
Insights into Network of Hot Spots of Aggregation in Nucleophosmin 1

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
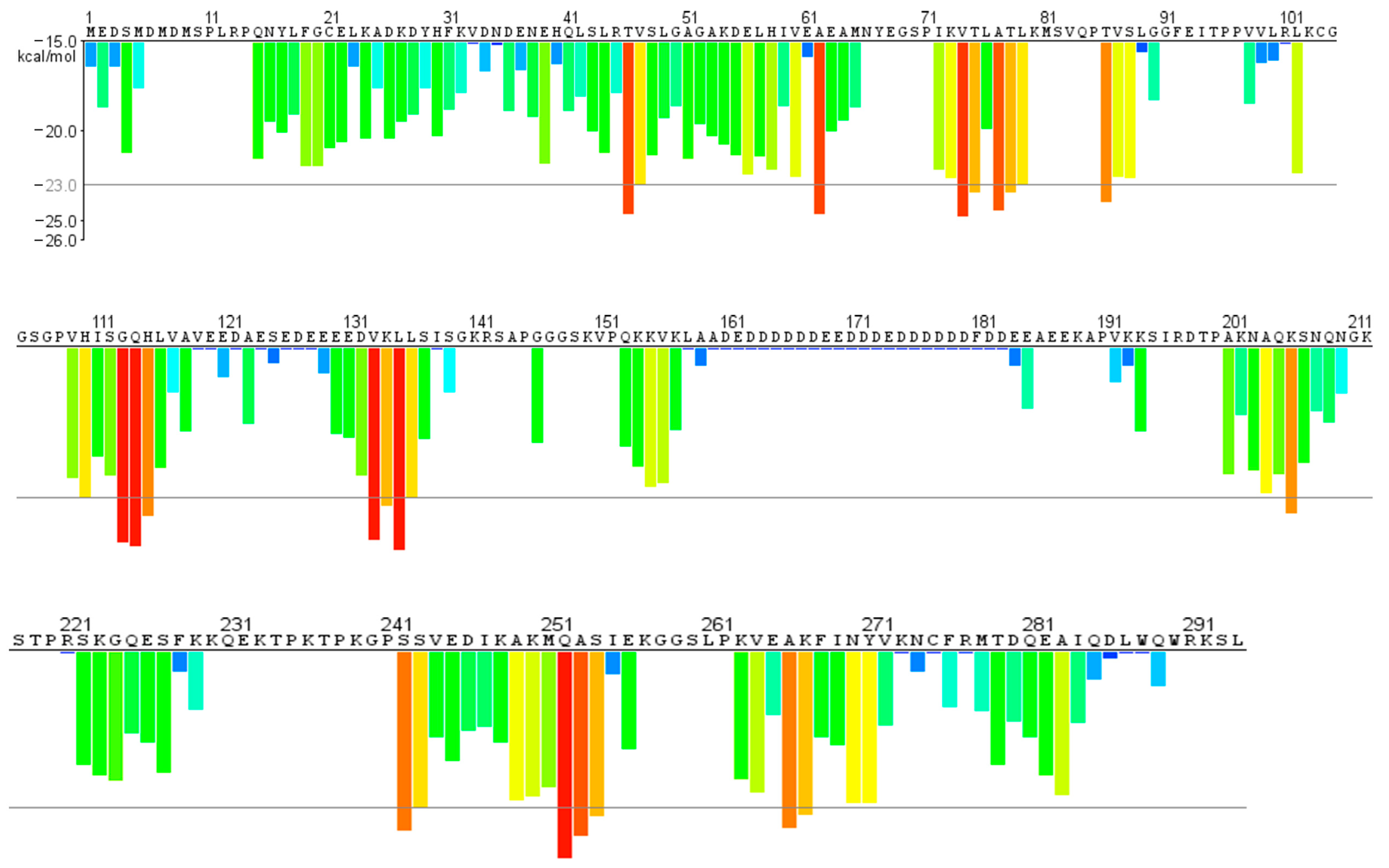
- Structure prediction and aggregation propensity through bioinformatic tools.
- β-sheet regions: ThT assay, conformational and SEM analyses of NPM141–65, NPM169–83 and NPM1107–120.
- Random region: ThT assay, conformational and SEM analyses of NPM184–93.
- Helical regions: ThT assay, conformational and SEM analyses of NPM1127–139 and NPM1242–259.
