Locus-Specific Isolation of the Nanog Chromatin Identifies Regulators Relevant to Pluripotency of Mouse Embryonic Stem Cells and Reprogramming of Somatic Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
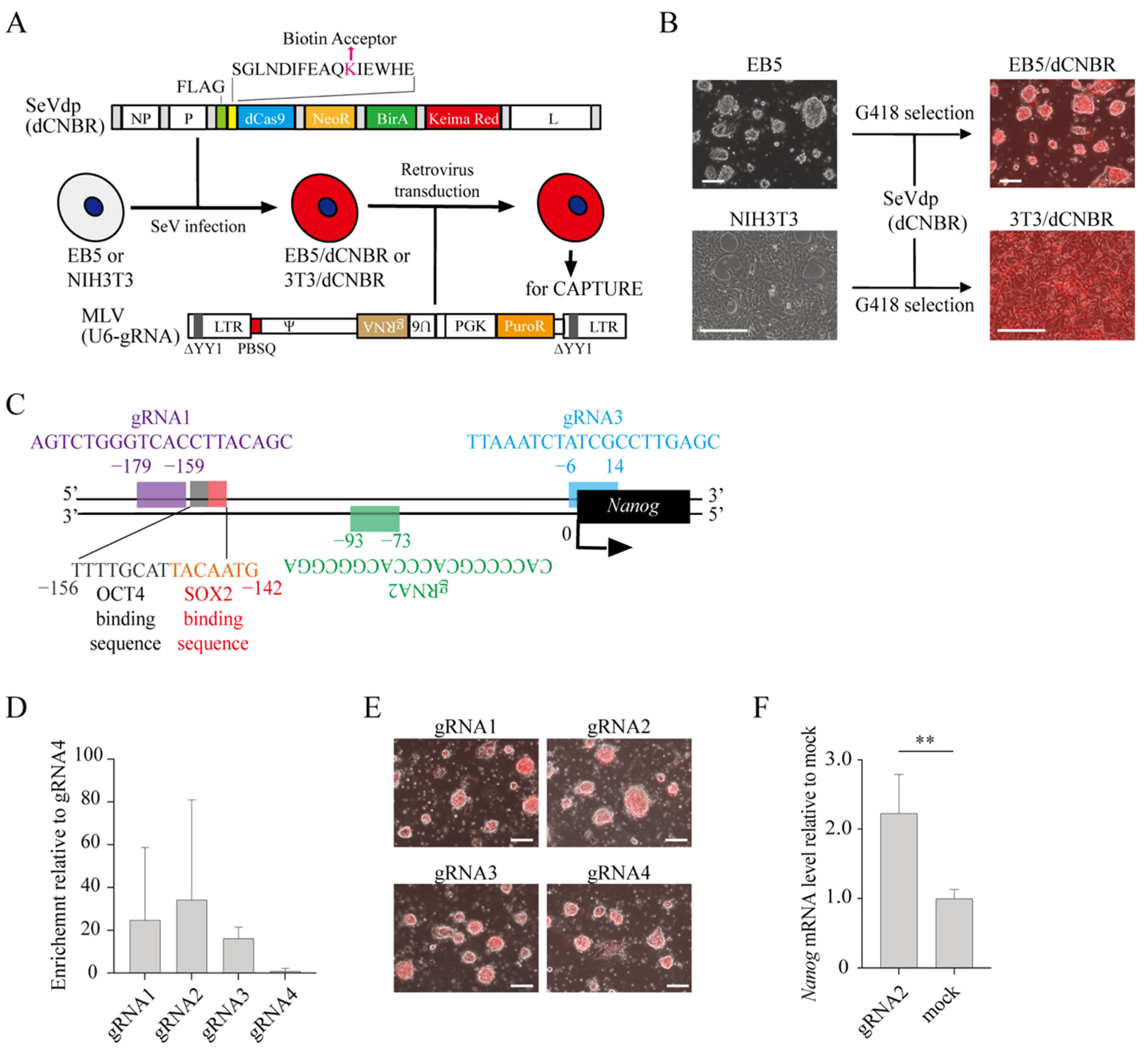
2.1. CAPTURE of the Nanog Promoter
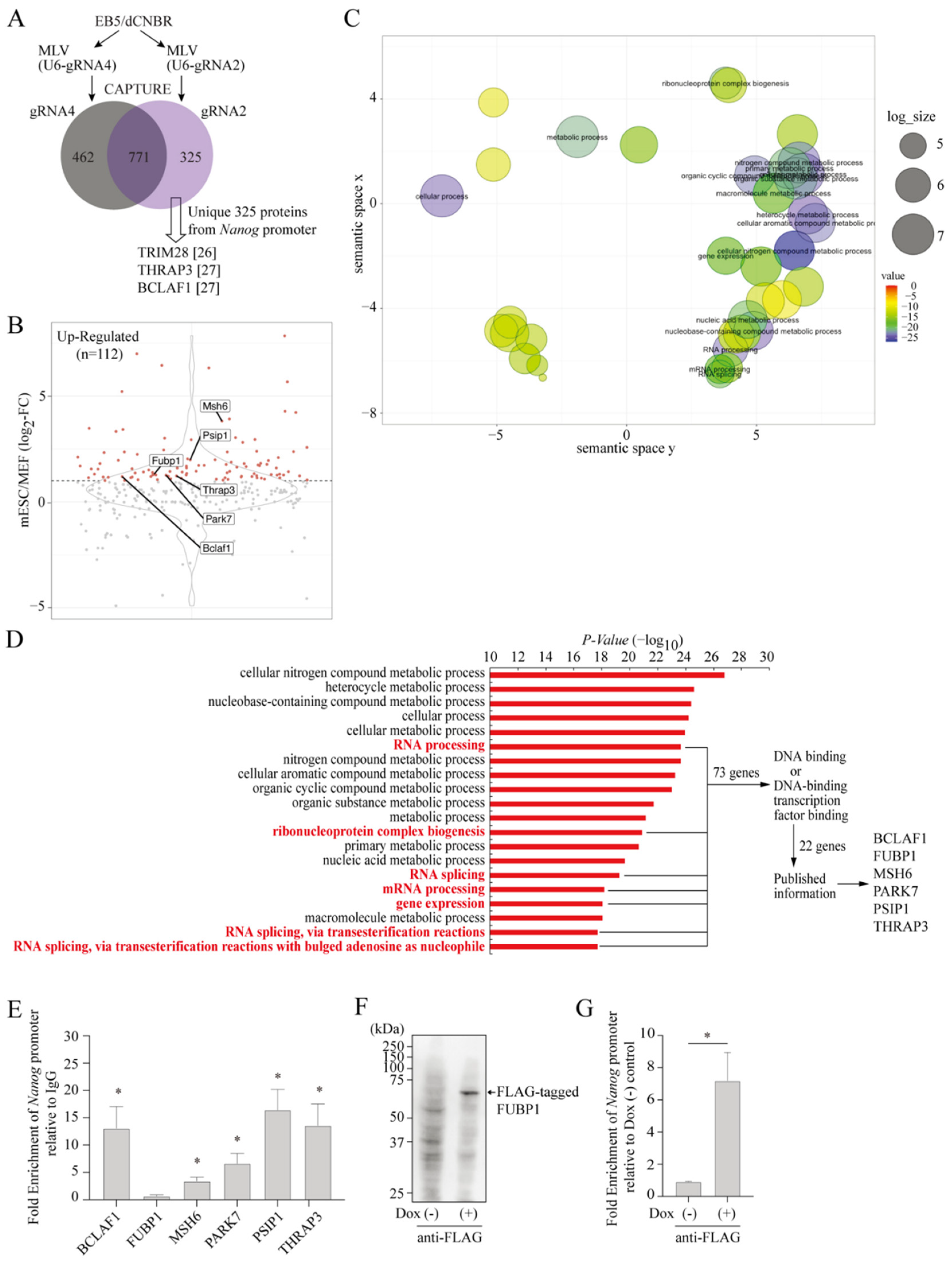
2.2. Isolation of Proteins Assembled on the Endogenous Nanog Chromatin
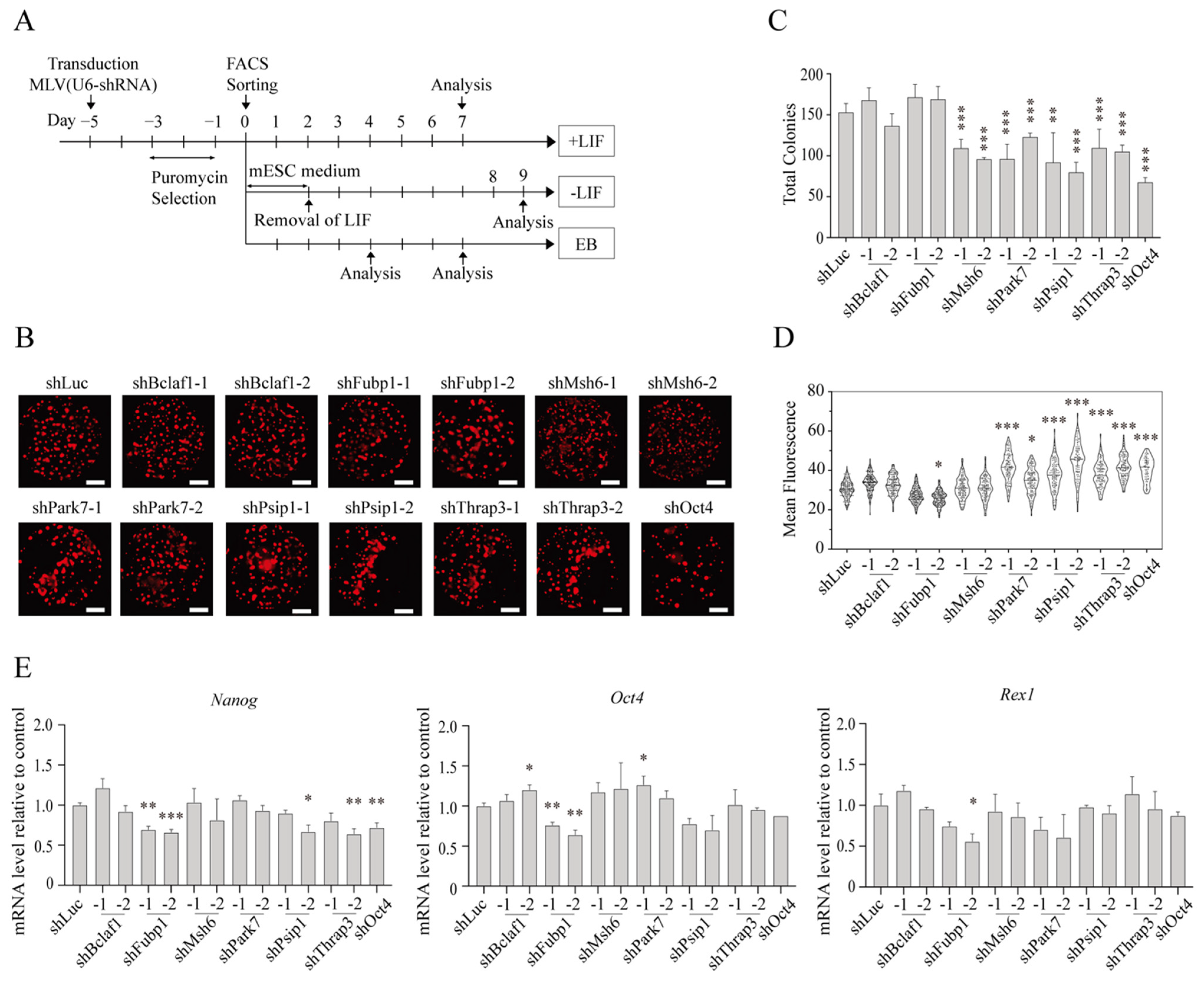
2.3. Effects of CAPTURE-Identified Proteins on mESC Pluripotency
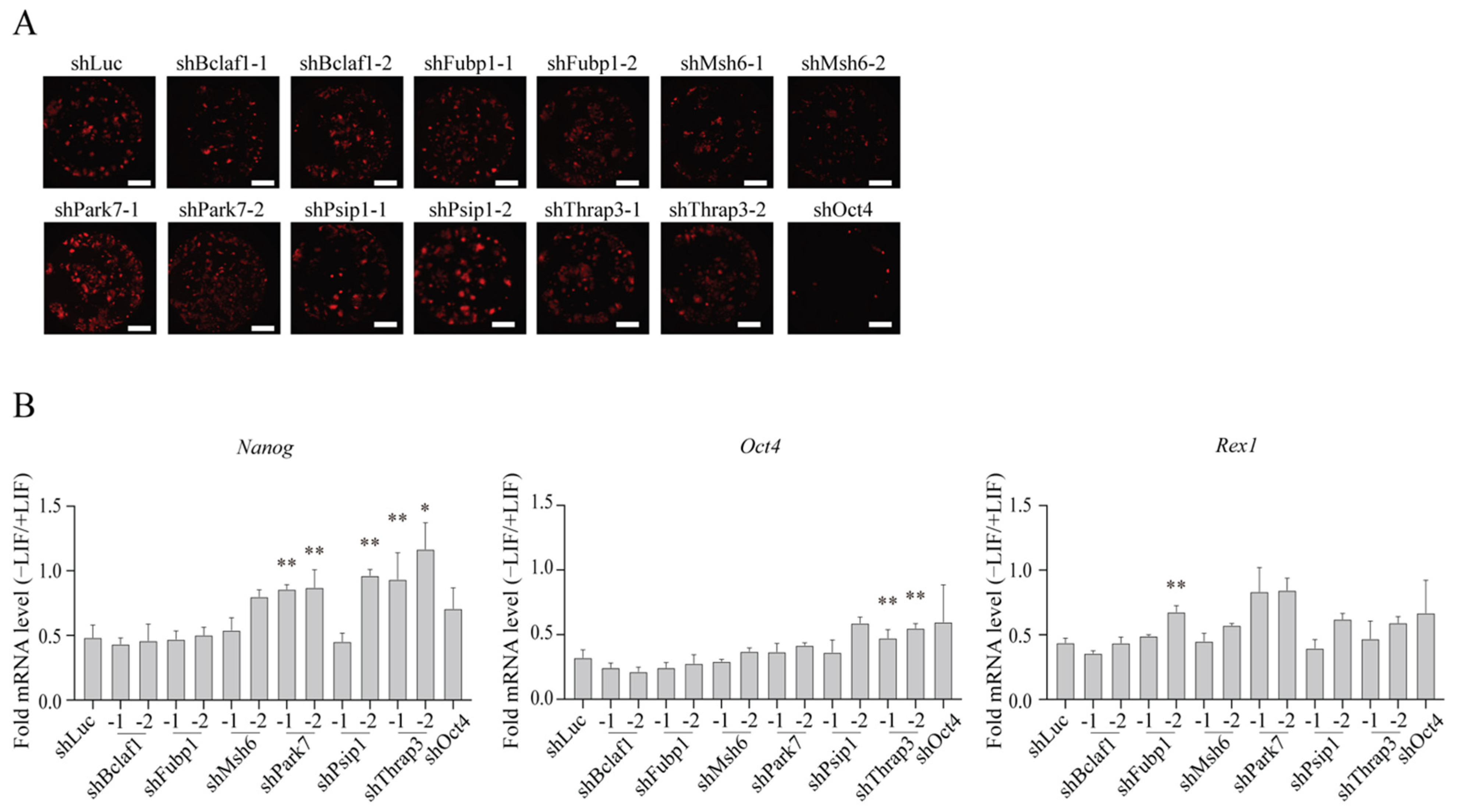
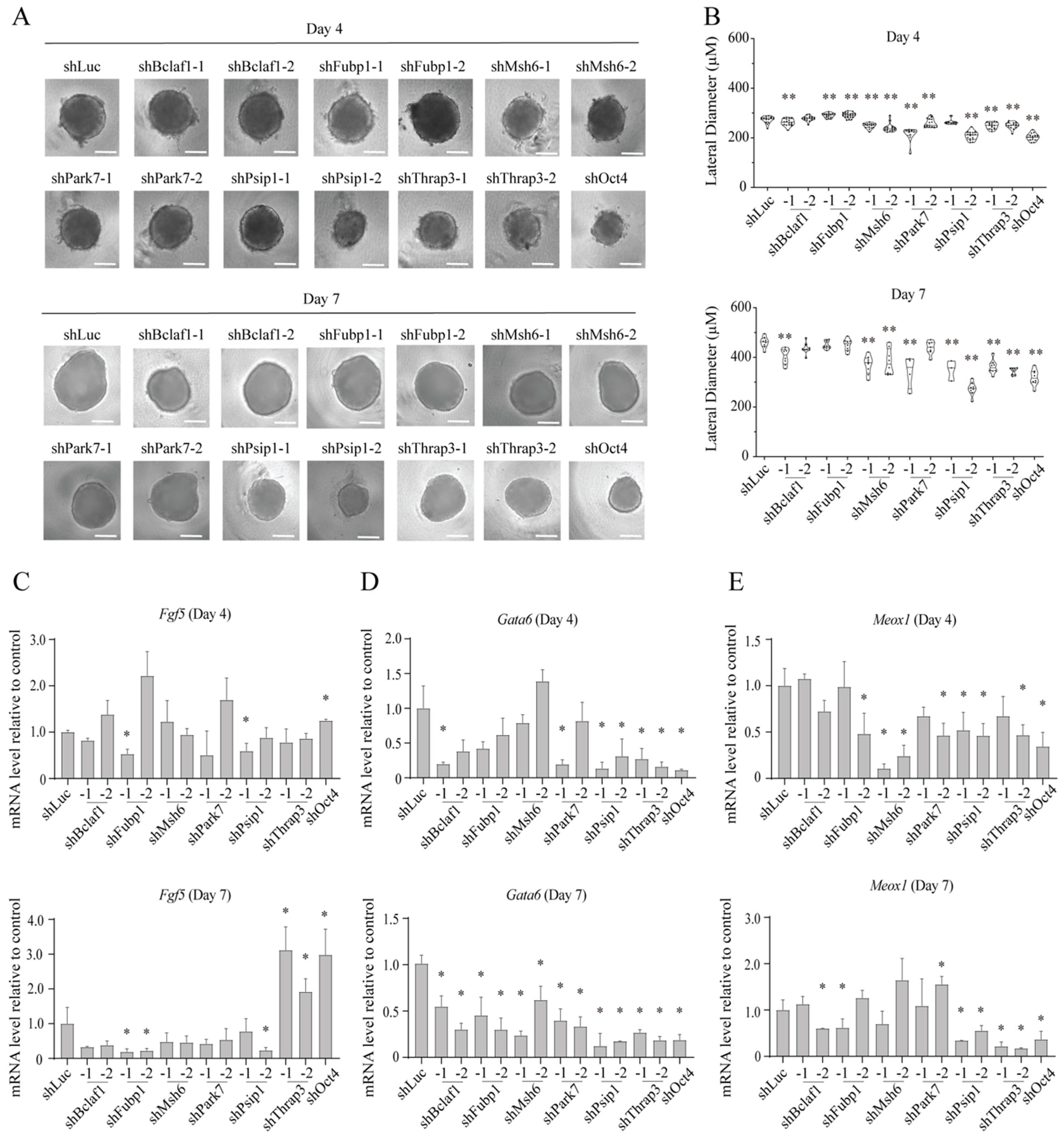
2.4. Roles for the Identified Proteins in mESC Differentiation
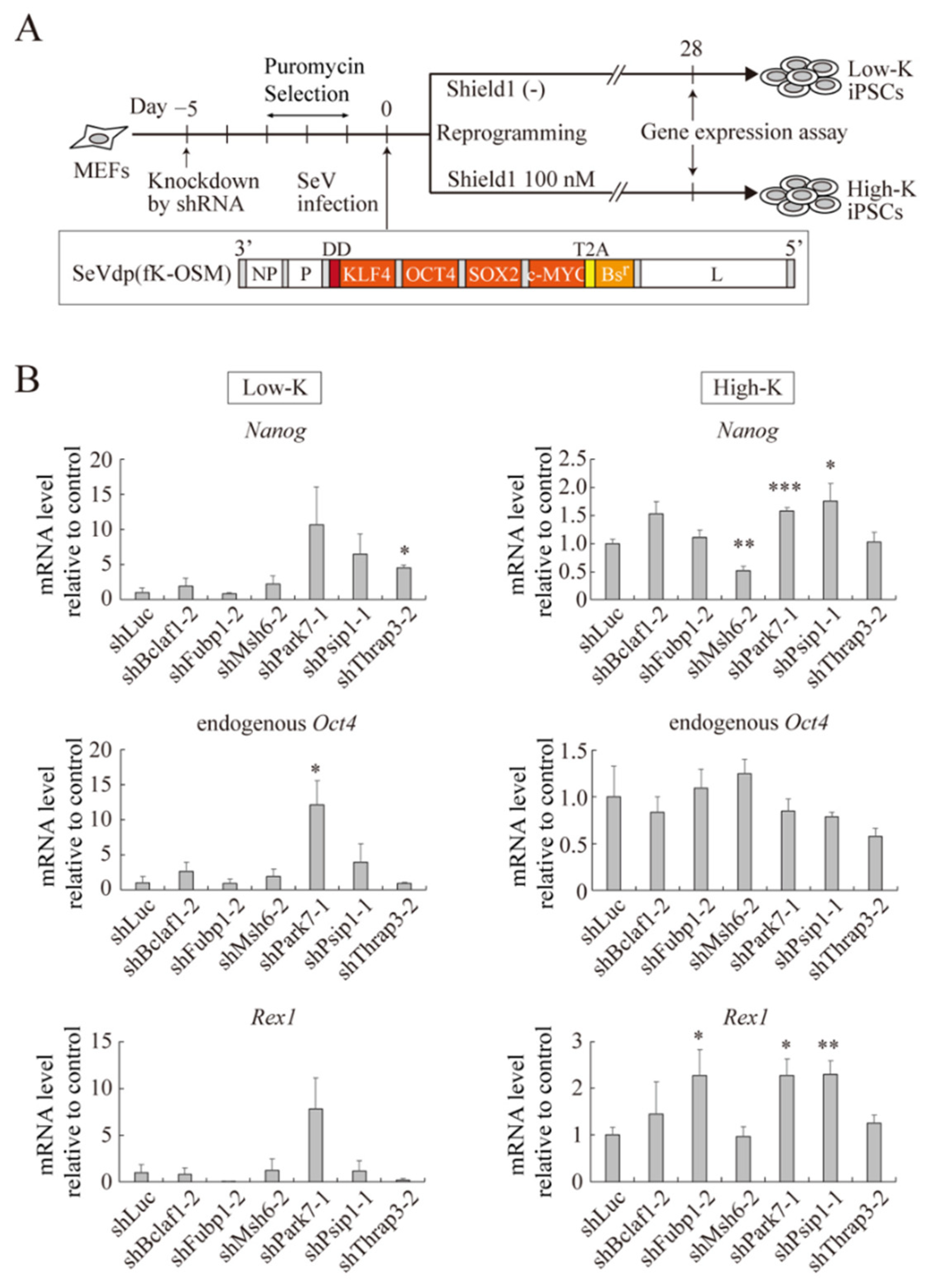
2.5. Roles for the Identified Proteins in Somatic Cell Reprogramming
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmid Construction
4.3. Production and Infection of Viral Vectors
4.4. CAPTURE of the Nanog Promoter
4.4.1. Isolation of Chromatin Containing the Nanog Promoter
4.4.2. CAPTURE-ChIP-qPCR
4.4.3. CAPTURE-Proteomics
4.4.4. Gene Ontology Analysis
4.5. Genome Editing
4.6. Cell Sorting and Image Analysis
4.7. Gene Expression Analysis
4.8. Somatic Cell Reprogramming
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evans, M.J.; Kaufman, M.H. Establishment in Culture of Pluripotential Cells from Mouse Embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R. Isolation of a Pluripotent Cell Line from Early Mouse Embryos Cultured in Medium Conditioned by Teratocarcinoma Stem Cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of Germline-Competent Induced Pluripotent Stem Cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef]
- Boyer, L.A.; Tong, I.L.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core Transcriptional Regulatory Circuitry in Human Embryonic Stem Cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Xue, K.; Ng, J.H.; Ng, H.H. Mapping the Networks for Pluripotency. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2011, 366, 2238–2246. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Gaspar-Maia, A.; Alajem, A.; Meshorer, E.; Ramalho-Santos, M. Open Chromatin in Pluripotency and Reprogramming. Nat. Rev. Mol. Cell Biol. 2011, 12, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Efroni, S.; Duttagupta, R.; Cheng, J.; Dehghani, H.; Hoeppner, D.J.; Dash, C.; Bazett-Jones, D.P.; Le Grice, S.; McKay, R.D.G.; Buetow, K.H.; et al. Global Transcription in Pluripotent Embryonic Stem Cells. Cell Stem Cell 2008, 2, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.; Dehghani, H.; Rugg-Gunn, P.; Fussner, E.; Rossant, J.; Bazett-Jones, D.P. Global Chromatin Architecture Reflects Pluripotency and Lineage Commitment in the Early Mouse Embryo. PLoS ONE 2010, 5, e10531. [Google Scholar] [CrossRef] [Green Version]
- Meshorer, E.; Yellajoshula, D.; George, E.; Scambler, P.J.; Brown, D.T.; Misteli, T. Hyperdynamic Plasticity of Chromatin Proteins in Pluripotent Embryonic Stem Cells. Dev. Cell 2006, 10, 105–116. [Google Scholar] [CrossRef]
- Hawkins, R.D.; Hon, G.C.; Lee, L.K.; Ngo, Q.; Lister, R.; Pelizzola, M.; Edsall, L.E.; Kuan, S.; Luu, Y.; Klugman, S.; et al. Distinct Epigenomic Landscapes of Pluripotent and Lineage-Committed Human Cells. Cell Stem Cell 2010, 6, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Tsankov, A.M.; Gu, H.; Akopian, V.; Ziller, M.J.; Donaghey, J.; Amit, I.; Gnirke, A.; Meissner, A. Transcription Factor Binding Dynamics during Human ES Cell Differentiation. Nature 2015, 518, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.R.; Jung, I.; Selvaraj, S.; Shen, Y.; Antosiewicz-Bourget, J.E.; Lee, A.Y.; Ye, Z.; Kim, A.; Rajagopal, N.; Xie, W.; et al. Chromatin Architecture Reorganization during Stem Cell Differentiation. Nature 2015, 518, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Déjardin, J.; Kingston, R.E. Purification of Proteins Associated with Specific Genomic Loci. Cell 2009, 136, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Fujii, H. Insertional Chromatin Immunoprecipitation: A Method for Isolating Specific Genomic Regions. J. Biosci. Bioeng. 2009, 108, 446–449. [Google Scholar] [CrossRef]
