
GSK3β Inhibition Is the Molecular Pivot That Underlies the Mir-210-Induced Attenuation of Intrinsic Apoptosis Cascade during Hypoxia


Abstract
:1. Introduction
2. Results
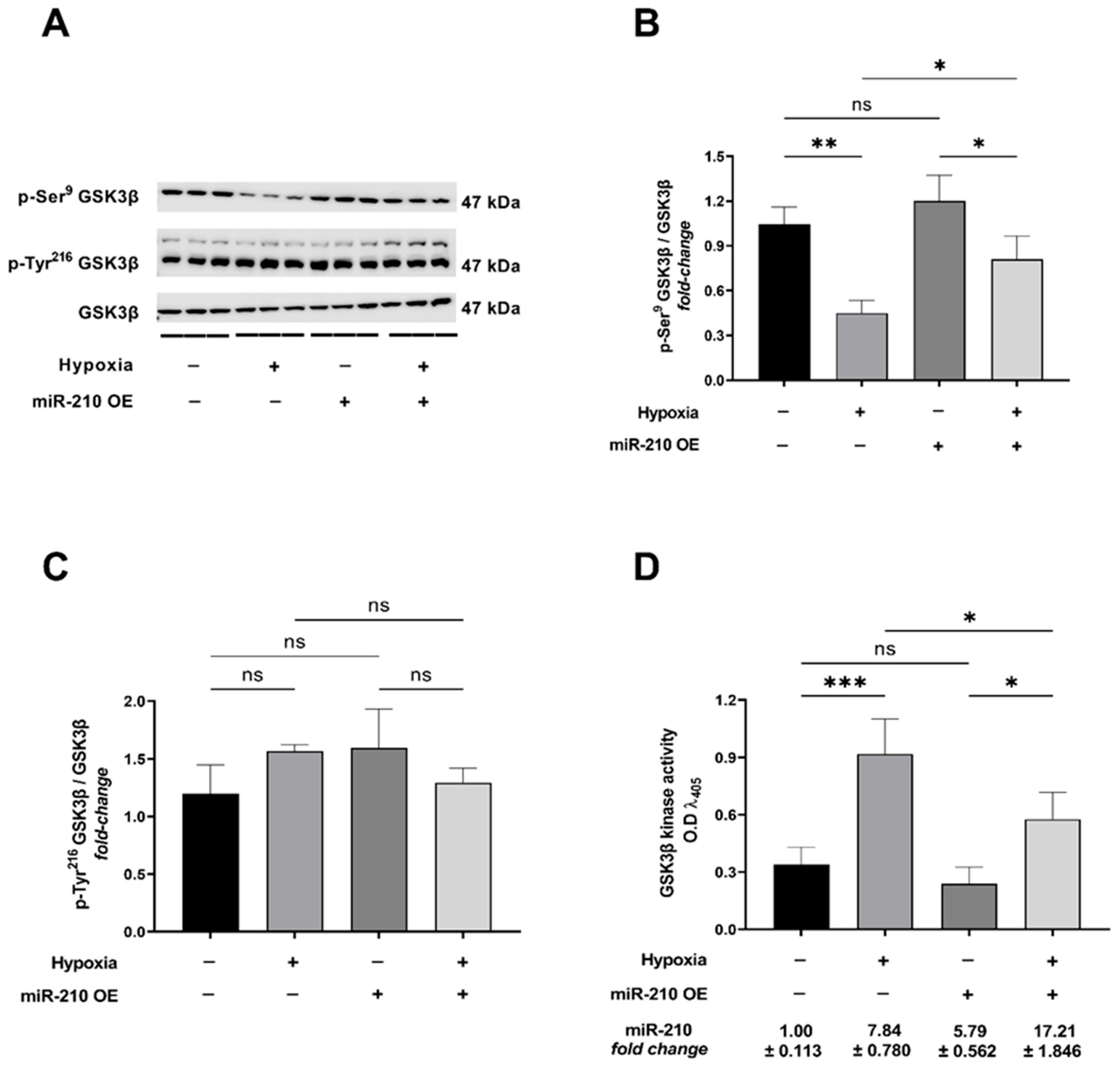
2.1. miR-210 Attenuates the Hypoxia-Induced GSK3β Activation
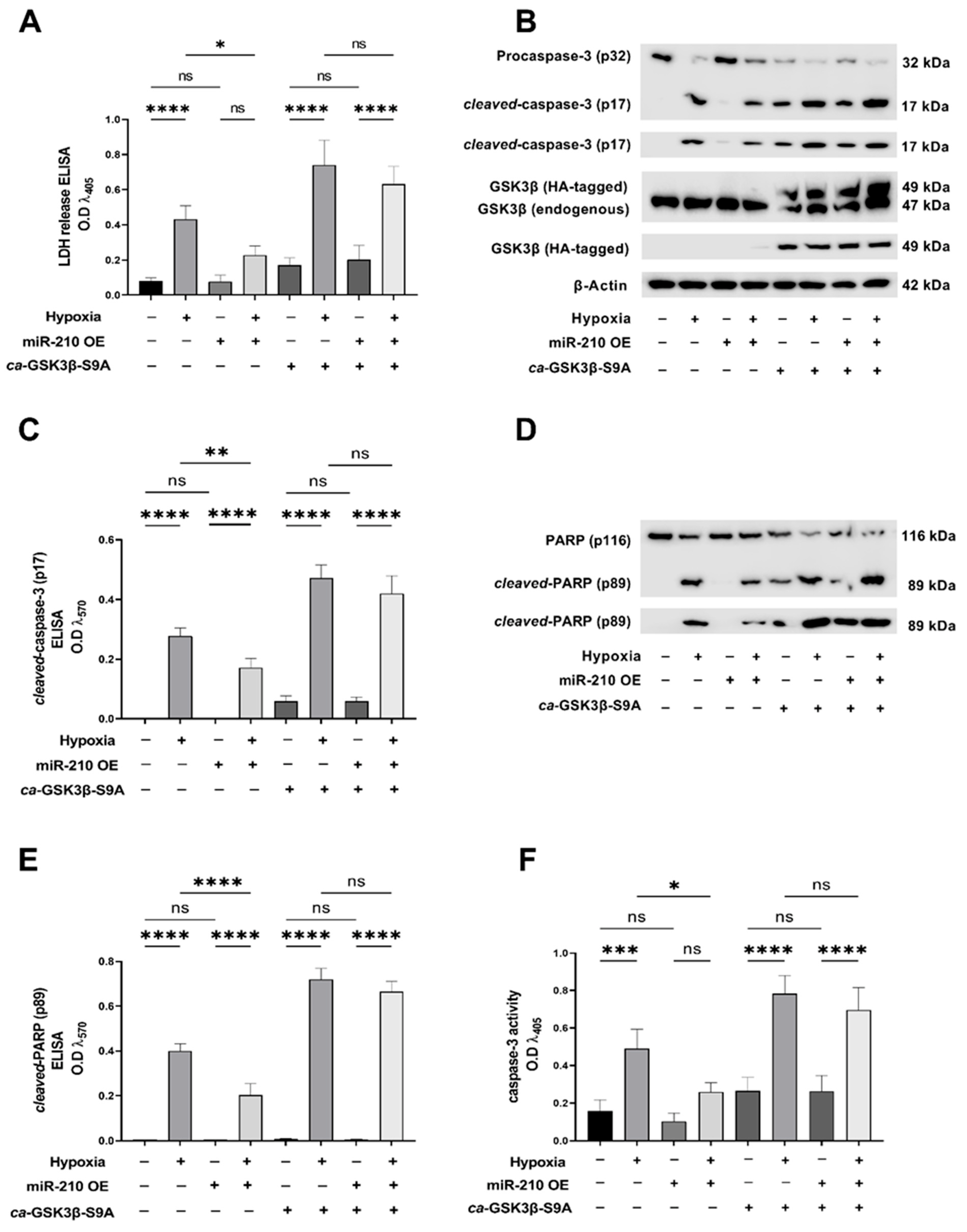
2.2. GSK3β Inhibition Is a Necessary Molecular Event for the miR-210-Induced Attenuation of Apoptotic Cell Death during Hypoxia
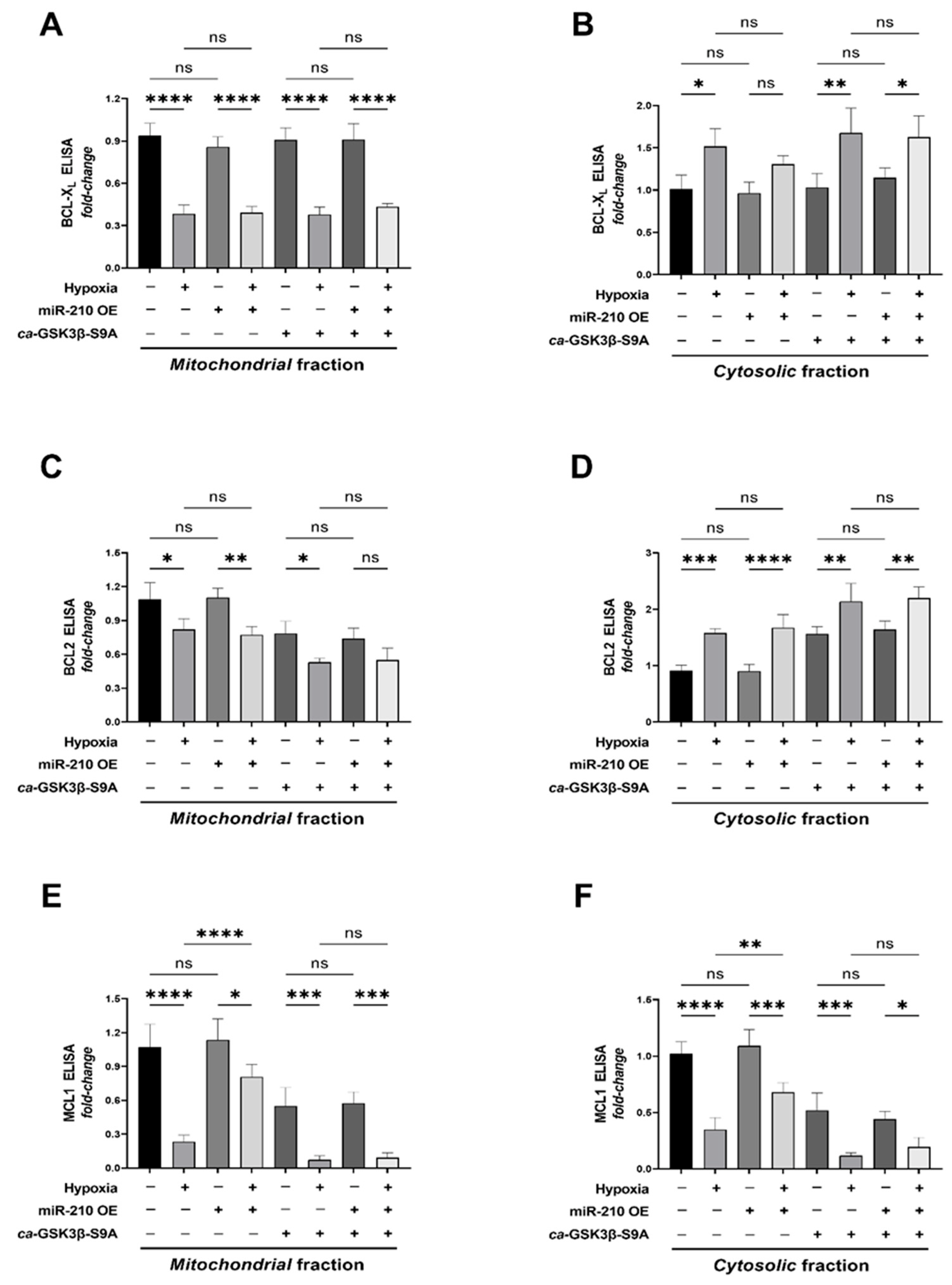
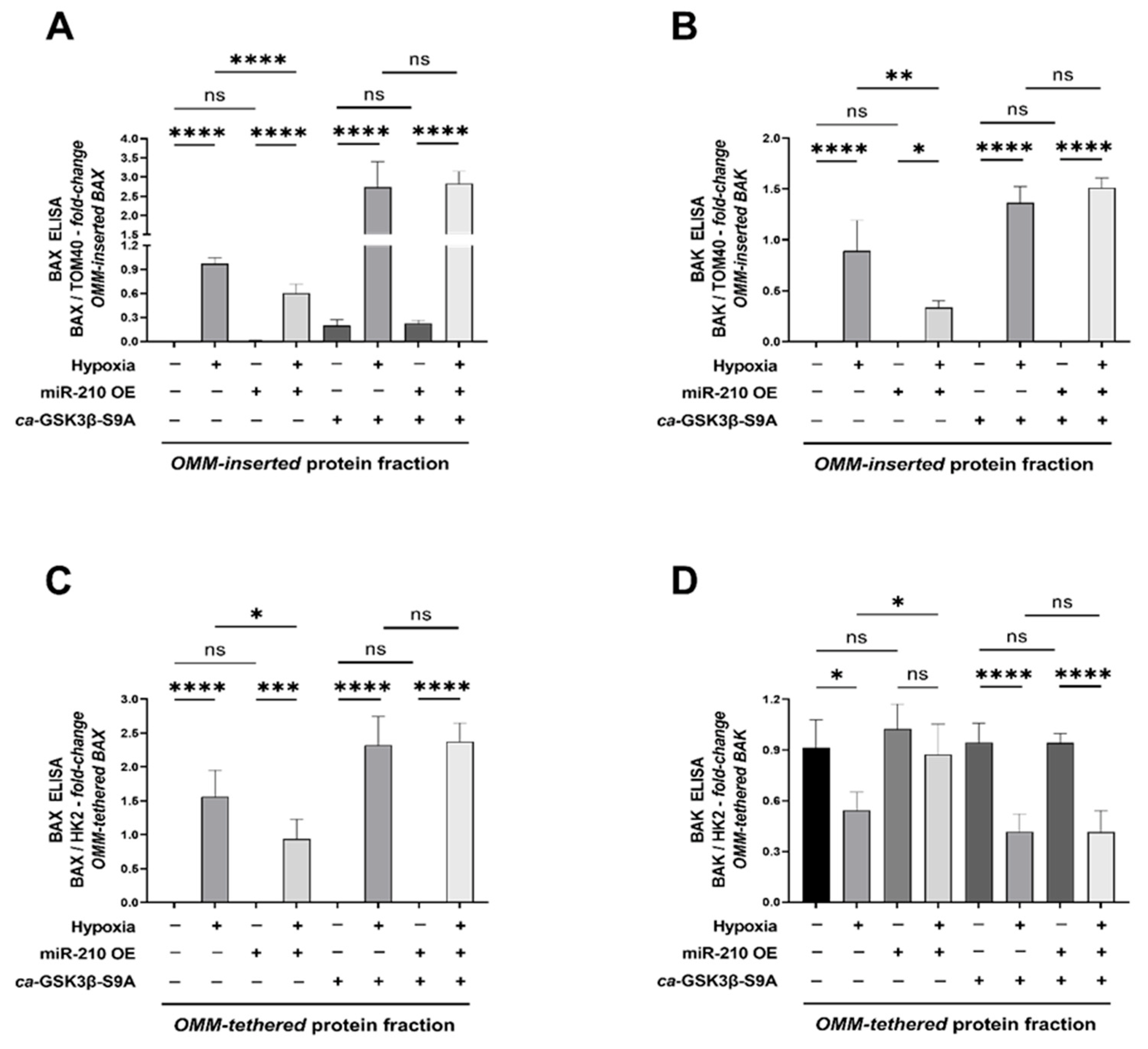
2.3. miR-210 Mitigates the Hypoxia-Induced Intrinsic Apoptotic Pathway through GSK3β Inhibition
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Western Blotting
4.3. Enzyme-Coupled miR-210 Hybridization Immunoassay
4.4. GSK3β Kinase Activity Assay
4.5. Lactate Dehydrogenase (LDH) Assay
4.6. Quantitative Measurement of Cleaved-Caspase-3, Cleaved-PARP, and Cleaved-DFF45
4.7. Caspase-3 Activity Assay
4.8. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
4.9. DFF40 (DFFβ, CAD) Endonuclease Activity Assay
4.10. Cytochrome C Release Assay