- Cellular effects of NPM1 fragments
3. Materials and Methods
3.1. Peptide Synthesis
3.2. Far-UV CD Spectroscopy
3.3. ThT Assay
3.4. SEM Analysis
3.5. NMR Experiments
3.6. Cells
3.7. Cell Viability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sawaya, M.R.; Hughes, M.P.; Rodriguez, J.A.; Riek, R.; Eisenberg, D.S. The expanding amyloid family: Structure, stability, function, and pathogenesis. Cell 2021, 184, 4857–4873. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 2011, 70, 532–540. [Google Scholar] [CrossRef] [Green Version]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Fandrich, M.; Fletcher, M.A.; Dobson, C.M. Amyloid fibrils from muscle myoglobin. Nature 2001, 410, 165–166. [Google Scholar] [CrossRef]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwald, J.; Kwiatkowski, W.; Riek, R. Peptide Amyloids in the Origin of Life. J. Mol. Biol. 2018, 430, 3735–3750. [Google Scholar] [CrossRef]
- Chapman, M.R.; Robinson, L.S.; Pinkner, J.S.; Roth, R.; Heuser, J.; Hammar, M.; Normark, S.; Hultgren, S.J. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006, 4, e6. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Kato, M.; Wu, L.C.; Lin, Y.; Ding, M.; Zhang, Y.; Yu, Y.; McKnight, S.L. The LC Domain of hnRNPA2 Adopts Similar Conformations in Hydrogel Polymers, Liquid-like Droplets, and Nuclei. Cell 2015, 163, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogler, T.O.; Wheeler, J.R.; Nguyen, E.D.; Hughes, M.P.; Britson, K.A.; Lester, E.; Rao, B.; Betta, N.D.; Whitney, O.N.; Ewachiw, T.E.; et al. TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature 2018, 563, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Beton, J.G.; Monistrol, J.; Wentink, A.; Johnston, E.C.; Roberts, A.J.; Bukau, B.; Hoogenboom, B.W.; Saibil, H.R. Cooperative Amyloid Fibre Binding and Disassembly by the Hsp70 disaggregase. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, A.; Falcon, B.; He, S.; Murzin, A.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.F.; Goedert, M.; Scheres, S.H.W.; et al. Cryo-EM structures of tau filaments fromAlzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Cui, W.; He, Z.; Shi, X.; Feng, K.; Ma, B.; Cai, Y.-D. Cooperativity among short amyloid stretches in long amyloidogenic sequences. PLoS ONE 2012, 7, e39369. [Google Scholar] [CrossRef] [Green Version]
- Mahler, H.C.; Friess, W.; Grauschopf, U.; Kiese, S. Protein aggregation: Pathways, induction factors and analysis. J. Pharm. Sci. 2009, 98, 2909–2934. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Nema, S.; Teagarden, D. Protein aggregation--pathways and influencing factors. Int. J. Pharm 2010, 390, 89–99. [Google Scholar] [CrossRef]
- Konstantoulea, K.; Guerreiro, P.; Ramakers, M.; Louros, N.; Aubrey, L.D.; Houben, B.; Michiels, E.; De Vleeschouwer, M.; Lampi, Y.; Ribeiro, L.F. Heterotypic Amyloid β interactions facilitate amyloid assembly and modify amyloid structure. EMBO J. 2022, 41, e108591. [Google Scholar] [CrossRef] [PubMed]
- Bhasne, K.; Sebastian, S.; Jain, N.; Mukhopadhyay, S. Synergistic amyloid switch triggered by early heterotypic oligomerization of intrinsically disordered α-synuclein and tau. J. Mol. Biol. 2018, 430, 2508–2520. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wu, N.; Guo, J.; Fan, Y. Prediction of amyloid fibril-forming segments based on a support vector machine. BMC Bioinform. 2009, 10 (Suppl. 1), S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Chen, H.; Lai, L. Identification of amyloid fibril-forming segments based on structure and residue-based statistical potential. Bioinformatics 2007, 23, 2218–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; Lopez de la Paz, M.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef]
