

Structural Basis of Cysteine Ligase MshC Inhibition by Cysteinyl-Sulfonamides

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results and Discussion
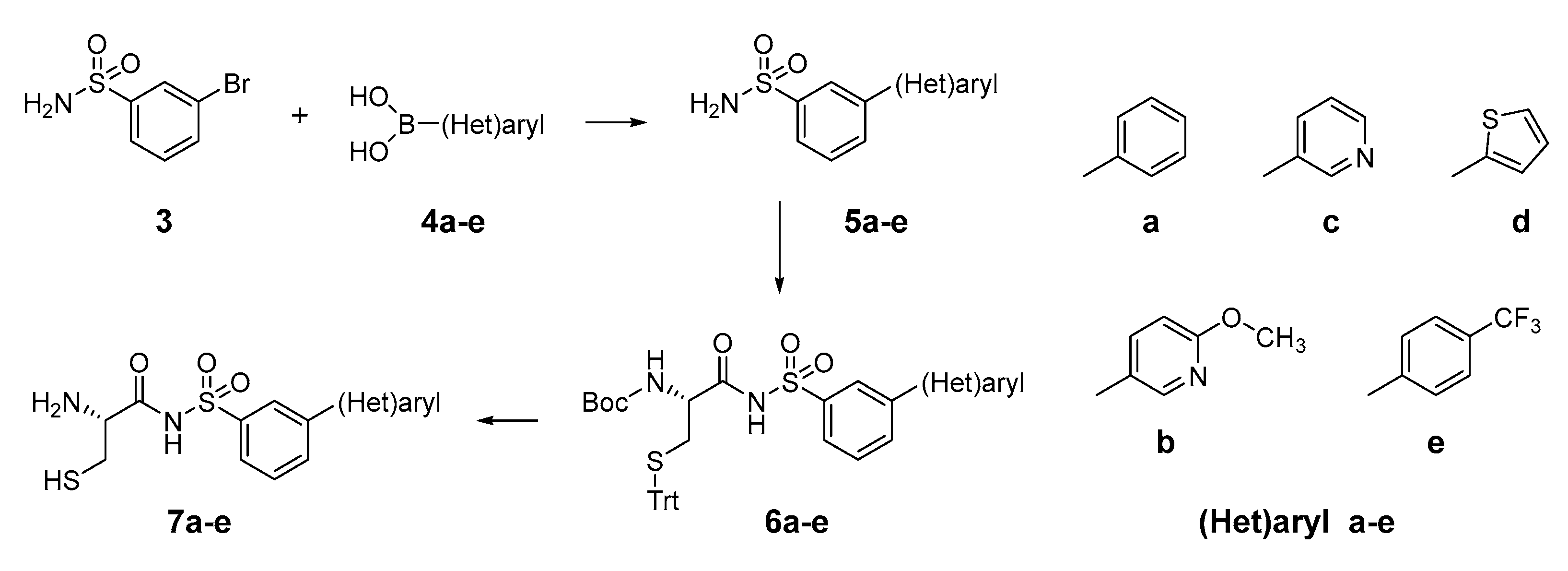
2.1. Design and Synthesis of Cysteinyl-Sulfonamides
2.2. Binding Affinity of Compounds against M. smegmatis MshC
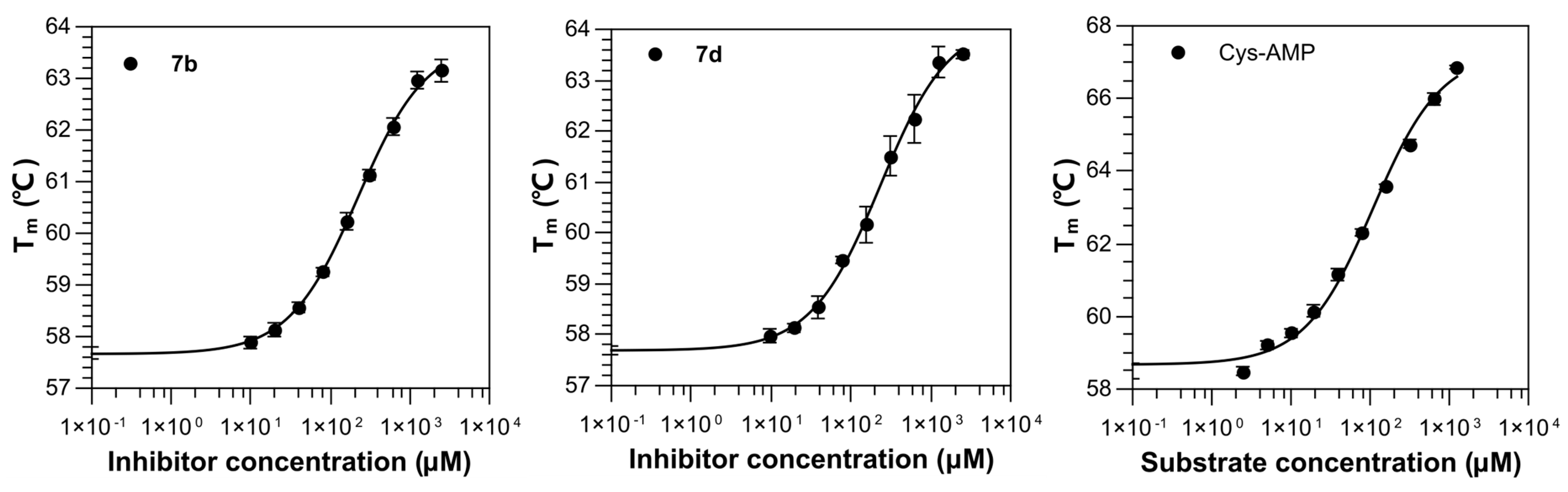
2.3. Structural Study on the Inhibitory Mechanism of Compounds 7b and 7d
2.4. Anti-Mycobacterium Activity of the Compounds
3. Materials and Methods
3.1. Reagents and Analytical Procedures
3.2. Chemical Synthesis and Analysis
3.2.1. Synthesis of Compounds 5a–e: General Procedure A
3.2.2. Synthesis of Compounds 6a–e: General Procedure B
3.2.3. Synthesis of Compounds 7a–e: General Procedure C
3.3. Cloning, Expression and Protein Purification
3.4. Crystallization
3.5. Data Collection and Structure Determination
3.6. Thermal Shift Assay
3.7. Antibacterial Activity Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO|Global Tuberculosis Report 2019. Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 25 February 2020).
- Pontali, E.; Raviglione, M.C.; Migliori, G.B.; The Writing Group Members of the Global TB Network Clinical Trials Committee. Regimens to Treat Multidrug-Resistant Tuberculosis: Past, Present and Future Perspectives. Eur. Respir. Rev. 2019, 28, 190035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, C.; Dheda, K.; Chesov, D.; Mandalakas, A.M.; Udwadia, Z.; Horsburgh, C.R. Management of Drug-Resistant Tuberculosis. Lancet 2019, 394, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Ruecker, N.; Jansen, R.; Trujillo, C.; Puckett, S.; Jayachandran, P.; Piroli, G.G.; Frizzell, N.; Molina, H.; Rhee, K.Y.; Ehrt, S. Fumarase Deficiency Causes Protein and Metabolite Succination and Intoxicates Mycobacterium Tuberculosis. Cell Chem. Biol. 2017, 24, 306–315. [Google Scholar] [CrossRef] [Green Version]
- Hillion, M.; Bernhardt, J.; Busche, T.; Rossius, M.; Maaß, S.; Becher, D.; Rawat, M.; Wirtz, M.; Hell, R.; Rückert, C.; et al. Monitoring Global Protein Thiol-Oxidation and Protein S-Mycothiolation in Mycobacterium Smegmatis under Hypochlorite Stress. Sci. Rep. 2017, 7, 1195. [Google Scholar] [CrossRef] [Green Version]
- Buchmeier, N.A.; Newton, G.L.; Koledin, T.; Fahey, R.C. Association of Mycothiol with Protection of Mycobacterium Tuberculosis from Toxic Oxidants and Antibiotics. Mol. Microbiol. 2003, 47, 1723–1732. [Google Scholar] [CrossRef]
