
Longevity-Associated Variant of BPIFB4 Confers Neuroprotection in the STHdh Cell Model of Huntington Disease
 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
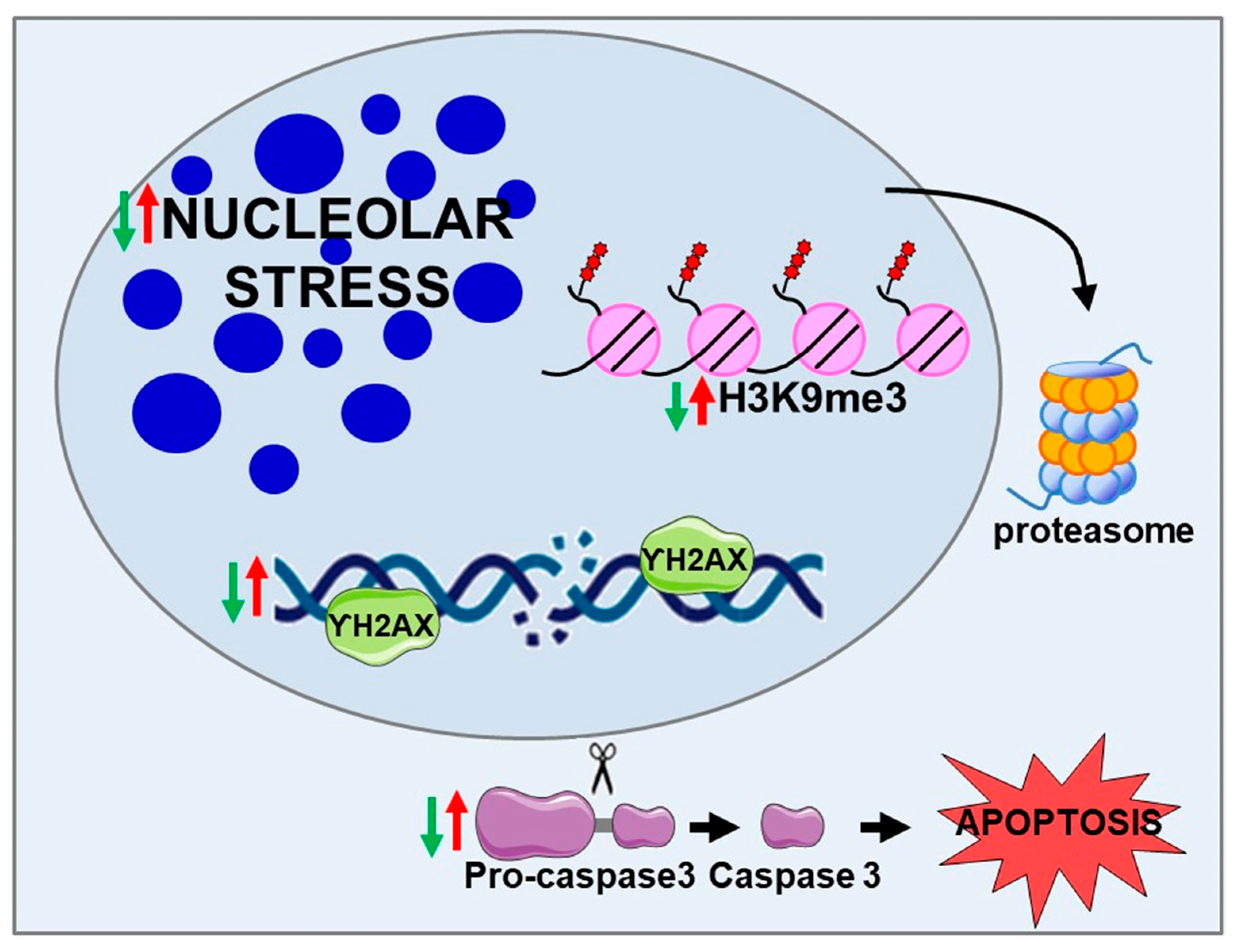
2.1. BPIFB4 Isoforms Confer Resistance to Nucleolar Stress
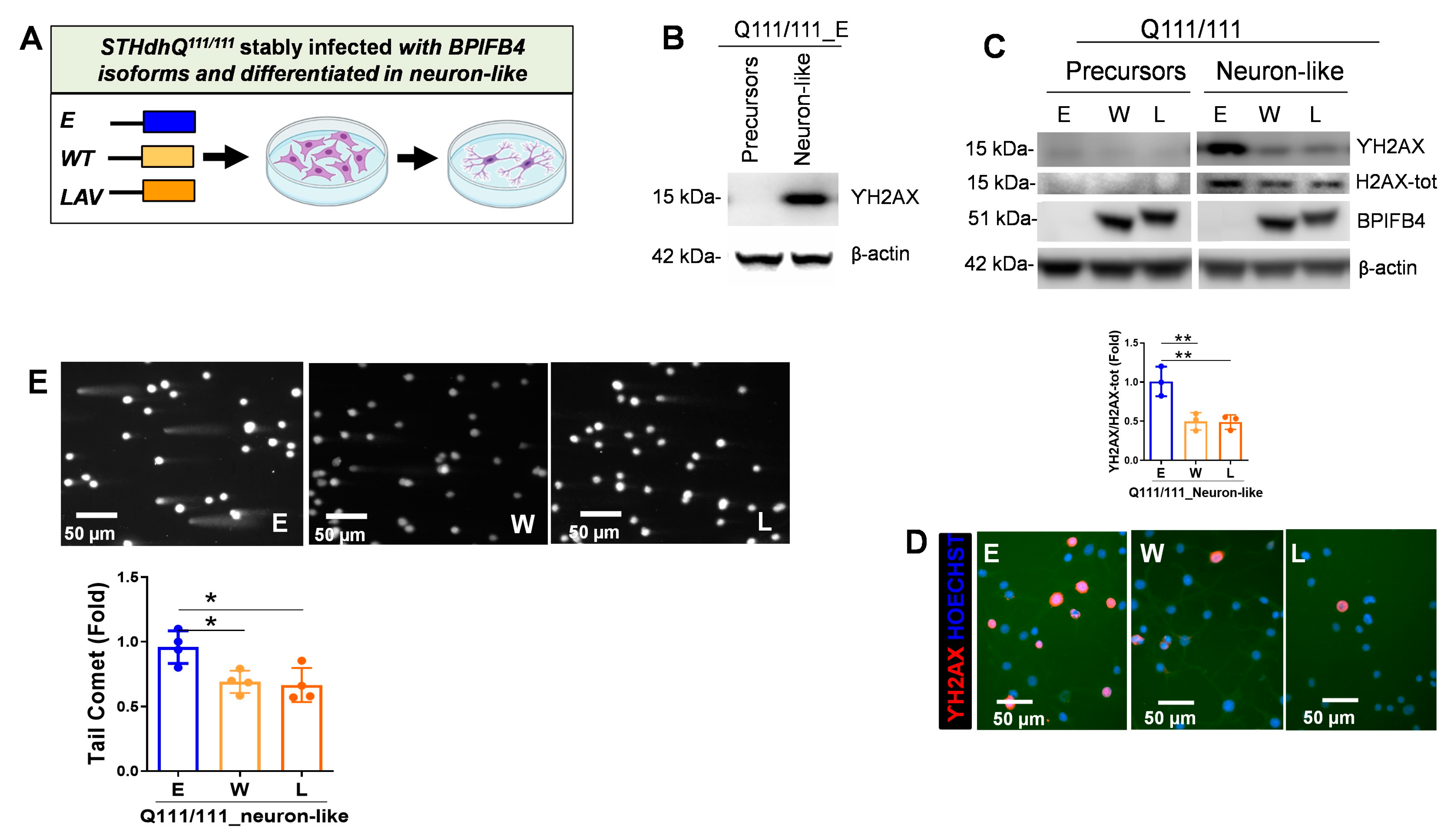
2.2. BPIFB4 Isoforms Decrease DNA Damage
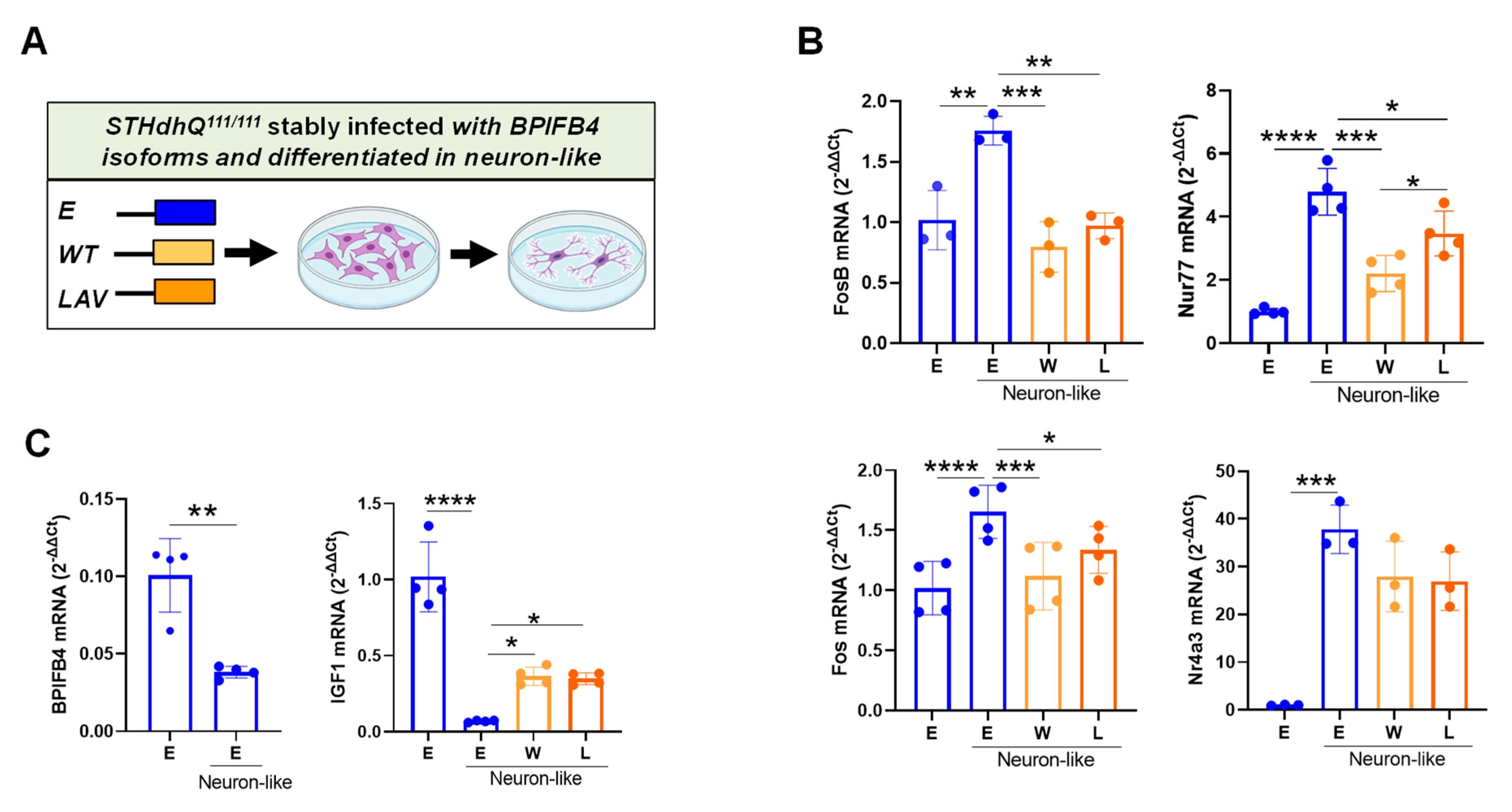
2.3. BPIFB4 Isoforms Partially Preserve Cellular Identity
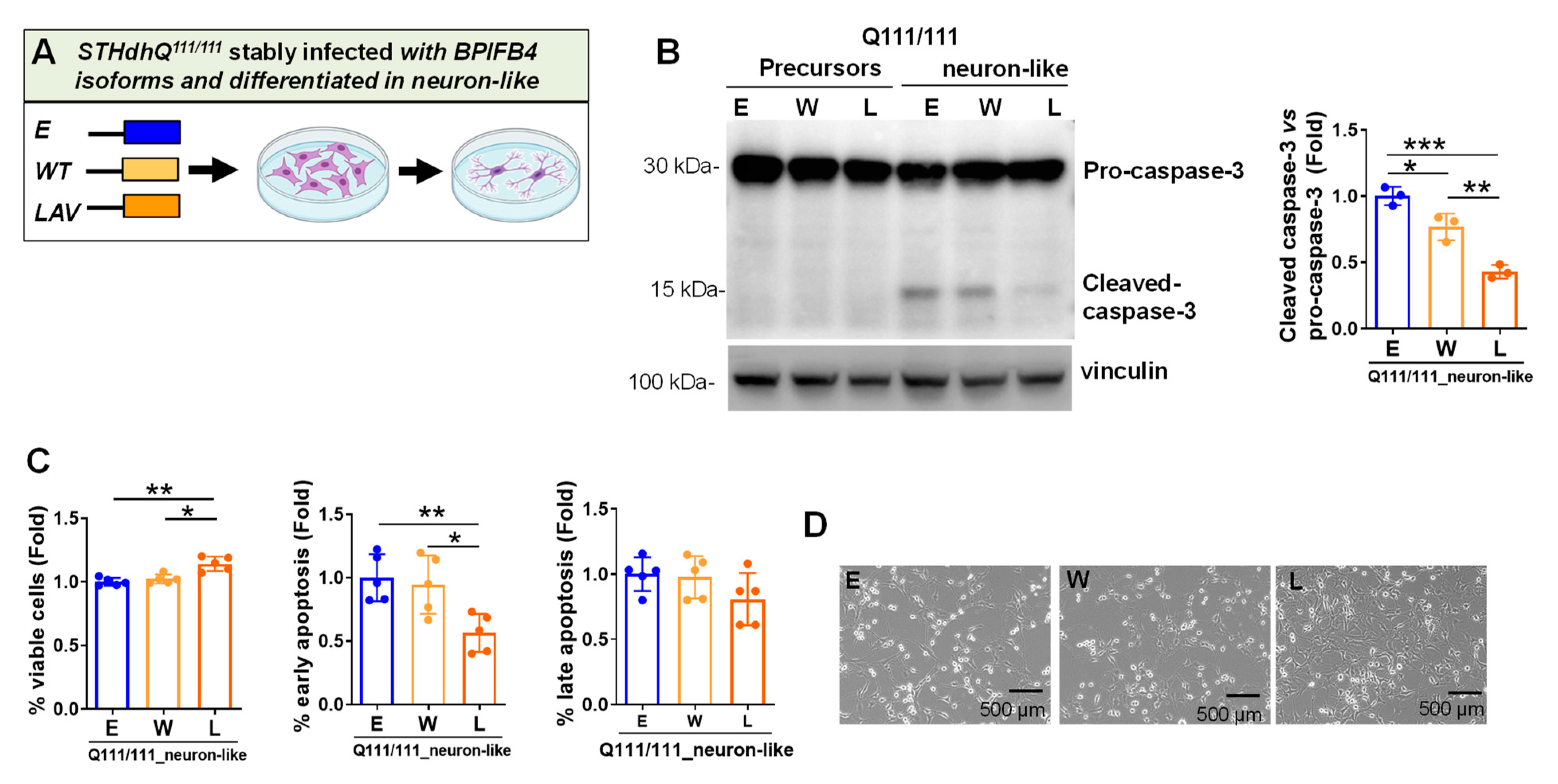
2.4. LAV-BPIFB4 Isoform Protects against Apoptosis through the Deregulation of Caspase Cascade
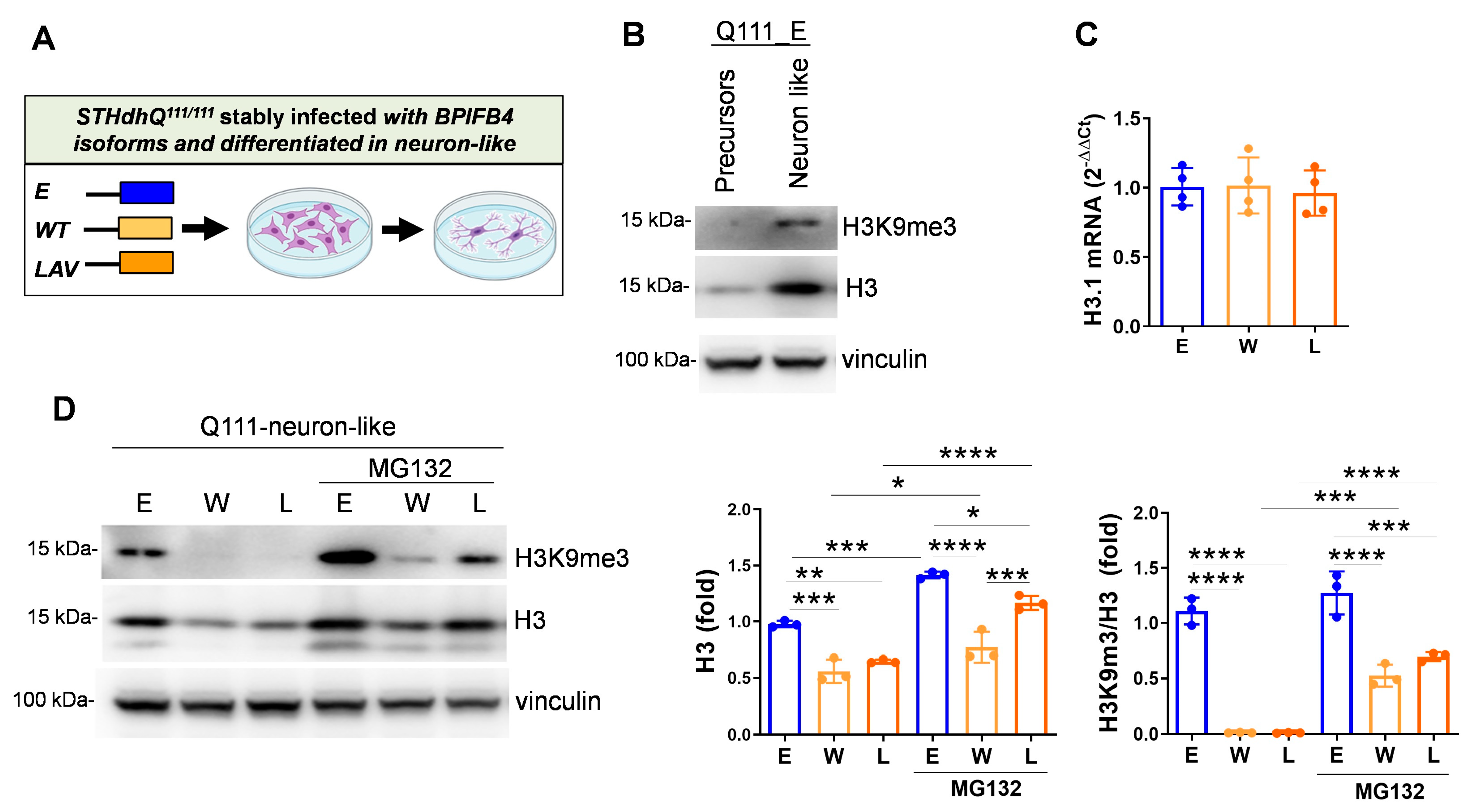
2.5. BPIFB4 Isoforms Decrease the Level of Trimethylated Histone 3 on Lysine 9 and Remodel Chromatin
- Nucleolar stress trigged by NCL sequestration in CAG RNA foci
- DNA damage and DNA repair dysfunction
- Caspase 3-dependent cell-death pathway
- H3K9me3-dependent heterochromatin condensation
- Decreases nucleolar stress
- Counteracts DNA damage and enhances DNA repair efficiency
- Inhibits the cleavage of pro-caspase 3, counteracting cell death in favor of cell viability
- Reduces the level of H3K9me3 through the ubiquitin–proteasome system
3. Discussion
3.1. LAV-BPIFB4 Counteracts the Nucleolar Stress
3.2. LAV-BPIFB4 Protects HD Neuron-like Cells from DNA Damage and Apoptosis via Inhibition of Caspase Signaling
3.3. BPIFB4 Isoforms Partially Counteract Neuronal Differentiation
3.4. BPIFB4 Reduces the Level of H3K9me3 in HD Neuron-like Cells
4. Material and Methods
4.1. Cell Maintenance, Treatment, and Differentiation
