
Quasispecies Analysis of SARS-CoV-2 of 15 Different Lineages during the First Year of the Pandemic Prompts Scratching under the Surface of Consensus Genome Sequences
 and
and
Abstract
:1. Introduction
2. Results
2.1. Quality and Lineages of the SARS-CoV-2 Genomes Analyzed
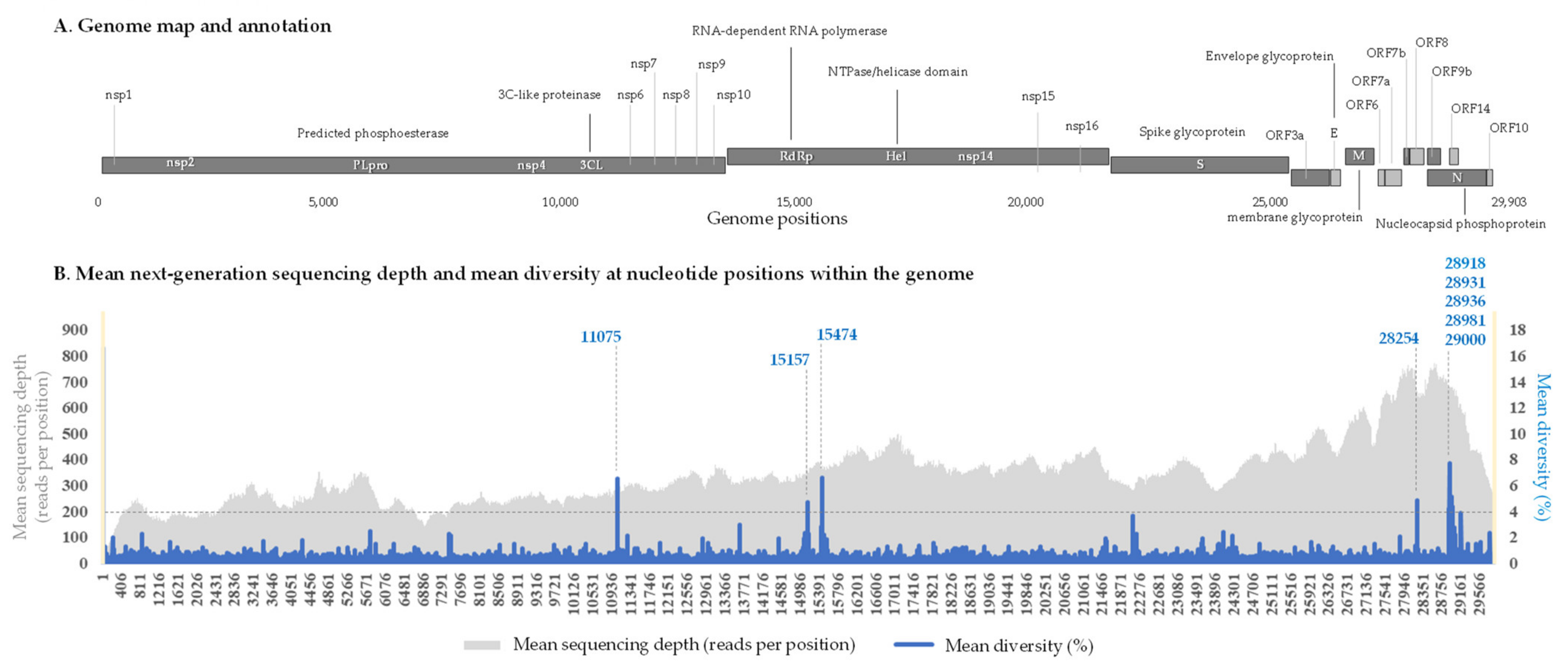
2.2. Nucleotide Diversity in the SARS-CoV-2 Genomes and Genes
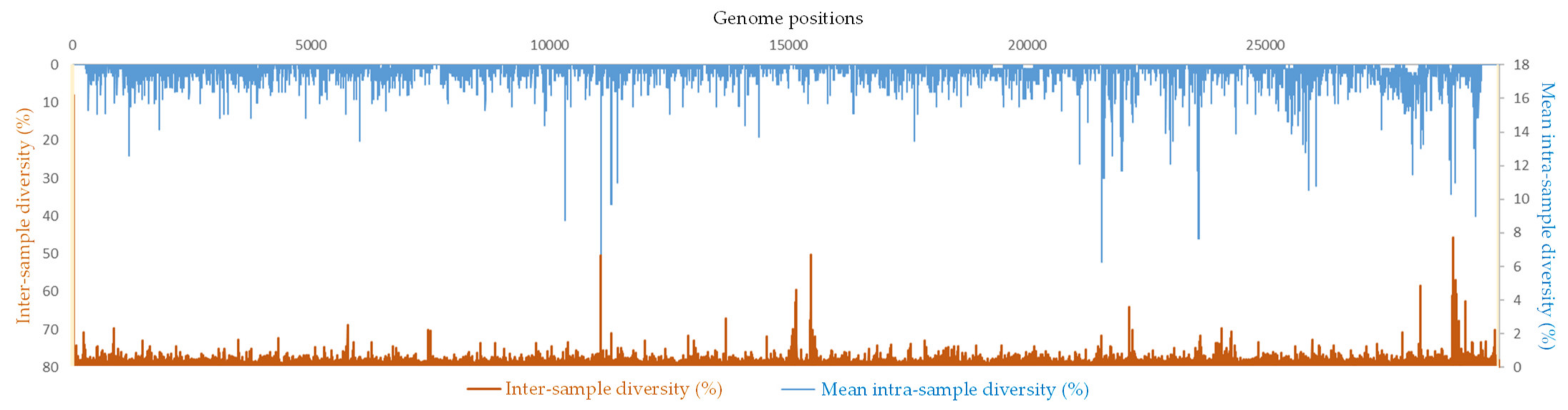
2.3. Hot Spots of Intra-Sample Genetic Diversity
2.4. Correlation between Intra-Sample and Inter-Sample Genetic Diversity in SARS-CoV-2 Genomes
2.5. Presence in Viral Quasispecies of Variant Hallmark Mutations of the Spike Gene
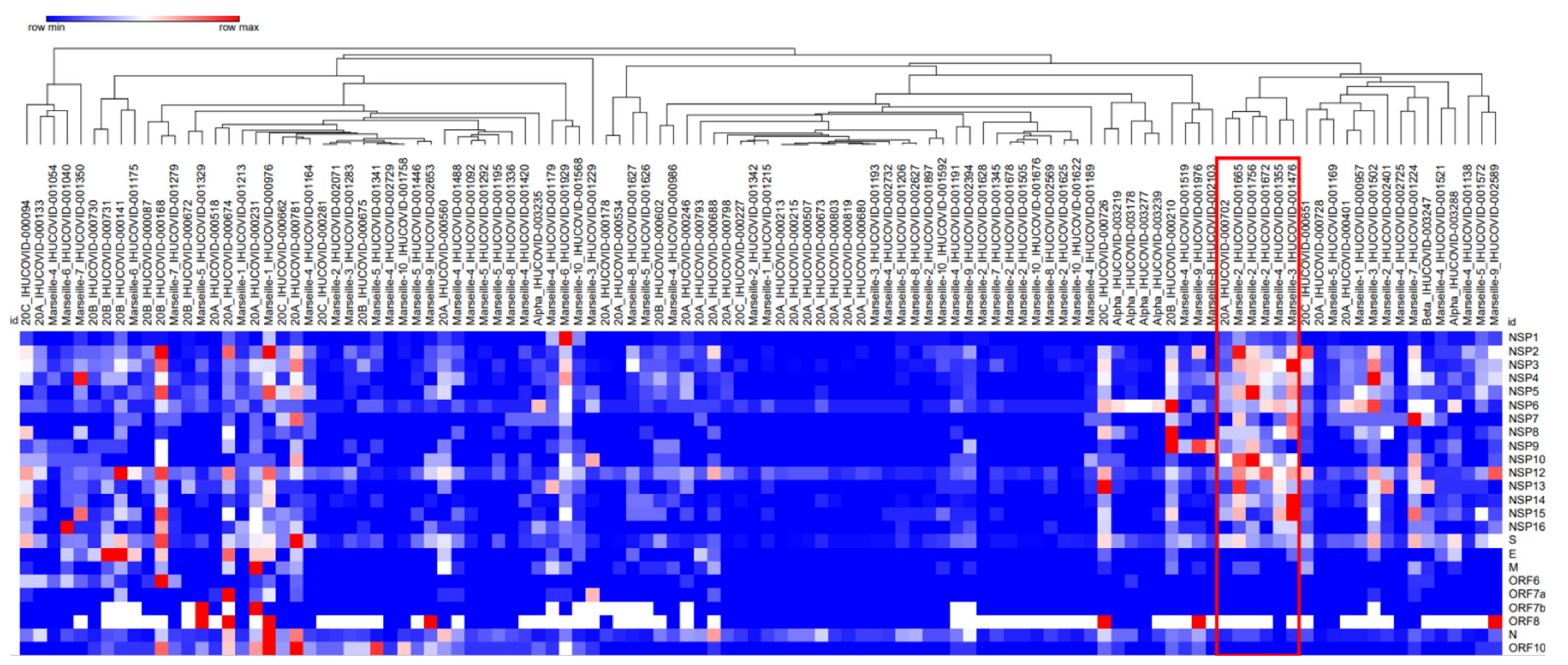
2.6. Intra-Sample Genetic Diversity for the Different SARS-CoV-2 Lineages and Variants
3. Discussion
4. Materials and Methods
4.1. Next-Generation SARS-CoV-2 Genome Sequencing Methods and Data
4.2. Detection and Characterization of Genetic Quasispecies
4.3. Analysis of SARS-CoV-2 Intra-Sample Genetic Diversity at the Genome and Gene Scales
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rabi, F.A.; Al Zoubi, M.S.; Kasasbeh, G.A.; Salameh, D.M.; Al-Nasser, A.D. SARS-CoV-2 and Coronavirus Disease 2019: What We Know So Far. Pathogens 2020, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020, 91, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Prates, E.T.; Garvin, M.R.; Pavicic, M.; Jones, P.; Shah, M.; Demerdash, O.; Amos, B.K.; Geiger, A.; Jacobson, D. Potential Pathogenicity Determinants Identified from Structural Proteomics of SARS-CoV and SARS-CoV-2. Mol. Biol. Evol. 2021, 38, 702–715. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, M. Newly Emerged Antiviral Strategies for SARS-CoV-2: From Deciphering Viral Protein Structural Function to the Development of Vaccines, Antibodies and Small Molecules. Int. J. Mol. Sci. 2022, 23, 6083. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, N.; Qi, W.; Li, T.; Zhang, Y.; Zhang, H.; Wu, J.; Zhu, Z.; Ai, J.; Qiu, C.; et al. Intra-host SARS-CoV-2 single-nucleotide variants emerged during the early stage of COVID-19 pandemic forecast population fixing mutations. J. Infect. 2022, 84, 722–746. [Google Scholar] [CrossRef]