- Nambi, S.; Long, J.E.; Mishra, B.B.; Baker, R.; Murphy, K.C.; Olive, A.J.; Nguyen, H.P.; Shaffer, S.A.; Sassetti, C.M. The Oxidative Stress Network of Mycobacterium Tuberculosis Reveals Coordination between Radical Detoxification Systems. Cell Host Microbe 2015, 17, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Holmgren, A. The Thioredoxin Antioxidant System. Free. Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Sareen, D.; Newton, G.L.; Fahey, R.C.; Buchmeier, N.A. Mycothiol Is Essential for Growth of Mycobacterium Tuberculosis Erdman. J. Bacteriol. 2003, 185, 6736–6740. [Google Scholar] [CrossRef]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes Required for Mycobacterial Growth Defined by High Density Mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef]
- Sareen, D.; Steffek, M.; Newton, G.L.; Fahey, R.C. ATP-Dependent L-Cysteine:1D-Myo-Inosityl 2-Amino-2-Deoxy-Alpha-D-Glucopyranoside Ligase, Mycothiol Biosynthesis Enzyme MshC, Is Related to Class I Cysteinyl-tRNA Synthetases. Biochemistry 2002, 41, 6885–6890. [Google Scholar] [CrossRef]
- Newberry, K.J. Structural Origins of Amino Acid Selection without Editing by Cysteinyl-tRNA Synthetase. EMBO J. 2002, 21, 2778–2787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, G.L.; Buchmeier, N.; La Clair, J.J.; Fahey, R.C. Evaluation of NTF1836 as an Inhibitor of the Mycothiol Biosynthetic Enzyme MshC in Growing and Non-Replicating Mycobacterium Tuberculosis. Bioorg. Med. Chem. 2011, 19, 3956–3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Lugo, M.-T.; Baker, H.; Shiloach, J.; Boshoff, H.; Bewley, C.A. Dequalinium, a New Inhibitor of Mycobacterium Tuberculosis Mycothiol Ligase Identified by High-Throughput Screening. J. Biomol. Screen. 2009, 14, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Ibba, M.; Soll, D. Aminoacyl-tRNA Synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef]
- Perona, J.J.; Hadd, A. Structural Diversity and Protein Engineering of the Aminoacyl-tRNA Synthetases. Biochemistry 2012, 51, 8705–8729. [Google Scholar] [CrossRef] [PubMed]
- Vondenhoff, G.H.M.; Van Aerschot, A. Aminoacyl-tRNA Synthetase Inhibitors as Potential Antibiotics. Eur. J. Med. Chem. 2011, 46, 5227–5236. [Google Scholar] [CrossRef]
- Francklyn, C.S.; Mullen, P. Progress and Challenges in Aminoacyl-tRNA Synthetase-Based Therapeutics. J. Biol. Chem. 2019, 294, 5365–5385. [Google Scholar] [CrossRef] [Green Version]
- Cochrane, R.V.K.; Norquay, A.K.; Vederas, J.C. Natural Products and Their Derivatives as tRNA Synthetase Inhibitors and Antimicrobial Agents. Med. Chem. Commun. 2016, 7, 1535–1545. [Google Scholar] [CrossRef]
