
Dynamic Epigenetic Changes during a Relapse and Recovery Cycle in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome
 , and
, and
Abstract
:1. Introduction
2. Results
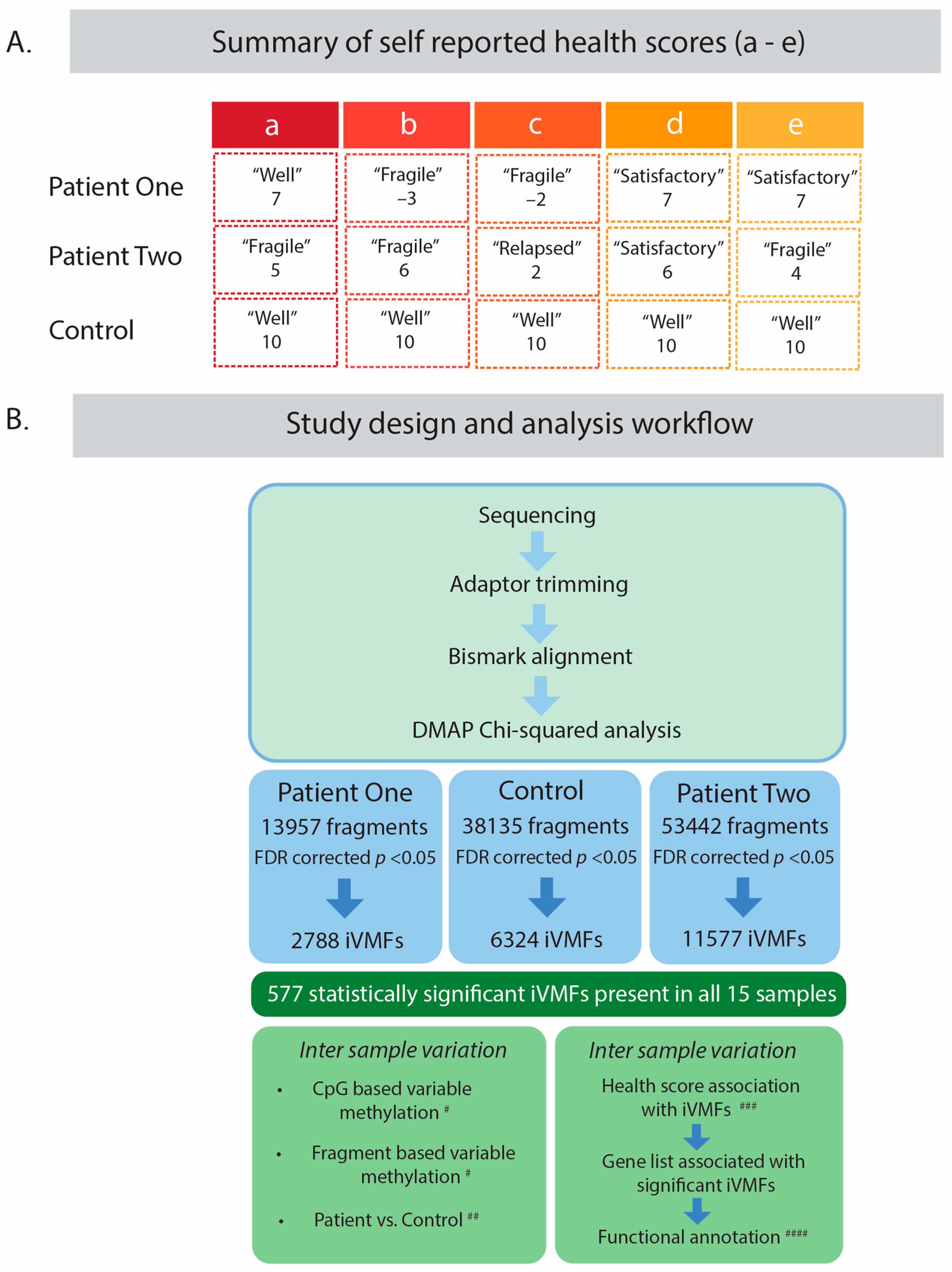
2.1. Study Design and Participants
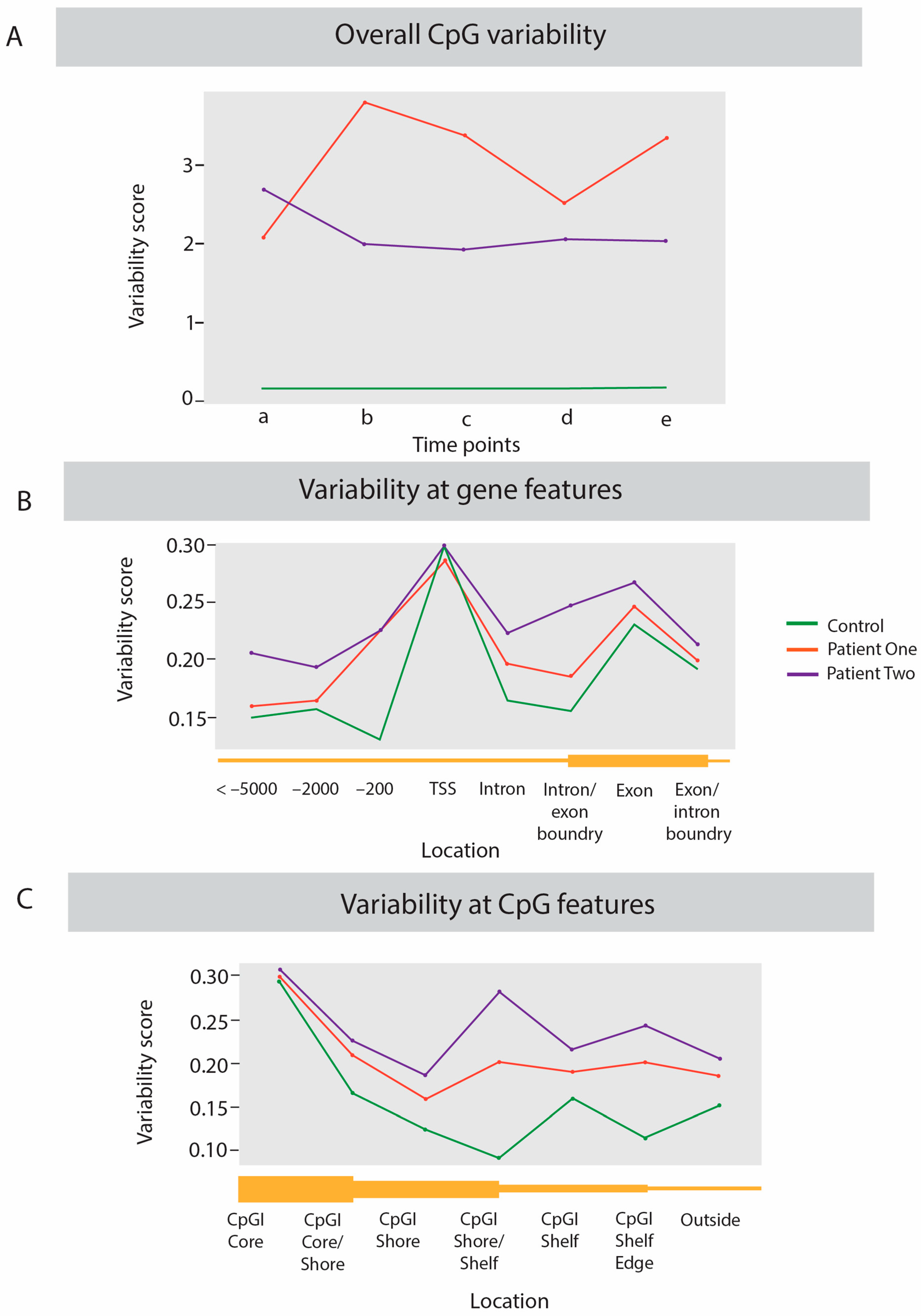
2.2. Dynamic Analysis of DNA Methylation Variation
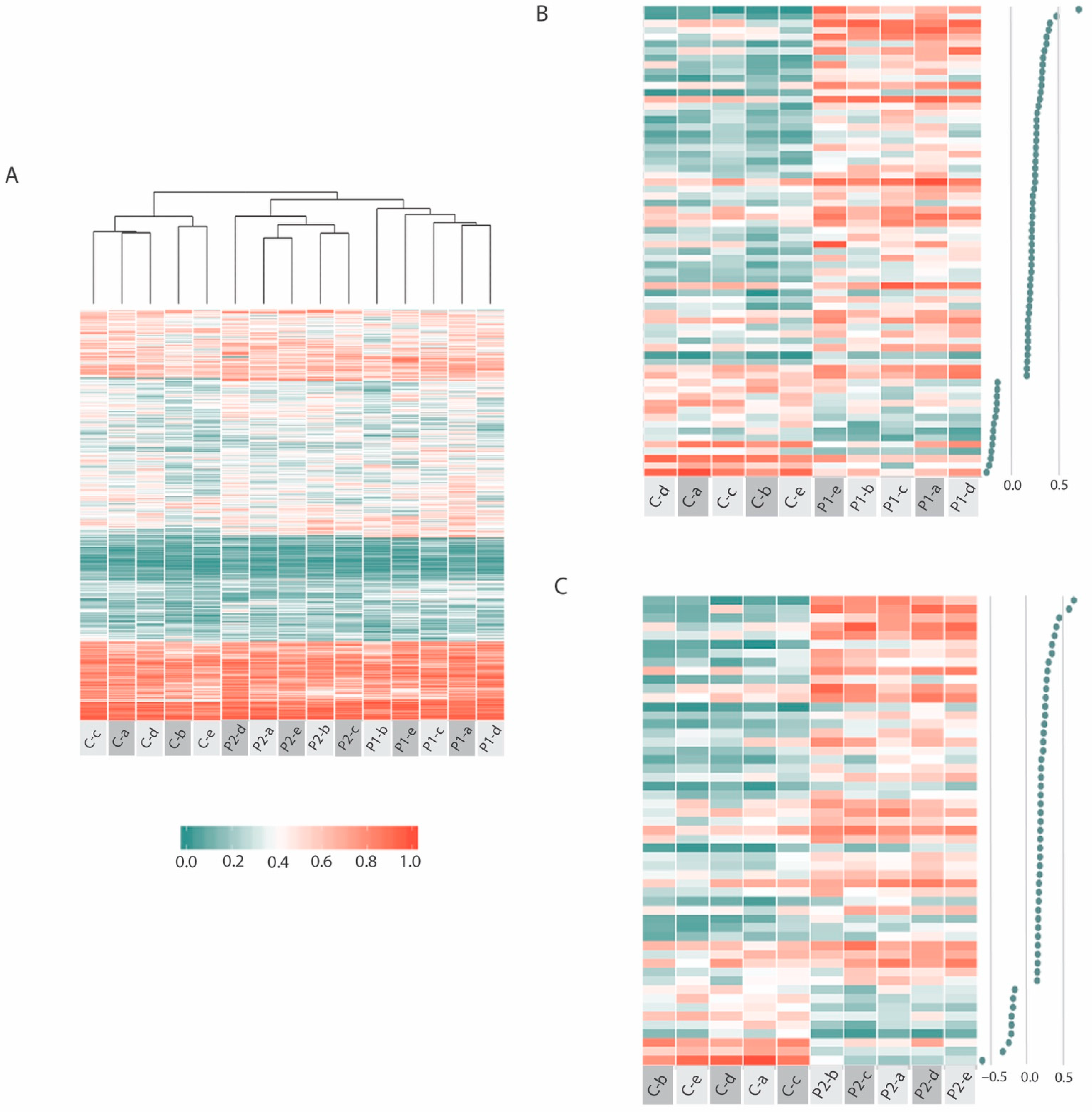
2.3. Common ME-iVMFs Methylation Patterns in Patients
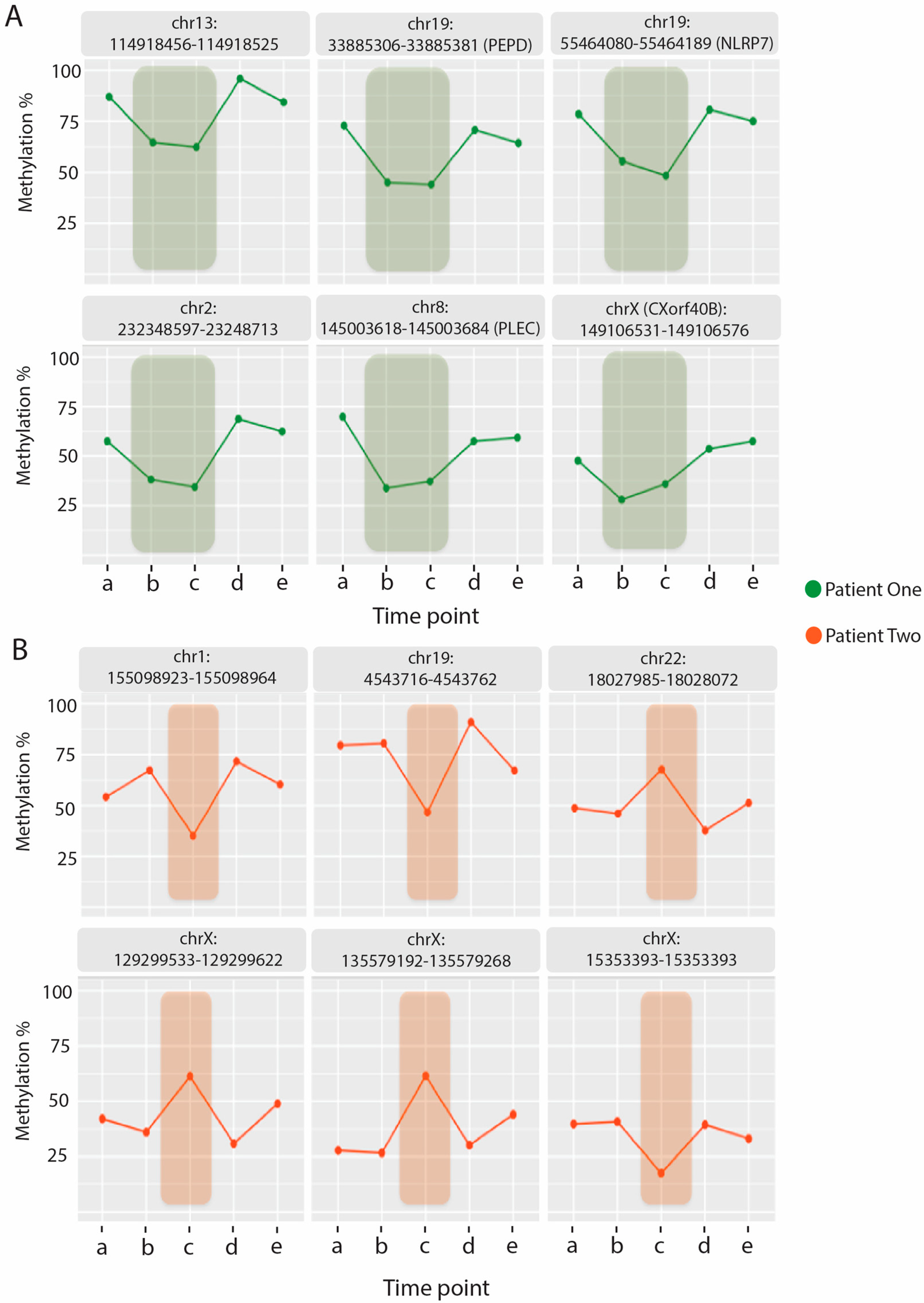
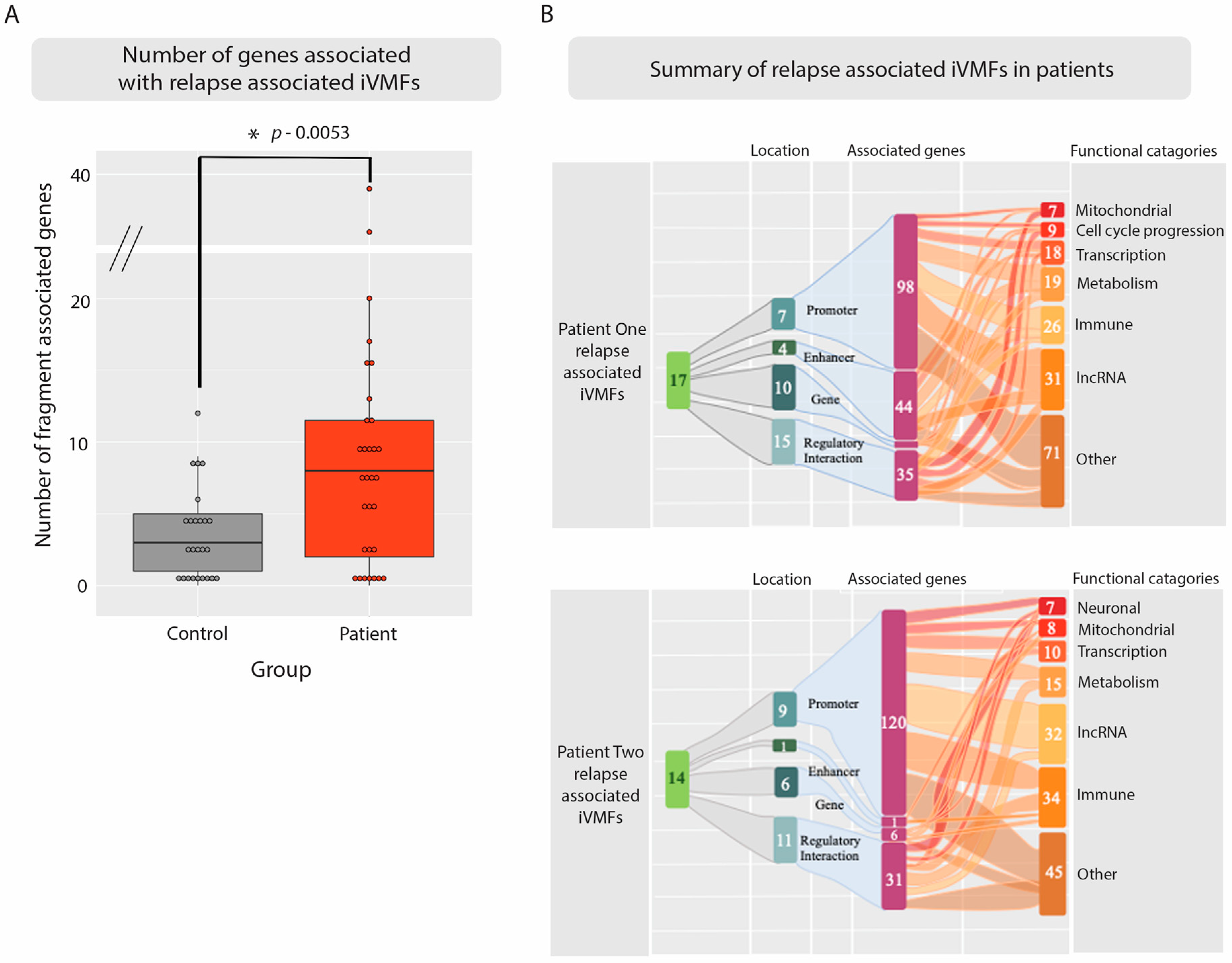
2.4. Identifying Methylation Pattern Associated with the Relapse Condition
2.5. Relapse Associated Methylation Signature Exhibits Striking Variation Compared to Control
2.6. Simulated Relapses for the Control Subject Identified Fewer Variable Methylated Genes Than the Patients
3. Discussion
3.1. Benefits of DNA Methylation for a Precision Investigation of ME/CFS
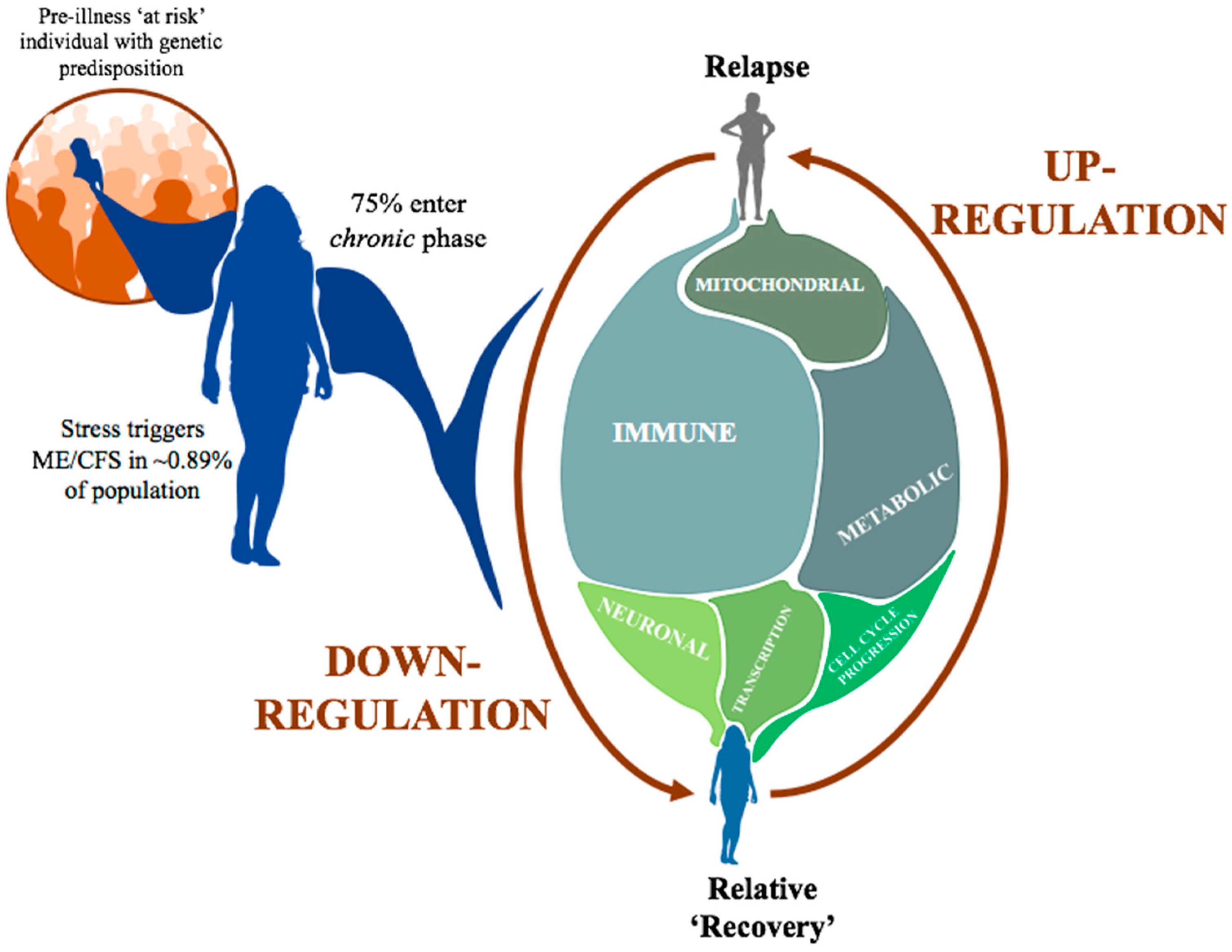
3.2. Inter-Individual Differences Indicate Increased Epigenetic Variation Linked to Disease Severity
3.3. Intra-Individual Variation Identifies Regulatory Regions
3.4. Immune and Inflammatory Changes Implicated in Relapse-Recovery Cycle
4. Materials and Methods
4.1. Cohort Recruitment
4.2. PBMC Isolation
4.3. DNA Extraction
4.4. Generating Methylation Map Using RRBS
4.5. High-Throughput Sequencing
4.6. DNA Methylation and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shepherd, C. ME/CFS/PVFS: An Exploration of the Key Clinical Issues; ME Association: Peterborough, UK, 2013; 52p. [Google Scholar]
- Lim, E.J.; Ahn, Y.C.; Jang, E.S.; Lee, S.W.; Lee, S.H.; Son, C.G. Systematic review and meta-analysis of the prevalence of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). J. Transl. Med. 2020, 18, 100. [Google Scholar] [CrossRef]
- Valdez, A.R.; Hancock, E.E.; Adebayo, S.; Kiernicki, D.J.; Proskauer, D.; Attewell, J.R.; Bateman, L.; DeMaria, A., Jr.; Lapp, C.W.; Rowe, P.C.; et al. Estimating Prevalence, Demographics, and Costs of ME/CFS Using Large Scale Medical Claims Data and Machine Learning. Front. Pediatr. 2018, 6, 412. [Google Scholar] [CrossRef] [Green Version]