- Domingo, E.; Escarmís, C.; Sevilla, N.; Moya, A.; Elena, S.F.; Quer, J.; Novella, I.S.; Holland, J.J. Basic concepts in RNA virus evolution. FASEB J. 1996, 10, 859–864. [Google Scholar] [CrossRef]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef]
- Wang, S.; Xu, X.; Wei, C.; Li, S.; Zhao, J.; Zheng, Y.; Liu, X.; Zeng, X.; Yuan, W.; Peng, S. Molecular evolutionary characteristics of SARS-CoV-2 emerging in the United States. J. Med. Virol. 2022, 94, 310–317. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [Green Version]
- Van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C.; Rambaut, A. Viral evolution and the emergence of SARS coronavirus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 1059–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E.V. Ongoing global and regional adaptive evolution of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2104241118. [Google Scholar] [CrossRef] [PubMed]
- Akaishi, T. Insertion-and-Deletion Mutations between the Genomes of SARS-CoV, SARS-CoV-2 and Bat Coronavirus RaTG13. Microbiol. Spectr. 2022, 10, e0071622. [Google Scholar] [CrossRef] [PubMed]
- Ignatieva, A.; Hein, J.; Jenkins, P.A. Ongoing Recombination in SARS-CoV-2 Revealed through Genealogical Reconstruction. Mol. Biol. Evol. 2022, 39, msac028. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.; et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 2021, 184, 5179–5188.e8. [Google Scholar] [CrossRef]
- Colson, P.; Fournier, P.E.; Delerce, J.; Million, M.; Bedotto, M.; Houhamdi, L.; Yahi, N.; Bayette, J.; Levasseur, A.; Fantini, J.; et al. Culture and identification of a “Deltamicron” SARS-CoV-2 in a three cases cluster in southern France. J. Med. Virol. 2022, 94, 3739–3749. [Google Scholar] [CrossRef]
- Andrés, C.; Garcia-Cehic, D.; Gregori, J.; Piñana, M.; Rodriguez-Frias, F.; Guerrero-Murillo, M.; Esperalba, J.; Rando, A.; Goterris, L.; Codina, M.G.; et al. Naturally occurring SARS-CoV-2 gene deletions close to the spike S1/S2 cleavage site in the viral quasispecies of COVID19 patients. Emerg. Microbes Infect. 2020, 9, 1900–1911. [Google Scholar] [CrossRef]