4.2. RNA Extraction and Quantitative Real-Time Analysis
4.3. Western Blotting
4.4. Immunofluorescence
4.5. Cell Viability
4.6. Comet Assay
4.7. Detection of Apoptosis by Flow-Cytometry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Wang, C.E.; Tydlacka, S.; Orr, A.L.; Yang, S.H.; Graham, R.K.; Hayden, M.R.; Li, S.; Chan, A.W.; Li, X.J. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington’s disease. Hum. Mol. Genet. 2008, 17, 2738–2751. [Google Scholar] [CrossRef] [Green Version]
- Leavitt, B.R.; van Raamsdonk, J.M.; Shehadeh, J.; Fernandes, H.; Murphy, Z.; Graham, R.K.; Wellington, C.L.; Raymond, L.A.; Hayden, M.R. Wild-type huntingtin protects neurons from excitotoxicity. J. Neurochem. 2006, 96, 1121–1129. [Google Scholar] [CrossRef]
- McKinstry, S.U.; Karadeniz, Y.B.; Worthington, A.K.; Hayrapetyan, V.Y.; Ozlu, M.I.; Serafin-Molina, K.; Risher, W.C.; Ustunkaya, T.; Dragatsis, I.; Zeitlin, S.; et al. Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J. Neurosci. 2014, 34, 9455–9472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragatsis, I.; Dietrich, P.; Ren, H.; Deng, Y.P.; Del Mar, N.; Wang, H.B.; Johnson, I.M.; Jones, K.R.; Reiner, A. Effect of early embryonic deletion of huntingtin from pyramidal neurons on the development and long-term survival of neurons in cerebral cortex and striatum. Neurobiol. Dis. 2018, 111, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Mehler, M.F.; Petronglo, J.R.; Arteaga-Bracho, E.E.; Gulinello, M.E.; Winchester, M.L.; Pichamoorthy, N.; Young, S.K.; DeJesus, C.D.; Ishtiaq, H.; Gokhan, S.; et al. Loss-of-Huntingtin in Medial and Lateral Ganglionic Lineages Differentially Disrupts Regional Interneuron and Projection Neuron Subtypes and Promotes Huntington’s Disease-Associated Behavioral, Cellular, and Pathological Hallmarks. J. Neurosci. 