- Chu, L.; Valencia, I.J.; Garvert, D.W.; Montoya, J.G. Onset Patterns and Course of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Pediatr. 2019, 7, 12. [Google Scholar] [CrossRef]
- Naviaux, R.K.; Naviaux, J.C.; Li, K.; Bright, A.T.; Alaynick, W.A.; Wang, L.; Baxter, A.; Nathan, N.; Anderson, W.; Gordon, E. Metabolic features of chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, E5472–E5480. [Google Scholar] [CrossRef] [Green Version]
- Missailidis, D.; Annesley, S.J.; Allan, C.Y.; Sanislav, O.; Lidbury, B.A.; Lewis, D.P.; Fisher, P.R. An isolated Complex V inefficiency and dysregulated mitochondrial function in immortalized lymphocytes from ME/CFS patients. Int. J. Mol. Sci. 2020, 21, 1074. [Google Scholar] [CrossRef] [Green Version]
- Sweetman, E.; Ryan, M.; Edgar, C.; MacKay, A.; Vallings, R.; Tate, W. Changes in the transcriptome of circulating immune cells of a New Zealand cohort with myalgic encephalomyelitis/chronic fatigue syndrome. Int. J. Immunopathol. Pharmacol. 2019, 33, 205873841882040. [Google Scholar] [CrossRef]
- Mackay, A.; Tate, W.P. A compromised paraventricular nucleus within a dysfunctional hypothalamus: A novel neuroinflammatory paradigm for ME/CFS. Int. J. Immunopathol. Pharmacol. 2018, 32, 2058738418812342. [Google Scholar] [CrossRef] [Green Version]
- Tate, W.; Walker, M.; Sweetman, E.; Helliwell, A.; Peppercorn, K.; Edgar, C.; Blair, A.; Chatterjee, A. Molecular mechanisms of neuroinflammation in ME/CFS and Long COVID to sustain disease and promote relapses. Front. Neurol. 2022, 13, 877772. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.G.; Holmes, T.H.; Anderson, J.N.; Maecker, H.T.; Rosenberg-Hasson, Y.; Valencia, I.J.; Chu, L.; Younger, J.W.; Tato, C.M.; Davis, M.M. Cytokine signature associated with disease severity in chronic fatigue syndrome patients. Proc. Natl. Acad. Sci. USA 2017, 114, E7150–F7158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, J.R.; Burke, B.; Petty, R.; Gough, J.; Fear, D.; Mattey, D.L.; Axford, J.S.; Dalgleish, A.G.; Nutt, D.J. Seven genomic subtypes of chronic fatigue syndrome/myalgic encephalomyelitis: A detailed analysis of gene networks and clinical phenotypes. J. Clin. Pathol. 2008, 61, 730–739. [Google Scholar] [CrossRef] [PubMed]