- McLean, G.; Kamil, J.; Lee, B.; Moore, P.; Schulz, T.F.; Muik, A.; Sahin, U.; Türeci, Ö.; Pather, S. The Impact of Evolving SARS-CoV-2 Mutations and Variants on COVID-19 Vaccines. mBio 2022, 13, e0297921. [Google Scholar] [CrossRef]
- Colson, P.; Fournier, P.E.; Chaudet, H.; Delerce, J.; Giraud-Gatineau, A.; Houhamdi, L.; Andrieu, C.; Brechard, L.; Bedotto, M.; Prudent, E.; et al. Analysis of SARS-CoV-2 Variants from 24,181 Patients Exemplifies the Role of Globalization and Zoonosis in Pandemics. Front. Microbiol. 2022, 12, 786233. [Google Scholar] [CrossRef]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. Molecular self-organization and the early stages of evolution. Q. Rev. Biophys. 1971, 4, 149–212. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. On the nature of virus quasispecies. Trends Microbiol. 1996, 4, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Flavell, R.A.; Weissmann, C. In vitro site-directed mutagenesis: Generation and properties of an infectious extracistronic mutant of bacteriophage Qβ. Gene 1976, 1, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andino, R.; Domingo, E. Viral quasispecies. Virology 2015, 479–480, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef] [Green Version]
- Tamalet, C.; Yahi, N.; Tourrès, C.; Colson, P.; Quinson, A.M.; Poizot-Martin, I.; Dhiver, C.; Fantini, J. Multidrug resistance genotypes (insertions in the beta3-beta4 finger subdomain and MDR mutations) of HIV-1 reverse transcriptase from extensively treated patients: Incidence and association with other resistance mutations. Virology 2000, 270, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; Du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Kuipers, J.; Batavia, A.A.; Jablonski, K.P.; Bayer, F.; Borgsmüller, N.; Dondi, A.; Drăgan, M.-A.; Ferreira, P.; Jahn, K.; Lamberti, L.; et al. Within-patient genetic diversity of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chen, C.; Nadeau, S.; Yared, M.; Voinov, P.; Xie, N.; Roemer, C.; Stadler, T. CoV-Spectrum: Analysis of Globally Shared SARS-CoV-2 Data to Identify and Characterize New Variants. Bioinformatics 2021, 38, 1735–1737. [Google Scholar] [CrossRef] [PubMed]
