
Comparative Transcriptome Analysis Unveils the Molecular Mechanism Underlying Sepal Colour Changes under Acidic pH Substratum in Hydrangea macrophylla

 , , and
, , and
Abstract
:1. Introduction
2. Results
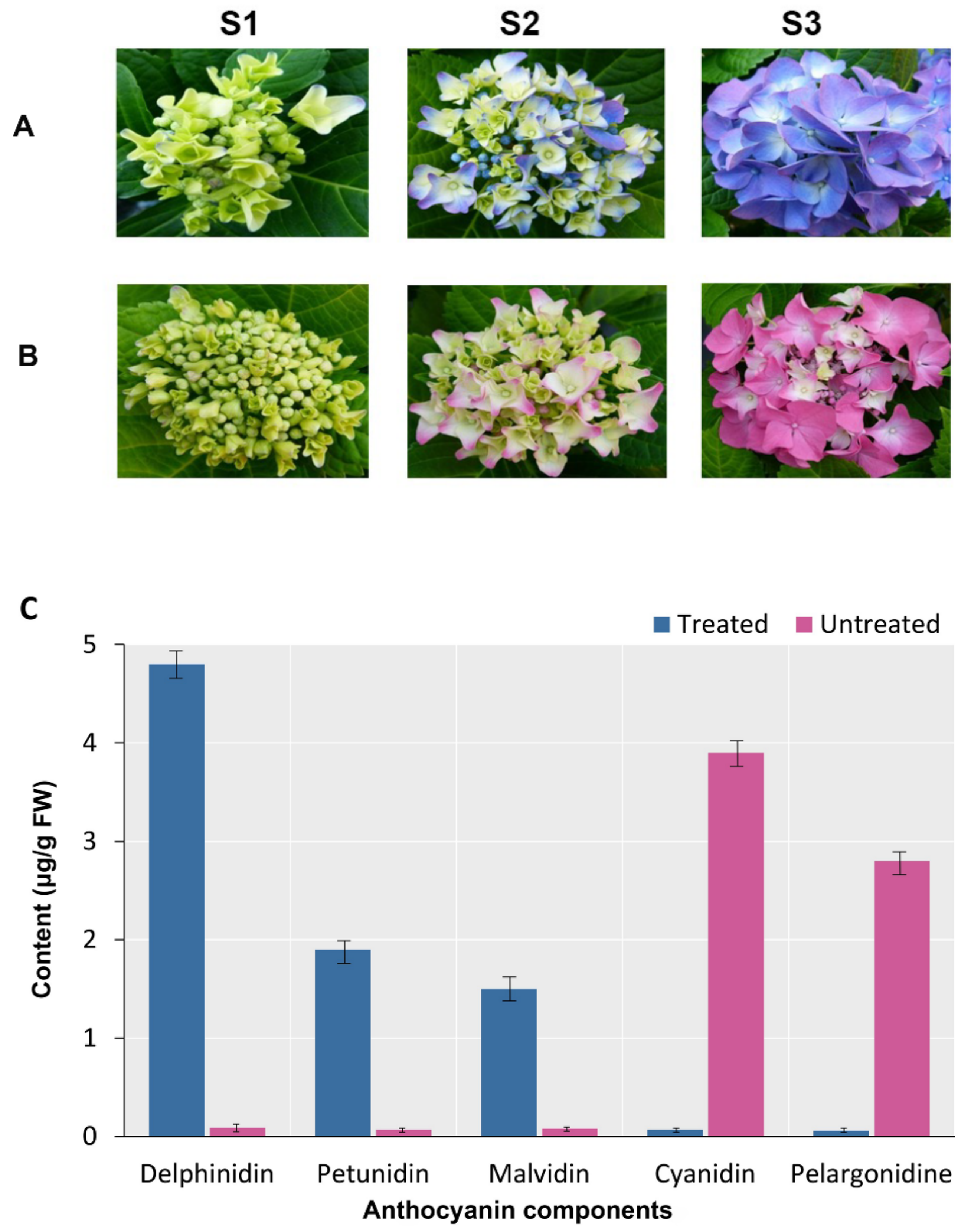
2.1. Change of Sepal Colour under Different Soil pH
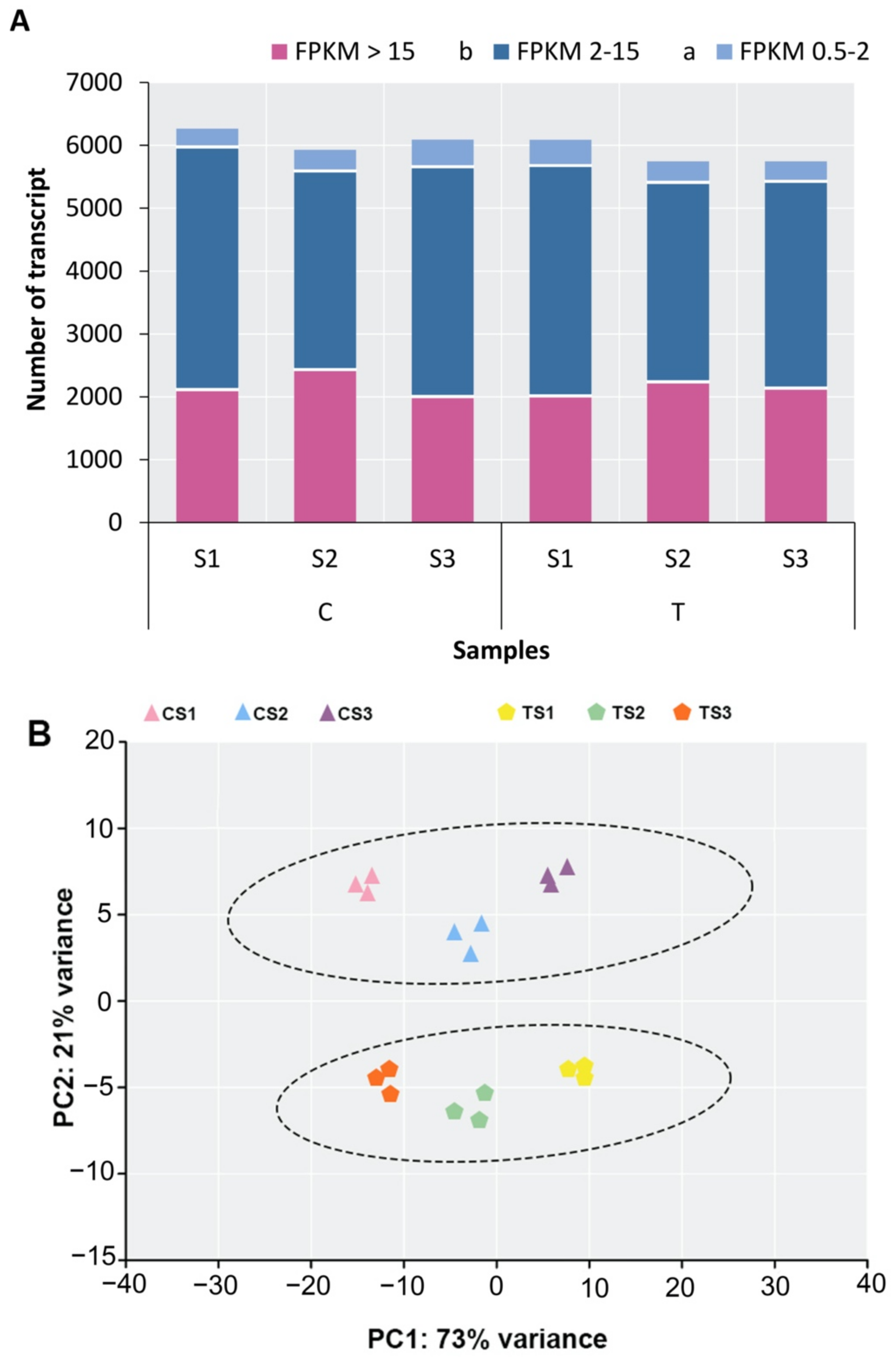
2.2. Transcriptome Sequencing, Annotation, and Analysis of DEGs
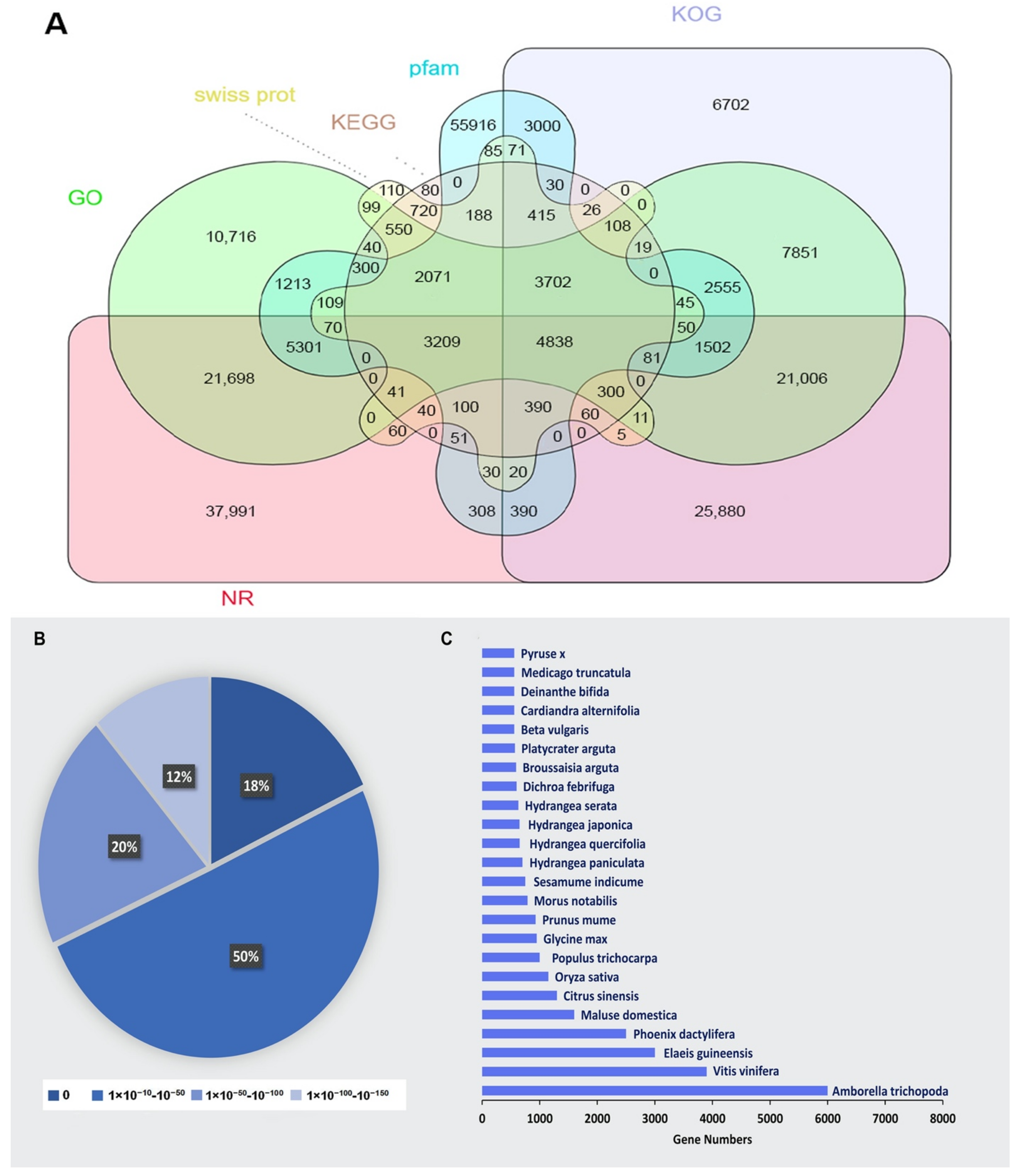
2.3. Functional Annotation
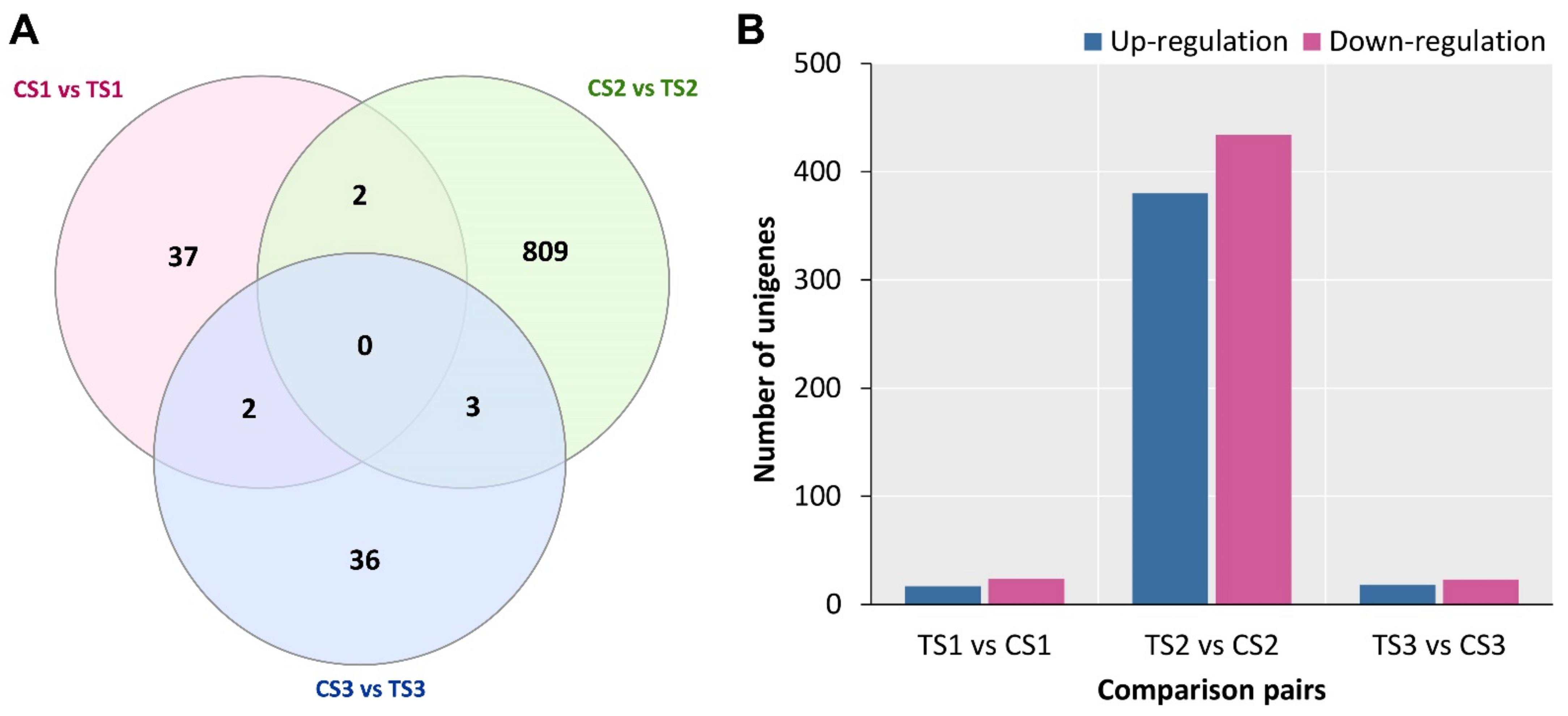
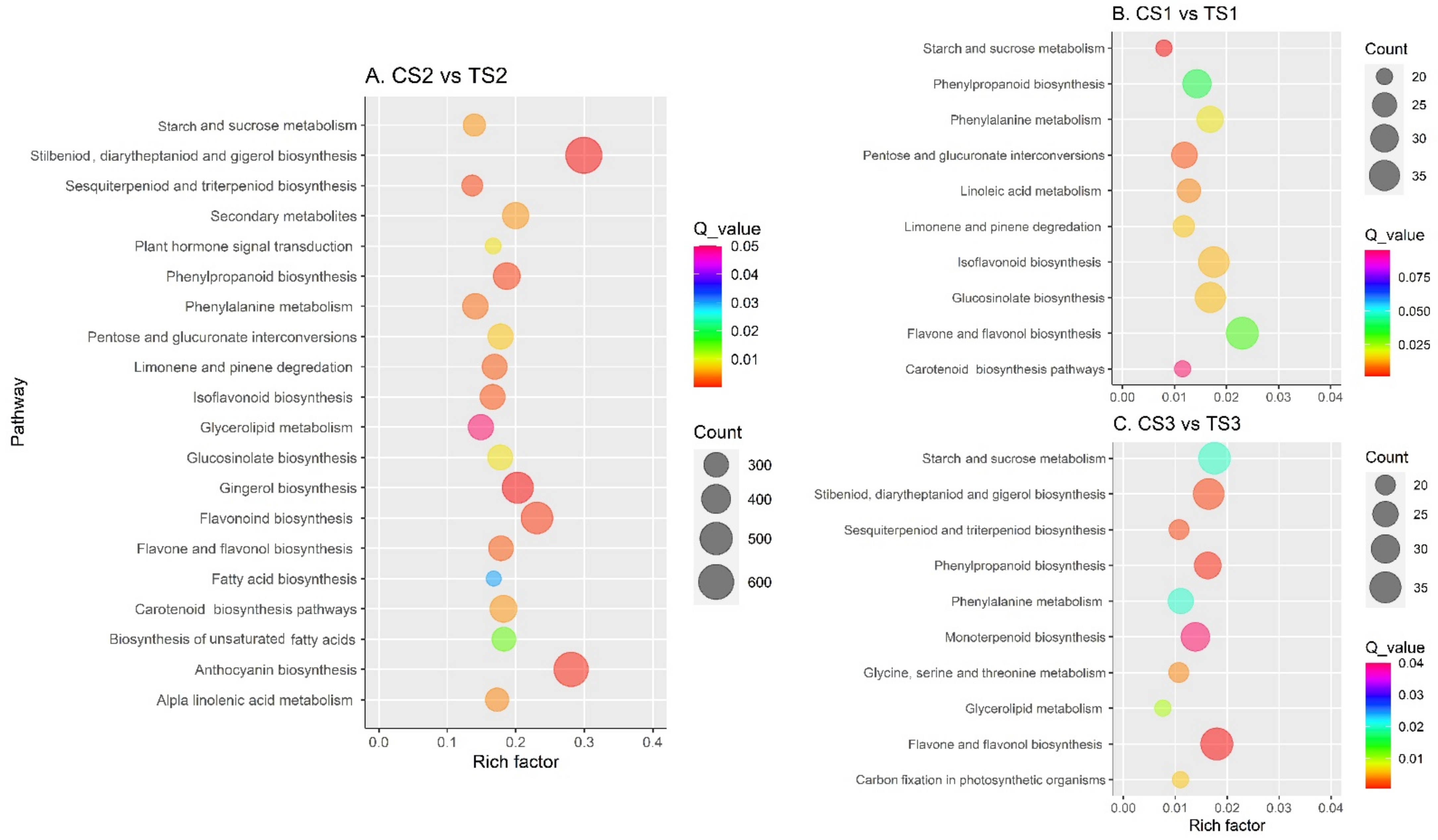
2.4. DEG Identification and Functional Enrichment Analysis
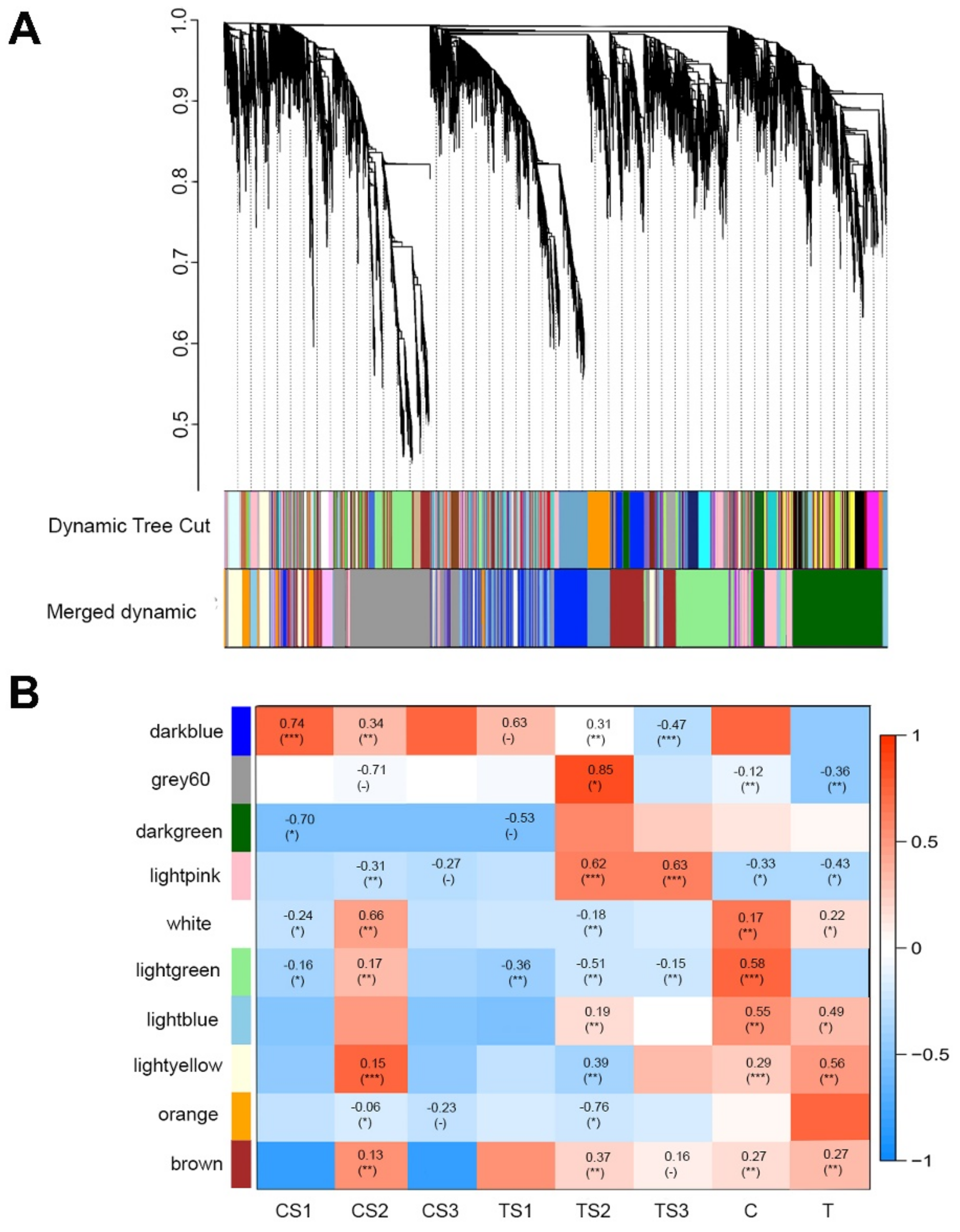
2.5. Identification of TFs and Establishment of Gene Co-Expression Network Analysis (WGCNA)
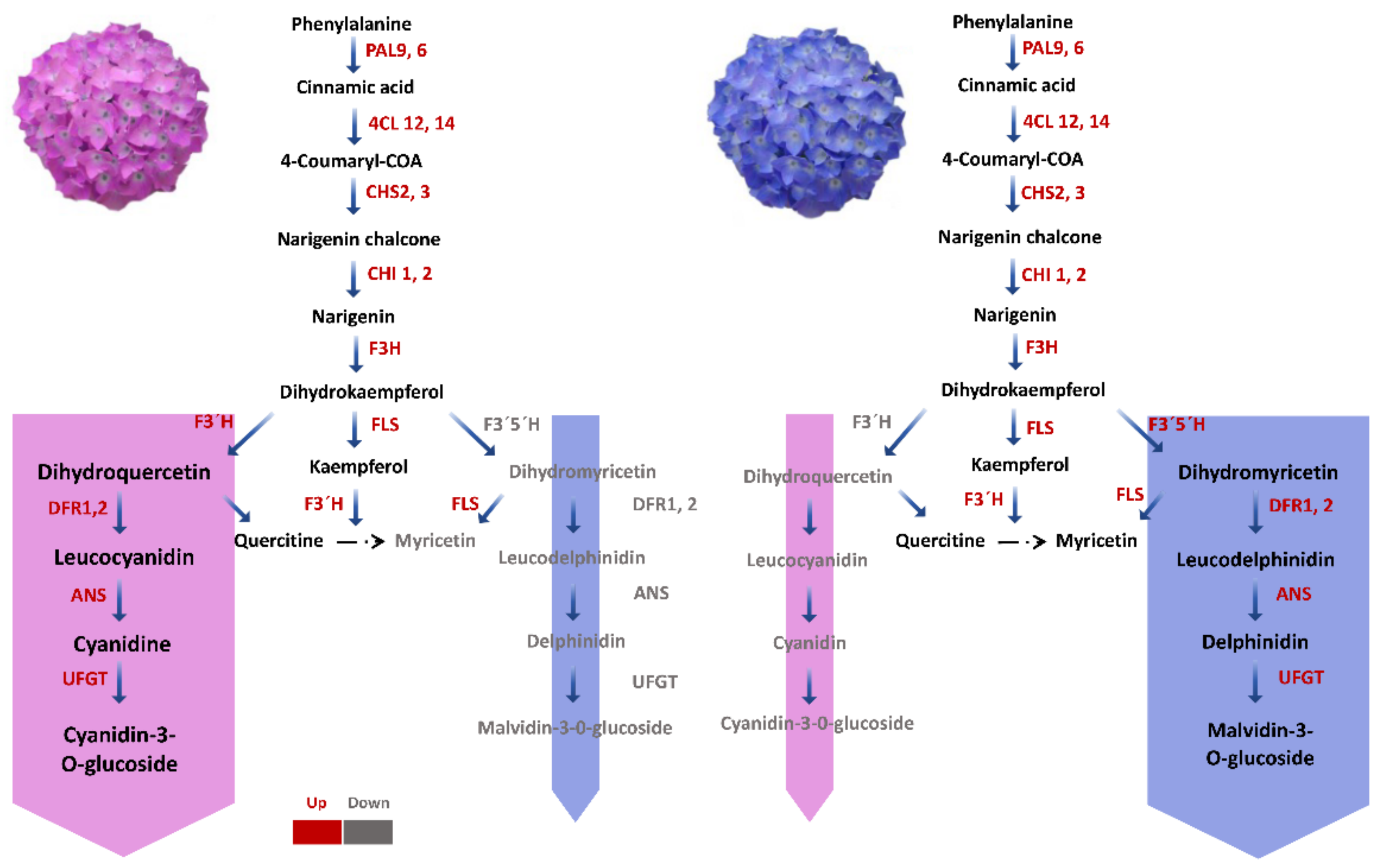
2.6. Candidates Accountable for the Gain of Blue Colour in Hydrangea with Pink-Coloured Flower
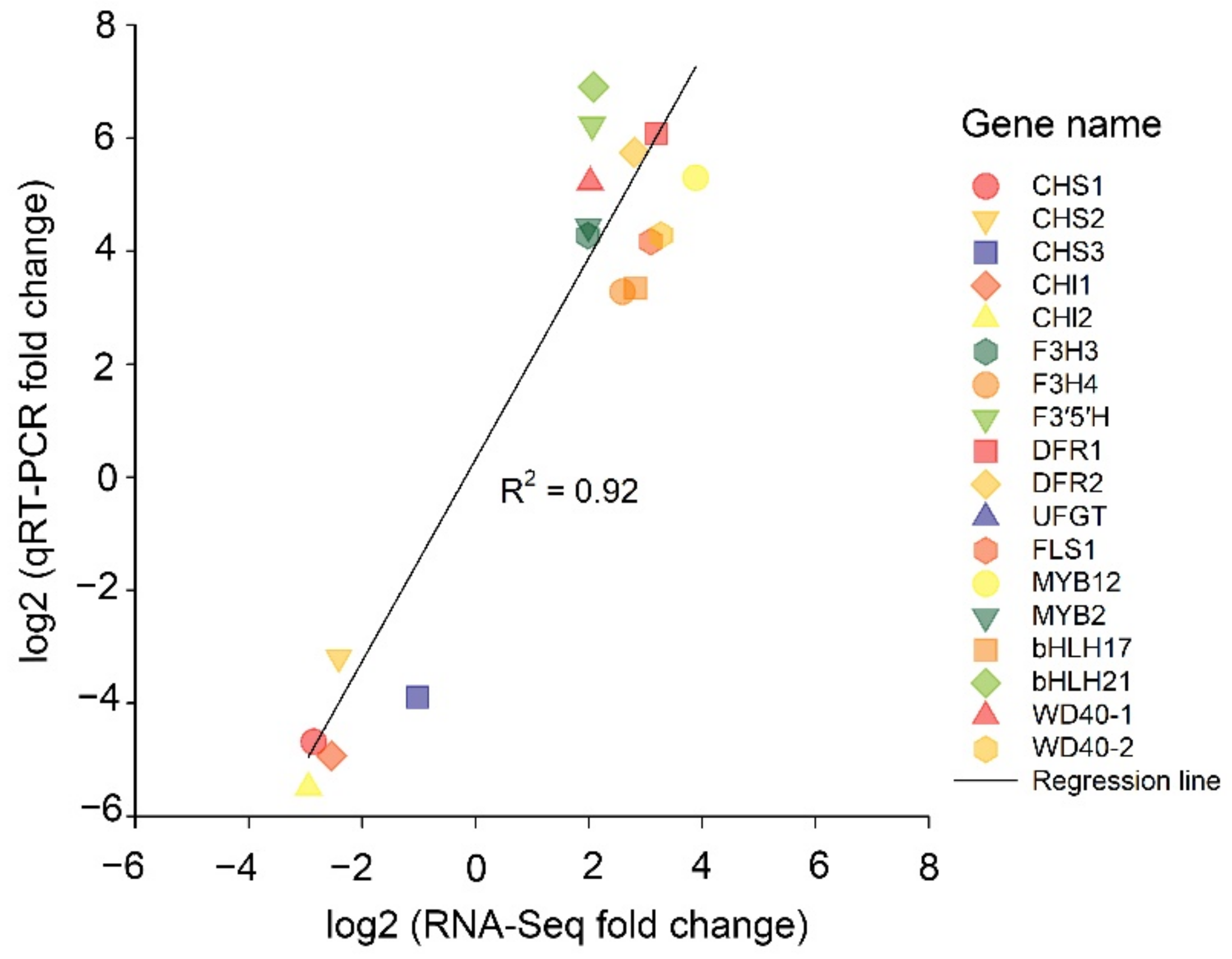
2.7. qRT–PCR of the Transcriptomic Data
3. Discussions
3.1. Comparison of Genes Involved in Flavonoid Biosynthesis in Hydrangeas Grown under Different pH Conditions
3.2. Identification of Hub Genes Related to Flower Formation by WGCNA
3.3. Identification of Transcription Factors Related to Flower Colour Transition
4. Materials and Methods
4.1. Plant Material
4.2. RNA Extraction, cDNA Library Creation, and Sequencing
4.3. Data Analysis
4.4. Expression Annotation
4.5. GO and KEGG Pathway Enrichment Analysis for Differentially Expressed Unigenes
4.6. Quantitative Reverse Transcription–Polymerase Chain Reaction-Based Validation
4.7. Estimation of Relative Pigment Content
4.8. Statistical Analyses
5. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Junker, R.R.; Parachnowitsch, A.L. Working towards a holistic view on flower traits—How floral scents mediate plant–animal interactions in concert with other floral characters. J. Indian Inst. Sci. 2015, 95, 43–68. [Google Scholar]
- Sapir, Y.; Gallagher, M.K.; Senden, E. What maintains flower colour variation within populations? Trends Ecol. Evol. 2021, 36, 507–519. [Google Scholar] [PubMed]
- Landi, M.; Tattini, M.; Gould, K.S. Multiple functional roles of anthocyanins in plant-environment interactions. Environ. Exp. Bot. 2015, 119, 4–17. [Google Scholar] [CrossRef]
- Sobral, M.; Neylan, I.P.; Narbona, E.; Dirzo, R. Transgenerational plasticity in flower color induced by caterpillars. Front. Plant Sci. 2021, 12, 617815. [Google Scholar] [PubMed]
- Johnson, I.M.; Edwards, T.J.; Johnson, S.D. Geographical Variation in Flower Color in the Grassland Daisy Gerbera aurantiaca: Testing for Associations with Pollinators and Abiotic Factors. Front. Ecol. Evol. 2021, 9, 676520. [Google Scholar] [CrossRef]
- Tilt, K. Extension Specialist and Professor; Horticulture Department, Auburn-University: Auburn, AL, USA, 2008. [Google Scholar]
- Noda, N. Recent advances in the research and development of blue flowers. Breed. Sci. 2018, 68, 17132. [Google Scholar] [CrossRef] [Green Version]
- De Smet, Y. Through the Fog: Evolutionary Insights Provide Novel Genus- and Species-Level Boundaries in Tribe Hydrangeeae and Genus Hydrangea. Ph.D. Dissertation, Ghent University, Ghent, Belgium, 2020. [Google Scholar]