- de Vega, W.C.; Vernon, S.D.; McGowan, P.O. DNA methylation modifications associated with chronic fatigue syndrome. PLoS ONE 2014, 9, e104757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vega, W.C.; Herrera, S.; Vernon, S.D.; McGowan, P.O. Epigenetic modifications and glucocorticoid sensitivity in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). BMC Med. Genom. 2017, 10, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, S.; de Vega, W.C.; Ashbrook, D.; Vernon, S.D.; McGowan, P.O. Genome-epigenome interactions associated with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Epigenetics 2018, 13, 1174–1190. [Google Scholar] [CrossRef] [Green Version]
- Brenu, E.W.; Staines, D.R.; Marshall-Gradnisek, S.M. Methylation Profile of CD4+ T Cells in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. J. Clin. Cell. Immunol. 2014, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Helliwell, A.M.; Sweetman, E.C.; Stockwell, P.A.; Edgar, C.D.; Chatterjee, A.; Tate, W.P. Changes in DNA methylation profiles of myalgic encephalomyelitis/chronic fatigue syndrome patients reflect systemic dysfunctions. Clin. Epigenetics 2020, 12, 167. [Google Scholar] [CrossRef]
- Trivedi, M.S.; Oltra, E.; Sarria, L.; Rose, N.; Beljanski, V.; Fletcher, M.A.; Klimas, N.G.; Nathanson, L. Identification of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome-associated DNA methylation patterns. PLoS ONE 2018, 13, e0201066. [Google Scholar] [CrossRef] [Green Version]
- de Vega, W.C.; Erdman, L.; Vernon, S.D.; Goldenberg, A.; McGowan, P.O. Integration of DNA methylation & health scores identifies subtypes in myalgic encephalomyelitis/chronic fatigue syndrome. Epigenomics 2018, 10, 539–557. [Google Scholar] [CrossRef]
- Gameiro, G.R.; Sinkunas, V.; Liguori, G.R.; Auler-Júnior, J.O.C. Precision Medicine: Changing the way we think about healthcare. Clinics 2018, 73, e723. [Google Scholar] [CrossRef]