- Nour, D.; Rafei, R.; Lamarca, A.P.; de Almeida, L.G.P.; Osman, M.; Ismail, M.B.; Mallat, H.; Berry, A.; Burfin, G.; Semanas, Q.; et al. The Role of Lebanon in the COVID-19 Butterfly Effect: The B.1.398 Example. Viruses 2022, 14, 1640. [Google Scholar] [CrossRef] [PubMed]
- Zinzula, L. Lost in deletion: The enigmatic ORF8 protein of SARS-CoV-2. Biochem. Biophys. Res. Commun. 2021, 538, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Gaurav, S.; Pandey, S.; Puvar, A.; Shah, T.; Joshi, M.; Joshi, C.; Kumar, S. Identification of unique mutations in SARS-CoV-2 strains isolated from India suggests its attenuated pathotype. bioRxiv 2020. [Google Scholar] [CrossRef]
- Dudas, G.; Hong, S.L.; Potter, B.I.; Calvignac-Spencer, S.; Niatou-Singa, F.S.; Tombolomako, T.B.; Fuh-Neba, T.; Vickos, U.; Ulrich, M.; Leendertz, F.H.; et al. Emergence and spread of SARS-CoV-2 lineage B.1.620 with variant of concern-like mutations and deletions. Nat. Commun. 2021, 12, 5769. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, D.; Zhang, L.; Sun, W.; Zhang, Z.; Chen, W.; Zhu, A.; Huang, Y.; Xiao, F.; Yao, J.; et al. Intra-host variation and evolutionary dynamics of SARS-CoV-2 populations in COVID-19 patients. Genome Med. 2021, 13, 30. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Colson, P.; Levasseur, A.; Gautret, P.; Fenollar, F.; Thuan Hoang, V.; Delerce, J.; Bitam, I.; Saile, R.; Maaloum, M.; Padane, A.; et al. Introduction into the Marseille geographical area of a mild SARS-CoV-2 variant originating from sub-Saharan Africa: An investigational study. Travel. Med. Infect. Dis. 2021, 40, 101980. [Google Scholar] [CrossRef]
- Hodcroft, E. CoVariants: SARS-CoV-2 Mutations and Variants of Interest. 2021. Available online: https://covariants.org/ (accessed on 30 September 2022).