4.11. Cellular Fractionation to Segregate the Cytosolic and Mitochondrial Compartments
4.12. Isolation of Heavy Mitochondrial Fractions for ELISA Immunoassays
4.13. Quantitative Measurement of Members of the BCL2 Family of Proteins by Sandwich ELISA
4.14. Isolation and Preparation of Alkali-Resistant OMM (Outer Mitochondrial Membrane): Inserted and Alkali-Soluble OMM-Tethered Protein Fractions for the Quantitative Measurement of OMM-Inserted and OMM-Tethered BAX and BAK
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abbate, A.; Melfi, R.; Patti, G.; Baldi, F.; D’Ambrosio, A.; Manzoli, A.; Baldi, A.; di Sciascio, G. Apoptosis in recent myocardial infarction. La Clin. Ter. 2000, 151, 247–251. [Google Scholar]
- Olivetti, G.; Abbi, R.; Quaini, F.; Kajstura, J.; Cheng, W.; Nitahara, J.A.; Quaini, E.; Di Loreto, C.; Beltrami, C.A.; Krajewski, S.; et al. Apoptosis in the Failing Human Heart. N. Eng. J. Med. 1997, 336, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Saraste, A.; Pulkki, K.; Kallajoki, M.; Henriksen, K.; Parvinen, M.; Voipio-Pulkki, L.