
Heparanase Increases Podocyte Survival and Autophagic Flux after Adriamycin-Induced Injury
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
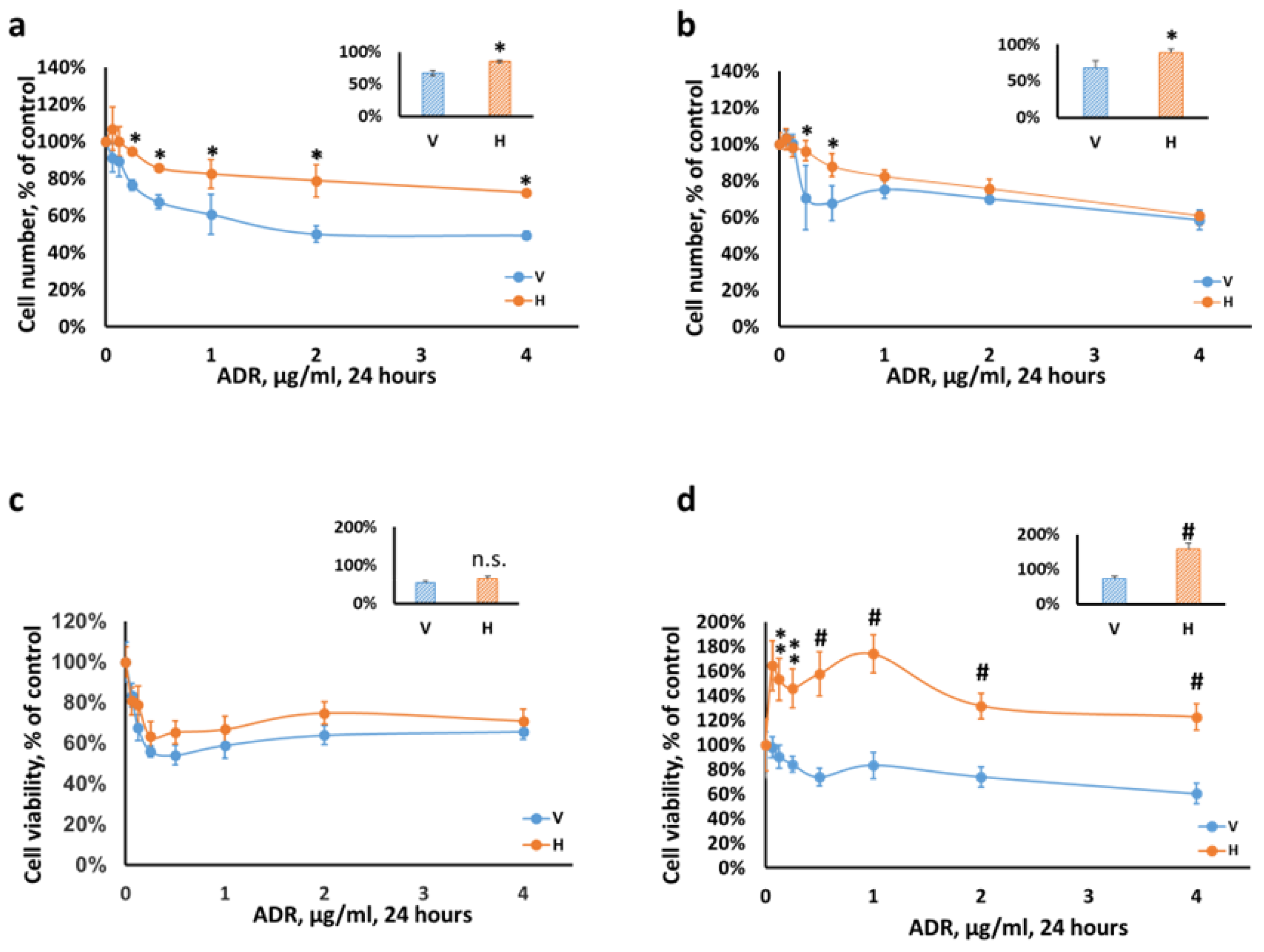
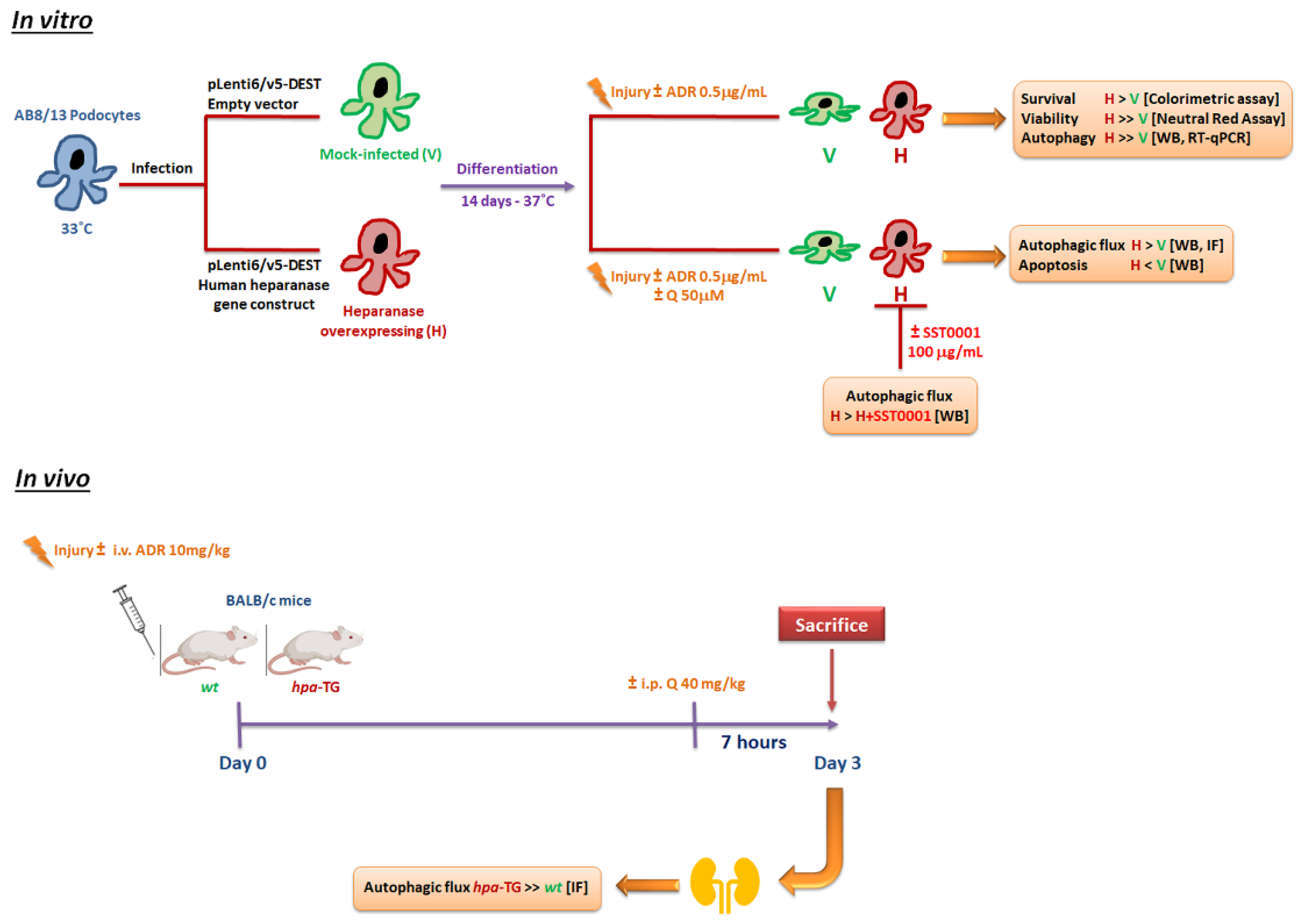
2.1. Podocytes Overexpressing Heparanase Are Resilient to Cellular Injury In Vitro
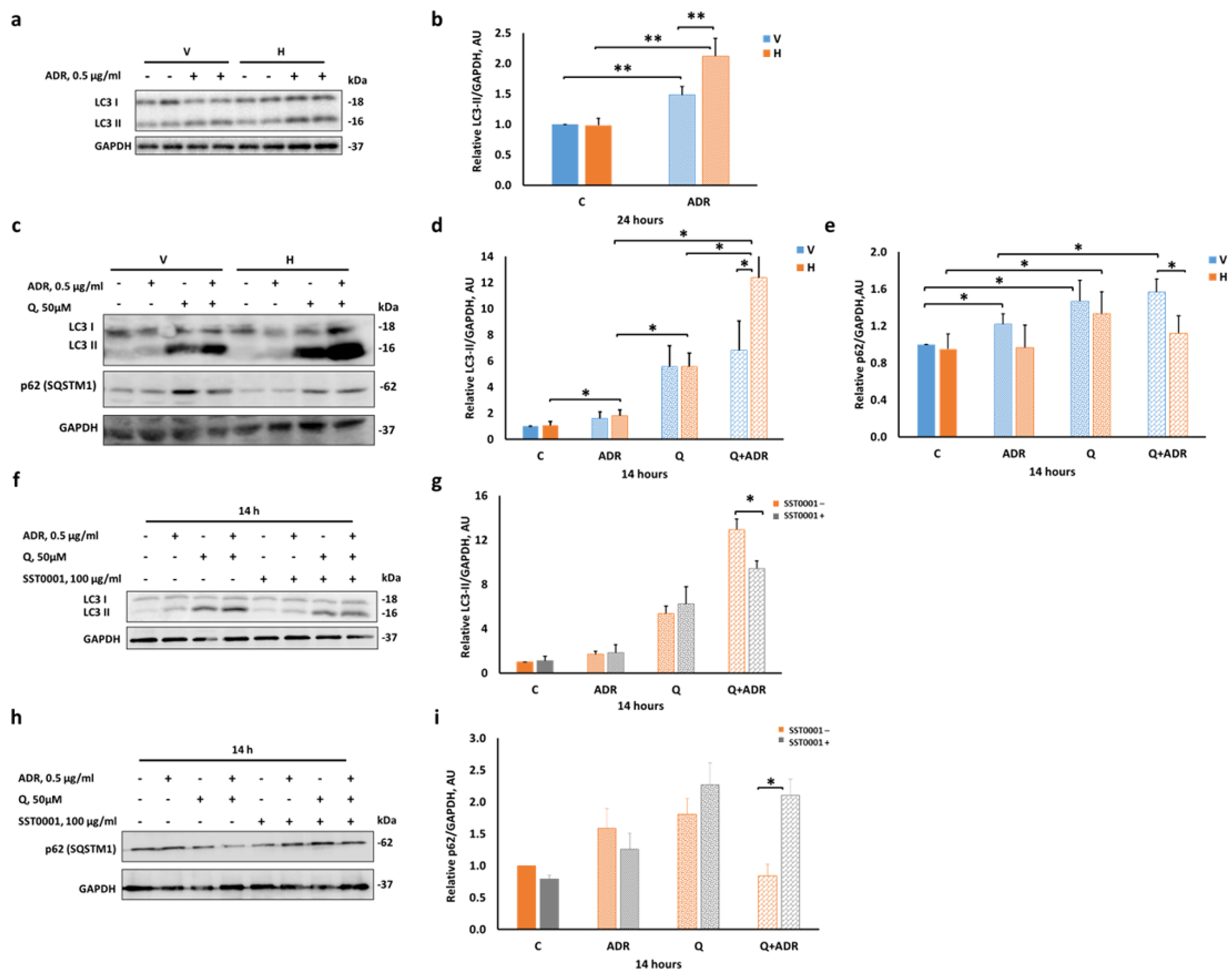
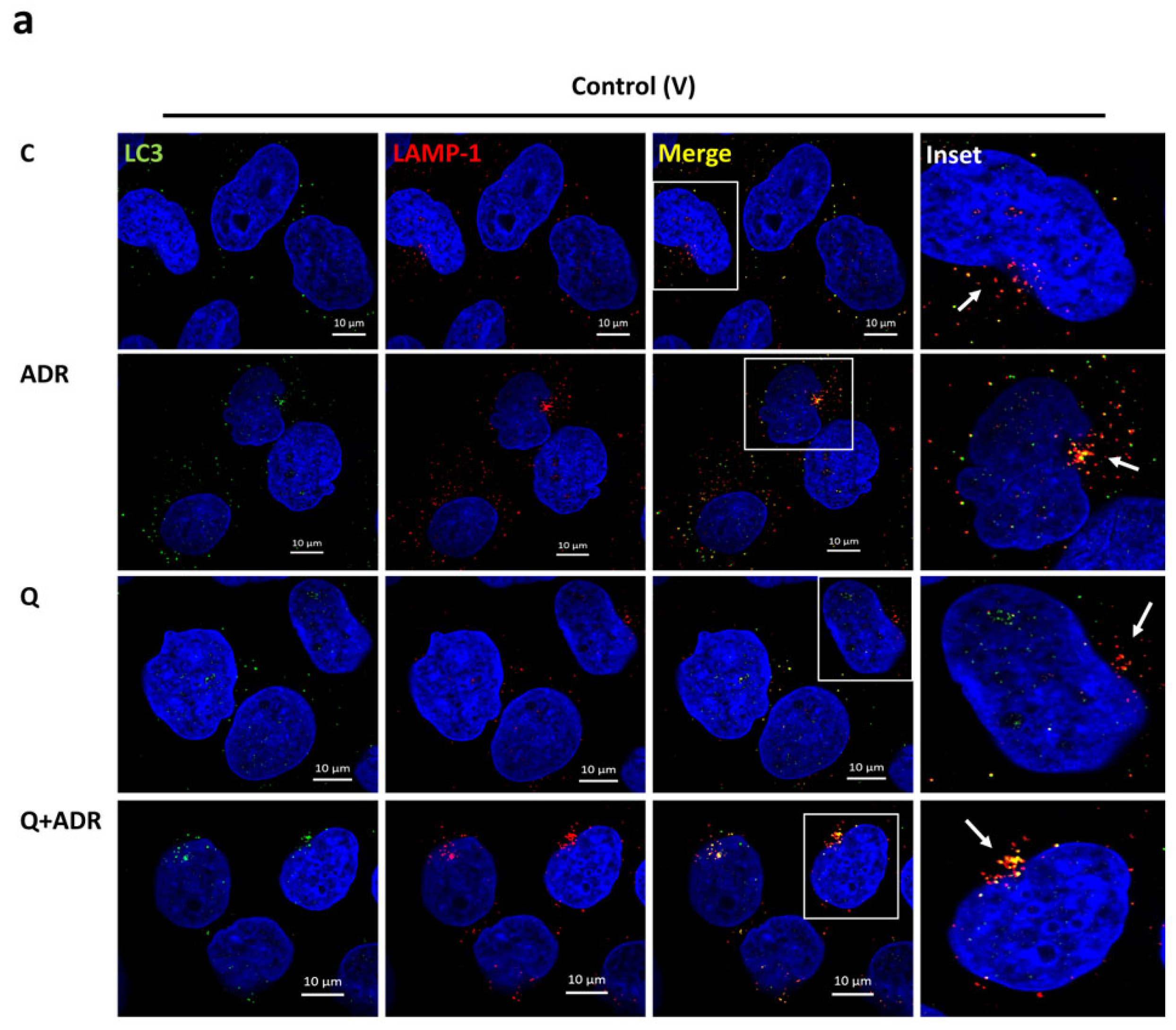
2.2. Heparanase Enhances Autophagic Flux following Podocyte Injury
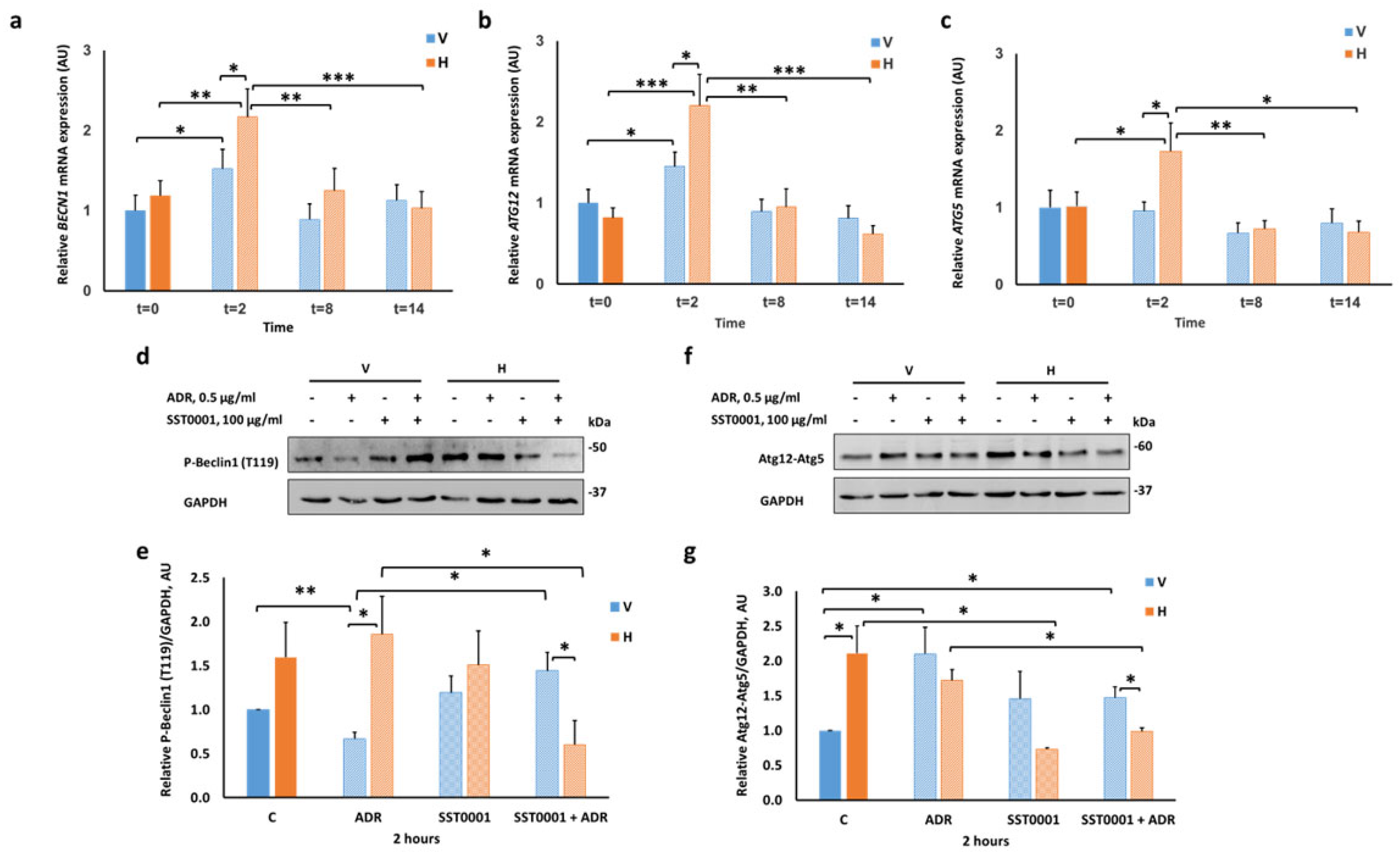
2.3. Heparanase Upregulates Autophagy Early after Induction of Injury
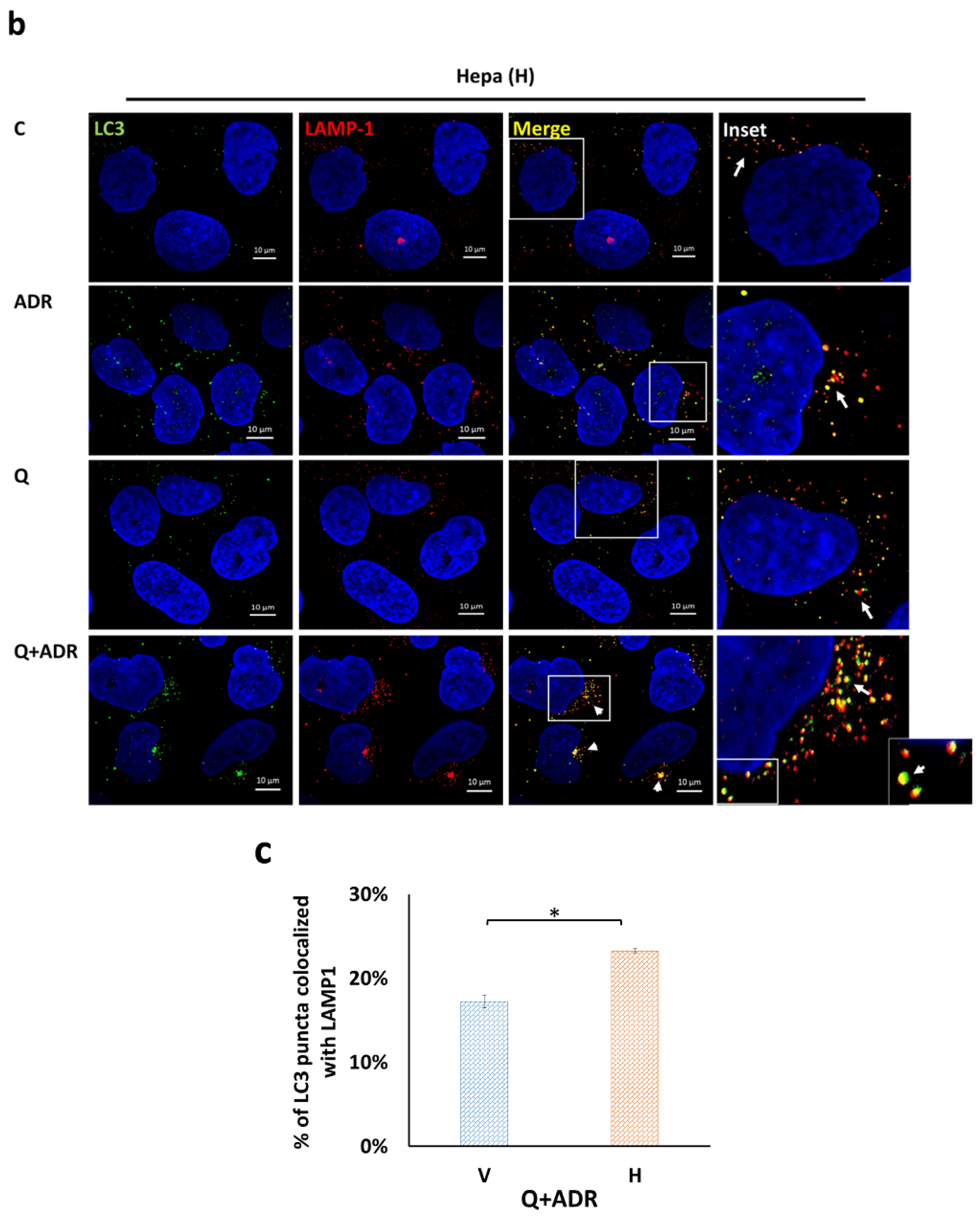
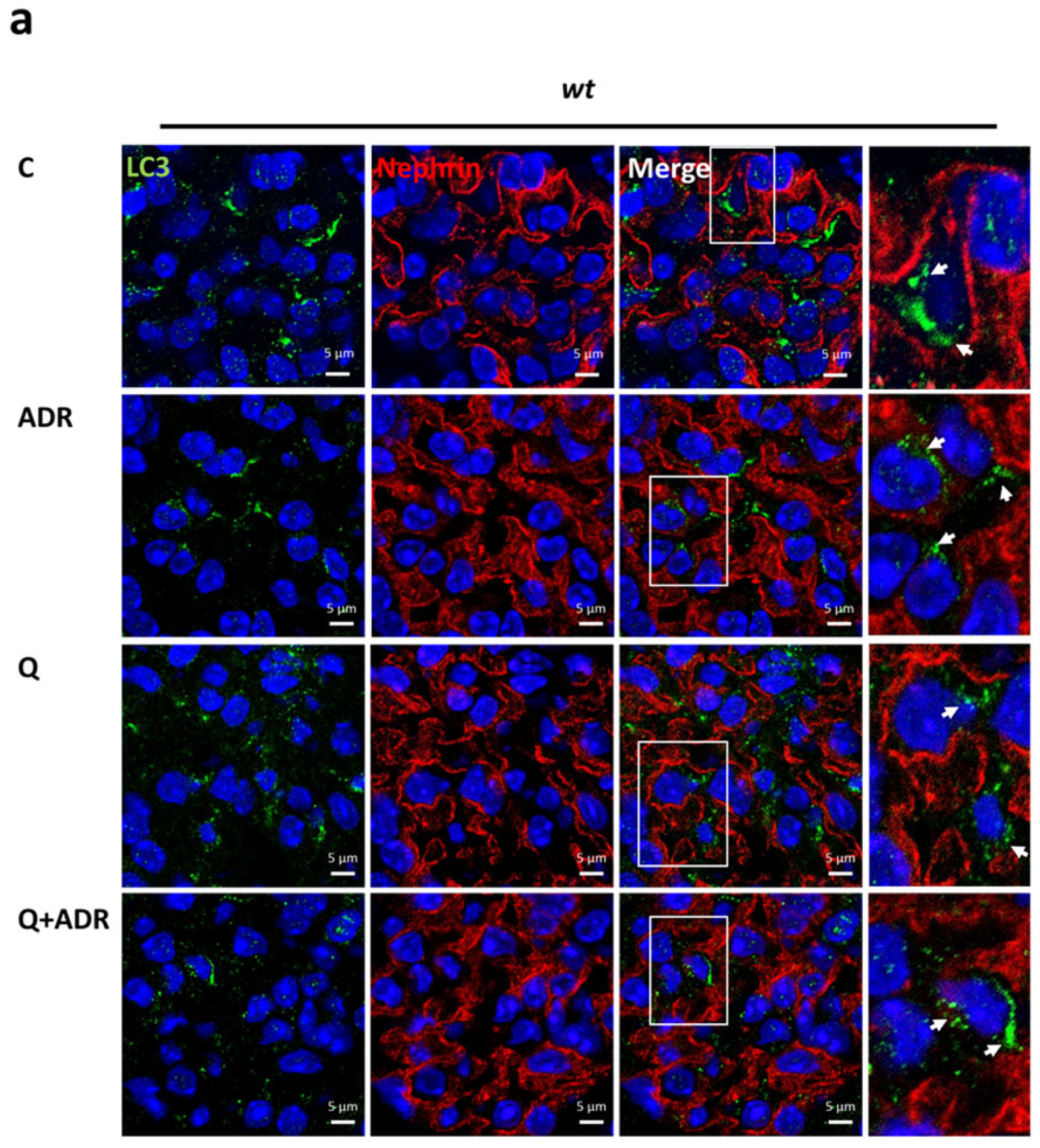
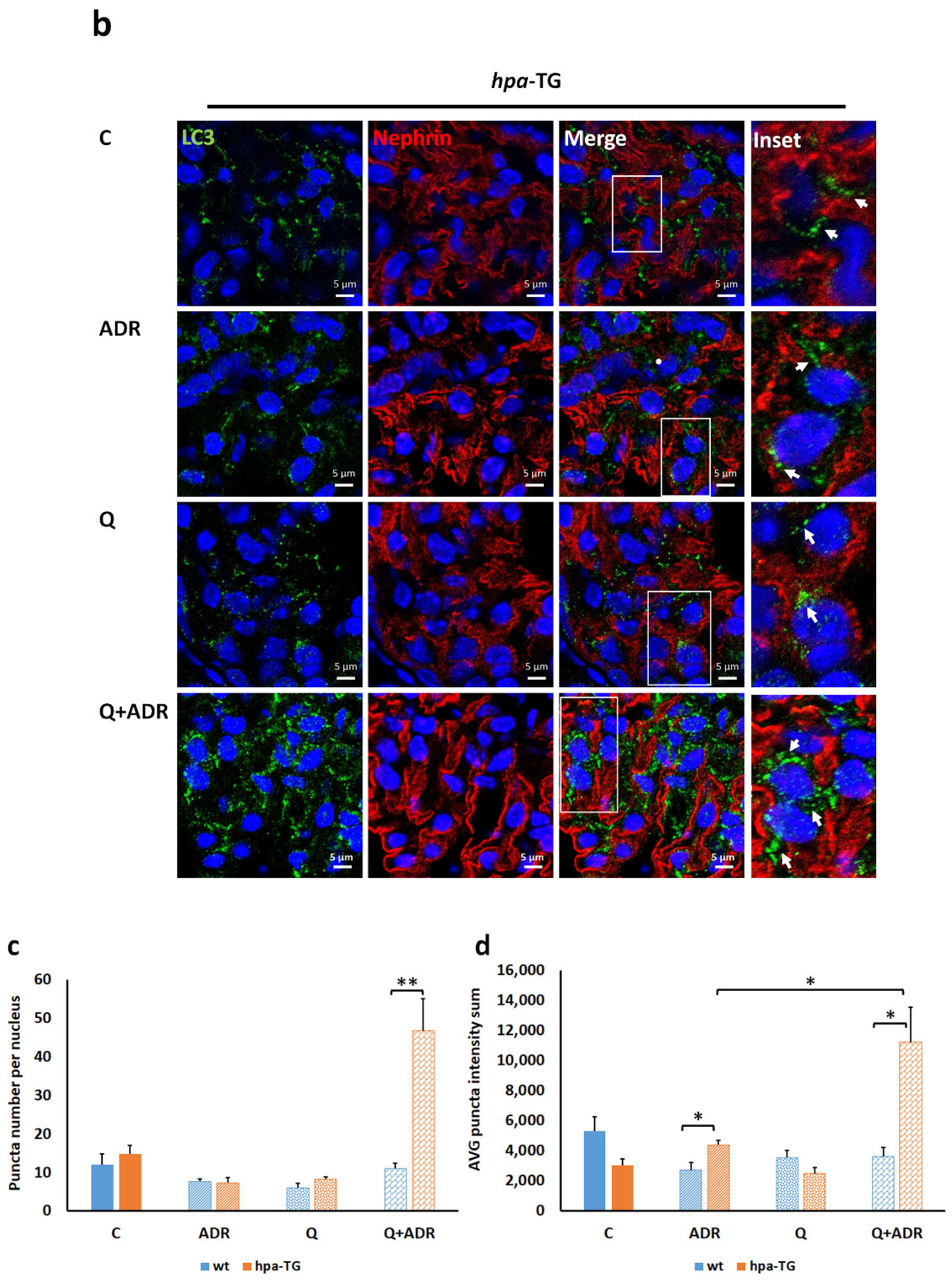
2.4. Heparanase Increases Autophagic Flux in Mice following Adriamycin-Induced Nephropathy
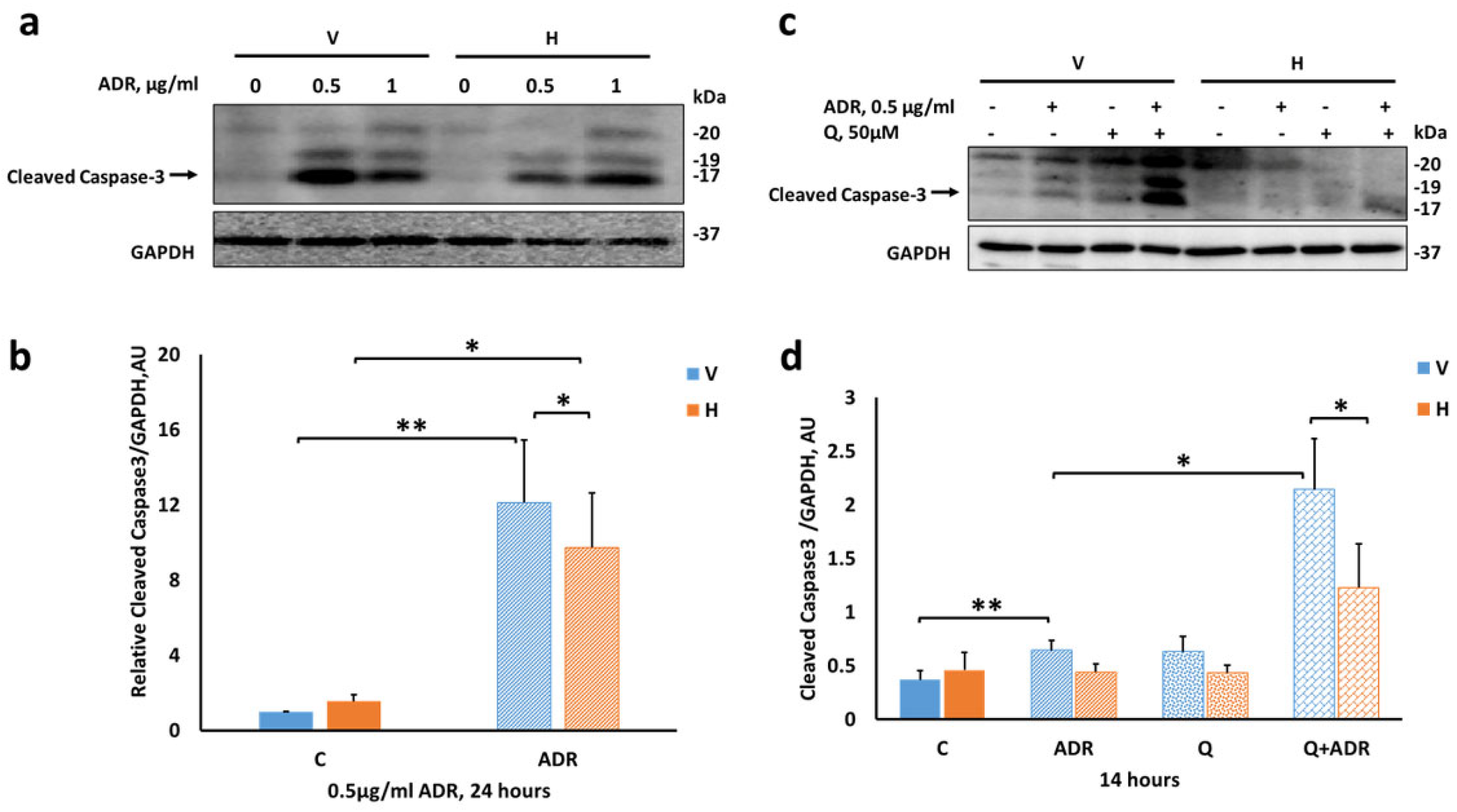
2.5. Heparanase Attenuates Apoptosis in Podocytes
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Heparanase Overexpression
4.2. Adriamycin-Induced Injury and Cell Survival
4.3. Cell Viability Assay
4.4. Experimental Animals
4.5. mRNA Expression Analysis
4.6. Western Blotting
4.7. Immunofluorescence
4.7.1. In Vivo
4.7.2. In Vitro
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hricik, D.E.; Chung-Park, M.; Sedor, J.R. Glomerulonephritis. N. Engl. J. Med. 1998, 339, 888–899. [Google Scholar] [CrossRef]
- Wiggins, R.C.; Alpers, C.E.; Holzman, L.B.; He, J.C.; Salant, D.J.; Chugh, S.S.; Natarajan, R.; Trachtman, H.; Brasile, L.; Star, R.A.; et al. Glomerular Disease: Looking beyond Pathology. Clin. J. Am. Soc. Nephrol. 2014, 9, 1138–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, H.; Yoshimura, A.; Kimata, K. The role of heparan sulfate in the glomerular basement membrane. Kidney Int. 2008, 73, 247–248. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Satchell, S.C.; Neal, C.R.; McKenzie, E.A.; Tooke, J.E.; Mathieson, P.W. Glomerular endothelial glycocalyx constitutes a barrier to protein permeability. J. Am. Soc. Nephrol. 