- Fournier, P.E.; Colson, P.; Levasseur, A.; Devaux, C.A.; Gautret, P.; Bedotto, M.; Delerce, J.; Brechard, L.; Pinault, L.; Lagier, J.C.; et al. Emergence and outcomes of the SARS-CoV-2 ‘Marseille-4’ variant. Int. J. Infect. Dis. 2021, 106, 228–236. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.J.; Long, S.W.; Christensen, P.A.; Olsen, R.J.; Olson, R.; Shukla, M.; Subedi, S.; Stevens, R.; Musser, J.M. Analysis of the ARTIC Version 3 and Version 4 SARS-CoV-2 Primers and Their Impact on the Detection of the G142D Amino Acid Substitution in the Spike Protein. Microbiol. Spectr. 2021, 9, e0180321. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, M.; Mloka, D.; Tovanabutra, S.; Sanders-Buell, E.; Hoffmann, O.; Maboko, L.; Mmbando, D.; Birx, D.L.; McCutchan, F.E.; Hoelscher, M. In-depth, longitudinal analysis of viral quasispecies from an individual triply infected with late-stage human immunodeficiency virus type 1, using a multiple PCR primer approach. J. Virol. 2005, 79, 8249–8261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracho, M.A.; García-Robles, I.; Jiménez, N.; Torres-Puente, M.; Moya, A.; González-Candelas, F. Effect of oligonucleotide primers in determining viral variability within hosts. Virol. J. 2004, 1, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itokawa, K.; Sekizuka, T.; Hashino, M.; Tanaka, R.; Kuroda, M. Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS ONE 2020, 15, e0239403. [Google Scholar] [CrossRef]
- Gao, R.; Zu, W.; Liu, Y.; Li, J.; Li, Z.; Wen, Y.; Wang, H.; Yuan, J.; Cheng, L.; Zhang, S.; et al. Quasispecies of SARS-CoV-2 revealed by single nucleotide polymorphisms (SNPs) analysis. Virulence 2021, 12, 1209–1226. [Google Scholar] [CrossRef]
- Han, L.; Zheng, Y.; Deng, J.; Nan, M.L.; Xiao, Y.; Zhuang, M.W.; Zhang, J.; Wang, W.; Gao, C.; Wang, P.H. SARS-CoV-2 ORF10 antagonizes STING-dependent interferon activation and autophagy. J. Med. Virol. 2022, 94, 5174–5188. [Google Scholar] [CrossRef]
- Armero, A.; Berthet, N.; Avarre, J.C. Intra-Host Diversity of SARS-Cov-2 Should Not Be Neglected: Case of the State of Victoria, Australia. Viruses 2021, 13, 133. [Google Scholar] [CrossRef]
- Sun, F.; Wang, X.; Tan, S.; Dan, Y.; Lu, Y.; Zhang, J.; Xu, J.; Tan, Z.; Xiang, X.; Zhou, Y.; et al. SARS-CoV-2 Quasispecies Provides an Advantage Mutation Pool for the Epidemic Variants. Microbiol. Spectr. 2021, 9, e0026121. [Google Scholar] [CrossRef]
- Quaranta, E.G.; Fusaro, A.; Giussani, E.; D’Amico, V.; Varotto, M.; Pagliari, M.; Giordani, M.T.; Zoppelletto, M.; Merola, F.; Antico, A.; et al. SARS-CoV-2 intra-host evolution during prolonged infection in an immunocompromised patient. Int. J. Infect. Dis. 2022, 122, 444–448. [Google Scholar] [CrossRef]