- Samain, M.-S.; Mendoza, C.G.; Salas, E.M.M. On Hydrangea peruviana, an endangered species from Ecuador, and Hydrangea oerstedii, very common in Costa Rica and Panama, and seven threatened Central and South American Hydrangeas, which have been confounded with these. PhytoKeys 2021, 171, 91. [Google Scholar]
- Wu, X.; Hulse-Kemp, A.M.; Wadl, P.A.; Smith, Z.; Mockaitis, K.; Staton, M.E.; Rinehart, T.A.; Alexander, L.W. Genomic Resource Development for Hydrangea (Hydrangea macrophylla (Thunb.) Ser.)—A Transcriptome Assembly and a High-Density Genetic Linkage Map. Horticulturae 2021, 7, 25. [Google Scholar]
- Anaya-Covarrubias, J.Y.; Larranaga, N.; Almaráz-Abarca, N.; Escoto-Delgadillo, M.; Rodríguez-Macías, R.; Torres-Morán, M.I. Hydrangea DNA methylation caused by pH substrate changes to modify sepal colour is detected by MSAP and ISSR markers. Agronomy 2019, 9, 871. [Google Scholar] [CrossRef] [Green Version]
- Waki, T.; Kodama, M.; Akutsu, M.; Namai, K.; Iigo, M.; Kurokura, T.; Yamamoto, T.; Nashima, K.; Nakayama, M.; Yagi, M. Development of DNA Markers Linked to Double-Flower and Hortensia Traits in Hydrangea macrophylla (Thunb.) Ser. Hortic. J. 2018, 87, 264–273. [Google Scholar]
- Wei, L.; Wang, C.; Liao, W. Hydrogen sulfide improves the vase life and quality of cut roses and chrysanthemums. J. Plant Growth Regul. 2021, 40, 2532–2547. [Google Scholar]
- Kikelly, J. How to Prune Hydrangea Macrophylla; West of Ireland. 2006. Available online: http://www.gardenplansireland.com/forum/about639.html (accessed on 13 February 2022).
- Robinson, G.M.; Robinson, R. A survey of anthocyanins. II. Biochem. J. 1932, 26, 1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Kariuda, M.; Itoi, H. Blueing of sepal colour of Hydrangea macrophylla. Phytochemistry 1985, 24, 2251–2254. [Google Scholar] [CrossRef]
- Sunil, L.; Shetty, N.P. Biosynthesis and regulation of anthocyanin pathway genes. Appl. Microbiol. Biotechnol. 2022, 106, 1783–1798. [Google Scholar] [CrossRef] [PubMed]
- Appelhagen, I.; Wulff-Vester, A.K.; Wendell, M.; Hvoslef-Eide, A.-K.; Russell, J.; Oertel, A.; Martens, S.; Mock, H.-P.; Martin, C.; Matros, A. Colour bio-factories: Towards scale-up production of anthocyanins in plant cell cultures. Metab. Eng. 2018, 48, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Oyama, K.-i.; Yoshida, K. Direct observation of hydrangea blue-complex composed of 3-O-glucosyldelphinidin, Al3+ and 5-O-acylquinic acid by ESI-mass spectrometry. Molecules 2018, 23, 1424. [Google Scholar] [PubMed] [Green Version]
- Yoshida, K.; Ito, D.; Miki, N.; Kondo, T. Single-cell analysis clarifies mosaic color development in purple hydrangea sepal. New Phytol. 2021, 229, 3549–3557. [Google Scholar] [CrossRef]
- Kar, D.; Pradhan, A.A.; Datta, S. The role of solute transporters in aluminum toxicity and tolerance. Physiol. Plant. 2021, 171, 638–652. [Google Scholar]
- Rahim, M.S.; Parveen, A.; Aggarwal, B.; Madhawan, A.; Kumar, P.; Kumar, V.; Rana, N.; Bansal, R.; Deshmukh, R.; Roy, J. Computational tools and approaches for aquaporin (AQP) research. In Metal and Nutrient Transporters in Abiotic Stress; Elsevier: Amsterdam, The Netherlands, 2021; pp. 1–32. [Google Scholar]
