition of glutamate activity [126]. As the EAAT-mediated glutamate uptake elevates an [Na+]i stronger than GATs, one can suggest that the GAT3- and/or EAAT-mediated [Na+]i increase reverse NCE, resulting in a near-membrane [Ca2+]i increase. Such local Ca2+ transients may open Best1 channels, leading to the GABA release (Figure 3C). Therefore, both GABA release pathways may be functional in parallel. Changes in intracellular ionic and GABA concentrations can modulate and/or inactivate one of them, potentiating in turn the remaining one.
5.4. Neuroglial Interaction and GABA Release from Astrocytes
Definitely not only changes in astrocytic activity or intracellular homeostasis determine the strength and direction of the GABA transport. In the hippocampus under resting conditions, the extracellular GABA has a mostly neuronal origin [41], and the high level [GABA]o keep GAT3 running in the uptake mode. Strongly elevated neuronal activity, such as during epileptic seizures, may increase the extracellular glutamate concentration and switch the GAT3 into release mode [21]. Physiological neuronal activity may result in the activation of astrocytic metabotropic receptors, including those for glutamate, ATP and GABA, resulting in local or global (waves) Ca2+ transients (for recent reviews, [130,131]. Such neuron activity-induced changes in [Ca2+]i can open Best1 channels, leading to a direct GABA release, or stimulate the GAT3 internalization, resulting in the reduced GABA uptake and indirect elevation in extracellular GABA level. Therefore, Best1 channels and GAT3 represent possible mechanisms of GABA release from astrocytes. Their (patho-) physiological contribution is determined as mainly neuroglial interactions at several levels.
6. Conclusions and Perspectives
In this review, we have summarized the experimental results reporting the GABA release under physiological and pathophysiological conditions. Increasing evidence demonstrates that astrocytes are able to synthesize GABA using different metabolic pathways. Intracellular GABA concentration may be as high as 10 mM. Given that the extracellular GABA concentration is in a low micromolar range, the transmembrane GABA gradient enables both Best1-channel- and GAT3-transporter-mediated GABA release. Indeed, the tonic GABA release from astrocytes has been demonstrated in the cortex, cerebellum, thalamus and striatum. Interestingly, in the cerebellum and thalamus, GABA is predominantly released via the Best1 channel, while in the cortex and striatum, the GAT3-mediated GABA release dominates. In diseases, the GABA release from astrocytes is typically facilitated and again, in some cases, the Best1-mediated release is potentiated, whereas, in other cases, the GAT3-mediated pathway plays the dominating role. The question why nature uses one or the other pathway remains elusive. The expression and trafficking of Best1 and GAT3 is modulated by several intracellular factors, including the intracellular GABA and Ca2+ concentration. In addition, the local intracellular Ca2+ concentration determines the Best1 conductance, while the intracellular Na+ concentration influences the GAT3 reversal potential. We suggest that the astrocytes possess both GABA release pathways. Local ionic signaling and GABA gradients, also as a consequence of neuronal activity, may play a decisive role to switch on/off a particular mechanism of GABA release. Elucidating the mechanisms governing GABA release may help to develop therapeutic tools for the treatments of pathophysiological conditions.
Funding
This work received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Allen, N.J. Astrocyte regulation of synaptic behavior. Annu Rev. Cell Dev. Biol. 2014, 30, 439–463. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Kofuji, P.; Araque, A. G-Protein-Coupled Receptors in Astrocyte-Neuron Communication. Neuroscience 2021, 456, 71–84. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Chvátal, A. NMDA Receptors in Astrocytes. Neurochem. Res. 2020, 45, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Rose, C.R.; Felix, L.; Zeug, A.; Dietrich, D.; Reiner, A.; Henneberger, C. Astroglial Glutamate Signaling and Uptake in the Hippocampus. Front. Mol. Neurosci. 2017, 10, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Feng, X.; Wang, Y.; Xia, X.; Zheng, J.C. Astrocytes: GABAceptive and GABAergic Cells in the Brain. Front. Cell Neurosci. 2022, 16, 892497. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.; Vanzulli, I.; Butt, A.M. A Central Role for ATP Signalling in Glial Interactions in the CNS. Curr. Drug Targets 2016, 17, 1829–1833. [Google Scholar] [CrossRef]
- Cornell-Bell, A.H.; Finkbeiner, S.M.; Cooper, M.S.; Smith, S.J. Glutamate induces calcium waves in cultured astrocytes: Long-range glial signaling. Science 1990, 247, 470–473. [Google Scholar] [CrossRef]