2019, 39, 1892–1909. [Google Scholar] [CrossRef] [Green Version]
- Abdelmohsen, K.; Gorospe, M. RNA-binding protein nucleolin in disease. RNA Biol. 2012, 9, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, H.; Lau, T.C.; Tsang, S.Y.; Lau, K.F.; Chan, H.Y. CAG expansion induces nucleolar stress in polyglutamine diseases. Proc. Natl. Acad. Sci. USA 2012, 109, 13428–13433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stack, E.C.; Del Signore, S.J.; Luthi-Carter, R.; Soh, B.Y.; Goldstein, D.R.; Matson, S.; Goodrich, S.; Markey, A.L.; Cormier, K.; Hagerty, S.W.; et al. Modulation of nucleosome dynamics in Huntington’s disease. Hum. Mol. Genet. 2007, 16, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.J.; Hyeon, S.J.; Kim, Y.; Lim, S.; Lee, M.Y.; Kim, J.; Londhe, A.M.; Gotina, L.; Kim, Y.; Pae, A.N.; et al. Modulation of SETDB1 activity by APQ ameliorates heterochromatin condensation, motor function, and neuropathology in a Huntington’s disease mouse model. J. Enzyme Inhib. Med. Chem. 2021, 36, 856–868. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, P.; De Cristofaro, T.; Affaitati, A.; Pizzulo, G.M.; Feliciello, A.; Criscuolo, C.; De Michele, G.; Filla, A.; Avvedimento, E.V.; Varrone, S. DNA damage induced by polyglutamine-expanded proteins. Hum. Mol Genet. 