2007, 18, 2885–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Wassenhove-McCarthy, D.; Yamaguchi, Y.; Holzman, L.; van Kuppevelt, T.H.; Orr, A.W.; Funk, S.; Woods, A.; McCarthy, K. Podocytes require the engagement of cell surface heparan sulfate proteoglycans for adhesion to extracellular matrices. Kidney Int. 2010, 78, 1088–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulett, M.D.; Freeman, C.; Hamdorf, B.J.; Baker, R.T.; Harris, M.J.; Parish, C.R. Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nat. Med. 1999, 5, 803–809. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Friedmann, Y.; Elkin, M.; Aingorn, H.; Atzmon, R.; Ishai-Michaeli, R.; Bitan, M.; Pappo, O.; Peretz, T.; Michal, I.; et al. Mammalian heparanase: Gene cloning, expression and function in tumor progression and metastasis. Nat. Med. 1999, 5, 793–802. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Ilan, N.; Naggi, A.; Casu, B. Heparanase: Structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Curr. Pharm. Des. 2007, 13, 2057–2073. [Google Scholar] [CrossRef]
- Shu, J.; Santulli, G. Heparanase in health and disease: The neglected housekeeper of the cell? Atherosclerosis 2019, 283, 124–126. [Google Scholar] [CrossRef]
- Rabelink, T.J.; van den Berg, B.M.; Garsen, M.; Wang, G.; Elkin, M.; van der Vlag, J. Heparanase: Roles in cell survival, extracellular matrix remodelling and the development of kidney disease. Nat. Rev. Nephrol. 2017, 13, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Garsen, M.; Rops, A.L.; Rabelink, T.J.; Berden, J.H.; van der Vlag, J. The role of heparanase and the endothelial glycocalyx in the development of proteinuria. Nephrol. Dial. Transplant. 2014, 29, 49–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, S.J.; Miner, J.H. Breaking down the barrier: Evidence against a role for heparan sulfate in glomerular permselectivity. J. Am. Soc. Nephrol. 2007, 18, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Rops, A.L.; van den Hoven, M.J.; Veldman, B.A.; Salemink, S.; Vervoort, G.; Elving, L.D.; Aten, J.; Wetzels, J.F.; van der Vlag, J.; Berden, J.H. Urinary heparanase activity in patients with Type 1 and Type 2 diabetes. Nephrol. Dial. Transplant. 2012, 27, 2853–2861. [Google Scholar] [CrossRef] [Green Version]
