
Mitochonic Acid 5 Improves Duchenne Muscular Dystrophy and Parkinson’s Disease Model of Caenorhabditis elegans

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Penetration and Homing Activity of MA-5 into Intact C. elegans Mitochondria
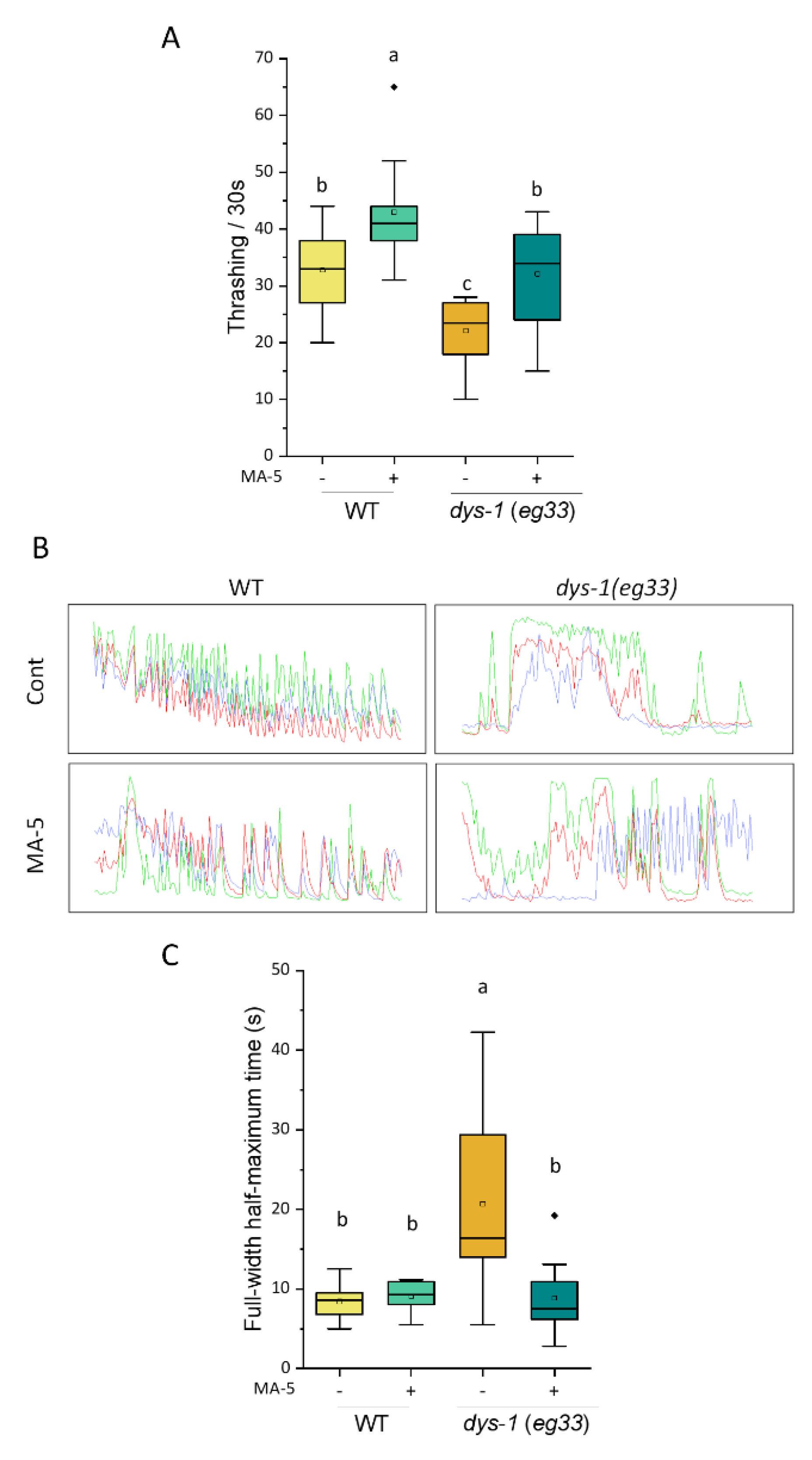
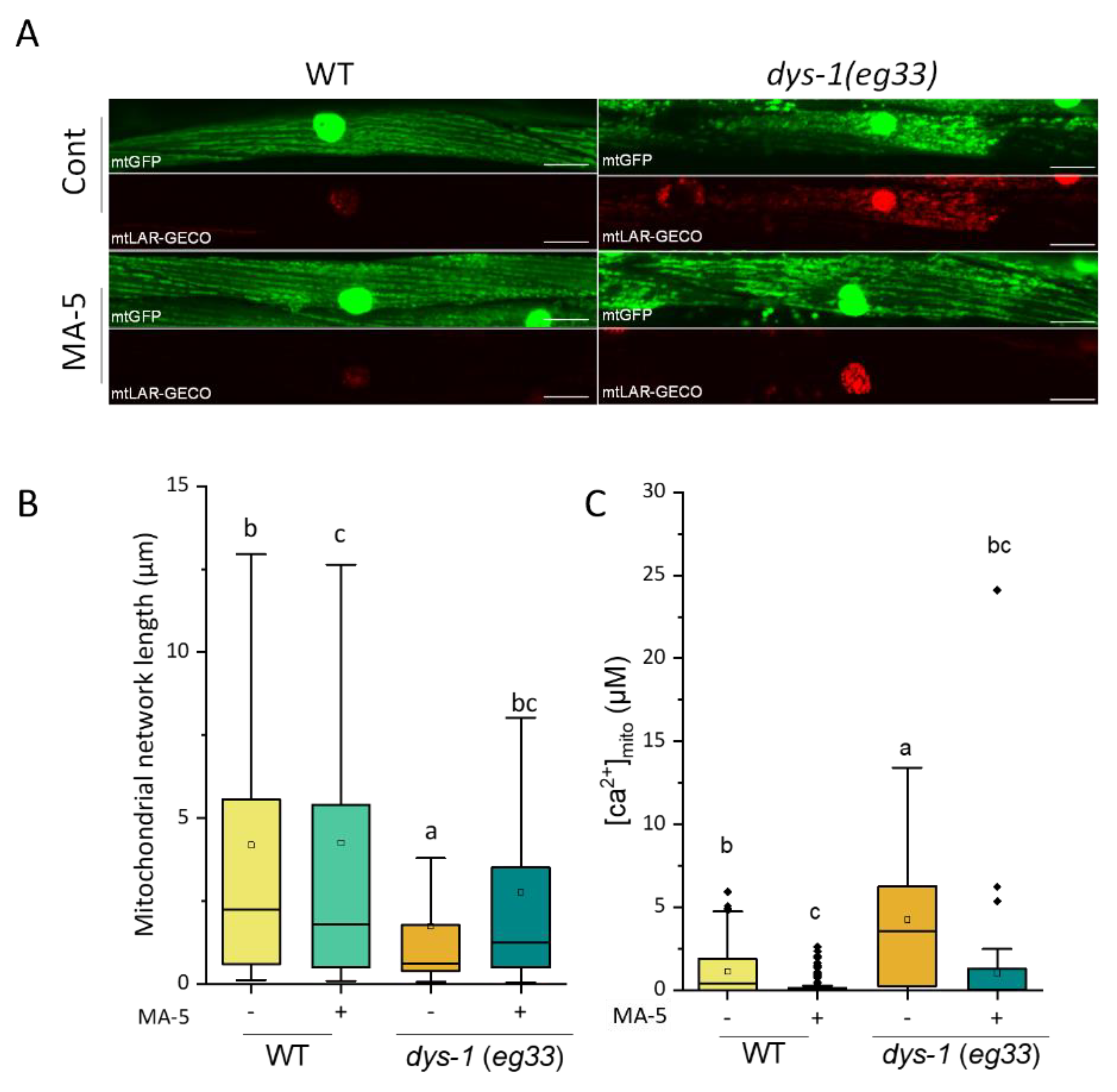
2.2. Alleviation of C. elegans DMD Model Symptom by MA-5
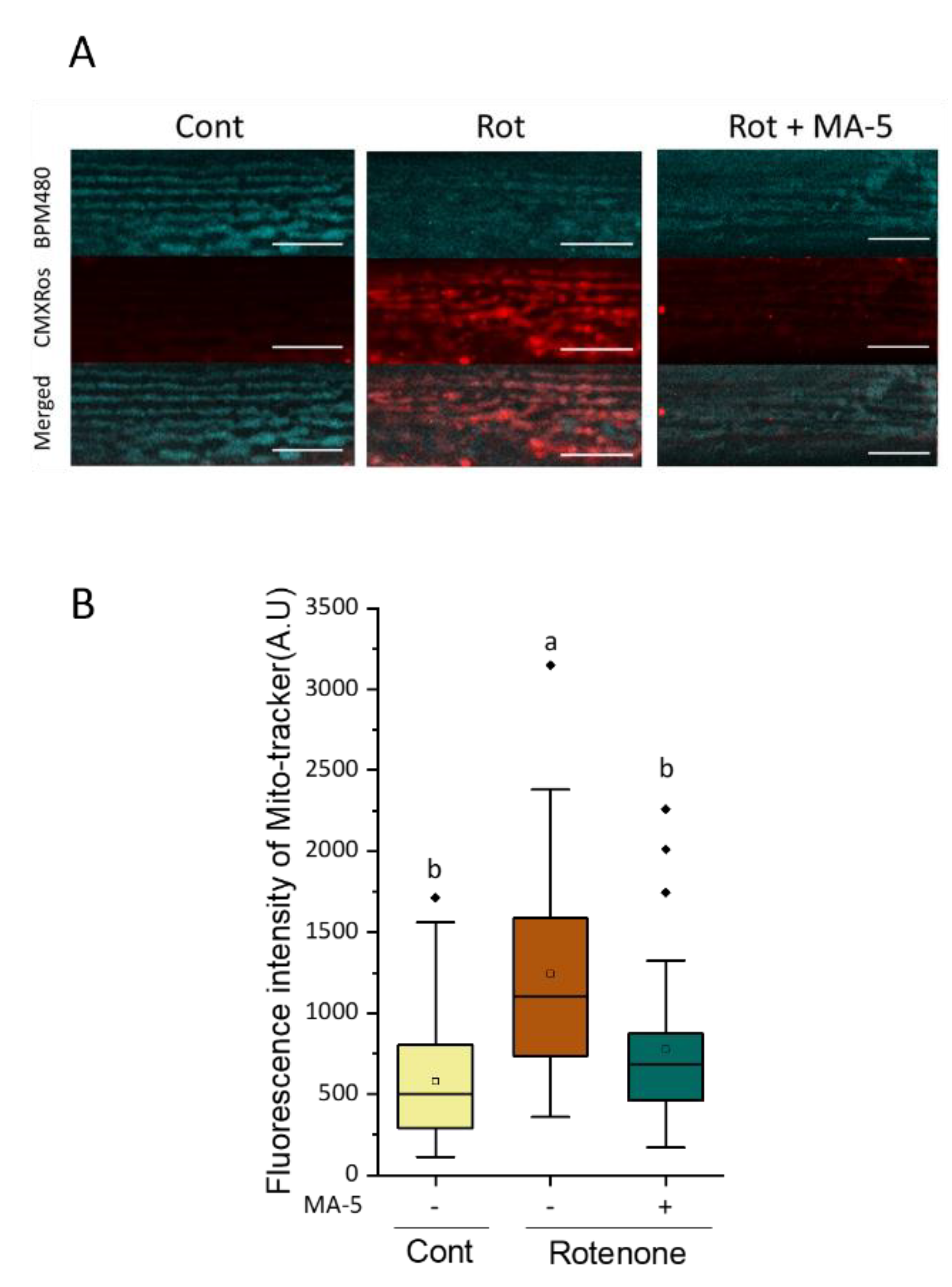
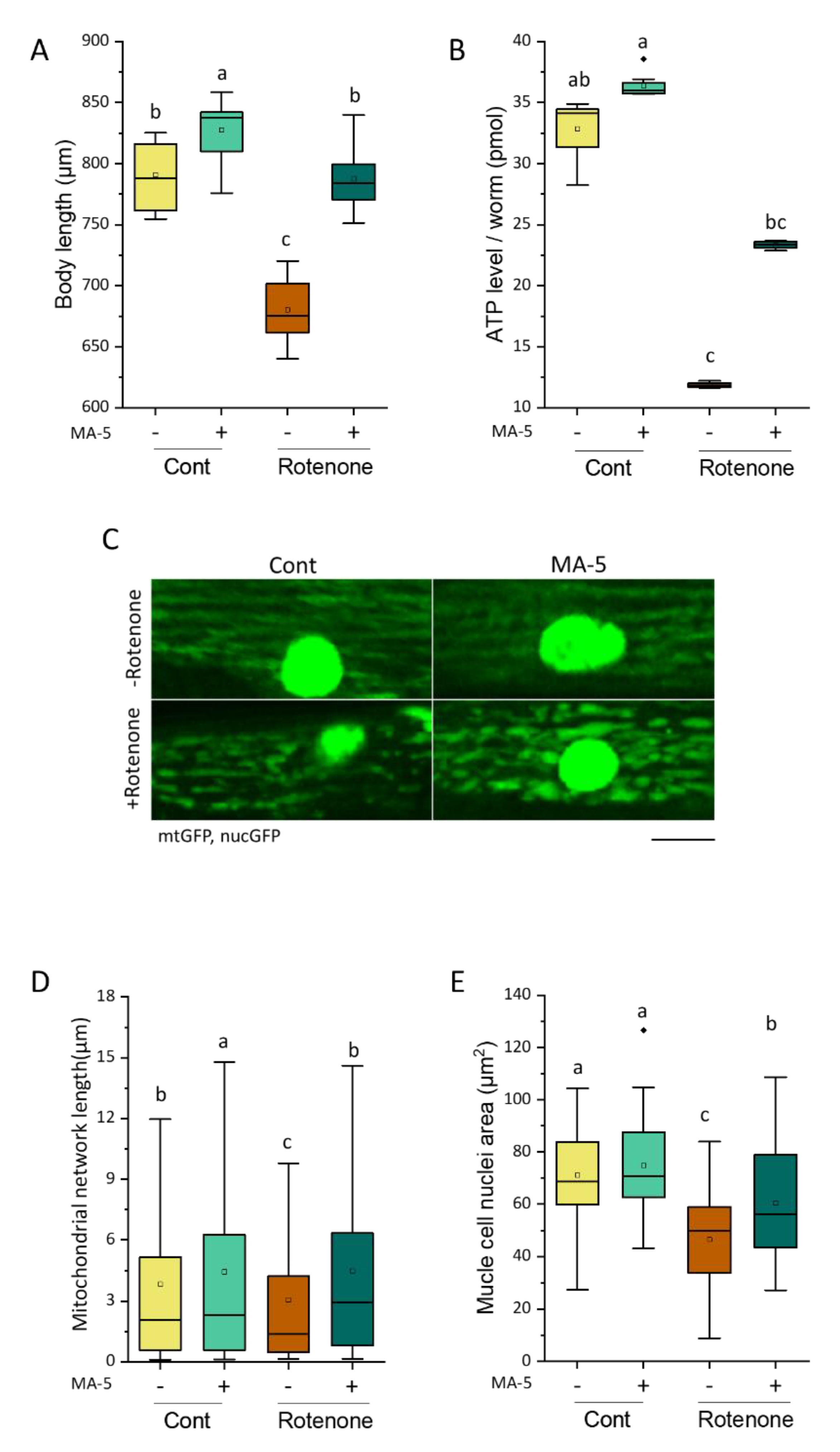
2.3. MA-5 Ameliorates Muscular Mitochondrial Perturbations with Rotenone Treatment
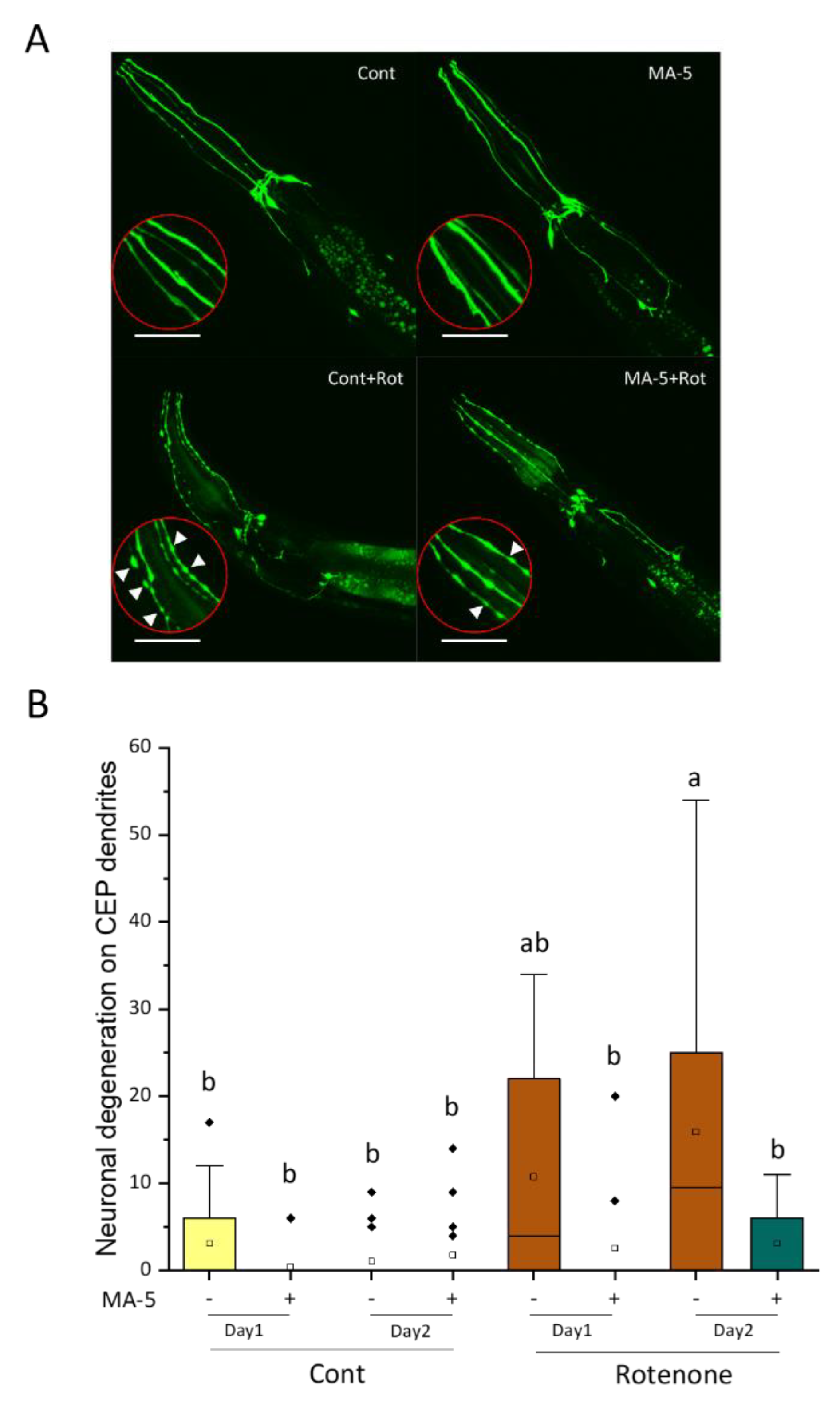
2.4. Alleviation of PD Model Progression by MA-5
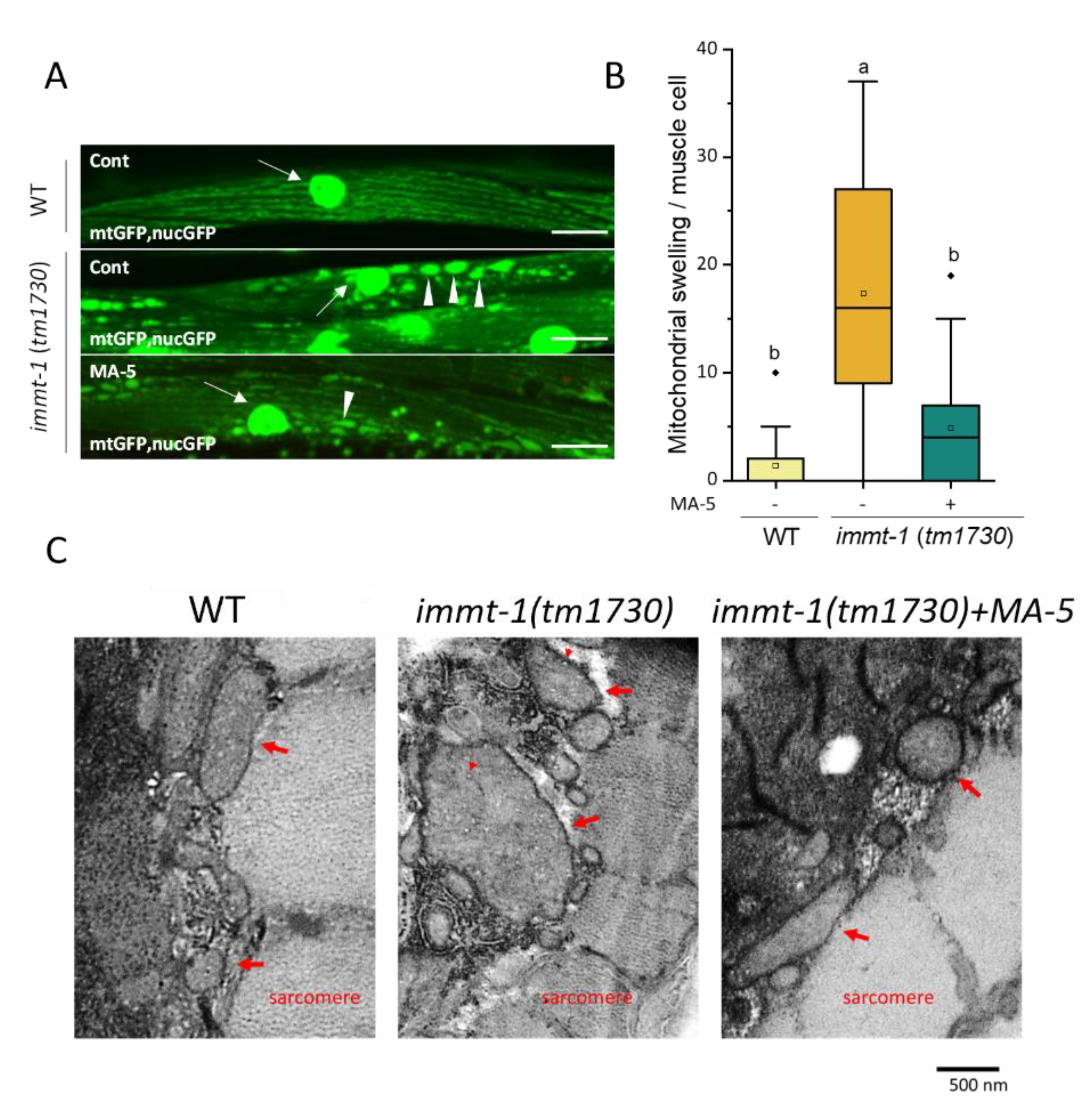
2.5. Suppression of Mitochondrial Swelling in C. elegans Mitofilin immt-1 Mutant with MA-5
3. Discussion
4. Materials and Methods
4.1. C. elegans Strains and Culture Conditions
4.2. BODIPY-Based Fluorescent-Conjugated MA-5 Contents in Mitochondria Assay
4.3. Measurement of Body Length
4.4. Thrashing Speed
4.5. Microscopic Imaging
4.6. Measurement of Mitochondrial Ca2+ Levels
4.7. Dopamine Neuron Degeneration Measurement
4.8. Mitochondrial ROS Measurement
4.9. ATP Detection
4.10. Analysis of mitofilin/immt-1gene Mutation Mitochondrial Morphology
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suzuki, T.; Yamaguchi, H.; Kikusato, M.; Matsuhashi, T.; Matsuo, A.; Sato, T.; Oba, Y.; Watanabe, S.; Minaki, D.; Saigusa, D.; et al. Mitochonic Acid 5 (MA-5), a Derivative of the Plant Hormone Indole-3-Acetic Acid, Improves Survival of Fibroblasts from Patients with Mitochondrial Diseases. Tohoku J. Exp. Med. 2015, 236, 225–232. [Google Scholar] [CrossRef]