2003, 12, 2301–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illuzzi, J.; Yerkes, S.; Parekh-Olmedo, H.; Kmiec, E.B. DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J. Neurosci. Res. 2009, 87, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Enokido, Y.; Tamura, T.; Ito, H.; Arumughan, A.; Komuro, A.; Shiwaku, H.; Sone, M.; Foulle, R.; Sawada, H.; Ishiguro, H.; et al. Mutant huntingtin impairs Ku70-mediated DNA repair. J. Cell Biol. 2010, 189, 425–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, F.; Carrizzo, A.; Ferrario, A.; Maciag, A.; Cattaneo, M.; Spinelli, C.C.; Montella, F.; Damato, A.; Ciaglia, E.; Puca, A.A. A Model of Evolutionary Selection: The Cardiovascular Protective Function of the Longevity Associated Variant of BPIFB4. Int. J. Mol. Sci. 2018, 19, 3229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, F.; Carrizzo, A.; Spinelli, C.C.; Ferrario, A.; Malovini, A.; Maciąg, A.; Damato, A.; Auricchio, A.; Spinetti, G.; Sangalli, E.; et al. Analysis Reveals a Longevity-Associated Protein Modulating Endothelial Function and Angiogenesis. Circ. Res. 2015, 117, 333–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, F.; Malovini, A.; Carrizzo, A.; Spinelli, C.C.; Ferrario, A.; Maciąg, A.; Madonna, M.; Bellazzi, R.; Milanesi, L.; Vecchione, C.; et al. Serum BPIFB4 levels classify health status in long-living individuals. Immun. Ageing 2015, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Puca, A.A.; Carrizzo, A.; Spinelli, C.; Damato, A.; Ambrosio, M.; Villa, F.; Ferrario, A.; Maciag, A.; Fornai, F.; Lenzi, P.; et al. Single systemic transfer of a human gene associated with exceptional