- Sehgal, P.B.; Westley, J.; Lerea, K.M.; DiSenso-Browne, S.; Etlinger, J.D. Biomolecular condensates in cell biology and virology: Phase-separated membraneless organelles (MLOs). Anal. Biochem. 2020, 597, 113691. [Google Scholar] [CrossRef]
- Linsenmeier, M.; Faltova, L.; Palmiero, U.C.; Seiffert, C.; Küffner, A.M.; Pinotsi, D.; Zhou, J.; Mezzenga, R.; Arosio, P. The interface of condensates of the hnRNPA1 low complexity domain promotes formation of amyloid fibrils. bioRxiv 2022. [Google Scholar] [CrossRef]
- Navarro, S.; Ventura, S. Computational methods to predict protein aggregation. Curr. Opin. Struct. Biol. 2022, 73, 102343. [Google Scholar] [CrossRef]
- Langenberg, T.; Gallardo, R.; van der Kant, R.; Louros, N.; Michiels, E.; Duran-Romaña, R.; Houben, B.; Cassio, R.; Wilkinson, H.; Garcia, T. Thermodynamic and evolutionary coupling between the native and amyloid state of globular proteins. Cell Rep. 2020, 31, 107512. [Google Scholar] [CrossRef]
- Lei, J.; Qi, R.; Wei, G.; Nussinov, R.; Ma, B. Self-aggregation and coaggregation of the p53 core fragment with its aggregation gatekeeper variant. Phys. Chem. Chem. Phys. 2016, 18, 8098–8107. [Google Scholar] [CrossRef]
- Colombo, E.; Marine, J.C.; Danovi, D.; Falini, B.; Pelicci, P.G. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 2002, 4, 529–533. [Google Scholar] [CrossRef]
- Alberti, S.; Carra, S. Nucleolus: A Liquid Droplet Compartment for Misbehaving Proteins. Curr. Biol. 2019, 29, R930–R932. [Google Scholar] [CrossRef]
- Lindstrom, M.S. NPM1/B23: A Multifunctional Chaperone in Ribosome Biogenesis and Chromatin Remodeling. Biochem. Res. Int. 2011, 2011, 195209. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.Y.; Liu, Q.R.; Borjigin, J.; Busch, H.; Rennert, O.M.; Tease, L.A.; Chan, P.K. Characterization of the cDNA encoding human nucleophosmin and studies of its role in normal and abnormal growth. Biochemistry 1989, 28, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, K.; Szebeni, A.; Olson, M.O. Mapping the functional domains of nucleolar protein B23. J. Biol. Chem. 2000, 275, 24451–24457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuwaki, M. The structure and functions of NPM1/Nucleophsmin/B23, a multifunctional nucleolar acidic protein. J. Biochem. 2008, 143, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Box, J.K.; Paquet, N.; Adams, M.N.; Boucher, D.; Bolderson, E.; O’Byrne, K.J.; Richard, D.J. Nucleophosmin: From structure and function to disease development. BMC Mol. Biol. 2016, 17, 19. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.H.; Kim, H.S.; Kang, J.Y.; Lee, B.I.; Ha, J.Y.; Yoon, H.J.; Lim, S.O.; Jung, G.; Suh, S.W. Crystal structure of human nucleophosmin-core reveals plasticity of the pentamer-pentamer interface. Proteins 2007, 69, 672–678. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Grace, C.R.; Buljan, M.; Yun, M.K.; Pytel, N.J.; Satumba, J.; Nourse, A.; Park, C.G.; Madan Babu, M.; White, S.W.; et al. Structural polymorphism in the N-terminal oligomerization domain of NPM1. Proc. Natl. Acad. Sci. USA 2014, 111, 4466–4471. [Google Scholar] [CrossRef] [Green Version]