- Bernardinelli, Y.; Magistretti, P.J.; Chatton, J.Y. Astrocytes generate Na+-mediated metabolic waves. Proc. Natl. Acad. Sci. USA 2004, 101, 14937–14942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parpura, V.; Basarsky, T.A.; Liu, F.; Jeftinija, K.; Jeftinija, S.; Haydon, P.G. Glutamate-mediated astrocyte-neuron signalling. Nature 1994, 369, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, C.; Papouin, T.; Oliet, S.H.; Rusakov, D.A. Long-term potentiation depends on release of D-serine from astrocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic purinergic signaling coordinates synaptic networks. Science 2005, 310, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Montana, V.; Malarkey, E.B.; Verderio, C.; Matteoli, M.; Parpura, V. Vesicular transmitter release from astrocytes. Glia 2006, 54, 700–715. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Anderson, C.M.; Keung, E.C.; Chen, Y.; Chen, Y.; Swanson, R.A. P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J. Neurosci. 2003, 23, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [CrossRef] [Green Version]
- Szatkowski, M.; Barbour, B.; Attwell, D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature 1990, 348, 443–446. [Google Scholar] [CrossRef]
- Attwell, D.; Barbour, B.; Szatkowski, M. Nonvesicular release of neurotransmitter. Neuron 1993, 11, 401–407. [Google Scholar] [CrossRef]
- Kimelberg, H.K.; Goderie, S.K.; Higman, S.; Pang, S.; Waniewski, R.A. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J. Neurosci. 1990, 10, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Wang, W.; Diez-Sampedro, A.; Richerson, G.B. Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron 2007, 56, 851–865. [Google Scholar] [CrossRef] [Green Version]
- Kinney, G.A. GAT-3 transporters regulate inhibition in the neocortex. J. Neurophysiol 2005, 94, 4533–4537. [Google Scholar] [CrossRef]
- Heja, L.; Barabas, P.; Nyitrai, G.; Kekesi, K.A.; Lasztoczi, B.; Toke, O.; Tarkanyi, G.; Madsen, K.; Schousboe, A.; Dobolyi, A.; et al. Glutamate uptake triggers transporter-mediated GABA release from astrocytes. PLoS ONE 2009, 4, e7153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Tsunenari, T.; Yau, K.W.; Nathans, J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc. Natl. Acad. Sci. USA 2002, 99, 4008–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Yoon, B.E.; Berglund, K.; Oh, S.J.; Park, H.; Shin, H.S.; Augustine, G.J.; Lee, C.J. Channel-mediated tonic GABA release from glia. Science 2010, 330, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, A.D.; Marmorstein, L.Y.; Rayborn, M.; Wang, X.; Hollyfield, J.G.; Petrukhin, K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2000, 97, 12758–12763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.A.; Guziewicz, K.E.; Lee, C.J.; Kalathur, R.C.; Pulido, J.S.; Marmorstein, L.Y.; Marmorstein, A.D. Bestrophin 1 and retinal disease. Prog. Retin. Eye Res. 2017, 58, 45–69. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Choi, J.; Yoon, B.E. Neuron-Glia Interactions in Neurodevelopmental Disorders. Cells 2020, 9, 2176. [Google Scholar] [CrossRef]
- Schousboe, A.; Westergaard, N.; Sonnewald, U.; Petersen, S.B.; Yu, A.C.; Hertz, L. Regulatory role of astrocytes for neuronal biosynthesis and homeostasis of glutamate and GABA. Prog. Brain Res. 1992, 94, 199–211. [Google Scholar]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model. Nat. Commun. 2014, 5, 4159. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Timmermann, A.; Henning, L.; Müller, H.; Steinhäuser, C.; Bedner, P. Astrocytic GABA Accumulation in Experimental Temporal Lobe Epilepsy. Front. Neurol. 2020, 11, 614923. [Google Scholar] [CrossRef]
- Otsuka, M.; Obata, K.; Miyata, Y.; Tanaka, Y. Measurement of gamma-aminobutyric acid in isolated nerve cells of cat central nervous system. J. Neurochem 1971, 18, 287–295. [Google Scholar] [CrossRef]
- Sacchettoni, S.A.; Benchaibi, M.; Sindou, M.; Belin, M.F.; Jacquemont, B. Glutamate-modulated production of GABA in immortalized astrocytes transduced by a glutamic acid decarboxylase-expressing retrovirus. Glia 1998, 22, 86–93. [Google Scholar] [CrossRef]
- Seiler, N.; Schmidt-Glenewinkel, T.; Sarhan, S. On the formation of gamma-aminobutyric acid from putrescine in brain. J. Biochem. 1979, 86, 277–278. [Google Scholar]
- Laschet, J.; Grisar, T.; Bureau, M.; Guillaume, D. Characteristics of putrescine uptake and subsequent GABA formation in primary cultured astrocytes from normal C57BL/6J and epileptic DBA/2J mouse brain cortices. Neuroscience 1992, 48, 151–157. [Google Scholar] [CrossRef]
- Kwak, H.; Koh, W.; Kim, S.; Song, K.; Shin, J.I.; Lee, J.M.; Lee, E.H.; Bae, J.Y.; Ha, G.E.; Oh, J.E.; et al. Astrocytes Control Sensory Acuity via Tonic Inhibition in the Thalamus. Neuron 2020, 108, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Min, J.O.; Kang, D.S.; Kim, Y.S.; Jung, G.H.; Park, H.J.; Kim, S.; An, H.; Kwon, J.; Kim, J.; et al. Control of motor coordination by astrocytic tonic GABA release through modulation of excitation/inhibition balance in cerebellum. Proc. Natl. Acad. Sci. USA 2018, 115, 5004–5009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Ju, Y.H.; Choi, J.W.; Song, H.J.; Jang, B.K.; Woo, J.; Chun, H.; Kim, H.J.; Shin, S.J.; Yarishkin, O.; et al. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci. Adv. 2019, 5, eaav0316. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.E.; Woo, J.; Chun, Y.E.; Chun, H.; Jo, S.; Bae, J.Y.; An, H.; Min, J.O.; Oh, S.J.; Han, K.S.; et al. Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition. J. Physiol. 2014, 592, 4951–4968. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [Green Version]