- Blom, T.J.; Piott, B.D. Florists’ hydrangea blueing with aluminum sulfate applications during forcing. HortScience 1992, 27, 1084. [Google Scholar]
- Hamid, R.; Jacob, F.; Marashi, H.; Rathod, V.; Tomar, R.S. Uncloaking lncRNA-meditated gene expression as a potential regulator of CMS in cotton (Gossypium hirsutum L.). Genomics 2020, 112, 3354–3364. [Google Scholar]
- Rathod, V.; Hamid, R.; Tomar, R.S.; Patel, R.; Padhiyar, S.; Kheni, J.; Thirumalaisamy, P.; Munshi, N.S. Comparative RNA-Seq profiling of a resistant and susceptible peanut (Arachis hypogaea) genotypes in response to leaf rust infection caused by Puccinia arachidis. 3 Biotech 2020, 10, 284. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Fan, H.; Yao, F.; Cao, X.; Liu, M.; Sun, J.; Liu, Y. Transcriptomics and targeted metabolomics profilings for elucidation of pigmentation in Lonicera japonica flowers at different developmental stages. Ind. Crop. Prod. 2020, 145, 111981. [Google Scholar]
- Luo, X.; Sun, D.; Wang, S.; Luo, S.; Fu, Y.; Niu, L.; Shi, Q.; Zhang, Y. Integrating full-length transcriptomics and metabolomics reveals the regulatory mechanisms underlying yellow pigmentation in tree peony (Paeonia suffruticosa Andr.) flowers. Hortic. Res. 2021, 8, 235. [Google Scholar] [PubMed]
- Jin, X.; Huang, H.; Wang, L.; Sun, Y.; Dai, S. Transcriptomics and metabolite analysis reveals the molecular mechanism of anthocyanin biosynthesis branch pathway in different Senecio cruentus cultivars. Front. Plant Sci. 2016, 7, 1307. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Lu, C.; Jiang, H.; Peng, J. Global transcriptome analysis reveals distinct aluminum-tolerance pathways in the Al-accumulating species Hydrangea macrophylla and marker identification. PLoS ONE 2015, 10, e0144927. [Google Scholar]
- Jiang, T.; Zhang, M.; Wen, C.; Xie, X.; Tian, W.; Wen, S.; Lu, R.; Liu, L. Integrated metabolomic and transcriptomic analysis of the anthocyanin regulatory networks in Salvia miltiorrhiza Bge. flowers. BMC Plant Biol. 2020, 20, 349. [Google Scholar]
- Cho, H.J.; Kim, G.H.; Choi, C. Differential gene expression and epigenetic analyses between striped and blushed skinned sports of ‘Fuji’apple. Sci. Hortic. 2020, 261, 108944. [Google Scholar] [CrossRef]
- Zhu, H.-H.; Yang, J.-X.; Xiao, C.-H.; Mao, T.-Y.; Zhang, J.; Zhang, H.-Y. Differences in flavonoid pathway metabolites and transcripts affect yellow petal colouration in the aquatic plant Nelumbo nucifera. BMC Plant Biol. 2019, 19, 277. [Google Scholar] [CrossRef]
- Love, M.; Anders, S.; Huber, W. Differential analysis of count data–the DESeq2 package. Genome Biol. 2014, 15, 1–21. [Google Scholar]
- Huang, P.; Lin, F.; Li, B.; Zheng, Y. Hybrid-transcriptome sequencing and associated metabolite analysis reveal putative genes involved in flower color difference in rose mutants. Plants 2019, 8, 267. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Lewis, D.; Shi, R.; McGhie, T.; Wang, L.; Arathoon, S.; Schwinn, K.; Davies, K.; Qian, X.; Zhang, H. The colour variations of flowers in wild Paeonia delavayi plants are determined by four classes of plant pigments. N. Z. J. Crop Hortic. Sci. 2022, 50, 69–84. [Google Scholar]