- Chaguza, C.; Hahn, A.M.; Petrone, M.E.; Zhou, S.; Ferguson, D.; Breban, M.I.; Pham, K.; Peña-Hernández, M.A.; Castaldi, C.; Hill, V.; et al. Accelerated SARS-CoV-2 intrahost evolution leading to distinct genotypes during chronic infection. medRxiv 2022. [Google Scholar] [CrossRef]
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef] [PubMed]
- Vellas, C.; Del Bello, A.; Debard, A.; Steinmeyer, Z.; Tribaudeau, L.; Ranger, N.; Jeanne, N.; Martin-Blondel, G.; Delobel, P.; Kamar, N.; et al. Influence of treatment with neutralizing monoclonal antibodies on the SARS-CoV-2 nasopharyngeal load and quasispecies. Clin. Microbiol. Infect. 2022, 28, 139.e5–139.e8. [Google Scholar] [CrossRef] [PubMed]
- Pondé, R.A.A. Physicochemical effect of the N501Y, E484K/Q, K417N/T, L452R and T478K mutations on the SARS-CoV-2 spike protein RBD and its influence on agent fitness and on attributes developed by emerging variants of concern. Virology 2022, 572, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136.e11. [Google Scholar] [CrossRef]
- Mansbach, R.A.; Chakraborty, S.; Nguyen, K.; Montefiori, D.C.; Korber, B.; Gnanakaran, S. The SARS-CoV-2 Spike variant D614G favors an open conformational state. Sci. Adv. 2021, 7, eabf3671. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Lubinski, B.; Fernandes, M.H.V.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional evaluation of the P681H mutation on the proteolytic activation of the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. iScience 2022, 25, 103589. [Google Scholar] [CrossRef]
- Van Cleemput, J.; Van Snippenberg, W.; Lambrechts, L.; Dendooven, A.; D’Onofrio, V.; Couck, L.; Trypsteen, W.; Vanrusselt, J.; Theuns, S.; Vereecke, N.; et al. Organ-specific genome diversity of replication-competent SARS-CoV-2. Nat. Commun. 2021, 12, 6612. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2022, 50, D161–D164. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissgerber, T.L.; Winham, S.J.; Heinzen, E.P.; Milin-Lazovic, J.S.; Garcia-Valencia, O.; Bukumiric, Z.; Savic, M.D.; Garovic, V.D.; Milic, N.M. Reveal, Don’t Conceal: Transforming Data Visualization to Improve Transparency. Circulation 2019, 140, 1506–1518. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Genes | Coordinates on the Genome GenBank Accession no. NC_045512.2 | Mean Diversity (%) | Number of Gene Positions Exhibiting a Significant (>4%) Diversity in Any of the 110 Samples | Number of Positions per 100 Nucleotides |
|---|---|---|---|---|
| ORF1ab | 266..21555 | 0.2 | 3123 | 0.15 |
| S | 21563..25384 | 0.22 | 1281 | 0.34 |
| ORF3a | 25393..26220 | 0.2 | 105 | 0.13 |
| E | 26245..26472 | 0.2 | 31 | 0.14 |
| M | 26523..27191 | 0.21 | 68 | 0.10 |