- Shafat, I.; Ilan, N.; Zoabi, S.; Vlodavsky, I.; Nakhoul, F. Heparanase levels are elevated in the urine and plasma of type 2 diabetes patients and associate with blood glucose levels. PLoS ONE 2011, 6, e17312. [Google Scholar] [CrossRef] [Green Version]
- Shafat, I.; Agbaria, A.; Boaz, M.; Schwartz, D.; Baruch, R.; Nakash, R.; Ilan, N.; Vlodavsky, I.; Weinstein, T. Elevated urine heparanase levels are associated with proteinuria and decreased renal allograft function. PLoS ONE 2012, 7, e44076. [Google Scholar] [CrossRef]
- Szymczak, M.; Kuzniar, J.; Klinger, M. The role of heparanase in diseases of the glomeruli. Arch. Immunol. Ther. Exp. 2010, 58, 45–56. [Google Scholar] [CrossRef]
- Levidiotis, V.; Freeman, C.; Tikellis, C.; Cooper, M.E.; Power, D.A. Heparanase is involved in the pathogenesis of proteinuria as a result of glomerulonephritis. J. Am. Soc. Nephrol. 2004, 15, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Levidiotis, V.; Kanellis, J.; Ierino, F.L.; Power, D.A. Increased expression of heparanase in puromycin aminonucleoside nephrosis. Kidney Int. 2001, 60, 1287–1296. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.; van den Hoven, M.; Rops, A.; Wijnhoven, T.; van den Heuvel, L.; Lensen, J.; van Kuppevelt, T.; van Goor, H.; van der Vlag, J.; Navis, G.; et al. Induction of glomerular heparanase expression in rats with adriamycin nephropathy is regulated by reactive oxygen species and the renin-angiotensin system. J. Am. Soc. Nephrol. 2006, 17, 2513–2520. [Google Scholar] [CrossRef]
- van den Hoven, M.J.; Waanders, F.; Rops, A.L.; Kramer, A.B.; van Goor, H.; Berden, J.H.; Navis, G.; van der Vlag, J. Regulation of glomerular heparanase expression by aldosterone, angiotensin II and reactive oxygen species. Nephrol. Dial. Transplant. 2009, 24, 2637–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levidiotis, V.; Freeman, C.; Tikellis, C.; Cooper, M.E.; Power, D.A. Heparanase inhibition reduces proteinuria in a model of accelerated anti-glomerular basement membrane antibody disease. Nephrology 2005, 10, 167–173. [Google Scholar] [CrossRef]