- de Melo, M.P.; Curi, T.C.P.; Miyasaka, C.K.; Palanch, A.C.; Curi, R. Effect of Indole Acetic Acid on Oxygen Metabolism in Cultured Rat Neutrophil. Gen. Pharmacol. Vasc. Syst. 1998, 31, 573–578. [Google Scholar] [CrossRef]
- Salopek-Sondi, B.; Piljac-Žegarac, J.; Magnus, V.; Kopjar, N. Free Radical–Scavenging Activity and DNA Damaging Potential of Auxins IAA and 2-Methyl-IAA Evaluated in Human Neutrophils by the Alkaline Comet Assay. J. Biochem. Mol. Toxicol. 2010, 24, 165–173. [Google Scholar] [CrossRef]
- Toyohara, T.; Akiyama, Y.; Suzuki, T.; Takeuchi, Y.; Mishima, E.; Tanemoto, M.; Momose, A.; Toki, N.; Sato, H.; Nakayama, M.; et al. Metabolomic Profiling of Uremic Solutes in CKD Patients. Hypertens. Res. 2010, 33, 944–952. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamaguchi, H.; Kikusato, M.; Hashizume, O.; Nagatoishi, S.; Matsuo, A.; Sato, T.; Kudo, T.; Matsuhashi, T.; Murayama, K.; et al. Mitochonic Acid 5 Binds Mitochondria and Ameliorates Renal Tubular and Cardiac Myocyte Damage. J. Am. Soc. Nephrol. 2016, 27, 1925–1932. [Google Scholar] [CrossRef]
- Matsuhashi, T.; Sato, T.; Kanno, S.I.; Suzuki, T.; Matsuo, A.; Oba, Y.; Kikusato, M.; Ogasawara, E.; Kudo, T.; Suzuki, K.; et al. Mitochonic Acid 5 (MA-5) Facilitates ATP Synthase Oligomerization and Cell Survival in Various Mitochondrial Diseases. EBioMedicine 2017, 20, 27–38. [Google Scholar] [CrossRef]
- Oikawa, Y.; Izumi, R.; Koide, M.; Hagiwara, Y.; Kanzaki, M.; Suzuki, N.; Kikuchi, K.; Matsuhashi, T.; Akiyama, Y.; Ichijo, M.; et al. Mitochondrial Dysfunction Underlying Sporadic Inclusion Body Myositis Is Ameliorated by the Mitochondrial Homing Drug MA-5. PLoS ONE 2020, 15, e0231064. [Google Scholar] [CrossRef]