-M. Apoptosis in Human Acute Myocardial Infarction. Circulation 1997, 95, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Veinot, J.P.; Gattinger, D.A.; Fliss, H. Early apoptosis in human myocardial infarcts. Hum. Pathol. 1997, 28, 485–492. [Google Scholar] [CrossRef]
- Baldi, A.; Abbate, A.; Bussani, R.; Patti, G.; Melfi, R.; Angelini, A.; Dobrina, A.; Rossiello, R.; Silvestri, F.; Baldi, F.; et al. Apoptosis and Post-infarction Left Ventricular Remodeling. J. Mol. Cell. Cardiol. 2002, 34, 165–174. [Google Scholar] [CrossRef]
- Narula, J.; Haider, N.; Virmani, R.; DiSalvo, T.G.; Kolodgie, F.D.; Hajjar, R.J.; Schmidt, U.; Semigran, M.J.; Dec, G.W.; Khaw, B.-A. Apoptosis in Myocytes in End-Stage Heart Failure. N. Eng. J. Med. 1996, 335, 1182–1189. [Google Scholar] [CrossRef]
- Li, Y.; Wang, C.; Li, T.; Ma, L.; Fan, F.; Jin, Y.; Shen, J. The whole transcriptome and proteome changes in the early stage of myocardial infarction. Cell Death Discov. 2019, 5, 73. [Google Scholar] [CrossRef]