- Miksad, R.A.; Samant, M.K.; Sarkar, S.; Abernethy, A.P. Small but Mighty: The Use of Real-World Evidence to Inform Precision Medicine. Clin. Pharmacol. Ther. 2019, 106, 87–90. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.K.; Vanderlinden, L.A.; Dong, F.; Carry, P.M.; Seifert, J.; Waugh, K.; Shorrosh, H.; Fibngerlin, T.; Frohnert, B.I.; Yang, I.V.; et al. Longitudinal DNA methylation differences precede type 1 diabetes. Sci. Rep. 2020, 10, 3721. [Google Scholar] [CrossRef]
- Eriksson, I.; Undén, A.-L.; Elofsson, S. Self-rated health. Comparisons between three different measures. Results from a population study. Int. J. Epidemiol. 2001, 30, 326–333. [Google Scholar] [CrossRef] [Green Version]
- Lim, U.; Song, M.A. Dietary and Lifestyle Factors of DNA Methylation. In Cancer Epigenetics. Methods in Molecular Biology; Dumitrescu, R., Verma, M., Eds.; Humana Press: Totowa, NJ, USA, 2012; Volume 863. [Google Scholar] [CrossRef]
- Maugeri, A.; Barchitta, M. How Dietary Factors Affect DNA Methylation: Lesson from Epidemiological Studies. Medicina 2020, 56, 374. [Google Scholar] [CrossRef]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Morison, I.M. Genome-scale DNA methylome and transcriptome profiling of human neutrophils. Sci. Data 2016, 3, 160019. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; MacAulay, E.C.; Ahn, A.; Ludgate, J.L.; Stockwell, P.A.; Weeks, R.J.; Parry, M.F.; Foster, T.J.; Knarston, I.M.; Eccles, M.R.; et al. Comparative assessment of DNA methylation patterns between reduced representation bisulfite sequencing and Sequenom EpiTyper methylation analysis. Epigenomics 2017, 9, 823–832. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Stockwell, P.A.; Weeks, R.J.; Morison, I.M. Technical Considerations for Reduced Representation Bisulfite Sequencing with Multiplexed Libraries. J. Biomed. Biotechnol. 2012, 2012, 741542. [Google Scholar] [CrossRef]
- Wang, Z.; Yin, J.; Zhou, W.; Bai, J.; Xie, Y.; Xu, K.; Zheng, X.; Xiao, J.; Zhou, L.; Qi, X.; et al. Complex impact of DNA methylation on transcriptional dysregulation across 22 human cancer types. Nucleic Acids Res. 2020, 48, 2287–2302. [Google Scholar] [CrossRef]