- Dvorzhak, A.; Myakhar, O.; Unichenko, P.; Kirmse, K.; Kirischuk, S. Estimation of ambient GABA levels in layer I of the mouse neonatal cortex in brain slices. J. Physiol. 2010, 588, 2351–2360. [Google Scholar] [CrossRef]
- Jensen, K.; Chiu, C.S.; Sokolova, I.; Lester, H.A.; Mody, I. GABA transporter-1 (GAT1)-deficient mice: Differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J. Neurophysiol. 2003, 90, 2690–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagelhus, E.A.; Ottersen, O.P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [PubMed]
- Walch, E.; Fiacco, T.A. Honey, I shrunk the extracellular space: Measurements and mechanisms of astrocyte swelling. Glia 2022, 70, 2013–2031. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-de la Paz, L.D.; Gulias-Cañizo, R. Glia as a key factor in cell volume regulation processes of the central nervous system. Front. Cell Neurosci. 2022, 16, 967496. [Google Scholar] [CrossRef]
- Osei-Owusu, J.; Yang, J.; Vitery, M.D.C.; Qiu, Z. Molecular Biology and Physiology of Volume-Regulated Anion Channel (VRAC). Curr. Top. Membr. 2018, 81, 177–203. [Google Scholar]
- Owji, A.P.; Wang, J.; Kittredge, A.; Clark, Z.; Zhang, Y.; Hendrickson, W.A.; Yang, T. Structures and gating mechanisms of human bestrophin anion channels. Nat. Commun. 2022, 13, 3836. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Chien, L.T.; Cui, Y.; Hartzell, H.C. The anion-selective pore of the bestrophins, a family of chloride channels associated with retinal degeneration. J. Neurosci. 2006, 26, 5411–5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrukhin, K.; Koisti, M.J.; Bakall, B.; Li, W.; Xie, G.; Marknell, T.; Sandgren, O.; Forsman, K.; Holmgren, G.; Andreasson, S.; et al. Identification of the gene responsible for Best macular dystrophy. Nat. Genet. 1998, 19, 241–247. [Google Scholar] [CrossRef]
- Singh Grewal, S.; Smith, J.J.; Carr, A.F. Bestrophinopathies: Perspectives on clinical disease, Bestrophin-1 function and developing therapies. Ther. Adv. Ophthalmol. 2021, 13, 2515841421997191. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Hartzell, H.C.; Yu, K. Bestrophins and retinopathies. Pflug. Arch. 2010, 460, 559–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Z.; Hartzell, H.C. Bestrophin Cl- channels are highly permeable to HCO3. Am. J. Physiol. Cell Physiol. 2008, 294, C1371-7. [Google Scholar] [CrossRef] [Green Version]
- Elorza-Vidal, X.; Gaitán-Peñas, H.; Estévez, R. Chloride Channels in Astrocytes: Structure, Roles in Brain Homeostasis and Implications in Disease. Int. J. Mol. Sci. 2019, 20, 1034. [Google Scholar] [CrossRef] [Green Version]
- Owji, A.P.; Kittredge, A.; Zhang, Y.; Yang, T. Structure and Function of the Bestrophin family of calcium-activated chloride channels. Channels 2021, 15, 604–623. [Google Scholar] [CrossRef]
- Yu, K.; Xiao, Q.; Cui, G.; Lee, A.; Hartzell, H.C. The best disease-linked Cl- channel hBest1 regulates Ca V 1 (L-type) Ca2+ channels via src-homology-binding domains. J. Neurosci. 2008, 28, 5660–5670. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Stanton, J.B.; Wu, J.; Yu, K.; Hartzell, H.C.; Peachey, N.S.; Marmorstein, L.Y.; Marmorstein, A.D. Suppression of Ca2+ signaling in a mouse model of Best disease. Hum. Mol. Genet. 2010, 19, 1108–1118. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Rao, Z.; Zhang, Z.; Zhou, J. Function of the GABAergic System in Diabetic Encephalopathy. Cell. Mol. Neurobiol. 2022. [Google Scholar] [CrossRef]
- Park, H.; Oh, S.J.; Han, K.S.; Woo, D.H.; Park, H.; Mannaioni, G.; Traynelis, S.F.; Lee, C.J. Bestrophin-1 encodes for the Ca2+-activated anion channel in hippocampal astrocytes. J. Neurosci. 2009, 29, 13063–13073. [Google Scholar] [CrossRef] [Green Version]
- Le Meur, K.; Mendizabal-Zubiaga, J.; Grandes, P.; Audinat, E. GABA release by hippocampal astrocytes. Front. Comput. Neurosci. 2012, 6, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Han, K.S.; Oh, S.J.; Jo, S.; Woo, J.; Yoon, B.E.; Lee, C.J. High glutamate permeability and distal localization of Best1 channel in CA1 hippocampal astrocyte. Mol. Brain 2013, 6, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, D.H.; Han, K.S.; Shim, J.W.; Yoon, B.E.; Kim, E.; Bae, J.Y.; Oh, S.J.; Hwang, E.M.; Marmorstein, A.D.; Bae, Y.C.; et al. TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell 2012, 151, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, B.E.; Jo, S.; Woo, J.; Lee, J.H.; Kim, T.; Kim, D.; Lee, C.J. The amount of astrocytic GABA positively correlates with the degree of tonic inhibition in hippocampal CA1 and cerebellum. Mol. Brain 2011, 4, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glykys, J.; Mody, I. The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J. Physiol. 2007, 582, 1163–1178. [Google Scholar] [CrossRef] [PubMed]