| ORF6 | 27202..27387 | 0.17 | 26 | 0.14 |
| ORF7a | 27394..27759 | 0.2 | 28 | 0.08 |
| ORF7b | 27756..27887 | 0.19 | 4 | 0.03 |
| ORF8 | 27894..28259 | 0.21 | 14 | 0.04 |
| N | 28274..29533 | 0.34 | 301 | 0.24 |
| ORF10 | 29558..29674 | 0.2 | 65 | 0.56 |
| Coordinates on the Genome GenBank Accession no. NC_045512.2 | Gene_Codon | Inter-Patient Diversity (%) | Mean Intra-Sample Diversity (%) | Nucleotide Position in Codon |
|---|---|---|---|---|
| 516 | ORF1a_84 | 11 | 1.21 | 1 |
| 517 | 11 | 1.06 | 2 | |
| 518 | ORF1a_85 | 13 | 1.25 | 1 |
| 519 | 13 | 1.11 | 2 | |
| 520 | 13 | 1.15 | 3 | |
| 521 | ORF1a_86 | 6 | 1.03 | 1 |
| 522 | 6 | 1.27 | 2 | |
| 867 | ORF1a_201 | 1 | 1.01 | 2 |
| 868 | 1 | 2.33 | 3 | |
| 873 | ORF1a_203 | 4 | 1.41 | 2 |
| 963 | ORF1a_233 | 1 | 1.12 | 2 |
| 1465 | ORF1a_400 | 3 | 1.61 | 3 |
| 1600 | ORF1a_445 | 7 | 1.05 | 3 |
| 3067 | ORF1a_934 | 1 | 1.12 | 3 |
| 3468 | ORF1a_1068 | 2 | 1.65 | 2 |
| 4318 | ORF1a_1351 | 1 | 1.76 | 3 |
| 5434 | ORF1a_1723 | 1 | 1.03 | 3 |
| 5743 | ORF1a_1826 | 1 | 1.24 | 3 |
| 5886 | ORF1a_1874 | 1 | 1.50 | 2 |
| 6268 | ORF1a_2001 | 2 | 1.51 | 3 |
| 6713 | ORF1a_2149 | 2 | 1.24 | 3 |
| 9044 | ORF1a_2927 | 1 | 1.10 | 1 |
| 9714 | ORF1a_3150 | 1 | 1.47 | 2 |
| 10037 | ORF1a_3258 | 3 | 1.23 | 1 |
| 11075 | ORF1a_3604 | 1 | 6.64 | 1 |
| 11117 | ORF1a_3618 | 5 | 1.02 | 1 |
| 11289 | ORF1a_3675 | 32 | 1.63 | 2 |
| 11290 | 32 | 1.57 | 3 | |
| 11291 | ORF1a_3676 | 37 | 1.57 | 1 |
| 11292 | 37 | 1.68 | 2 | |
| 11293 | 37 | 2.03 | 3 | |
| 11294 | ORF1a_3677 | 37 | 1.73 | 1 |
| 11295 | 37 | 1.67 | 2 | |
| 11296 | 37 | 1.72 | 3 | |
| 11997 | ORF1a_3911 | 1 | 1.57 | 2 |
| 15156 | ORF1b_572 | 1 | 1.14 | 2 |
| 15157 | 1 | 3.87 | 3 | |
| 15173 | ORF1b_578 | 1 | 2.18 | 1 |
| 15491 | ORF1b_684 | 1 | 1.52 | 1 |
| 15492 | 1 | 1.01 | 2 | |
| 15500 | ORF1b_687 | 1 | 1.15 | 1 |
| 15501 | 1 | 1.11 | 2 | |
| 15576 | ORF1b_712 | 1 | 1.11 | 2 |
| 17152 | ORF1b_1237 | 1 | 1.34 | 3 |
| 17514 | ORF1b_1358 | 1 | 1.01 | 2 |
| 18314 | ORF1b_1625 | 1 | 1.14 | 1 |
| 18354 | ORF1b_1638 | 2 | 1.21 | 2 |
| 18482 | ORF1b_1681 | 6 | 1.25 | 1 |
| 19477 | ORF1b_2012 | 2 | 1.31 | 3 |
| 21243 | ORF1b_2601 | 1 | 1.05 | 2 |
| 21492 | ORF1b_2684 | 1 | 1.23 | 2 |
| 21765 | S_68 | 4 | 1.19 | 2 |
| 22214 | S_218 | 3 | 2.25 | 1 |
| 22216 | 3 | 1.13 | 3 | |
| 22218 | S_219 | 1 | 1.38 | 2 |
| 22219 | 1 | 1.15 | 3 | |
| 22645 | S_361 | 1 | 1.03 | 3 |
| 23531 | S_657 | 3 | 1.18 | 1 |
| 23534 | S_658 | 1 | 1.09 | 1 |
| 23622 | S_687 | 2 | 1.16 | 2 |
| 23642 | S_694 | 3 | 1.88 | 1 |
| 23652 | S_697 | 1 | 1.14 | 2 |
| 24038 | S_826 | 2 | 1.20 | 1 |
| 24089 | S_843 | 1 | 2.30 | 1 |
| 24365 | S_935 | 1 | 1.29 | 1 |
| 25620 | ORF3a_76 | 2 | 1.26 | 3 |