- van den Hoven, M.J.; Rops, A.L.; Bakker, M.A.; Aten, J.; Rutjes, N.; Roestenberg, P.; Goldschmeding, R.; Zcharia, E.; Vlodavsky, I.; van der Vlag, J.; et al. Increased expression of heparanase in overt diabetic nephropathy. Kidney Int. 2006, 70, 2100–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garsen, M.; Lenoir, O.; Rops, A.L.; Dijkman, H.B.; Willemsen, B.; van Kuppevelt, T.H.; Rabelink, T.J.; Berden, J.H.; Tharaux, P.L.; van der Vlag, J. Endothelin-1 Induces Proteinuria by Heparanase-Mediated Disruption of the Glomerular Glycocalyx. J. Am. Soc. Nephrol. 2016, 27, 3545–3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, Y.P.; Tay, Y.C.; Harris, D.C. Progressive adriamycin nephropathy in mice: Sequence of histologic and immunohistochemical events. Kidney Int. 2000, 58, 1797–1804. [Google Scholar] [CrossRef] [Green Version]
- Garsen, M.; Benner, M.; Dijkman, H.B.; van Kuppevelt, T.H.; Li, J.P.; Rabelink, T.J.; Vlodavsky, I.; Berden, J.H.; Rops, A.L.; Elkin, M.; et al. Heparanase Is Essential for the Development of Acute Experimental Glomerulonephritis. Am. J. Pathol. 2016, 186, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, R.; Rubinstein, A.M.; Gil, N.; Hermano, E.; Li, J.P.; van der Vlag, J.; Atzmon, R.; Meirovitz, A.; Elkin, M. Role of heparanase-driven inflammatory cascade in pathogenesis of diabetic nephropathy. Diabetes 2014, 63, 4302–4313. [Google Scholar] [CrossRef] [Green Version]
- Assady, S.; Alter, J.; Axelman, E.; Zohar, Y.; Sabo, E.; Litvak, M.; Kaplan, M.; Ilan, N.; Vlodavsky, I.; Abassi, Z. Nephroprotective effect of heparanase in experimental nephrotic syndrome. PLoS ONE 2015, 10, e0119610. [Google Scholar] [CrossRef]
- Assady, S.; Benzing, T.; Kretzler, M.; Skorecki, K.L. Glomerular podocytes in kidney health and disease. Lancet 2019, 393, 856–858. [Google Scholar] [CrossRef]
- Saleem, M.A. One hundred ways to kill a podocyte. Nephrol. Dial. Transplant. 2015, 30, 1266–1271. [Google Scholar] [CrossRef]
- Ni, L.; Saleem, M.; Mathieson, P.W. Podocyte culture: Tricks of the trade. Nephrology 2012, 17, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Hartleben, B.; Godel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Kobler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartleben, B.; Wanner, N.; Huber, T.B. Autophagy in glomerular health and disease. Semin. Nephrol. 2014, 34, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Zcharia, E.; Jia, J.; Zhang, X.; Baraz, L.; Lindahl, U.; Peretz, T.; Vlodavsky, I.; Li, J.P. Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. PLoS ONE 2009, 4, e5181. [Google Scholar] [CrossRef] [Green Version]