- Semyanov, A.; Walker, M.C.; Kullmann, D.M. GABA uptake regulates cortical excitability via cell type-specific tonic inhibition. Nat. Neurosci. 2003, 6, 484–490. [Google Scholar] [CrossRef]
- Oh, S.J.; Lee, C.J. Distribution and Function of the Bestrophin-1 (Best1) Channel in the Brain. Exp. Neurobiol. 2017, 26, 113–121. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Koh, W.; Kang, S.; Nam, M.H.; Lee, C.J. Differential Proximity of Perisynaptic Astrocytic Best1 at the Excitatory and Inhibitory Tripartite Synapses in APP/PS1 and MAOB-KO Mice Revealed by Lattice Structured Illumination Microscopy. Exp. Neurobiol. 2021, 30, 213–221. [Google Scholar] [CrossRef]
- Kristensen, A.S.; Andersen, J.; Jorgensen, T.N.; Sorensen, L.; Eriksen, J.; Loland, C.J.; Stromgaard, K.; Gether, U. SLC6 neurotransmitter transporters: Structure, function, and regulation. Pharmacol. Rev. 2011, 63, 585–640. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.A. Calcium-independent release of GABA from isolated horizontal cells of the toad retina. J. Physiol. 1982, 323, 211–227. [Google Scholar] [CrossRef] [Green Version]
- Gordon-Weeks, P.R.; Lockerbie, R.O.; Pearce, B.R. Uptake and release of [3H]GABA by growth cones isolated from neonatal rat brain. Neurosci. Lett. 1984, 52, 205–210. [Google Scholar] [CrossRef]
- Gaspary, H.L.; Wang, W.; Richerson, G.B. Carrier-mediated GABA release activates GABA receptors on hippocampal neurons. J. Neurophysiol. 1998, 80, 270–281. [Google Scholar] [CrossRef] [Green Version]
- Kirmse, K.; Kirischuk, S. Ambient GABA constrains the strength of GABAergic synapses at Cajal-Retzius cells in the developing visual cortex. J. Neurosci. 2006, 26, 4216–4227. [Google Scholar] [CrossRef] [Green Version]
- Minelli, A.; Brecha, N.C.; Karschin, C.; DeBiasi, S.; Conti, F. GAT-1, a high-affinity GABA plasma membrane transporter, is localized to neurons and astroglia in the cerebral cortex. J. Neurosci. 1995, 15, 7734–7746. [Google Scholar] [CrossRef]
- Minelli, A.; DeBiasi, S.; Brecha, N.C.; Zuccarello, L.V.; Conti, F. GAT-3, a high-affinity GABA plasma membrane transporter, is localized to astrocytic processes, and it is not confined to the vicinity of GABAergic synapses in the cerebral cortex. J. Neurosci. 1996, 16, 6255–6264. [Google Scholar] [CrossRef] [Green Version]
- Heja, L.; Nyitrai, G.; Kekesi, O.; Dobolyi, A.; Szabo, P.; Fiath, R.; Ulbert, I.; Pal-Szenthe, B.; Palkovits, M.; Kardos, J. Astrocytes convert network excitation to tonic inhibition of neurons. BMC. Biol. 2012, 10, 26. [Google Scholar] [CrossRef]
- Chatton, J.Y.; Pellerin, L.; Magistretti, P.J. GABA uptake into astrocytes is not associated with significant metabolic cost: Implications for brain imaging of inhibitory transmission. Proc. Natl. Acad. Sci. USA 2003, 100, 12456–12461. [Google Scholar] [CrossRef] [Green Version]
- Rose, C.R.; Ransom, B.R. Intracellular sodium homeostasis in rat hippocampal astrocytes. J. Physiol. 1996, 491, 291–305. [Google Scholar] [CrossRef]
- Unichenko, P.; Myakhar, O.; Kirischuk, S. Intracellular Na+ concentration influences short-term plasticity of glutamate transporter-mediated currents in neocortical astrocytes. Glia 2012, 60, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Langer, J.; Rose, C.R. Synaptically induced sodium signals in hippocampal astrocytes in situ. J. Physiol. 2009, 587, 5859–5877. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Backus, K.H.; Schachner, M. Gamma-Aminobutyric acid opens Cl-channels in cultured astrocytes. Brain Res. 1987, 404, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kimelberg, H.K. Active accumulation and exchange transport of chloride in astroglial cells in culture. Biochim. Biophys. Acta 1981, 646, 179–184. [Google Scholar] [CrossRef]
- Unichenko, P.; Dvorzhak, A.; Kirischuk, S. Transporter-mediated replacement of extracellular glutamate for GABA in the developing murine neocortex. Eur. J. Neurosci. 2013, 38, 3580–3588. [Google Scholar] [CrossRef]
- Yu, X.; Taylor, A.M.W.; Nagai, J.; Golshani, P.; Evans, C.J.; Coppola, G.; Khakh, B.S. Reducing Astrocyte Calcium Signaling In Vivo Alters Striatal Microcircuits and Causes Repetitive Behavior. Neuron 2018, 99, 1170–1187. [Google Scholar] [CrossRef] [Green Version]
- Nusser, Z.; Mody, I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J. Neurophysiol. 2002, 87, 2624–2628. [Google Scholar] [CrossRef]
- Kersanté, F.; Rowley, S.C.; Pavlov, I.; Gutièrrez-Mecinas, M.; Semyanov, A.; Reul, J.M.; Walker, M.C.; Linthorst, A.C. A functional role for both -aminobutyric acid (GABA) transporter-1 and GABA transporter-3 in the modulation of extracellular GABA and GABAergic tonic conductances in the rat hippocampus. J. Physiol. 2013, 591, 2429–2441. [Google Scholar] [CrossRef]