- Zhang, P.; Ma, S. Recent Development of Leucyl-tRNA Synthetase Inhibitors as Antimicrobial Agents. Medchemcomm 2019, 10, 1329–1341. [Google Scholar] [CrossRef]
- Lee, E.-Y.; Kim, S.; Kim, M.H. Aminoacyl-tRNA Synthetases, Therapeutic Targets for Infectious Diseases. Biochem. Pharmacol. 2018, 154, 424–434. [Google Scholar] [CrossRef]
- Pang, L.; Weeks, S.D.; Van Aerschot, A. Aminoacyl-tRNA Synthetases as Valuable Targets for Antimicrobial Drug Discovery. Int. J. Mol. Sci. 2021, 22, 1750. [Google Scholar] [CrossRef] [PubMed]
- Bouz, G.; Zitko, J. Inhibitors of Aminoacyl-tRNA Synthetases as Antimycobacterial Compounds: An up-to-Date Review. Bioorg. Chem. 2021, 110, 104806. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Hilgers, M.T.; Cunningham, M.L.; Borchardt, A.; Locke, J.B.; Abraham, S.; Haley, G.; Kwan, B.P.; Hall, C.; Hough, G.W.; et al. Identification of Bacteria-Selective Threonyl-tRNA Synthetase Substrate Inhibitors by Structure-Based Design. J. Med. Chem. 2013, 56, 1748–1760. [Google Scholar] [CrossRef]
- Charlton, M.H.; Aleksis, R.; Saint-Leger, A.; Gupta, A.; Loza, E.; Ribas de Pouplana, L.; Kaula, I.; Gustina, D.; Madre, M.; Lola, D.; et al. N-Leucinyl Benzenesulfonamides as Structurally Simplified Leucyl-tRNA Synthetase Inhibitors. ACS Med. Chem. Lett. 2018, 9, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, K.; Matejtschuk, P.; Thelwell, C.; Burns, C.J. Differential Scanning Fluorimetry: Rapid Screening of Formulations That Promote the Stability of Reference Preparations. J. Pharm. Biomed. Anal. 2013, 77, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, L.W.; Fan, F.; Vetting, M.W.; Blanchard, J.S. The 1.6 Å Crystal Structure of Mycobacterium Smegmatis MshC: The Penultimate Enzyme in the Mycothiol Biosynthetic Pathway. Biochemistry 2008, 47, 13326–13335. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- De Ruysscher, D.; Pang, L.; Lenders, S.M.G.; Cappoen, D.; Cos, P.; Rozenski, J.; Strelkov, S.V.; Weeks, S.D.; Van Aerschot, A. Synthesis and Structure-Activity Studies of Novel Anhydrohexitol-Based Leucyl-tRNA Synthetase Inhibitors. Eur. J. Med. Chem. 2021, 211, 113021. [Google Scholar] [CrossRef]
- Weeks, S.D.; Drinker, M.; Loll, P.J. Ligation Independent Cloning Vectors for Expression of SUMO Fusions. Protein Expr. Purif. 2007, 53, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; De Graef, S.; Nautiyal, M.; Pang, L.; Gadakh, B.; Froeyen, M.; Van Mellaert, L.; Strelkov, S.V.; Weeks, S.D.; Van Aerschot, A. Family-Wide Analysis of Aminoacyl-Sulfamoyl-3-Deazaadenosine Analogues as Inhibitors of Aminoacyl-tRNA Synthetases. Eur. J. Med. Chem. 2018, 148, 384–396. [Google Scholar] [CrossRef]