- Vila, M.C.; Rayavarapu, S.; Hogarth, M.W.; van der Meulen, J.H.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.J.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria Mediate Cell Membrane Repair and Contribute to Duchenne Muscular Dystrophy. Cell Death Differ. 2016, 24, 330–342. [Google Scholar] [CrossRef]
- Hattori, N.; Mizuno, Y. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103. [Google Scholar] [CrossRef]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2021, 8, 1704. [Google Scholar] [CrossRef]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in Mdx Mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Campbell, K.A.; Fox, D.J.; Matthews, D.J.; Valdez, R. Corticosteroid Treatments in Males with Duchenne Muscular Dystrophy: Treatment Duration and Time to Loss of Ambulation. J. Child. Neurol 2015, 30, 1275. [Google Scholar] [CrossRef] [PubMed]
- Iarkov, A.; Barreto, G.E.; Grizzell, J.A.; Echeverria, V. Strategies for the Treatment of Parkinson’s Disease: Beyond Dopamine. Front. Aging Neurosci. 2020, 12, 4. [Google Scholar] [CrossRef]
- Gaffney, C.J.; Pollard, A.; Barratt, T.F.; Constantin-Teodosiu, D.; Greenhaff, P.L.; Szewczyk, N.J. Greater Loss of Mitochondrial Function with Ageing Is Associated with Earlier Onset of Sarcopenia in C. Elegans. Aging 2018, 10, 3382–3396. [Google Scholar] [CrossRef]
- Mack, H.I.D.; Heimbucher, T.; Murphy, C.T. DISEASE The Nematode Caenorhabditis Elegans as a Model for Aging Research. Drug Discov. Today Dis. Models 2018, 27, 3–13. [Google Scholar] [CrossRef]
- Culetto, E.; Sattelle, D.B. A Role for Caenorhabditis Elegans in Understanding the Function and Interactions of Human Disease Genes. Hum. Mol. Genet. 2000, 9, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Kaletta, T.; Hengartner, M.O. Finding Function in Novel Targets: C. Elegans as a Model Organism. Nat. Rev. Drug Discov. 2006, 5, 387–399. [Google Scholar] [CrossRef]