- Savtchenko, L.; Megalogeni, M.; Rusakov, D.A.; Walker, M.C.; Pavlov, I. Synaptic GABA release prevents GABA transporter type-1 reversal during excessive network activity. Nat. Commun. 2015, 6, 6597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, C.R.; Ransom, B.R. Regulation of intracellular sodium in cultured rat hippocampal neurones. J. Physiol. 1997, 499, 573–587. [Google Scholar] [CrossRef]
- Kirmse, K.; Dvorzhak, A.; Kirischuk, S.; Grantyn, R. GABA transporter 1 tunes GABAergic synaptic transmission at output neurons of the mouse neostriatum. J. Physiol. 2008, 586, 5665–5678. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.M.; Doig, N.M.; Brimblecombe, K.R.; Lopes, E.F.; Siddorn, R.E.; Threlfell, S.; Connor-Robson, N.; Bengoa-Vergniory, N.; Pasternack, N.; Wade-Martins, R.; et al. GABA uptake transporters support dopamine release in dorsal striatum with maladaptive downregulation in a parkinsonism model. Nat. Commun. 2020, 11, 4958. [Google Scholar] [CrossRef] [PubMed]
- Kirmse, K.; Kirischuk, S.; Grantyn, R. Role of GABA transporter 3 in GABAergic synaptic transmission at striatal output neurons. Synapse 2009, 63, 921–929. [Google Scholar] [CrossRef]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef]
- Andersen, J.V.; Schousboe, A.; Verkhratsky, A. Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: Integration of the glutamate/GABA-glutamine cycle. Prog Neurobiol. 2022, 217, 102331. [Google Scholar] [CrossRef]
- Aldabbagh, Y.; Islam, A.; Zhang, W.; Whiting, P.; Ali, A.B. Alzheimer’s Disease Enhanced Tonic Inhibition is Correlated With Upregulated Astrocyte GABA Transporter-3/4 in a Knock-In APP Mouse Model. Front. Pharmacol. 2022, 13, 822499. [Google Scholar] [CrossRef] [PubMed]
- Yarishkin, O.; Lee, J.; Jo, S.; Hwang, E.M.; Lee, C.J. Disinhibitory Action of Astrocytic GABA at the Perforant Path to Dentate Gyrus Granule Neuron Synapse Reverses to Inhibitory in Alzheimer’s Disease Model. Exp. Neurobiol. 2015, 24, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brawek, B.; Chesters, R.; Klement, D.; Müller, J.; Lerdkrai, C.; Hermes, M.; Garaschuk, O. A bell-shaped dependence between amyloidosis and GABA accumulation in astrocytes in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 61, 187–197. [Google Scholar] [CrossRef]
- Pandit, S.; Neupane, C.; Woo, J.; Sharma, R.; Nam, M.H.; Lee, G.S.; Yi, M.H.; Shin, N.; Kim, D.W.; Cho, H.; et al. Bestrophin1-mediated tonic GABA release from reactive astrocytes prevents the development of seizure-prone network in kainate-injected hippocampi. Glia 2020, 68, 1065–1080. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Behrens, P.F.; Franz, P.; Woodman, B.; Lindenberg, K.S.; Landwehrmeyer, G.B. Impaired glutamate transport and glutamate-glutamine cycling: Downstream effects of the Huntington mutation. Brain 2002, 125, 1908–1922. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.R.; Walker, A.G.; Fowler, S.C.; von Hörsten, S.; Riess, O.; Johnson, M.A.; Rebec, G.V. Dysregulation of coordinated neuronal firing patterns in striatum of freely behaving transgenic rats that model Huntington’s disease. Neurobiol. Dis. 2010, 37, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Skotte, N.H.; Andersen, J.V.; Santos, A.; Aldana, B.I.; Willert, C.W.; Nørremølle, A.; Waagepetersen, H.S.; Nielsen, M.L. Integrative Characterization of the R6/2 Mouse Model of Huntington’s Disease Reveals Dysfunctional Astrocyte Metabolism. Cell Rep. 2018, 23, 2211–2224. [Google Scholar] [CrossRef]
- Lee, W.; Reyes, R.C.; Gottipati, M.K.; Lewis, K.; Lesort, M.; Parpura, V.; Gray, M. Enhanced Ca2+-dependent glutamate release from astrocytes of the BACHD Huntington’s disease mouse model. Neurobiol. Dis. 2013, 58, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.T.; Chang, Y.G.; Chern, Y. Insights into GABA(A)ergic system alteration in Huntington’s disease. Open Biol. 2018, 8, 180165. [Google Scholar] [CrossRef] [Green Version]
- Wojtowicz, A.M.; Dvorzhak, A.; Semtner, M.; Grantyn, R. Reduced tonic inhibition in striatal output neurons from Huntington mice due to loss of astrocytic GABA release through GAT-3. Front. Neural. Circuits 2013, 7, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Diaz-Castro, B.; Looger, L.L.; Khakh, B.S. Dysfunctional Calcium and Glutamate Signaling in Striatal Astrocytes from Huntington’s Disease Model Mice. J. Neurosci. 2016, 36, 3453–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, A.D.; Jackson, W.S.; King, O.D.; Lindquist, S. The power of automated high-resolution behavior analysis revealed by its application to mouse models of Huntington’s and prion diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Canitano, R.; Palumbi, R. Excitation/Inhibition Modulators in Autism Spectrum Disorder: Current Clinical Research. Front. Neurosci. 2021, 15, 753274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Mao, X.; Zhu, C.; Zou, X.; Peng, F.; Yang, W.; Li, B.; Li, G.; Ge, T.; Cui, R. GABAergic System Dysfunction in Autism Spectrum Disorders. Front. Cell Dev. Biol. 2021, 9, 781327. [Google Scholar] [CrossRef] [PubMed]
- Yizhar, O.; Fenno, L.E.; Prigge, M.; Schneider, F.; Davidson, T.J.; O’Shea, D.J.; Sohal, V.S.; Goshen, I.; Finkelstein, J.; Paz, J.T.; et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 2011, 477, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Rubenstein, J.L.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef]