- Roy, S.; Khanna, S.; Kuhn, N.E.; Rink, C.; Williams, W.T.; Zweier, J.L.; Sen, C.K. Transcriptome analysis of the ischemia-reperfused remodeling myocardium: Temporal changes in inflammation and extracellular matrix. Physiol. Genom. 2006, 25, 364–374. [Google Scholar] [CrossRef]
- Datta, K.; Basak, T.; Varshney, S.; Sengupta, S.; Sarkar, S. Quantitative proteomic changes during post myocardial infarction remodeling reveals altered cardiac metabolism and Desmin aggregation in the infarct region. J. Proteom. 2017, 152, 283–299. [Google Scholar] [CrossRef]
- Lu, D.; Xia, Y.; Chen, Z.; Chen, A.; Wu, Y.; Jia, J.; Sun, A.; Zou, Y.; Qian, J.; Ge, J. Cardiac Proteome Profiling in Ischemic and Dilated Cardiomyopathy Mouse Models. Front. Physiol. 2019, 10, 750. [Google Scholar] [CrossRef]
- Simon, M.C.; Liu, L.; Barnhart, B.C.; Young, R.M. Hypoxia-Induced Signaling in the Cardiovascular System. Annu. Rev. Physiol. 2008, 70, 51–71. [Google Scholar] [CrossRef] [PubMed]
- Colombe, A.-S.; Pidoux, G. Cardiac Camp-Pka Signaling Compartmentalization in Myocardial Infarction. Cells 2021, 10, 922. [Google Scholar] [CrossRef] [PubMed]