| 25979 | ORF3a_196 | 3 | 1.66 | 2 |
| 26390 | E_49 | 2 | 1.01 | 2 |
| 26433 | E_63 | 1 | 1.31 | 3 |
| 27870 | ORF7b_37 | 5 | 2.10 | 3 |
| 28215 | ORF8_108 | 1 | 1.38 | 1 |
| 28251 | ORF8_120 | 11 | 1.15 | 1 |
| 28252 | ORF8_120 | 11 | 1.01 | 2 |
| 28253 | 11 | 1.17 | 3 | |
| 28254 | ORF8_121 | 22 | 4.84 | 1 |
| 20918 | N_215 | 4 | 4.27 | 3 |
| 28920 | N_216 | 2 | 2.70 | 2 |
| 28921 | 2 | 1.24 | 3 | |
| 28922 | N_217 | 2 | 1.10 | 1 |
| 28923 | 2 | 1.69 | 2 | |
| 28924 | 2 | 2.42 | 3 | |
| 28926 | N_218 | 1 | 1.52 | 2 |
| 28927 | 1 | 3.04 | 3 | |
| 28929 | N_219 | 1 | 1.87 | 2 |
| 28931 | N_220 | 5 | 4.20 | 1 |
| 28933 | 5 | 1.23 | 3 | |
| 28949 | N_226 | 1 | 1.12 | 1 |
| 28952 | N_227 | 1 | 1.56 | 1 |
| 28954 | 1 | 2.84 | 3 | |
| 28959 | N_229 | 3 | 1.49 | 2 |
| 28962 | N_230 | 1 | 1.77 | 2 |
| 28967 | N_232 | 5 | 1.31 | 1 |
| 28974 | N_234 | 31 | 1.92 | 2 |
| 28976 | N_235 | 6 | 1.07 | 1 |
| 28979 | N_236 | 1 | 1.33 | 1 |
| 28981 | 1 | 5.20 | 3 | |
| 28985 | N_238 | 7 | 2.52 | 1 |
| 28987 | 7 | 1.46 | 3 | |
| 28989 | N_239 | 1 | 2.27 | 2 |
| 28994 | N_241 | 1 | 1.96 | 1 |
| 28997 | N_242 | 1 | 2.36 | 1 |
| 29000 | N_243 | 3 | 4.36 | 3 |
| 29004 | N_244 | 3 | 1.45 | 2 |
| 29010 | N_246 | 1 | 1.11 | 2 |
| 29014 | N_247 | 1 | 1.08 | 3 |
| 29021 | N_250 | 2 | 1.62 | 1 |
| 29024 | N_251 | 1 | 1.12 | 1 |
| 29029 | N_252 | 4 | 1.93 | 3 |
| 29035 | N_254 | 2 | 1.42 | 3 |
| 29039 | N_256 | 2 | 1.69 | 1 |
| 29041 | 2 | 2.43 | 3 | |
| 29049 | N_259 | 1 | 2.77 | 2 |
| 29057 | N_262 | 1 | 1.55 | 1 |
| 29059 | 1 | 2.23 | 3 | |
| 29072 | N_267 | 1 | 1.13 | 1 |
| 29325 | N_351 | 1 | 1.49 | 2 |
| 29336 | N_355 | 1 | 1.37 | 1 |
| 29514 | N_414 | 7 | 1.25 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bader, W.; Delerce, J.; Aherfi, S.; La Scola, B.; Colson, P. Quasispecies Analysis of SARS-CoV-2 of 15 Different Lineages during the First Year of the Pandemic Prompts Scratching under the Surface of Consensus Genome Sequences. Int. J. Mol. Sci. 2022, 23, 15658. https://doi.org/10.3390/ijms232415658
Bader W, Delerce J, Aherfi S, La Scola B, Colson P. Quasispecies Analysis of SARS-CoV-2 of 15 Different Lineages during the First Year of the Pandemic Prompts Scratching under the Surface of Consensus Genome Sequences. International Journal of Molecular Sciences. 2022; 23(24):15658. https://doi.org/10.3390/ijms232415658
Chicago/Turabian StyleBader, Wahiba, Jeremy Delerce, Sarah Aherfi, Bernard La Scola, and Philippe Colson. 2022. "Quasispecies Analysis of SARS-CoV-2 of 15 Different Lineages during the First Year of the Pandemic Prompts Scratching under the Surface of Consensus Genome Sequences" International Journal of Molecular Sciences 23, no. 24: 15658. https://doi.org/10.3390/ijms232415658