- Murphy, S.K.; Adigun, A.; Huang, Z.; Overcash, F.; Wang, F.; Jirtle, R.L.; Schildkraut, J.S.; Murtha, A.P.; Iversen, E.S.; Hoyo, C. Gender-specific methylation differences in relation to prenatal exposure to cigarette smoke. Gene 2012, 494, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Pheby, D.; Saffron, L. Risk factors for severe ME/CFS. Res. Artic. Biol. Med. 2009, 1, 50–74. [Google Scholar] [CrossRef]
- Tang, J.; Xiong, Y.; Zhou, H.-H.; Chen, X.-P. DNA methylation and personalized medicine. J. Clin. Pharm. Ther. 2014, 39, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Sweetman, E.; Kleffmann, T.; Edgar, C.; de Lange, M.; Vallings, R.; Tate, W. A SWATH-MS analysis of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome peripheral blood mononuclear cell proteomes reveals mitochondrial dysfunction. J. Transl. Med. 2020, 18, 365. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, S.; Brenu, E.; Johnston, S.; Nguyen, T.; Huth, T.; Ramos, S. Longitudinal analysis of immune abnormalities in varying severities of Chronic Fatigue Syndrome/Myalgic Encephalomyelitis patients. J. Transl. Med. 2015, 13, 299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, K.; Pollitt, R.J.; Middleton, B. Human liver long-chain 3-hydroxyacyl-coenzyme A dehydrogenase is a multifunctional membrane-bound beta-oxidation enzyme of mitochondria. Biochem. Biophys. Res. Commun. 1992, 183, 443–448. [Google Scholar] [CrossRef]
- Fluge, Ø.; Mella, O.; Bruland, O.; Risa, K.; Dyrstad, S.E.; Alme, K.; Rekeland, I.G.; Sapkota, D.; Rosland, G.V.; Fossa, A.; et al. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight 2016, 1, e89376. [Google Scholar] [CrossRef] [Green Version]
- Carruthers, B.M.; van de Sande, M.I.; De Meirleir, K.L.; Klimas, N.G.; Broderick, G.; Mitchell, T.; Staines, D.; S Powles, A.C.P.; Speight, N.; Vallings, R.; et al. Myalgic encephalomyelitis: International Consensus Criteria. J. Intern. Med. 2011, 270, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Ludgate, J.L.; Wright, J.; Stockwell, P.A.; Morison, I.M.; Eccles, M.R.; Chatterjee, A. A streamlined method for analysing genome-wide DNA methylation patterns from low amounts of FFPE DNA. BMC Med. Genom. 2017, 10, 54. [Google Scholar] [CrossRef] [Green Version]