- Mitrea, D.M.; Kriwacki, R.W. On the relationship status for Arf and NPM1—It’s complicated. FEBS J. 2018, 285, 828–831. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Cika, J.A.; Stanley, C.B.; Nourse, A.; Onuchic, P.L.; Banerjee, P.R.; Phillips, A.H.; Park, C.G.; Deniz, A.A.; Kriwacki, R.W. Self-interaction of NPM1 modulates multiple mechanisms of liquid-liquid phase separation. Nat. Commun. 2018, 9, 842. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Signal. 2016, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrea, D.M.; Cika, J.A.; Guy, C.S.; Ban, D.; Banerjee, P.R.; Stanley, C.B.; Nourse, A.; Deniz, A.A.; Kriwacki, R.W. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. eLife 2016, 5, e13571. [Google Scholar] [CrossRef]
- Grummitt, C.G.; Townsley, F.M.; Johnson, C.M.; Warren, A.J.; Bycroft, M. Structural consequences of nucleophosmin mutations in acute myeloid leukemia. J. Biol. Chem. 2008, 283, 23326–23332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.W.; Lee, S.B.; Kim, C.K.; Lee, K.H.; Cho, S.W.; Ahn, J.Y. Lysine 263 residue of NPM/B23 is essential for regulating ATP binding and B23 stability. FEBS Lett. 2008, 582, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Marasco, D.; Ruggiero, A.; Vascotto, C.; Poletto, M.; Scognamiglio, P.L.; Tell, G.; Vitagliano, L. Role of mutual interactions in the chemical and thermal stability of nucleophosmin NPM1 domains. Biochem. Biophys. Res. Commun. 2013, 430, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Di Natale, C.; Scognamiglio, P.L.; Cascella, R.; Cecchi, C.; Russo, A.; Leone, M.; Penco, A.; Relini, A.; Federici, L.; Di Matteo, A.; et al. Nucleophosmin contains amyloidogenic regions that are able to form toxic aggregates under physiological conditions. FASEB J. 2015, 29, 3689–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scognamiglio, P.L.; Di Natale, C.; Leone, M.; Cascella, R.; Cecchi, C.; Lirussi, L.; Antoniali, G.; Riccardi, D.; Morelli, G.; Tell, G.; et al. Destabilisation, aggregation, toxicity and cytosolic mislocalisation of nucleophosmin regions associated with acute myeloid leukemia. Oncotarget 2016, 7, 59129–59143. [Google Scholar] [CrossRef] [Green Version]
- Di Natale, C.; Florio, D.; Di Somma, S.; Di Matteo, A.; Federici, L.; Netti, P.A.; Morelli, G.; Malfitano, A.M.; Marasco, D. Proteostasis unbalance of nucleophosmin 1 in Acute Myeloid Leukemia: An aggregomic perspective. Int. J. Biol. Macromol. 2020, 164, 3501–3507. [Google Scholar] [CrossRef]
- Di Natale, C.; Natale, C.F.; Florio, D.; Netti, P.A.; Morelli, G.; Ventre, M.; Marasco, D. Effects of surface nanopatterning on internalization and amyloid aggregation of the fragment 264-277 of Nucleophosmin 1. Colloids Surf. B Biointerfaces 2021, 197, 111439. [Google Scholar] [CrossRef]
- La Manna, S.; Florio, D.; Di Natale, C.; Scognamiglio, P.L.; Sibillano, T.; Netti, P.A.; Giannini, C.; Marasco, D. Type F mutation of nucleophosmin 1 Acute Myeloid Leukemia: A tale of disorder and aggregation. Int. J. Biol. Macromol. 2021, 188, 207–214. [Google Scholar] [CrossRef]
- La Manna, S.; Florio, D.; Di Natale, C.; Napolitano, F.; Malfitano, A.M.; Netti, P.A.; De Benedictis, I.; Marasco, D. Conformational consequences of NPM1 rare mutations: An aggregation perspective in Acute Myeloid Leukemia. Bioorg. Chem. 2021, 113, 104997. [Google Scholar] [CrossRef]
- La Manna, S.; Florio, D.; Di Natale, C.; Lagreca, E.; Sibillano, T.; Giannini, C.; Marasco, D. Type C mutation of nucleophosmin 1 acute myeloid leukemia: Consequences of intrinsic disorder. Biochim. Biophys. Acta Gen. Subj. 2022, 1866, 130173. [Google Scholar] [CrossRef] [PubMed]