- Pang, L.; Zanki, V.; Strelkov, S.V.; Van Aerschot, A.; Gruic-Sovulj, I.; Weeks, S.D. Partitioning of the Initial Catalytic Steps of Leucyl-tRNA Synthetase Is Driven by an Active Site Peptide-Plane Flip. Commun. Biol. 2022, 5, 883. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W. Stable Expression Clones and Auto-Induction for Protein Production in E. Coli. Methods Mol. Biol. 2014, 1091, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data Processing and Analysis with the AutoPROC Toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Gore, S.; Sanz García, E.; Hendrickx, P.M.S.; Gutmanas, A.; Westbrook, J.D.; Yang, H.; Feng, Z.; Baskaran, K.; Berrisford, J.M.; Hudson, B.P.; et al. Validation of Structures in the Protein Data Bank. Structure 2017, 25, 1916–1927. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Conc. (µM) | Tm (°C) 1 | ∆Tm (°C) | Tm (°C; + 1mM TCEP) 5 | ∆Tm (°C) 6 | EC50 |
|---|---|---|---|---|---|---|
| Control | / | 58.47 ± 0.07 | / | / | / | / |
| DMSO 10% (v/v) | 55.09 ± 0.98 | −3.38 2 | / | / | / | |
| EDO 10% (v/v) | 57.99 ± 0.04 | −0.48 3 | 57.91 ± 0.05 | / | / | |
 7a | 1000 | 57.56 ± 0.16 | −0.43 4 | 57.46 ± 0.26 | −0.45 | / |
| 100 | 58.05 ± 0.12 | +0.06 4 | 58.00 ± 0.09 | +0.09 | ||
 7b | 1000 | 58.08 ± 0.23 | +0.09 4 | 60.99 ± 0.23 | +3.08 | 219.29 ± 1.27 |
| 100 | 57.98 ± 0.08 | −0.01 4 | 59.15 ± 0.05 | +1.24 | ||
 7c | 1000 | 58.20 ± 0.14 | +0.21 4 | 58.50 ± 0.15 | +0.59 | / |
| 100 | 58.01 ± 0.07 | +0.02 4 | 57.90 ± 0.01 | −0.01 | ||
 7d | 1000 | 58.98 ± 0.22 | +0.99 4 | 61.66 ± 0.29 | +3.75 | 230.60 ± 2.16 |
| 100 | 57.94 ± 0.09 | −0.05 4 | 59.01 ± 0.22 | +1.10 | ||
 7e | 1000 | 58.13 ± 0.14 | +0.14 4 | 58.67 ± 0.17 | +0.76 | / |
| 100 | 57.76 ± 0.08 | −0.23 4 | 57.76 ± 0.03 | −0.15 | ||
| L-Cys | 10,000 | 61.56 ± 0.06 | +3.09 4 | / | / | / |
| 1000 | 59.20 ± 0.07 | +0.73 4 | / | / | ||
| ATP | 10,000 | 59.82 ± 0.18 | +1.35 4 | / | / | / |
| 1000 | 58.20 ± 0.03 | −0.27 4 | / | / | ||
| Cys-AMP | 1000 | 66.55 ± 0.13 | +8.08 4 | / | / | 106.64 ± 1.34 |
| 100 | 62.23 ± 0.13 | +3.76 4 | / | / |
| MshC (apo) | MshC-Compound 7b | MshC-Compound 7d | |
|---|---|---|---|
| PDB Code | 8HFM | 8HFN | 8HFO |
| Data collection | |||
| Resolution range (Å) | 48.32–2.41 (2.50–2.41) | 43.68–1.98 (2.05–1.98) | 55.03–2.773 (2.87–2.77) |
| Space group | P 41 2 2 | P 61 | P 61 |
| Unit cell | |||
| a, b, c (Å) | 69.81 69.81 236.01 | 166.02 166.02 51.36 | 168.11 168.11 52.30 |