- Rajabi, A.; Fahmideh, L.; Keykhasaber, M.; Omran, V.G. Genetic engineering of novel yellow color african violet (Saintpaulia ionantha) produced by accumulation of Aureusidin 6-O-glucoside. Biol. Proced. Online 2022, 24, 3. [Google Scholar] [PubMed]
- Sun, J.; Qiu, C.; Ding, Y.; Wang, Y.; Sun, L.; Fan, K.; Gai, Z.; Dong, G.; Wang, J.; Li, X. Fulvic acid ameliorates drought stress-induced damage in tea plants by regulating the ascorbate metabolism and flavonoids biosynthesis. BMC Genom. 2020, 21, 411. [Google Scholar] [CrossRef] [PubMed]
- Yonekura-Sakakibara, K.; Higashi, Y.; Nakabayashi, R. The origin and evolution of plant flavonoid metabolism. Front. Plant Sci. 2019, 10, 943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Li, M.; Dai, S. Ectopic expression of multiple chrysanthemum (Chrysanthemum × morifolium) R2R3-MYB transcription factor genes regulates anthocyanin accumulation in tobacco. Genes 2019, 10, 777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Wang, Y.; Ren, L.; Shi, Q.; Zheng, B.; Miao, K.; Guo, X. Overexpression of Ps-CHI1, a homologue of the chalcone isomerase gene from tree peony (Paeonia suffruticosa), reduces the intensity of flower pigmentation in transgenic tobacco. Plant Cell Tissue Organ Cult. (PCTOC) 2014, 116, 285–295. [Google Scholar] [CrossRef]
- Peng, Y.; Lin-Wang, K.; Cooney, J.M.; Wang, T.; Espley, R.V.; Allan, A.C. Differential regulation of the anthocyanin profile in purple kiwifruit (Actinidia species). Hortic. Res. 2019, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, X.; Cai, X.; Shan, X.; Gao, R.; Yang, S.; Han, T.; Wang, S.; Wang, L.; Gao, X. Dihydroflavonol 4-reductase genes from Freesia hybrida play important and partially overlapping roles in the biosynthesis of flavonoids. Front. Plant Sci. 2017, 8, 428. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.-W.; Zhang, N.; Jiang, C.-Y.; Wu, S.-X.; Feng, X.-Y.; Feng, X.-Y. Exploring the genes involved in biosynthesis of dihydroquercetin and dihydromyricetin in Ampelopsis grossedentata. Sci. Rep. 2021, 11, 15596. [Google Scholar] [CrossRef]
- Zhou, C.; Mei, X.; Rothenberg, D.O.N.; Yang, Z.; Zhang, W.; Wan, S.; Yang, H.; Zhang, L. Metabolome and transcriptome analysis reveals putative genes involved in anthocyanin accumulation and coloration in white and pink tea (Camellia sinensis) flower. Molecules 2020, 25, 190. [Google Scholar]
- Shimada, N.; Sasaki, R.; Sato, S.; Kaneko, T.; Tabata, S.; Aoki, T.; Ayabe, S.-I. A comprehensive analysis of six dihydroflavonol 4-reductases encoded by a gene cluster of the Lotus japonicus genome. J. Exp. Bot. 2005, 56, 2573–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.T.; Ryu, S.; Yi, H.; Shin, B.; Cheong, H.; Choi, G. Alteration of a single amino acid changes the substrate specificity of dihydroflavonol 4-reductase. Plant J. 2001, 25, 325–333. [Google Scholar] [PubMed]
- Tanaka, Y.; Brugliera, F. Flower colour and cytochromes P450. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Ke, H.; Keting, H.; Qiaoyan, X.; Silan, D. Flower colour modification of chrysanthemum by suppression of F3’H and overexpression of the exogenous Senecio cruentus F3′5′H gene. PLoS ONE 2013, 8, e74395. [Google Scholar]