- De Santis, A.; La Manna, S.; Krauss, I.R.; Malfitano, A.M.; Novellino, E.; Federici, L.; De Cola, A.; Di Matteo, A.; D’Errico, G.; Marasco, D. Nucleophosmin-1 regions associated with acute myeloid leukemia interact differently with lipid membranes. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 967–978. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Scognamiglio, P.L.; Roviello, V.; Borbone, F.; Florio, D.; Di Natale, C.; Bigi, A.; Cecchi, C.; Cascella, R.; Giannini, C.; et al. The acute myeloid leukemia-associated Nucleophosmin 1 gene mutations dictate amyloidogenicity of the C-terminal domain. FEBS J. 2019, 286, 2311–2328. [Google Scholar] [CrossRef] [Green Version]
- La Manna, S.; Roviello, V.; Scognamiglio, P.L.; Diaferia, C.; Giannini, C.; Sibillano, T.; Morelli, G.; Novellino, E.; Marasco, D. Amyloid fibers deriving from the aromatic core of C-terminal domain of nucleophosmin 1. Int. J. Biol. Macromol. 2019, 122, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Di Natale, C.; La Manna, S.; Malfitano, A.M.; Di Somma, S.; Florio, D.; Scognamiglio, P.L.; Novellino, E.; Netti, P.A.; Marasco, D. Structural insights into amyloid structures of the C-terminal region of nucleophosmin 1 in type A mutation of acute myeloid leukemia. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 637–644. [Google Scholar] [CrossRef]
- Florio, D.; Roviello, V.; La Manna, S.; Napolitano, F.; Maria Malfitano, A.; Marasco, D. Small molecules enhancers of amyloid aggregation of C-terminal domain of Nucleophosmin 1 in acute myeloid leukemia. Bioorg. Chem. 2022, 127, 106001. [Google Scholar] [CrossRef] [PubMed]
- Scognamiglio, P.L.; Di Natale, C.; Leone, M.; Poletto, M.; Vitagliano, L.; Tell, G.; Marasco, D. G-quadruplex DNA recognition by nucleophosmin: New insights from protein dissection. Biochim. Biophys. Acta 2014, 1840, 2050–2059. [Google Scholar] [CrossRef]
- La Manna, S.; Florio, D.; Panzetta, V.; Roviello, V.; Netti, P.A.; Di Natale, C.; Marasco, D. Hydrogelation tunability of bioinspired short peptides. Soft Matter 2022, 18, 8418–8426. [Google Scholar] [CrossRef]
- Melnikov, S.; Mailliot, J.; Rigger, L.; Neuner, S.; Shin, B.S.; Yusupova, G.; Dever, T.E.; Micura, R.; Yusupov, M. Molecular insights into protein synthesis with proline residues. EMBO Rep. 2016, 17, 1776–1784. [Google Scholar] [CrossRef]
- Galzitskaya, O.V.; Selivanova, O.M.; Gorbunova, E.Y.; Mustaeva, L.G.; Azev, V.N.; Surin, A.K. Mechanism of Amyloid Gel Formation by Several Short Amyloidogenic Peptides. Nanomaterials 2021, 11, 3129. [Google Scholar] [CrossRef] [PubMed]
- Pfister, J.A.; D’Mello, S.R. Regulation of Neuronal Survival by Nucleophosmin 1 (NPM1) Is Dependent on Its Expression Level, Subcellular Localization, and Oligomerization Status. J. Biol. Chem. 2016, 291, 20787–20797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griesinger, C.; Otting, G.; Wuthrich, K.; Ernst, R.R. Clean Tocsy for H-1 Spin System-Identification in Macromolecules. J. Am. Chem. Soc. 1988, 110, 7870–7872. [Google Scholar] [CrossRef]
- Kumar, A.; Ernst, R.R.; Wuthrich, K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 1980, 95, 1–6. [Google Scholar] [CrossRef]
- Bartels, C.; Xia, T.H.; Billeter, M.; Guntert, P.; Wuthrich, K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 1995, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Kneller, D.G. SPARKY 3, University of California, San Francisco. Available online: https://www.cgl.ucsf.edu/home/sparky/ (accessed on 30 October 2022).