- Aida, T.; Yoshida, J.; Nomura, M.; Tanimura, A.; Iino, Y.; Soma, M.; Bai, N.; Ito, Y.; Cui, W.; Aizawa, H.; et al. Astroglial glutamate transporter deficiency increases synaptic excitability and leads to pathological repetitive behaviors in mice. Neuropsychopharmacology 2015, 40, 1569–1579. [Google Scholar] [CrossRef]
- Kirischuk, S.; Kettenmann, H.; Verkhratsky, A. Na+/Ca2+ exchanger modulates kainate-triggered Ca2+ signaling in Bergmann glial cells in situ. FASEB J. 1997, 11, 566–572. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K. Astroglia and Obsessive Compulsive Disorder. Adv. Neurobiol. 2021, 26, 139–149. [Google Scholar] [PubMed]
- Treiman, D.M. GABAergic mechanisms in epilepsy. Epilepsia 2001, 42 (Suppl. S3), 8–12. [Google Scholar] [CrossRef] [PubMed]
- Akyuz, E.; Polat, A.K.; Eroglu, E.; Kullu, I.; Angelopoulou, E.; Paudel, Y.N. Revisiting the role of neurotransmitters in epilepsy: An updated review. Life Sci. 2021, 265, 118826. [Google Scholar] [CrossRef] [PubMed]
- Richter, D.; Luhmann, H.J.; Kilb, W. Intrinsic activation of GABA(A) receptors suppresses epileptiform activity in the cerebral cortex of immature mice. Epilepsia 2010, 51, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.H.; Reddy, D.S. Genetic and Molecular Regulation of Extrasynaptic GABA-A Receptors in the Brain: Therapeutic Insights for Epilepsy. J. Pharm. Exp. Ther. 2018, 364, 180–197. [Google Scholar] [CrossRef] [PubMed]
- Maljevic, S.; Møller, R.S.; Reid, C.A.; Pérez-Palma, E.; Lal, D.; May, P.; Lerche, H. Spectrum of GABAA receptor variants in epilepsy. Curr. Opin. Neurol. 2019, 32, 183–190. [Google Scholar] [CrossRef]
- Lie, M.E.K.; Al-Khawaja, A.; Damgaard, M.; Haugaard, A.S.; Schousboe, A.; Clarkson, A.N.; Wellendorph, P. Glial GABA Transporters as Modulators of Inhibitory Signalling in Epilepsy and Stroke. Adv. Neurobiol. 2017, 16, 137–167. [Google Scholar]
- Kovács, Z.; Skatchkov, S.N.; Veh, R.W.; Szabó, Z.; Németh, K.; Szabó, P.T.; Kardos, J.; Héja, L. Critical Role of Astrocytic Polyamine and GABA Metabolism in Epileptogenesis. Front. Cell Neurosci. 2021, 15, 787319. [Google Scholar] [CrossRef]
- Kovács, Z.; Skatchkov, S.N.; Szabó, Z.; Qahtan, S.; Méndez-González, M.P.; Malpica-Nieves, C.J.; Eaton, M.J.; Kardos, J.; Héja, L. Putrescine Intensifies Glu/GABA Exchange Mechanism and Promotes Early Termination of Seizures. Int. J. Mol. Sci. 2022, 23, 8191. [Google Scholar] [CrossRef]
- de Lanerolle, N.C.; Kim, J.H.; Robbins, R.J.; Spencer, D.D. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989, 495, 387–395. [Google Scholar] [CrossRef]
- Buckmaster, P.S.; Abrams, E.; Wen, X. Seizure frequency correlates with loss of dentate gyrus GABAergic neurons in a mouse model of temporal lobe epilepsy. J. Comp. Neurol. 2017, 525, 2592–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Righes Marafiga, J.; Vendramin Pasquetti, M.; Calcagnotto, M.E. GABAergic interneurons in epilepsy: More than a simple change in inhibition. Epilepsy Behav. 2021, 121, 106935. [Google Scholar] [CrossRef] [PubMed]
- Kirischuk, S.; Parpura, V.; Verkhratsky, A. Sodium dynamics: Another key to astroglial excitability? Trends Neurosci. 2012, 35, 497–506. [Google Scholar] [CrossRef]
- Felix, L.; Delekate, A.; Petzold, G.C.; Rose, C.R. Sodium Fluctuations in Astroglia and Their Potential Impact on Astrocyte Function. Front. Physiol. 2020, 11, 871. [Google Scholar] [CrossRef]
- Chatton, J.Y.; Magistretti, P.J.; Barros, L.F. Sodium signaling and astrocyte energy metabolism. Glia 2016, 64, 1667–1676. [Google Scholar] [CrossRef]
- Boddum, K.; Jensen, T.P.; Magloire, V.; Kristiansen, U.; Rusakov, D.A.; Pavlov, I.; Walker, M.C. Astrocytic GABA transporter activity modulates excitatory neurotransmission. Nat. Commun. 2016, 7, 13572. [Google Scholar] [CrossRef]
- Kirischuk, S.; Moller, T.; Voitenko, N.; Kettenmann, H.; Verkhratsky, A. ATP-induced cytoplasmic calcium mobilization in Bergmann glial cells. J. Neurosci. 1995, 15, 7861–7871. [Google Scholar] [CrossRef] [PubMed]
- King, C.M.; Bohmbach, K.; Minge, D.; Delekate, A.; Zheng, K.; Reynolds, J.; Rakers, C.; Zeug, A.; Petzold, G.C.; Rusakov, D.A.; et al. Local Resting Ca2+ Controls the Scale of Astroglial Ca2+ Signals. Cell Rep. 2020, 30, 3466–3477. [Google Scholar] [CrossRef] [Green Version]
- Barakat, L.; Bordey, A. GAT-1 and reversible GABA transport in Bergmann glia in slices. J. Neurophysiol. 2002, 88, 1407–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, D.; Semyanov, A.; Genazzani, A.; Verkhratsky, A. Calcium signaling in neuroglia. Int. Rev. Cell Mol. Biol. 2021, 362, 1–53. [Google Scholar]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Several pathways of GABA synthesis in astrocytes. (1) Glutamate decarboxylase (GAD) can convert glutamate into GABA [28]. (2,3) GABA can be synthesized from putrescine, a monoamine, via (2) Diamine Oxydase (DAO) and Aldehyde Dehydrogenase 1a1 (Aldh1a1, [34]) or (3) Monoamine oxidase B (MAO-B, [35]).