- Nass, R.; Hall, D.H.; Miller, D.M.; Blakely, R.D. Neurotoxin-Induced Degeneration of Dopamine Neurons in Caenorhabditis Elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 3264–3269. [Google Scholar] [CrossRef]
- Maulik, M.; Mitra, S.; Bult-Ito, A.; Taylor, B.E.; Vayndorf, E.M. Behavioral Phenotyping and Pathological Indicators of Parkinson’s Disease in C. Elegans Models. Front. Genet. 2017, 8, 77. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, Z.; Klaunig, J.E. Caenorhabditis Elegans Neuron Degeneration and Mitochondrial Suppression Caused by Selected Environmental Chemicals. Int. J. Biochem. Mol. Biol. 2013, 4, 191. [Google Scholar]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic Systemic Pesticide Exposure Reproduces Features of Parkinson’s Disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of Toxicity in Rotenone Models of Parkinson’s Disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef] [PubMed]
- Xiong, N.; Long, X.; Xiong, J.; Jia, M.; Chen, C.; Huang, J.; Ghoorah, D.; Kong, X.; Lin, Z.; Wang, T. Mitochondrial Complex I Inhibitor Rotenone-Induced Toxicity and Its Potential Mechanisms in Parkinson’s Disease Models. Crit. Rev. Toxicol. 2012, 42, 613–632. [Google Scholar] [CrossRef] [PubMed]
- Aliagan, A.D.I.; Ahwazi, M.D.; Tombo, N.; Feng, Y.; Bopassa, J.C. Parkin Interacts with Mitofilin to Increase Dopaminergic Neuron Death in Response to Parkinson’s Disease-Related Stressors. Am. J. Transl. Res. 2020, 12, 7542. [Google Scholar]
- Cooper, J.F.; Dues, D.J.; Spielbauer, K.K.; Machiela, E.; Senchuk, M.M.; van Raamsdonk, J.M. Delaying Aging Is Neuroprotective in Parkinson’s Disease: A Genetic Analysis in C. Elegans Models. Npj. Parkinson Dis. 2015, 1, 1–12. [Google Scholar] [CrossRef]