- van den Hoven, M.J.; Wijnhoven, T.J.; Li, J.P.; Zcharia, E.; Dijkman, H.B.; Wismans, R.G.; Rops, A.L.; Lensen, J.F.; van den Heuvel, L.P.; van Kuppevelt, T.H.; et al. Reduction of anionic sites in the glomerular basement membrane by heparanase does not lead to proteinuria. Kidney Int. 2008, 73, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Zcharia, E.; Metzger, S.; Chajek-Shaul, T.; Aingorn, H.; Elkin, M.; Friedmann, Y.; Weinstein, T.; Li, J.P.; Lindahl, U.; Vlodavsky, I. Transgenic expression of mammalian heparanase uncovers physiological functions of heparan sulfate in tissue morphogenesis, vascularization, and feeding behavior. FASEB J. 2004, 18, 252–263. [Google Scholar] [CrossRef]
- Finer, G.; Schnaper, H.W.; Kanwar, Y.S.; Liang, X.; Lin, H.Y.; Hayashida, T. Divergent roles of Smad3 and PI3-kinase in murine adriamycin nephropathy indicate distinct mechanisms of proteinuria and fibrogenesis. Kidney Int. 2012, 82, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.W.; Harris, D.C. Adriamycin nephropathy: A model of focal segmental glomerulosclerosis. Nephrology 2011, 16, 30–38. [Google Scholar] [CrossRef]
- Jeansson, M.; Bjorck, K.; Tenstad, O.; Haraldsson, B. Adriamycin alters glomerular endothelium to induce proteinuria. J. Am. Soc. Nephrol. 2009, 20, 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giroux, L.; Smeesters, C.; Boury, F.; Faure, M.P.; Jean, G. Adriamycin and adriamycin-DNA nephrotoxicity in rats. Lab. Invest. 1984, 50, 190–196. [Google Scholar] [PubMed]
- Mallipattu, S.K.; Horne, S.J.; D’Agati, V.; Narla, G.; Liu, R.; Frohman, M.A.; Dickman, K.; Chen, E.Y.; Ma’ayan, A.; Bialkowska, A.B.; et al. Kruppel-like factor 6 regulates mitochondrial function in the kidney. J. Clin. Invest. 2015, 125, 1347–1361. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Xuan, X.; Che, R.; Ding, G.; Zhao, M.; Bai, M.; Jia, Z.; Huang, S.; Zhang, A. Dysfunction of the PGC-1alpha-mitochondria axis confers adriamycin-induced podocyte injury. Am. J. Physiol. Renal Physiol. 