- Al Momani, S.; Rodger, E.; Stockwell, P.A.; Eccles, M.R.; Chatterjee, A. Generating Sequencing-Based DNA Methylation Maps from Low DNA Input Samples. In Chromatin. Methods in Molecular Biology; Horsfield, J., Marsman, J., Eds.; Humana: New York, NY, USA, 2022; Volume 2458. [Google Scholar] [CrossRef]
- Stockwell, P.A.; Chatterjee, A.; Rodger, E.J.; Morison, I.M. DMAP: Differential methylation analysis package for RRBS and WGBS data. Bioinformatics 2014, 30, 1814–1822. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Morison, I.M. Comparison of alignment software for genome-wide bisulphite sequence data. Nucleic Acids Res. 2012, 40, e79. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Rodger, E.J.; Ahn, A.; Stockwell, P.A.; Parry, M.; Motwani, J.; Gallagher, S.; Shklovskaya, E.; Tiffen, J.; Eccles, M.R.; et al. Marked Global DNA Hypomethylation Is Associated with Constitutive PD-L1 Expression in Melanoma. iScience 2018, 4, 312–325. [Google Scholar] [CrossRef]
- Chatterjee, A.; Macaulay, E.C.; Rodger, E.; Stockwell, P.A.; Parry, M.F.; Roberts, H.E.; Slatter, T.L.; Hung, N.A.; Devenish, C.J.; Morison, I.M. Placental hypomethylation is more pronounced in genomic Loci Devoid of retroelements. G3 Genes Genomes Genet. 2016, 6, 1911–1921. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Duncan, E.J.; Parry, M.F.; Weeks, R.J.; Morison, I. Genome-wide DNA methylation map of human neutrophils reveals widespread inter-individual epigenetic variation. Sci. Rep. 2015, 5, 17328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snel, B.; Lehmann, G.; Bork, P.; Huynen, M.A. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 15, 3442–3444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Stein, T.I.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 2016, 1.30.1–1.30.33. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Frag | Chr | Start | End | Position | GeneID | Genehancer | Regulatory Interactions | P1-a | P1-b | P1-c | P1-d | P1-e |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 19 | 33,885,306 | 33,885,381 | On Intron | PEPD | GH09J033388 | CEBPG:PEPD | 78 | 45 | 44 | 71 | 64 |
| 2 | X | 150,565,438 | 150,565,527 | On Intron | VMA21 | GH0XJ151395 | VMA21 | 50 | 33 | 31 | 45 | 49 |