- McDiarmid, T.A.; Au, V.; Loewen, A.D.; Liang, J.; Mizumoto, K.; Moerman, D.G.; Rankin, C.H. CRISPR-Cas9 Human Gene Replacement and Phenomic Characterization in Caenorhabditis Elegans to Understand the Functional Conservation of Human Genes and Decipher Variants of Uncertain Significance. DMM Dis. Models Mech. 2018, 11, dmm036517. [Google Scholar] [CrossRef]
- Vicencio, J.; Cerón, J. A Living Organism in Your CRISPR Toolbox: Caenorhabditis Elegans Is a Rapid and Efficient Model for Developing CRISPR-Cas Technologies. CRISPR J. 2021, 4, 32–42. [Google Scholar] [CrossRef]
- Artal-Sanz, M.; de Jong, L.; Tavernarakis, N. Caenorhabditis Elegans: A Versatile Platform for Drug Discovery. Biotechnol. J. 2006, 1, 1405–1418. [Google Scholar] [CrossRef]
- O’Reilly, L.P.; Luke, C.J.; Perlmutter, D.H.; Silverman, G.A.; Pak, S.C.C. Elegans in High-Throughput Drug Discovery. Adv. Drug Deliv. Rev. 2014, 69–70, 247–253. [Google Scholar] [CrossRef]
- Sudevan, S.; Takiura, M.; Kubota, Y.; Higashitani, N.; Cooke, M.; Ellwood, R.A.; Etheridge, T.; Szewczyk, N.J.; Higashitani, A. Mitochondrial Dysfunction Causes Ca2+ Overload and ECM Degradation–Mediated Muscle Damage in C. Elegans. FASEB J. 2019, 8, 9540–9550. [Google Scholar] [CrossRef]
- Ellwood, R.A.; Hewitt, J.E.; Torregrossa, R.; Philp, A.M.; Hardee, J.P.; Hughes, S.; van de Klashorst, D.; Gharahdaghi, N.; Anupom, T.; Slade, L.; et al. Mitochondrial Hydrogen Sulfide Supplementation Improves Health in the C. Elegans Duchenne Muscular Dystrophy Model. Proc. Natl. Acad. Sci. USA 2021, 118, e2018342118. [Google Scholar] [CrossRef] [PubMed]