- Fujita, T.; Asano, Y.; Ohtsuka, J.; Takada, Y.; Saito, K.; Ohki, R.; Fujii, H. Identification of Telomere-Associated Molecules by Engineered DNA-Binding Molecule-Mediated Chromatin Immunoprecipitation (EnChIP). Sci. Rep. 2013, 3, 3171. [Google Scholar] [CrossRef] [Green Version]
- Fujita, T.; Fujii, H. Efficient Isolation of Specific Genomic Regions and Identification of Associated Proteins by Engineered DNA-Binding Molecule-Mediated Chromatin Immunoprecipitation (EnChIP) using CRISPR. Biochem. Biophys. Res. Commun. 2013, 439, 132–136. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.A.; Wright, J.; Peckner, R.; Kalish, B.T.; Zhang, F.; Carr, S.A. Discovery of Proteins Associated with a Predefined Genomic Locus via dCas9-APEX-Mediated Proximity Labeling. Nat. Methods 2018, 15, 437–439. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Chen, Y.; Li, M.; Zhou, F.; Li, K.; Cao, H.; Ni, M.; Liu, Y.; Gu, Z.; et al. In Situ Capture of Chromatin Interactions by Biotinylated dCas9. Cell 2017, 170, 1028–1043.e19. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.; Sano, M.; Ohtaka, M.; Furuta, B.; Umemura, Y.; Nakajima, Y.; Ikehara, Y.; Kobayashi, T.; Segawa, H.; Takayasu, S.; et al. Development of Defective and Persistent Sendai Virus Vector: A Unique Gene Delivery/Expression System Ideal for Cell Reprogramming. J. Biol. Chem. 2011, 286, 4760–4771. [Google Scholar] [CrossRef]
- Bui, P.L.L.; Nishimura, K.; Seminario Mondejar, G.; Kumar, A.; Aizawa, S.; Murano, K.; Nagata, K.; Hayashi, Y.; Fukuda, A.; Onuma, Y.; et al. Template Activating Factor-I Alpha Regulates Retroviral Silencing during Reprogramming. Cell Rep. 2019, 29, 1909–1922.