| 3 | 19 | 55,464,080 | 55,464,189 | On Intron | NLRP7 | GH19J054952 | NLRP2 | 78 | 55 | 48 | 81 | 75 |
| 4 | 7 | 5,741,705 | 5,741,780 | On Intron | NF216 | GH07J005687 | ACTB:CCZ1:RNF216:USP42 | 86 | 60 | 74 | 84 | 88 |
| 5 | X | 135,579,269 | 135,579,310 | On Intron | HTATSF1 | - | - | 39 | 28 | 22 | 46 | 40 |
| 6 | X | 152,908,188 | 152,908,279 | On Intron | DUSP9 | - | DUSP9 | 46 | 18 | 30 | 37 | 45 |
| 7 | 17 | 45,925,149 | 45,925,204 | On Exon | SP6 | GH17J047846 | SP2:CDK5RAP3: OSBPL7:SCRN2 | 46 | 28 | 29 | 46 | 44 |
| 8 | X | 23,761,294 | 23,761,378 | On Exon | ACOT9 | GH0XJ023741 | ACOT9 | 37 | 18 | 18 | 34 | 37 |
| 9 | 8 | 145,003,618 | 145,003,684 | On Exon | PLEC | GH08J143914 | ZC3H3:EEF1D:PLEC | 70 | 34 | 37 | 58 | 59 |
| 10 | X | 149,106,531 | 149,106,576 | On Exon | CXorf40B | GH0XJ149937 | INC00B94:CXorf40B | 48 | 28 | 36 | 54 | 58 |
| 11 | 22 | 42,316,243 | 42,316,306 | Intergenic | - | GH22J041918 | WBP2NL:CYP2D8P:CENPM:CYPSD6:TNFRSF13C | 44 | 30 | 27 | 44 | 44 |
| 12 | 1 | 17,199,256 | 17,199,369 | Intergenic | - | - | NECAP2:CROCC | 55 | 35 | 44 | 59 | 57 |
| 13 | 2 | 232,348,597 | 232,348,713 | Intergenic | - | - | NMUR1:NCL | 57 | 38 | 34 | 69 | 62 |
| 14 | 13 | 114,918,456 | 114,918,525 | Intergenic | - | - | CDC16:UPF31: RASA3 | 87 | 65 | 62 | 96 | 84 |
| 15 | 2 | 26,521,360 | 26,521,433 | Intergenic | - | GH02J026298 | HADHB:HADHA: ADGRF3 | 53 | 38 | 35 | 60 | 53 |
| 16 | 3 | 10,334,731 | 10,334,778 | Intergenic | - | GH03J010291 | GHRLOS:GHRL | 33 | 13 | 22 | 40 | 41 |
| 17 | 15 | 22,095,431 | 22,095,475 | Intergenic | - | - | - | 51 | 40 | 34 | 51 | 56 |
| Frag | Chr | Start | End | Location | GeneID | Genehancer | Regulatory Interactions | P2-a | P2-b | P2-c | P2-d | P2-e |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 19 | 4,543,716 | 4,543,762 | On Intron | SEMA6B | GH19J004539 | YJU2:PLIN5:SEMA6B:LRG1 | 80 | 81 | 47 | 91 | 67 |
| 2 | X | 15,353,393 | 15,353,501 | On Intron | PIGA | GH0XJ015333 | ZRSR2:PIGA | 40 | 41 | 18 | 39 | 33 |
| 3 | 22 | 17,640,812 | 17,640,923 | On Intron | CECR5 | GH22J017157 | HDHD5 | 69 | 82 | 60 | 80 | 71 |