- Weinkove, D.; Zavagno, G. Applying C. Elegans to the Industrial Drug Discovery Process to Slow Aging. Front. Aging 2021, 2, 740582. [Google Scholar] [CrossRef] [PubMed]
- Klapper, M.; Ehmke, M.; Palgunow, D.; Böhme, M.; Matthäus, C.; Bergner, G.; Dietzek, B.; Popp, J.; Döring, F. Fluorescence-Based Fixative and Vital Staining of Lipid Droplets in Caenorhabditis Elegans Reveal Fat Stores Using Microscopy and Flow Cytometry Approaches. J. Lipid Res. 2011, 52, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Higashitani, A.; Teranishi, M.; Nakagawa, Y.; Itoh, Y.; Sudevan, S.; Szewczyk, N.J.; Kubota, Y.; Abe, T.; Kobayashi, T. Increased Mitochondrial Ca2+ Contributes to Health Decline with Age and Duchene Muscular Dystrophy in C. Elegans. bioRxiv 2022. [Google Scholar] [CrossRef]
- Ellwood, R.A.; Piasecki, M.; Szewczyk, N.J. Caenorhabditis Elegans as a Model System for Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2021, 22, 4891. [Google Scholar] [CrossRef]
- Epstein, F.H.; Ptacek, L.J.; Johnson, K.J.; Griggs, R.C. Genetics and Physiology of the Myotonic Muscle Disorders. N. Engl. J. Med. 1993, 328, 482–489. [Google Scholar] [CrossRef]
- Kurihara, T. New Classification and Treatment for Myotonic Disorders. Intern. Med. 2005, 44, 1027–1032. [Google Scholar] [CrossRef]
- Hewitt, J.E.; Pollard, A.K.; Lesanpezeshki, L.; Deane, C.S.; Gaffney, C.J.; Etheridge, T.; Szewczyk, N.J.; Vanapalli, S.A. Muscle Strength Deficiency and Mitochondrial Dysfunction in a Muscular Dystrophy Model of Caenorhabditis Elegans and Its Functional Response to Drugs. DMM Dis. Models Mech. 2018, 11, dmm036137. [Google Scholar] [CrossRef]
- Mun, J.Y.; Lee, T.H.; Kim, J.H.; Yoo, B.H.; Bahk, Y.Y.; Koo, H.S.; Han, S.S. Caenorhabditis Elegans Mitofilin Homologs Control the Morphology of Mitochondrial Cristae and Influence Reproduction and Physiology. J. Cell. Physiol. 2010, 224, 748–756. [Google Scholar] [CrossRef]