- Ishiguro, K.; Taniguchi, M.; Tanaka, Y. Functional analysis of Antirrhinum kelloggii flavonoid 3′-hydroxylase and flavonoid 3′, 5′-hydroxylase genes; critical role in flower color and evolution in the genus Antirrhinum. J. Plant Res. 2012, 125, 451–456. [Google Scholar] [CrossRef]
- Hopkins, R.; Rausher, M.D. Identification of two genes causing reinforcement in the Texas wildflower Phlox drummondii. Nature 2011, 469, 411–414. [Google Scholar] [CrossRef]
- Peng, J.; Xue, C.; Dong, X.; Zeng, C.; Wu, Y.; Cao, F. Gene cloning and analysis of the pattern of expression of the transcription factor HymMYB2 related to blue flower formation in hydrangea macrophylla. Euphytica 2021, 217, 115. [Google Scholar] [CrossRef]
- Rosati, C.; Simoneau, P.; Treutter, D.; Poupard, P.; Cadot, Y.; Cadic, A.; Duron, M. Erratum: Engineering of flower color in forsythia by expression of two independently-transformed dihydroflavonol 4-reductase and anthocyanidin synthase genes of flavonoid pathway. Mol. Breed. 2004, 13, 209–210. [Google Scholar] [CrossRef] [Green Version]
- Kogawa, K.; Kato, N.; Kazuma, K.; Noda, N.; Suzuki, M. Purification and characterization of UDP-glucose: Anthocyanin 3′, 5′-O-glucosyltransferase from Clitoria ternatea. Planta 2007, 226, 1501–1509. [Google Scholar]
- Lin, R.-C. Evolution and Genetics of Floral Color Polymorphisms in Clarkia gracilis ssp. Sonomensis and Erythronium umbilicatum. Ph.D. Thesis, Duke University, Durham, NC, USA, 2020. [Google Scholar]
- Duan, H.-R.; Wang, L.-R.; Cui, G.-X.; Zhou, X.-H.; Duan, X.-R.; Yang, H.-S. Identification of the regulatory networks and hub genes controlling alfalfa floral pigmentation variation using RNA-sequencing analysis. BMC Plant Biol. 2020, 20, 110. [Google Scholar]
- Wang, Q.; Zeng, X.; Song, Q.; Sun, Y.; Feng, Y.; Lai, Y. Identification of key genes and modules in response to Cadmium stress in different rice varieties and stem nodes by weighted gene co-expression network analysis. Sci. Rep. 2020, 10, 9525. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, J.; Zhu, Y.; Ni, S.; Chen, J.; Feng, X.; Zhang, Y.; Li, S.; Zhu, H.; Wen, Y. De novo assembly of the Camellia nitidissima transcriptome reveals key genes of flower pigment biosynthesis. Front. Plant Sci. 2017, 8, 1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Cheng, D.; Yang, Y.; Zhang, G.; Qin, M.; Chen, J.; Chen, Y.; Jiang, M. Co-expression network analysis of the transcriptomes of rice roots exposed to various cadmium stresses reveals universal cadmium-responsive genes. BMC Plant Biol. 2017, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cui, Y.; Vainstein, A.; Chen, S.; Ma, H. Regulation of fig (Ficus carica L.) fruit color: Metabolomic and transcriptomic analyses of the flavonoid biosynthetic pathway. Front. Plant Sci. 2017, 8, 1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S. Transcriptional control of flavonoid biosynthesis: Fine-tuning of the MYB-bHLH-WD40 (MBW) complex. Plant Signal. Behav. 2014, 9, e27522. [Google Scholar] [PubMed]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB–bHLH–WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef]