longevity halts the progression of atherosclerosis and inflammation in ApoE knockout mice through a CXCR4-mediated mechanism. Eur. Heart J. 2020, 41, 2487–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, Z.; Avolio, E.; Thomas, A.C.; Faulkner, A.; Beltrami, A.P.; Cervellin, C.; Carrizzo, A.; Maciag, A.; Gu, Y.; Ciaglia, E.; et al. Transfer of a human gene variant associated with exceptional longevity improves cardiac function in obese type 2 diabetic mice through induction of the SDF-1/CXCR4 signalling pathway. Eur. J. Heart Fail. 2020, 22, 1568–1581. [Google Scholar] [CrossRef]
- Malavolta, M.; Dato, S.; Villa, F.; Rango, F.; Iannone, F.; Ferrario, A.; Maciag, A.; Ciaglia, E.; D’amato, A.; Carrizzo, A.; et al. LAV-BPIFB4 associates with reduced frailty in humans and its transfer prevents frailty progression in old mice. Aging 2019, 11, 6555–6568. [Google Scholar] [CrossRef]
- Di Pardo, A.; Ciaglia, E.; Cattaneo, M.; Maciag, A.; Montella, F.; Lopardo, V.; Ferrario, A.; Villa, F.; Madonna, M.; Amico, E.; et al. The longevity-associated variant of BPIFB4 improves a CXCR4-mediated striatum-microglia crosstalk preventing disease progression in a mouse model of Huntington’s disease. Cell Death Dis. 2020, 11, 546. [Google Scholar] [CrossRef]
- Ciaglia, E.; Lopardo, V.; Montella, F.; Carrizzo, A.; Di Pietro, P.; Malavolta, M.; Giacconi, R.; Orlando, F.; Cattaneo, M.; Madeddu, P.; et al. Transfer of the longevity-associated variant of BPIFB4 gene rejuvenates immune system and vasculature by a reduction of CD38+ macrophages and NAD+ decline. Cell Death Dis. 2022, 13, 86. [Google Scholar] [CrossRef]