2014, 306, F1410–F1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boya, P. Lysosomal function and dysfunction: Mechanism and disease. Antioxid. Redox Signal. 2012, 17, 766–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Banu, K.; Ni, Z.; Leventhal, J.S.; Menon, M.C. Podocyte Autophagy in Homeostasis and Disease. J. Clin. Med. 2021, 10, 1184. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 489–508. [Google Scholar] [CrossRef]
- Liu, W.J.; Gan, Y.; Huang, W.F.; Wu, H.-l.; Zhang, X.-q.; Zheng, H.J.; Liu, H.-f. Lysosome restoration to activate podocyte autophagy: A new therapeutic strategy for diabetic kidney disease. Cell Death Dis. 2019, 10, 806. [Google Scholar] [CrossRef] [Green Version]
- Bork, T.; Liang, W.; Yamahara, K.; Lee, P.; Tian, Z.; Liu, S.; Schell, C.; Thedieck, K.; Hartleben, B.; Patel, K.; et al. Podocytes maintain high basal levels of autophagy independent of mtor signaling. Autophagy 2020, 16, 1932–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Zhang, L.; Liu, Y.; Livingston, M.J.; Chen, J.K.; Nahman, N.S., Jr.; Liu, F.; Dong, Z. Autophagy is activated to protect against podocyte injury in adriamycin-induced nephropathy. Am. J. Physiol. Renal Physiol. 2017, 313, F74–F84. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetser, A.; Levy-Adam, F.; Kaplan, V.; Gingis-Velitski, S.; Bashenko, Y.; Schubert, S.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Processing and activation of latent heparanase occurs in lysosomes. J. Cell Sci. 2004, 117, 2249–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetser, A.; Bashenko, Y.; Edovitsky, E.; Levy-Adam, F.; Vlodavsky, I.; Ilan, N. Heparanase induces vascular endothelial growth factor expression: Correlation with p38 phosphorylation levels and Src activation. Cancer Res. 2006, 66, 1455–1463. [Google Scholar] [CrossRef] [Green Version]
- Koganti, R.; Suryawanshi, R.; Shukla, D. Heparanase, cell signaling, and viral infections. Cell Mol. Life Sci. 2020, 77, 5059–5077. [Google Scholar] [CrossRef]