- Head, B.P.; Zulaika, M.; Ryazantsev, S.; van der Bliek, A.M. A Novel Mitochondrial Outer Membrane Protein, MOMA-1, that Affects Cristae Morphology in Caenorhabditis Elegans. Mol. Biol. Cell 2011, 22, 831–841. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Poláková, E.; Janíček, R.; Shirokova, N. Mitochondrial Dysfunctions during Progression of Dystrophic Cardiomyopathy. Cell Calcium 2015, 58, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.J.; Rodriguez, A.; Flatt, K.M.; Ray, S.; Schuler, A.; Rodemoyer, B.; Veerappan, V.; Cuciarone, K.; Kullman, A.; Lim, C.; et al. Physical Exertion Exacerbates Decline in the Musculature of an Animal Model of Duchenne Muscular Dystrophy. Proc. Natl. Acad. Sci. USA 2019, 116, 3508–3517. [Google Scholar] [CrossRef] [PubMed]
- Nogami, K.; Maruyama, Y.; Sakai-Takemura, F.; Motohashi, N.; Elhussieny, A.; Imamura, M.; Miyashita, S.; Ogawa, M.; Noguchi, S.; Tamura, Y.; et al. Pharmacological Activation of SERCA Ameliorates Dystrophic Phenotypes in Dystrophin-Deficient Mdx Mice. Hum. Mol. Genet. 2021, 30, 1006–1019. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen Peroxide: A Metabolic by-Product or a Common Mediator of Ageing Signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Brenner, S. The Genetics of Caenorabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsienb, R.Y. A New Generation of Ca2+ Indicators with Greatly Improved Fluorescence Properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Wu, J.; Prole, D.L.; Shen, Y.; Lin, Z.; Gnanasekaran, A.; Liu, Y.; Chen, L.; Zhou, H.; Chen, S.R.W.; Usachev, Y.M.; et al. Red Fluorescent Genetically Encoded Ca2+ Indicators for Use in Mitochondria and Endoplasmic Reticulum. Biochem. J. 2014, 464, 13–22. [Google Scholar] [CrossRef]