- Nesi, N.; Debeaujon, I.; Jond, C.; Stewart, A.J.; Jenkins, G.I.; Caboche, M.; Lepiniec, L.c. The TRANSPARENT TESTA16 locus encodes the ARABIDOPSIS BSISTER MADS domain protein and is required for proper development and pigmentation of the seed coat. Plant Cell 2002, 14, 2463–2479. [Google Scholar] [CrossRef]
- Gou, J.-Y.; Felippes, F.F.; Liu, C.-J.; Weigel, D.; Wang, J.-W. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 2011, 23, 1512–1522. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, M. A novel R2R3-MYB transcription factor regulates light-mediated floral and vegetative anthocyanin pigmentation patterns in Lilium regale. Mol. Breed. 2016, 36, 3. [Google Scholar]
- Zhang, Y.; Cao, G.; Qu, L.-J.; Gu, H. Characterization of Arabidopsis MYB transcription factor gene AtMYB17 and its possible regulation by LEAFY and AGL15. J. Genet. Genom. 2009, 36, 99–107. [Google Scholar] [CrossRef]
- David, P.-C.L.; Camilo, L.-A.J.; Farid, R.-E.J.; Felipe, M.-M.J.; Stephanie, P.C.; Julio, R.-R.; Janeth, M.-C.F.; Carlos, S.-R.J.; Ana, D.-A.L.; Santiago, L.-P.H. Effect of domestic wastewater as co-substrate on biological stain wastewater treatment using fungal/Bacterial consortia in pilot plant and greenhouse reuse. J. Water Resour. Prot. 2018, 10, 369–393. [Google Scholar]
- Thoppurathu, F.J.; Ghorbanzadeh, Z.; Vala, A.K.; Hamid, R.; Joshi, M. Unravelling the treasure trove of drought-responsive genes in wild-type peanut through transcriptomics and physiological analyses of root. Funct. Integr. Genom. 2022, 22, 215–233. [Google Scholar]
- Jeon, J.; Bong, S.J.; Park, J.S.; Park, Y.-K.; Arasu, M.V.; Al-Dhabi, N.A.; Park, S.U. De novo transcriptome analysis and glucosinolate profiling in watercress (Nasturtium officinale R. Br.). BMC Genom. 2017, 18, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.R.; Bakshi, S.; Dubey, K.; Hasija, S.; Rai, G.K.; Jain, N.; Jambhulkar, S.; Singh, B.; Singh, G.P.; Praveen, S. Insight into the Mechanisms of Terminal HS-Tolerance in Wheat Mutant with Improved Nutritional Quality through De Novo Transcriptome Sequencing. 2021. Available online: https://www.researchsquare.com/article/rs-241832/v1 (accessed on 25 February 2021).
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Preced. 2010. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [PubMed] [Green Version]
- Khan, I.A.; Cao, K.; Guo, J.; Li, Y.; Wang, Q.; Yang, X.; Wu, J.; Fang, W.; Wang, L. Identification of key gene networks controlling anthocyanin biosynthesis in peach flower. Plant Sci. 2022, 316, 111151. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Dong, X.; Xue, C.; Liu, Z.; Cao, F. Exploring the molecular mechanism of blue flower color formation in Hydrangea macrophylla cv.“Forever Summer”. Front. Plant Sci. 2021, 101, 585665. [Google Scholar]
- Zheng, Y.; Chen, Y.; Liu, Z.; Wu, H.; Jiao, F.; Xin, H.; Zhang, L.; Yang, L. Important Roles of Key Genes and Transcription Factors in Flower Color Differences of Nicotiana alata. Genes 2021, 12, 1976. [Google Scholar] [CrossRef]
- Shen, Q.; Zhu, T.; Wu, C.; Xu, Y.; Li, C. Ultrasonic-assisted extraction of zeaxanthin from Lycium barbarum L. with composite solvent containing ionic liquid: Experimental and theoretical research. J. Mol. Liq. 2022, 347, 118265. [Google Scholar] [CrossRef]