- He, Y.Q.; Sutcliffe, E.L.; Bunting, K.L.; Li, J.; Goodall, K.J.; Poon, I.K.; Hulett, M.D.; Freeman, C.; Zafar, A.; McInnes, R.L.; et al. The endoglycosidase heparanase enters the nucleus of T lymphocytes and modulates H3 methylation at actively transcribed genes via the interplay with key chromatin modifying enzymes. Transcription 2012, 3, 130–145. [Google Scholar] [CrossRef] [Green Version]
- Nadir, Y.; Brenner, B.; Zetser, A.; Ilan, N.; Shafat, I.; Zcharia, E.; Goldshmidt, O.; Vlodavsky, I. Heparanase induces tissue factor expression in vascular endothelial and cancer cells. J. Thromb. Haemost. 2006, 4, 2443–2451. [Google Scholar] [CrossRef]
- Gingis-Velitski, S.; Zetser, A.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J. Biol. Chem. 2004, 279, 23536–23541. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Kaplan, V.; Doweck, I.; Naroditsky, I.; Vlodavsky, I.; Ilan, N. Heparanase augments epidermal growth factor receptor phosphorylation: Correlation with head and neck tumor progression. Cancer Res. 2008, 68, 10077–10085. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Tang, B.; Wang, S.; Tang, L.; Wen, D.; Vlodavsky, I.; Yang, S.M. Non-enzymatic heparanase enhances gastric tumor proliferation via TFEB-dependent autophagy. Oncogenesis 2022, 11, 49. [Google Scholar] [CrossRef]
- Gurkar, A.U.; Chu, K.; Raj, L.; Bouley, R.; Lee, S.H.; Kim, Y.B.; Dunn, S.E.; Mandinova, A.; Lee, S.W. Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat. Commun. 2013, 4, 2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalckvar, E.; Berissi, H.; Eisenstein, M.; Kimchi, A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 2009, 5, 720–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.F.; Gu, C.J.; Wang, Q.; Xu, L.; Chen, J.; Zhou, W.; Zhou, Z.; Zhao, S.J.; Li, L.W.; Kong, F.Q.; et al. The protective effort of GPCR kinase 2-interacting protein-1 in neurons via promoting Beclin1-Parkin induced mitophagy at the early stage of spinal cord ischemia-reperfusion injury. FASEB J. 2020, 34, 2055–2074. [Google Scholar] [CrossRef] [Green Version]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A. Apoptosis and autophagy: Regulatory connections between two supposedly different processes. Apoptosis 2008, 13, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Pulinilkunnil, T.; Flibotte, S.; Nislow, C.; Vlodavsky, I.; Hussein, B.; Rodrigues, B. Heparanase protects the heart against chemical or ischemia/reperfusion injury. J. Mol. Cell Cardiol. 