| α, β, γ (°) | 90 90 90 | 90 90 120 | 90 90 120 |
| Unique reflections | 23,525 (2275) | 56,710 (5394) | 21,800 (2176) |
| Multiplicity | 26.2 (26.6) | 10.2 (10.6) | 20.1 (20.7) |
| Completeness (%) | 99.9 (100.0) | 99.9 (99.9) | 99.9 (99.9) |
| Mean I/σ (I) | 15.3 (1.6) | 14.1 (2.6) | 19.0 (2.3) |
| Wilson B-factor (Å2) | 56.4 | 41.2 | 92.4 |
| Rmerge | 0.167 (1.995) | 0.086 (1.160) | 0.098 (1.438) |
| Rmeas | 0.171 (2.034) | 0.090 (1.219) | 0.100 (1.474) |
| Rpim | 0.033 (0.393) | 0.029 (0.373) | 0.023 (0.323) |
| CC1/2 | 0.998 (0.760) | 0.998 (0.886) | 0.993 (0.889) |
| Refinement | |||
| Reflections used for refinement | 23,514 (2276) | 53,801 (5391) | 21,784 (2174) |
| Rwork | 0.207 (0.298) | 0.199 (0.490) | 0.213 (0.409) |
| Rfree | 0.238 (0.359) | 0.223 (0.460) | 0.258 (0.356) |
| Number of non-H atoms | 3197 | 3473 | 3214 |
| Macromolecules | 3130 | 3200 | 3181 |
| Inhibitor | / | 27 | 23 |
| Solvent | 65 | 246 | 10 |
| RMS bonds (Å) | 0.004 | 0.004 | 0.005 |
| RMS angles (°) | 0.66 | 0.77 | 0.89 |
| Ramachandran favoured (%) | 98.25 | 99.27 | 95.38 |
| Ramachandran allowed (%) | 1.75 | 0.73 | 4.38 |
| Average B-factor (Å2) | 65.9 | 53.64 | 109.07 |
| Protein | 66.06 | 53.55 | 109.27 |
| Inhibitor | / | 46.85 | 82.91 |
| Solvent | 57.98 | 55.63 | 105.87 |
| Compound | pIC50 a | ||
|---|---|---|---|
| Mtb b | Mab c | ||
| WT d | Δmtr::Hyg e | WT | |
| 7a | <3.9 | <3.9 | <3.9 |
| 7b | <3.9 | <3.9 | <3.9 |
| 7c | <3.9 | <3.9 | <3.9 |
| 7d | <3.9 | <3.9 | <3.9 |
| 7e | <3.9 | <3.9 | <3.9 |
| f MXF | 8.1 | 6.3 | 5.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, L.; Lenders, S.; Osipov, E.M.; Weeks, S.D.; Rozenski, J.; Piller, T.; Cappoen, D.; Strelkov, S.V.; Van Aerschot, A. Structural Basis of Cysteine Ligase MshC Inhibition by Cysteinyl-Sulfonamides. Int. J. Mol. Sci. 2022, 23, 15095. https://doi.org/10.3390/ijms232315095
Pang L, Lenders S, Osipov EM, Weeks SD, Rozenski J, Piller T, Cappoen D, Strelkov SV, Van Aerschot A. Structural Basis of Cysteine Ligase MshC Inhibition by Cysteinyl-Sulfonamides. International Journal of Molecular Sciences. 2022; 23(23):15095. https://doi.org/10.3390/ijms232315095
Chicago/Turabian StylePang, Luping, Stijn Lenders, Evgenii M. Osipov, Stephen D. Weeks, Jef Rozenski, Tatiana Piller, Davie Cappoen, Sergei V. Strelkov, and Arthur Van Aerschot. 2022. "Structural Basis of Cysteine Ligase MshC Inhibition by Cysteinyl-Sulfonamides" International Journal of Molecular Sciences 23, no. 23: 15095. https://doi.org/10.3390/ijms232315095