- Florio, D.; Malfitano, A.M.; Di Somma, S.; Mugge, C.; Weigand, W.; Ferraro, G.; Iacobucci, I.; Monti, M.; Morelli, G.; Merlino, A.; et al. Platinum(II) O,S Complexes Inhibit the Aggregation of Amyloid Model Systems. Int. J. Mol. Sci. 2019, 20, 829. [Google Scholar] [CrossRef] [Green Version]
- Manna, S.; Florio, D.; Iacobucci, I.; Napolitano, F.; Benedictis, I.; Malfitano, A.M.; Monti, M.; Ravera, M.; Gabano, E.; Marasco, D. A Comparative Study of the Effects of Platinum (II) Complexes on beta-Amyloid Aggregation: Potential Neurodrug Applications. Int. J. Mol. Sci. 2021, 22, 3015. [Google Scholar] [CrossRef]
- Di Matteo, A.; Franceschini, M.; Paiardini, A.; Grottesi, A.; Chiarella, S.; Rocchio, S.; Di Natale, C.; Marasco, D.; Vitagliano, L.; Travaglini-Allocatelli, C.; et al. Structural investigation of nucleophosmin interaction with the tumor suppressor Fbw7gamma. Oncogenesis 2017, 6, e379. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragments | Sequences | pI | Net Charge at Neutral pH | Conformation |
|---|---|---|---|---|
| 41–65 | Ac-Q41LSLRTVSLGAGAKDELHIVEAEAM65-NH2 | 4.52 | −1.9 | β-sheet |
| 69–83 | Ac-G69SPIKVTLATLKMSV83-NH2 | 14 | 2 | β-sheet |
| 84–93 | Ac-84QPTVSLGGFE93-NH2 | 0 | −1 | random |
| 107–120 | Ac-G107PVHISGQHLVAVE120-NH2 | 6.05 | −0.8 | β-sheet |
| 127–139 | Ac-D127EEEEDVKLLSIS139-NH2 | 3.27 | −5 | α-helix |
| 242–259 | Ac-S242SVEDIKAKMQASIEKGG259-NH2 | 7.36 | 0 | α-helix |
| Time (h) | Helix | Beta | Turn | Others | |
|---|---|---|---|---|---|
| NPM141–65 | 0 | 0.0 | 30.8 | 25.5 | 43.7 |
| 2 | 0.1 | 27.9 | 21.7 | 50.3 | |
| 21 | 0.6 | 28.2 | 21.0 | 50.3 | |
| NPM169–83 | 0 | 9.1 | 35.3 | 13.6 | 42.0 |
| 2 | 3.6 | 49.7 | 9.9 | 36.8 | |
| 4 | 0.0 | 34.9 | 18.5 | 46.7 | |
| 30 | 0.0 | 35.4 | 17.9 | 46.8 | |
| NPM184–93 | 0 | 7.0 | 27.3 | 22.9 | 42.7 |
| 2 | 5.7 | 25.6 | 20.4 | 48.2 | |
| 21 | 7.2 | 27.3 | 21.8 | 43.7 | |
| 24 | 7.0 | 24.3 | 19.9 | 48.8 | |
| NPM1107–120 | 0 | 5.8 | 25.4 | 19.7 | 49.1 |
| 2 | 3.9 | 27.9 | 19.1 | 49.1 | |
| 4 | 2.9 | 31.2 | 18.2 | 47.7 | |
| 28 | 2.5 | 34.3 | 17.2 | 45.9 | |
| 30 | 0.2 | 36.4 | 16.0 | 47.4 | |
| NPM1127–139 | 0 | 0.5 | 32.3 | 19.5 | 47.6 |
| 3 | 0.0 | 32.7 | 19.4 | 47.9 | |
| 4 | 0.0 | 33.2 | 19.4 | 47.4 | |
| 24 | 0.0 | 34.2 | 19.1 | 46.7 | |
| 28 | 0.0 | 34.6 | 18.8 | 46.6 | |
| NPM1242–259 | 0 | 11.5 | 24.1 | 16.8 | 47.6 |
| 4 | 9.0 | 25.5 | 17.3 | 48.2 | |
| 24 | 3.5 | 29.6 | 18.0 | 48.9 | |
| 30 | 0.1 | 35.3 | 16.9 | 47.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florio, D.; La Manna, S.; Di Natale, C.; Leone, M.; Mercurio, F.A.; Napolitano, F.; Malfitano, A.M.; Marasco, D. Insights into Network of Hot Spots of Aggregation in Nucleophosmin 1. Int. J. Mol. Sci. 2022, 23, 14704. https://doi.org/10.3390/ijms232314704
Florio D, La Manna S, Di Natale C, Leone M, Mercurio FA, Napolitano F, Malfitano AM, Marasco D. Insights into Network of Hot Spots of Aggregation in Nucleophosmin 1. International Journal of Molecular Sciences. 2022; 23(23):14704. https://doi.org/10.3390/ijms232314704
Chicago/Turabian StyleFlorio, Daniele, Sara La Manna, Concetta Di Natale, Marilisa Leone, Flavia Anna Mercurio, Fabiana Napolitano, Anna Maria Malfitano, and Daniela Marasco. 2022. "Insights into Network of Hot Spots of Aggregation in Nucleophosmin 1" International Journal of Molecular Sciences 23, no. 23: 14704. https://doi.org/10.3390/ijms232314704