- Spinelli, C.C.; Carrizzo, A.; Ferrario, A.; Villa, F.; Damato, A.; Ambrosio, M.; Madonna, M.; Frati, G.; Fucile, S.; Sciaccaluga, M.; et al. LAV-BPIFB4 isoform modulates eNOS signalling through Ca2+/PKC-alpha-dependent mechanism. Cardiovasc. Res. 2017, 113, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Trettel, F.; Rigamonti, D.; Hilditch-Maguire, P.; Wheeler, V.C.; Sharp, A.H.; Persichetti, F.; Cattaneo, E.; MacDonald, M.E. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 2000, 9, 2799–2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, R.Y.; Pederson, T. Connecting the nucleolus to the cell cycle and human disease. FASEB J. 2014, 28, 3290–32963. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, E.; Parisot, P.; Pinto-Monteiro, C.; de Walque, R.; De Vleeschouwer, C.; Lafontaine, D.L. Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress. Nat. Commun. 2016, 7, 11390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariharan, N.; Sussman, M.A. Stressing on the nucleolus in cardiovascular disease. Biochim. Biophys. Acta 2014, 1842, 798–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [Green Version]
- Supeno, N.E.; Pati, S.; Hadi, R.A.; Ghani, A.R.; Mustafa, Z.; Abdullah, J.M.; Idris, F.M.; Han, X.; Jaafar, H. IGF-1 acts as controlling switch for long-term proliferation and maintenance of EGF/FGF-responsive striatal neural stem cells. Int. J. Med. Sci. 2013, 10, 522–531. [Google Scholar] [CrossRef]