| 4 | 14 | 105,936,238 | 105,936,292 | On Exon | MTA1 | GH14J105464 | IGHGP:CDCA4:CRIP2:MTA1:ENSG00000257270 | 84 | 80 | 63 | 87 | 76 |
| 5 | 22 | 18,027,985 | 18,028,072 | On Exon | CECR2 | - | - | 49 | 46 | 68 | 38 | 51 |
| 6 | X | 102,565,776 | 102,565,848 | Intron–Exon Boundary | BEX2 | GH0XJ103310 | BEX2 | 42 | 45 | 26 | 43 | 41 |
| 7 | 1 | 155,098,923 | 155,098,964 | Intergenic | - | GH01J155123 | DAP3:CLK2:DPM3:GBAP1:THBS3:EFNA1 | 54 | 67 | 35 | 72 | 60 |
| 8 | 3 | 10,334,731 | 10,334,778 | Intergenic | - | GH03J010291 | GHRLOS:GHRL | 49 | 56 | 32 | 61 | 41 |
| 9 | 9 | 38,687,682 | 38,687,760 | Intergenic | - | - | - | 61 | 62 | 39 | 65 | 42 |
| 10 | 7 | 100,882,140 | 100,882,220 | Intergenic | - | GH07J101231 | FIS1:CLDN15 | 80 | 80 | 65 | 90 | 75 |
| 11 | 2 | 219,233,608 | 219,233,704 | Intergenic | - | GH02J218366 | AAMP:SCL11A1:TMBIM1:CATIP | 49 | 55 | 38 | 62 | 52 |
| 12 | 6 | 170,403,979 | 170,404,085 | Intergenic | - | - | WDR27 | 62 | 66 | 81 | 63 | 70 |
| 13 | X | 129,299,533 | 129,299,622 | Intergenic | - | GH0XJ130164 | ELF4:AIMF1:ZNF280C | 42 | 36 | 61 | 31 | 49 |
| 14 | X | 135,579,192 | 135,579,268 | Intergenic | - | - | - | 28 | 27 | 61 | 30 | 44 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helliwell, A.M.; Stockwell, P.A.; Edgar, C.D.; Chatterjee, A.; Tate, W.P. Dynamic Epigenetic Changes during a Relapse and Recovery Cycle in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Int. J. Mol. Sci. 2022, 23, 11852. https://doi.org/10.3390/ijms231911852
Helliwell AM, Stockwell PA, Edgar CD, Chatterjee A, Tate WP. Dynamic Epigenetic Changes during a Relapse and Recovery Cycle in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. International Journal of Molecular Sciences. 2022; 23(19):11852. https://doi.org/10.3390/ijms231911852
Chicago/Turabian StyleHelliwell, Amber M., Peter A. Stockwell, Christina D. Edgar, Aniruddha Chatterjee, and Warren P. Tate. 2022. "Dynamic Epigenetic Changes during a Relapse and Recovery Cycle in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome" International Journal of Molecular Sciences 23, no. 19: 11852. https://doi.org/10.3390/ijms231911852