Figure 1.
Several pathways of GABA synthesis in astrocytes. (1) Glutamate decarboxylase (GAD) can convert glutamate into GABA [28]. (2,3) GABA can be synthesized from putrescine, a monoamine, via (2) Diamine Oxydase (DAO) and Aldehyde Dehydrogenase 1a1 (Aldh1a1, [34]) or (3) Monoamine oxidase B (MAO-B, [35]).

Figure 2.
Possible pathways of GABA release from astrocytes. (A) GABA can be released via a Ca2+-activated anion channel (for example Bestrophin 1, Best1, [23]). The driving force for GABA release is the transmembrane GABA gradient. Best1 opening and conductance is controlled by [Ca2+]i. Note that Best1 is also permeable for Cl− and glutamate. (B) GABA transporter (GAT3) operating in reverse mode can release GABA [20]. In addition to the GABA gradient, the transmembrane gradients of Na+ (quadratic impact) and Cl− determine the electromotive driving force and, in turn, the direction of GABA transport. (C) The GAT reversal potentials depend on the [GABA]o. To calculate GAT reversal potential, following values have been taken: [Na+]o = 140 mM, [Cl−]i = 135 mM, [Na+]i = 15 mM, [Cl−]i = 40 mM, and [GABA]i = 1 mM. The reported [GABA]o of 0.2 µM in the cortex [40] and 0.8 µM in the hippocampus [41] favor the GAT3-mediated release under resting conditions in the former (GATrev ~ −70 mV) but not in the latter (GATrev ~ −35 mV). (D) To change the direction of GAT-mediated transport in the hippocampus, either [Na+]i has to be increased to about 25–30 mM (gray arrow; for instance, as a result of EAAT-mediated glutamate uptake [21]) or [GABA]i, it has to be elevated to about 4 mM (dashed arrow) [28].
Figure 2.
Possible pathways of GABA release from astrocytes. (A) GABA can be released via a Ca2+-activated anion channel (for example Bestrophin 1, Best1, [23]). The driving force for GABA release is the transmembrane GABA gradient. Best1 opening and conductance is controlled by [Ca2+]i. Note that Best1 is also permeable for Cl− and glutamate. (B) GABA transporter (GAT3) operating in reverse mode can release GABA [20]. In addition to the GABA gradient, the transmembrane gradients of Na+ (quadratic impact) and Cl− determine the electromotive driving force and, in turn, the direction of GABA transport. (C) The GAT reversal potentials depend on the [GABA]o. To calculate GAT reversal potential, following values have been taken: [Na+]o = 140 mM, [Cl−]i = 135 mM, [Na+]i = 15 mM, [Cl−]i = 40 mM, and [GABA]i = 1 mM. The reported [GABA]o of 0.2 µM in the cortex [40] and 0.8 µM in the hippocampus [41] favor the GAT3-mediated release under resting conditions in the former (GATrev ~ −70 mV) but not in the latter (GATrev ~ −35 mV). (D) To change the direction of GAT-mediated transport in the hippocampus, either [Na+]i has to be increased to about 25–30 mM (gray arrow; for instance, as a result of EAAT-mediated glutamate uptake [21]) or [GABA]i, it has to be elevated to about 4 mM (dashed arrow) [28].

Figure 3.
Best1- and/or GAT3-mediated GABA release from astrocytes. (A) Upon elevated extracellular glutamate concentrations, EAAT-mediated glutamate uptake increases [Na+]I and switches GAT into reverse mode [21] (B) High [Ca2+]i opens Best1 channels. Elevated [GABA]o supports the uptake mode of GATs. Lowering of [Ca2+]i or Best1 blockade results in [GABA]o reduction and switches GAT into reverse mode (indirect in [34]). (C) Glutamate (EAAT) and/or GABA (GAT) uptake switch Na+-Ca2+ exchange (NCE) in reverse mode. Local [Ca2+]I increase opens Best1 and enables GABA release. Note that local [Ca2+]I increase may also affect GAT trafficking [81].
Figure 3.
Best1- and/or GAT3-mediated GABA release from astrocytes. (A) Upon elevated extracellular glutamate concentrations, EAAT-mediated glutamate uptake increases [Na+]I and switches GAT into reverse mode [21] (B) High [Ca2+]i opens Best1 channels. Elevated [GABA]o supports the uptake mode of GATs. Lowering of [Ca2+]i or Best1 blockade results in [GABA]o reduction and switches GAT into reverse mode (indirect in [34]). (C) Glutamate (EAAT) and/or GABA (GAT) uptake switch Na+-Ca2+ exchange (NCE) in reverse mode. Local [Ca2+]I increase opens Best1 and enables GABA release. Note that local [Ca2+]I increase may also affect GAT trafficking [81].