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pannell, D.; Ellis, J. Silencing of Gene Expression: Implications for Design of Retrovirus Vectors. Rev. Med. Virol. 2001, 11, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Nichols, J.; Theunissen, T.W.; Guo, G.; van Oosten, A.L.; Barrandon, O.; Wray, J.; Yamanaka, S.; Chambers, I.; Smith, A. Nanog Is the Gateway to the Pluripotent Ground State. Cell 2009, 138, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Mashiko, D.; Fujihara, Y.; Satouh, Y.; Miyata, H.; Isotani, A.; Ikawa, M. Generation of Mutant Mice by Pronuclear Injection of Circular Plasmid Expressing Cas9 and Single Guided RNA. Sci. Rep. 2013, 3, 3355. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Kim, J.; Xu, Q.; Leng, Y.; Orkin, S.H.; Elledge, S.J. A Genome-wide RNAi Screen Identifies a New Transcriptional Module Required for Self-Renewal. Genes Dev. 2009, 23, 837–848. [Google Scholar] [CrossRef] [Green Version]
- Seki, Y.; Kurisaki, A.; Watanabe-Susaki, K.; Nakajima, Y.; Nakanishi, M.; Arai, Y.; Shiota, K.; Sugino, H.; Asashima, M. TIF1beta Regulates the Pluripotency of Embryonic Stem Cells in a Phosphorylation-Dependent Manner. Proc. Natl. Acad. Sci. USA 2010, 107, 10926–10931. [Google Scholar] [CrossRef] [Green Version]
- Knaupp, A.S.; Mohenska, M.; Larcombe, M.R.; Ford, E.; Lim, S.M.; Wong, K.; Chen, J.; Firas, J.; Huang, C.; Liu, X.; et al. TINC—A Method to Dissect Regulatory Complexes at Single-Locus Resolution— Reveals an Extensive Protein Complex at the Nanog Promoter. Stem Cell Rep. 2020, 15, 1246–1259. [Google Scholar] [CrossRef]
- Chronis, C.; Fiziev, P.; Papp, B.; Butz, S.; Bonora, G.; Sabri, S.; Ernst, J.; Plath, K. Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell 2017, 168, 442–459.e20. [Google Scholar] [CrossRef] [Green Version]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO::TermFinder—Open Source Software for Accessing Gene Ontology Information and Finding Significantly Enriched Gene Ontology Terms Associated with a List of Genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Vohhodina, J.; Barros, E.M.; Savage, A.L.; Liberante, F.G.; Manti, L.; Bankhead, P.; Cosgrove, N.; Madden, A.F.; Harkin, D.P.; Savage, K.I. The RNA Processing Factors THRAP3 and BCLAF1 Promote the DNA Damage Response through Selective mRNA Splicing and Nuclear Export. Nucleic Acids Res. 2017, 45, 12816–12833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, R.; Bazar, L.; Michelotti, G.; Tomonaga, T.; Krutzsch, H.; Avigan, M.; Levens, D. A Sequence-Specific, Single-Strand Binding Protein Activates the Far Upstream Element of c-myc and Defines a New DNA-Binding Motif. Genes Dev. 1994, 8, 465–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wang, Z.; Zhou, X.; Cheng, Y.; Xie, Z.; Manley, J.L.; Feng, Y. Far Upstream Element-Binding Protein 1 and RNA Secondary Structure Both Mediate Second-Step Splicing Repression. Proc. Natl. Acad. Sci. USA 2013, 110, E2687–E2695. [Google Scholar] [CrossRef] [Green Version]
- Vasseur, S.; Afzal, S.; Tardivel-Lacombe, J.; Park, D.S.; Iovanna, J.L.; Mak, T.W. DJ-1/PARK7 Is an Important Mediator of Hypoxia-Induced Cellular Responses. Proc. Natl. Acad. Sci. USA 2009, 106, 1111–1116. [Google Scholar] [CrossRef] [Green Version]
- Ge, H.; Si, Y.; Roeder, R.G. Isolation of cDNAs Encoding Novel Transcription Coactivators P52 and P75 Reveals an Alternate Regulatory Mechanism of Transcriptional Activation. EMBO J. 1998, 17, 6723–6729. [Google Scholar] [CrossRef] [Green Version]
- Pradeepa, M.M.; Sutherland, H.G.; Ule, J.; Grimes, G.R.; Bickmore, W.A. Psip1/Ledgf P52 Binds Methylated Histone H3K36 and Splicing Factors and Contributes to the Regulation of Alternative Splicing. PLoS Genet. 2012, 8, e1002717. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Xu, H.; Faiola, F.; Ma’Ayan, A.; Wang, J. Oct4 Links Multiple Epigenetic Pathways to the Pluripotency Network. Cell Res. 2012, 22, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Pardo, M.; Lang, B.; Yu, L.; Prosser, H.; Bradley, A.; Babu, M.M.; Choudhary, J. An Expanded Oct4 Interaction Network: Implications for Stem Cell Biology, Development, and Disease. Cell Stem Cell 2010, 6, 382–395. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Park, K.M.; Gontarz, P.; Zhang, B.; Pan, J.; McKenzie, Z.; Fischer, L.A.; Dong, C.; Dietmann, S.; Xing, X.; et al. OCT4 Cooperates with Distinct ATP-Dependent Chromatin Remodelers in Naïve and Primed Pluripotent States in Human. Nat. Commun. 2021, 12, 5123. [Google Scholar] [CrossRef]
- Nishimura, K.; Kato, T.; Chen, C.; Oinam, L.; Shiomitsu, E.; Ayakawa, D.; Ohtaka, M.; Fukuda, A.; Nakanishi, M.; Hisatake, K. Manipulation of KLF4 Expression Generates iPSCs Paused at Successive Stages of Reprogramming. Stem Cell Rep. 2014, 3, 915–929. [Google Scholar] [CrossRef]
- Banaszynski, L.A.; Chen, L.C.; Maynard-Smith, L.A.; Ooi, A.G.; Wandless, T.J. A Rapid, Reversible, and Tunable Method to Regulate Protein Function in Living Cells Using Synthetic Small Molecules. Cell 2006, 126, 995–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, M.; Déjardin, J. Locus-Specific Chromatin Isolation. Nat. Rev. Mol. Cell Biol. 2020, 21, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, Y.; Zhang, Y.; Liu, Y.; Liu, N.; Botten, G.A.; Cao, H.; Orkin, S.H.; Zhang, M.Q.; Xu, J. Multiplexed Capture of Spatial Configuration and Temporal Dynamics of Locus-Specific 3D Chromatin by Biotinylated dCas9. Genome Biol. 2020, 21, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Brug, M.P.; Blackinton, J.; Chandran, J.; Hao, L.Y.; Lal, A.; Mazan-Mamczarz, K.; Martindale, J.; Xie, C.; Ahmad, R.; Thomas, K.J.; et al. RNA Binding Activity of the Recessive Parkinsonism Protein DJ-1 Supports Involvement in Multiple Cellular Pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 10244–10249. [Google Scholar] [CrossRef] [Green Version]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Carlos Izpisua Belmonte, J. Deconstructing the Pluripotency Gene Regulatory Network. Nat. Cell Biol. 2018, 20, 382–392. [Google Scholar] [CrossRef]