- Kobayashi, J.; Fujimoto, H.; Sato, J.; Hayashi, I.; Burma, S.; Matsuura, S.; Chen, D.J.; Komatsu, K. Nucleolin participates in DNA double-strand break-induced damage response through MDC1-dependent pathway. PLoS ONE 2012, 7, e49245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, M.; Derheimer, F.A.; Tait-Mulder, J.; Kastan, M.B. Nucleolin mediates nucleosome disruption critical for DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2013, 110, 16874–16879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianov, L.; De Both, M.; Chawla, M.K.; Rani, A.; Kennedy, A.J.; Piras, I.; Day, J.J.; Siniard, A.; Kumar, A.; Sweatt, J.D.; et al. Hippocampal Transcriptomic Profiles: Subfield Vulnerability to Age and Cognitive Impairment. Front. Aging Neurosci. 2017, 9, 383. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, K.; Kim, K.; Yi, S.J. The role of histone modifications: From neurodevelopment to neurodiseases. Signal Transduct. Target. Ther. 2022, 7, 217. [Google Scholar]
- Köhler, F.; Bormann, F.; Raddatz, G.; Gutekunst, J.; Corless, S.; Musch, T.; Lonsdorf, A.S.; Erhardt, S.; Lyko, F.; Rodríguez-Paredes, M. Epigenetic deregulation of lamina-associated domains in Hutchinson-Gilford progeria syndrome. Genome Med. 2020, 12, 46. [Google Scholar] [CrossRef]
- Kaneda, R.; Takada, S.; Yamashita, Y.; Choi, Y.L.; Nonaka-Sarukawa, M.; Soda, M.; Misawa, Y.; Isomura, T.; Shimada, K.; Mano, H. Genome-wide histone methylation profile for heart failure. Genes Cells 2009, 14, 69–77. [Google Scholar] [CrossRef]