- Saini, R.K.; Keum, Y.-S. Carotenoid extraction methods: A review of recent developments. Food Chem. 2018, 240, 90–103. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length Range | Contigs | Unigenes |
|---|---|---|
| 200–300 | 120,191 (31.13%) | 48,868 (26.20%) |
| 300–500 | 92,335 (27%) | 60,448 (32.41%) |
| 500–1000 | 72,233 (21.11%) | 48,027 (25.75%) |
| 1000–2000+ | 57,309 (16.75%) | 29,134 (15.62%) |
| Total number | 342,068 | 186,477 |
| Total length | 845,487,394 | 94,130,265 |
| N50 length | 903 | 794 |
| Mean length | 624 | 618 |
| Gene Ontology Term | Cluster Frequency | Genome Frequency | p-Value | Corrected p-Value |
|---|---|---|---|---|
| BP: Pigment biosynthetic process (GO:0046148) | 31 out of 207, 14.97584% | 24 out of 298, 8.05369% | 6.61 × 10−9 | 0.000000 |
| BP: Metabolic process (GO:0008152) | 4 out of 207, 1.67687% | 2 out of 298, 0.67111% | 5.62 × 10−5 | 0.000622 |
| BP: Developmental process (GO:0032502) | 2 out of 207, 0.96618% | 1 out of 298, 0.33557% | 4.33 ×10−5 | 0.000008 |
| BP: Anthocyanin process (GO:0046283) | 3 out of 207, 1.44927% | 2 out of 298, 0.671114% | 3.45 × 10−5 | 0.000018 |
| BP: Flavonoid biosynthetic process (GO: 0009813) | 4 out of 207, 1.93236% | 1 out of 298, 0.33557% | 6.89 × 10−5 | 0.00173 |
| BP: Chalcone biosynthetic process (GO:0016210) | 6 out of 207, 2.89855% | 3 out of 298, 1.00671% | 5.22 × 10−5 | 0.000000 |
| MF: Catalytic activity (GO:0003824) | 3 out of 237, 1.26533% | 1 out of 339, 0.29498% | 7.86 × 10−5 | 0.004689 |
| MF: Binding (GO:0005488) | 6 out of 237, 2.53145% | 3 out of 339, 0.83454% | 4.34 × 10−5 | 0.00173 |
| MF: Electron carrier activity (GO:0009055) | 9 out of 237, 3.79746% | 5 out of 339, 1.47579% | 5.76 × 10−4 | 0.002882 |
| MF: Transcription factor activity (GO:0003700) | 10 out of 237, 4.219400% | 6 out of 339, 1.7699% | 2.14 × 10−4 | 0.005734 |
| CC: Intracellular (GO:0005622) | 5 out of 177 2.82475% | 1 out of 259 0.38610% | 3.65 × 10−5 | 0.009393 |
| CC: Organelle (GO:0020037) | 3 out of 177 1.69491% | 2 out of 259 0.77220% | 6.23 × 10−1 | 0.000062 |
| Name | C-S1 | C-S2 | C-S3 | T-S1 | T-S2 | T-S3 |
|---|---|---|---|---|---|---|
| CHS1 PB.10727.1|chr7:5288756-5290374 | 51.98 | 43.18 | 62.38 | 13.43 | 14.85 | 11.29 |
| CHS2 PB.10728.1|chr7:5301940-5316126 | 3.11 | 4.51 | 3.11 | 15.29 | 7.91 | 12.44 |
| CHS3 PB.10728.2|chr7:5301941-5316113 | 4.14 | 5.512 | 2.77 | 16.92 | 8.93 | 13.34 |
| CHS4 PB.10728.3|chr7:5301944-5316192 | 2.16 | 4.15 | 3.10 | 15.34 | 7.13 | 11.24 |
| 4CL12 PB.405.4|chr1:8532291-8600510 | 15.32 | 17.61 | 15.33 | 31.62 | 21.21 | 24.34 |
| 4CL14 PB.4253.1|chr3:23869958-23873008 | 13.11 | 18.21 | 19.33 | 24.12 | 49.11 | 33.14 |
| 4CL18 PB.5838.1|chr4:349590-353192 | 12.12 | 18.16 | 15.13 | 34.17 | 46.21 | 36.15 |
| CHR1 PB.8347.2|chr5:1481568-1493170 | 11.33 | 15.71 | 13.23 | 32.12 | 29.41 | 33.91 |
| CHR3 PB.8348.1|chr5:1481568-1487857 | 13.10 | 18.71 | 13.13 | 35.62 | 29.21 | 26.34 |
| CHI1 PB.2128.1|chr1:52317616-52318844 | 7.97 | 8.52 | 9.46 | 10.72 | 11.10 | 15.23 |
| CHI2 PB.2129.1|chr1:52333401-52334681 | 21.12 | 22.81 | 28.53 | 11.22 | 12.90 | 13.01 |