- Gabut, M.; Samavarchi-Tehrani, P.; Wang, X.; Slobodeniuc, V.; O’Hanlon, D.; Sung, H.K.; Alvarez, M.; Talukder, S.; Pan, Q.; Mazzoni, E.O.; et al. An Alternative Splicing Switch Regulates Embryonic Stem Cell Pluripotency and Reprogramming. Cell 2011, 147, 132–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, A.; Blelloch, R.H. Regulation of MicroRNA Function in Somatic Stem Cell Proliferation and Differentiation. Nat. Rev. Mol. Cell Biol. 2014, 15, 565–576. [Google Scholar] [CrossRef]
- Fatica, A.; Bozzoni, I. Long Non-Coding RNAs: New Players in Cell Differentiation and Development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef]
- Tahmasebi, S.; Amiri, M.; Sonenberg, N. Translational Control in Stem Cells. Front. Genet. 2019, 9, 709. [Google Scholar] [CrossRef]
- Li, D.; Kishta, M.S.; Wang, J. Regulation of Pluripotency and Reprogramming by RNA Binding Proteins. Curr. Top. Dev. Biol. 2020, 138, 113–138. [Google Scholar] [PubMed]
- Tahmasebi, S.; Jafarnejad, S.M.; Tam, I.S.; Gonatopoulos-Pournatzis, T.; Matta-Camacho, E.; Tsukumo, Y.; Yanagiya, A.; Li, W.; Atlasi, Y.; Caron, M.; et al. Control of Embryonic Stem Cell Self-Renewal and Differentiation via Coordinated Alternative Splicing and Translation of YY2. Proc. Natl. Acad. Sci. USA 2016, 113, 12360–12367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Zhao, W.; Olson, S.D.; Prabhakara, K.S.; Zhou, X. Alternative Splicing Links Histone Modifications to Stem Cell Fate Decision. Genome Biol. 2018, 19, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider-Lunitz, V.; Ruiz-Orera, J.; Hubner, N.; van Heesch, S. Multifunctional RNA-Binding Proteins Influence MRNA Abundance and Translational Efficiency of Distinct Sets of Target Genes. PLoS Comput. Biol. 2021, 17, e1009658. [Google Scholar] [CrossRef]