- Papait, R.; Cattaneo, P.; Kunderfranco, P.; Greco, C.; Carullo, P.; Guffanti, A.; Viganò, V.; Stirparo, G.G.; Latronico, M.V.; Hasenfuss, G.; et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 20164–20169. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.Y.; Lee, J.; Hyeon, S.J.; Cho, H.; Hwang, Y.J.; Shin, J.Y.; McKee, A.C.; Kowall, N.W.; Kim, J.I.; Stein, T.D.; et al. Epigenome signatures landscaped by histone H3K9me3 are associated with the synaptic dysfunction in Alzheimer’s disease. Aging Cell 2020, 19, e13153. [Google Scholar] [CrossRef]
- Song, W.; Zsindely, N.; Faragó, A.; Marsh, J.L.; Bodai, L. Systematic genetic interaction studies identify histone demethylase Utx as potential target for ameliorating Huntington’s disease. Hum. Mol. Genet. 2018, 27, 649–666. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cattaneo, M.; Maciag, A.; Milella, M.S.; Ciaglia, E.; Bruno, A.; Puca, A.A. Longevity-Associated Variant of BPIFB4 Confers Neuroprotection in the STHdh Cell Model of Huntington Disease. Int. J. Mol. Sci. 2022, 23, 15313. https://doi.org/10.3390/ijms232315313
Cattaneo M, Maciag A, Milella MS, Ciaglia E, Bruno A, Puca AA. Longevity-Associated Variant of BPIFB4 Confers Neuroprotection in the STHdh Cell Model of Huntington Disease. International Journal of Molecular Sciences. 2022; 23(23):15313. https://doi.org/10.3390/ijms232315313
Chicago/Turabian StyleCattaneo, Monica, Anna Maciag, Maria Serena Milella, Elena Ciaglia, Antonino Bruno, and Annibale Alessandro Puca. 2022. "Longevity-Associated Variant of BPIFB4 Confers Neuroprotection in the STHdh Cell Model of Huntington Disease" International Journal of Molecular Sciences 23, no. 23: 15313. https://doi.org/10.3390/ijms232315313