2019, 131, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, F.; Lal, N.; Chiu, A.P.; Wan, A.; Jia, J.; Bierende, D.; Flibotte, S.; Sinha, S.; Asadi, A.; et al. Heparanase Overexpression Induces Glucagon Resistance and Protects Animals From Chemically Induced Diabetes. Diabetes 2017, 66, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Vlodavsky, I.; Goldshmidt, O.; Zcharia, E.; Atzmon, R.; Rangini-Guatta, Z.; Elkin, M.; Peretz, T.; Friedmann, Y. Mammalian heparanase: Involvement in cancer metastasis, angiogenesis and normal development. Semin. Cancer Biol. 2002, 12, 121–129. [Google Scholar] [CrossRef]
- Oliver, M.H.; Harrison, N.K.; Bishop, J.E.; Cole, P.J.; Laurent, G.J. A rapid and convenient assay for counting cells cultured in microwell plates: Application for assessment of growth factors. J. Cell Sci. 1989, 92 Pt 3, 513–518. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu-Tayeh Suleiman, H.; Said, S.; Ali Saleh, H.; Gamliel-Lazarovich, A.; Haddad, E.; Minkov, I.; Zohar, Y.; Ilan, N.; Vlodavsky, I.; Abassi, Z.; et al. Heparanase Increases Podocyte Survival and Autophagic Flux after Adriamycin-Induced Injury. Int. J. Mol. Sci. 2022, 23, 12691. https://doi.org/10.3390/ijms232012691
Abu-Tayeh Suleiman H, Said S, Ali Saleh H, Gamliel-Lazarovich A, Haddad E, Minkov I, Zohar Y, Ilan N, Vlodavsky I, Abassi Z, et al. Heparanase Increases Podocyte Survival and Autophagic Flux after Adriamycin-Induced Injury. International Journal of Molecular Sciences. 2022; 23(20):12691. https://doi.org/10.3390/ijms232012691
Chicago/Turabian StyleAbu-Tayeh Suleiman, Hanan, Shereen Said, Haya Ali Saleh, Aviva Gamliel-Lazarovich, Eyas Haddad, Irina Minkov, Yaniv Zohar, Neta Ilan, Israel Vlodavsky, Zaid Abassi, and et al. 2022. "Heparanase Increases Podocyte Survival and Autophagic Flux after Adriamycin-Induced Injury" International Journal of Molecular Sciences 23, no. 20: 12691. https://doi.org/10.3390/ijms232012691