- Aizawa, S.; Nishimura, K.; Mondejar, G.S.; Kumar, A.; Bui, P.L.; Tran, Y.T.H.; Kuno, A.; Muratani, M.; Kobayashi, S.; Nabekura, T.; et al. Early Reactivation of Clustered Genes on the Inactive X Chromosome during Somatic Cell Reprogramming. Stem Cell Rep. 2022, 17, 53–67. [Google Scholar] [CrossRef]
- Nishimura, K.; Ishiwata, H.; Sakuragi, Y.; Hayashi, Y.; Fukuda, A.; Hisatake, K. Live-Cell Imaging of Subcellular Structures for Quantitative Evaluation of Pluripotent Stem Cells. Sci. Rep. 2019, 9, 1777. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.; Aizawa, S.; Nugroho, F.L.; Shiomitsu, E.; Tran, Y.T.H.; Bui, P.L.; Borisova, E.; Sakuragi, Y.; Takada, H.; Kurisaki, A.; et al. A Role for KLF4 in Promoting the Metabolic Shift via TCL1 during Induced Pluripotent Stem Cell Generation. Stem Cell Rep. 2017, 8, 787–801. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burramsetty, A.K.; Nishimura, K.; Kishimoto, T.; Hamzah, M.; Kuno, A.; Fukuda, A.; Hisatake, K. Locus-Specific Isolation of the Nanog Chromatin Identifies Regulators Relevant to Pluripotency of Mouse Embryonic Stem Cells and Reprogramming of Somatic Cells. Int. J. Mol. Sci. 2022, 23, 15242. https://doi.org/10.3390/ijms232315242
Burramsetty AK, Nishimura K, Kishimoto T, Hamzah M, Kuno A, Fukuda A, Hisatake K. Locus-Specific Isolation of the Nanog Chromatin Identifies Regulators Relevant to Pluripotency of Mouse Embryonic Stem Cells and Reprogramming of Somatic Cells. International Journal of Molecular Sciences. 2022; 23(23):15242. https://doi.org/10.3390/ijms232315242
Chicago/Turabian StyleBurramsetty, Arun Kumar, Ken Nishimura, Takumi Kishimoto, Muhammad Hamzah, Akihiro Kuno, Aya Fukuda, and Koji Hisatake. 2022. "Locus-Specific Isolation of the Nanog Chromatin Identifies Regulators Relevant to Pluripotency of Mouse Embryonic Stem Cells and Reprogramming of Somatic Cells" International Journal of Molecular Sciences 23, no. 23: 15242. https://doi.org/10.3390/ijms232315242