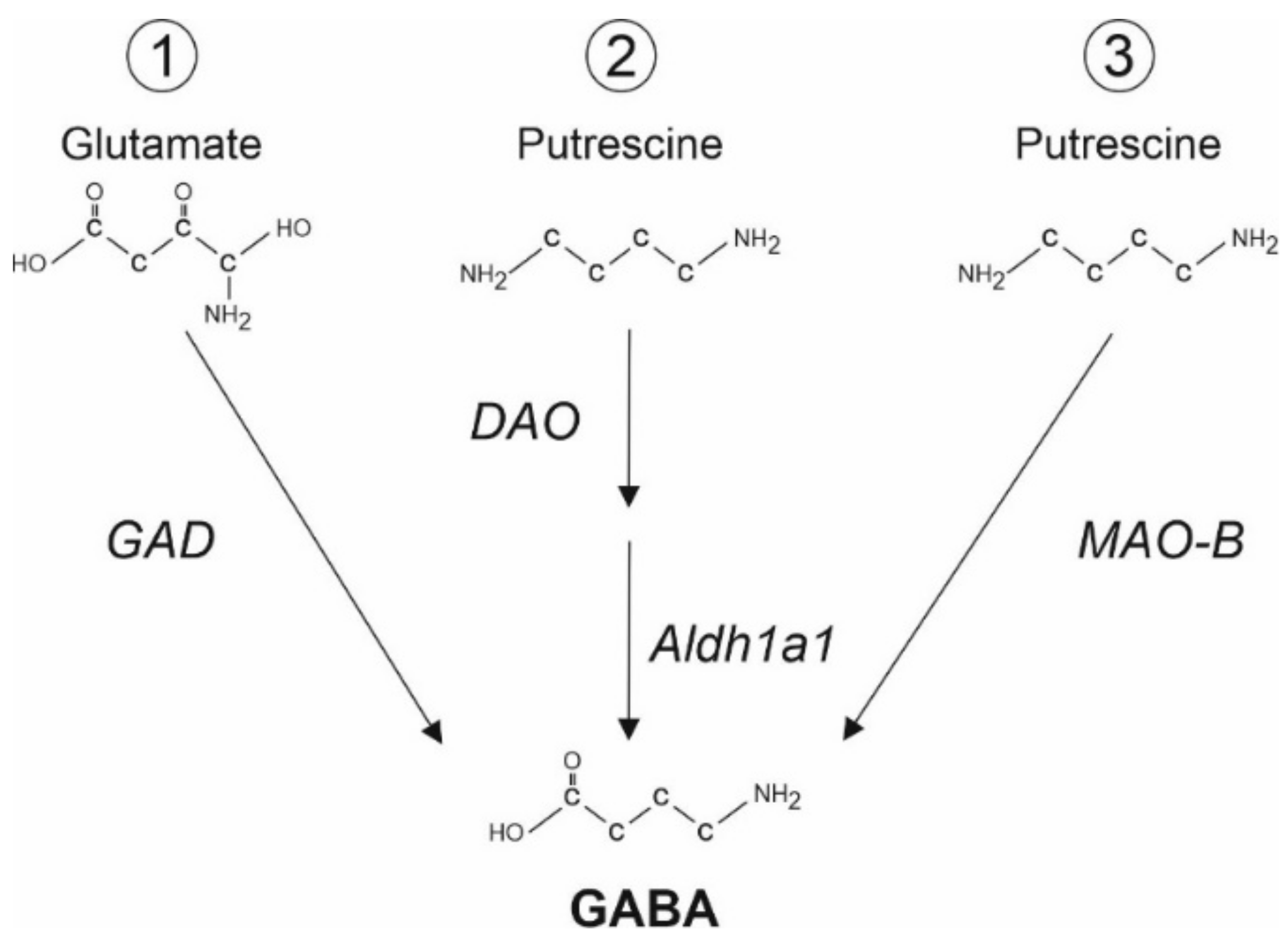{kind=link}
{kind=link}
{kind=link}
Table 1.
Best1-channel- and GAT3-mediated GABA release in mouse models.
| Brain Region | Disease Model | Best1 | GAT3 | References |
|---|---|---|---|---|
| Cerebellum | WT | + | [23] | |
| Thalamus | WT | + | [34] | |
| Cerebral Cortex | WT | + | [20,76] | |
| Striatum | WT | + | [101] | |
| Hippocampus | 5xFAD (AD) | [28] | ||
| Hippocampus | APPNL−F/NL−F knock-in (AD) | + | [91] | |
| Hippocampus | APP/PS1 (AD) | + | [35] | |
| Hippocampus | APP/PS1 (AD) | + | [35] | |
| Striatum | Z-Q175-KI and R2/6 (HD) | + (reduced) | [101] | |
| Hippocampus | Kainate model (epilepsy) | + | [94] | |
| Hippocampus | WAG/Rij (epilepsy) | + | [118] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kilb, W.; Kirischuk, S. GABA Release from Astrocytes in Health and Disease. Int. J. Mol. Sci. 2022, 23, 15859. https://doi.org/10.3390/ijms232415859
AMA Style
Kilb W, Kirischuk S. GABA Release from Astrocytes in Health and Disease. International Journal of Molecular Sciences. 2022; 23(24):15859. https://doi.org/10.3390/ijms232415859
Chicago/Turabian StyleKilb, Werner, and Sergei Kirischuk. 2022. "GABA Release from Astrocytes in Health and Disease" International Journal of Molecular Sciences 23, no. 24: 15859. https://doi.org/10.3390/ijms232415859
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.