- Sudevan, S.; Muto, K.; Higashitani, N.; Hashizume, T.; Higashibata, A.; Ellwood, R.A.; Deane, C.S.; Rahman, M.; Vanapalli, S.A.; Etheridge, T.; et al. Loss of Physical Contact in Space Alters the Dopamine System in C. Elegans. iScience 2022, 25, 103762. [Google Scholar] [CrossRef]
- Momma, K.; Homma, T.; Isaka, R.; Sudevan, S.; Higashitani, A. Heat-Induced Calcium Leakage Causes Mitochondrial. Genetics 2017, 206, 1985–1994. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MA-5 Final Concentraation | Median Lifespan (Days) b |
|---|---|
| control (0 μM) | 13.6 ± 0.6 |
| 5 μM | 15.3 ± 0.5 |
| 10 μM | 14.8 ± 0.4 |
| 20 μM | 14.8 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Nagasawa, S.; Muto, K.; Ueda, M.; Suzuki, C.; Abe, T.; Higashitani, A. Mitochonic Acid 5 Improves Duchenne Muscular Dystrophy and Parkinson’s Disease Model of Caenorhabditis elegans. Int. J. Mol. Sci. 2022, 23, 9572. https://doi.org/10.3390/ijms23179572
Wu X, Nagasawa S, Muto K, Ueda M, Suzuki C, Abe T, Higashitani A. Mitochonic Acid 5 Improves Duchenne Muscular Dystrophy and Parkinson’s Disease Model of Caenorhabditis elegans. International Journal of Molecular Sciences. 2022; 23(17):9572. https://doi.org/10.3390/ijms23179572
Chicago/Turabian StyleWu, Xintong, Satoi Nagasawa, Kasumi Muto, Maiko Ueda, Chitose Suzuki, Takaaki Abe, and Atsushi Higashitani. 2022. "Mitochonic Acid 5 Improves Duchenne Muscular Dystrophy and Parkinson’s Disease Model of Caenorhabditis elegans" International Journal of Molecular Sciences 23, no. 17: 9572. https://doi.org/10.3390/ijms23179572