| FLS1 PB.4570.1|chr3:32763532-32765066 | 23.14 | 28.17 | 21.01 | 53.44 | 68.57 | 78.11 |
| FLS2 PB.4570.2|chr3:32763692-32765066 | 14.32 | 19.61 | 16.33 | 44.62 | 39.21 | 36.34 |
| F3′H PB.7377.2|chr4:42376257-42378956 | 5.10 | 7.16 | 3.29 | 16.83 | 14.56 | 13.01 |
| F3′5′H PB.4084.1|chr3:12278062-12281867 | 13.01 | 12.59 | 11.74 | 30.45 | 36.18 | 35.16 |
| F3′H4 PB.7478.2|chr4:42392721-42394930 | 12.61 | 11.19 | 17.14 | 39.15 | 33.12 | 31.14 |
| FOMT PB.7235.1|chr4:37833577-37835915 | 53.11 | 43.17 | 51.21 | 15.16 | 19.20 | 17.31 |
| ANR PB.13205.1|chr8:31320120-31332067 | 10.14 | 12.23 | 11.32 | 24.15 | 22.11 | 21.34 |
| ANS2 PB.8440.1|chr5:3196923-3199377 | 16.12 | 16.80 | 14.24 | 35.11 | 36.70 | 34.15 |
| ANS3 PB.8440.2|chr5:3196930-3199422 | 19.15 | 17.14 | 18.07 | 25.81 | 23.55 | 22.65 |
| UFGT1 PB.2783.1|chr2:13439523-13441448 | 21.65 | 28.19 | 26.13 | 35.19 | 39.54 | 37.32 |
| UFGT2 PB.2783.2|chr2:13439659-13441448 | 10.22 | 16.36 | 13.41 | 25.41 | 24.33 | 29.17 |
| PAL5 PB.11849.17|chr7:40943081-40945873 | 26.13 | 21.75 | 22.98 | 45.21 | 42.76 | 49.81 |
| PAL6 PB.916.1|chr1:28183235-28188005 | 14.23 | 11.10 | 13.43 | 36.29 | 44.11 | 35.18 |
| PAL9 PB.11849.2|chr7:40942885-40959392 | 23.14 | 24.15 | 25.18 | 39.22 | 32.16 | 39.41 |
| PAL11 PB.11849.8|chr7:40942885-40959918 | 24.13 | 22.09 | 23.59 | 50.19 | 54.23 | 55.68 |
| DFR1 PB.339.2|chr1:7156508-7160534 | 22.12 | 23.15 | 26.19 | 40.28 | 44.13 | 45.18 |
| DFR2 PB.340.1|chr1:7164081-7167125 | 20.11 | 21.89 | 22.55 | 39.11 | 36.23 | 39.90 |
| UA3GT PB.6187.1|chr4:11023032-11024681 | 14.13 | 12.09 | 13.59 | 20.18 | 24.23 | 25.78 |
| PIP2 PB.9675.6|chr5:39486362-39487950 | 22.11 | 22.19 | 21.49 | 33.29 | 34.22 | 35.24 |
| UYP75A PB.9676.5|chr5:39463191-39464819 | 14.12 | 12.49 | 13.45 | 22.11 | 21.55 | 23.19 |
| UYP75A PB.9848.1|chr5:43311230-43312833 | 13.11 | 11.19 | 13.69 | 21.29 | 24.55 | 22.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahmati, R.; Hamid, R.; Ghorbanzadeh, Z.; Jacob, F.; Azadi, P.; Zeinalabedini, M.; Karimi Farsad, L.; Kazemi, M.; Ebrahimi, M.A.; Shahinnia, F.; et al. Comparative Transcriptome Analysis Unveils the Molecular Mechanism Underlying Sepal Colour Changes under Acidic pH Substratum in Hydrangea macrophylla. Int. J. Mol. Sci. 2022, 23, 15428. https://doi.org/10.3390/ijms232315428
Rahmati R, Hamid R, Ghorbanzadeh Z, Jacob F, Azadi P, Zeinalabedini M, Karimi Farsad L, Kazemi M, Ebrahimi MA, Shahinnia F, et al. Comparative Transcriptome Analysis Unveils the Molecular Mechanism Underlying Sepal Colour Changes under Acidic pH Substratum in Hydrangea macrophylla. International Journal of Molecular Sciences. 2022; 23(23):15428. https://doi.org/10.3390/ijms232315428
Chicago/Turabian StyleRahmati, Razieh, Rasmieh Hamid, Zahra Ghorbanzadeh, Feba Jacob, Pezhman Azadi, Mehrshad Zeinalabedini, Laleh Karimi Farsad, Mehrbano Kazemi, Mohammad Ali Ebrahimi, Fahimeh Shahinnia, and et al. 2022. "Comparative Transcriptome Analysis Unveils the Molecular Mechanism Underlying Sepal Colour Changes under Acidic pH Substratum in Hydrangea macrophylla" International Journal of Molecular Sciences 23, no. 23: 15428. https://doi.org/10.3390/ijms232315428