- Stanton, L.W.; Garrard, L.J.; Damm, D.; Garrick, B.L.; Lam, A.; Kapoun, A.M.; Zheng, Q.; Protter, A.A.; Schreiner, G.F.; White, R.T. Altered Patterns of Gene Expression in Response to Myocardial Infarction. Circ. Res. 2000, 86, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, L.-L.; Cao, X.; Qi, L.-C.; Wei, G.-Q.; Liu, J.-Y.; Yan, S.-J.; Liu, J.-G.; Li, X.-Q. Bioinformatics analysis of time series gene expression in left ventricle (LV) with acute myocardial infarction (AMI). Gene 2014, 543, 259–267. [Google Scholar] [CrossRef]
- Zimmermann, M.; Beer, L.; Ullrich, R.; Lukovic, D.; Simader, E.; Traxler, D.; Wagner, T.; Nemec, L.; Altenburger, L.; Zuckermann, A.; et al. Analysis of region specific gene expression patterns in the heart and systemic responses after experimental myocardial ischemia. Oncotarget 2017, 8, 60809–60825. [Google Scholar] [CrossRef]
- Sun, T.; Dong, Y.-H.; Du, W.; Shi, C.-Y.; Wang, K.; Tariq, M.-A.; Wang, J.-X.; Li, P.-F. The Role of MicroRNAs in Myocardial Infarction: From Molecular Mechanism to Clinical Application. Int. J. Mol. Sci. 2017, 18, 745. [Google Scholar] [CrossRef]
- Fiedler, J.; Thum, T. MicroRNAs in Myocardial Infarction. Arter. Thromb. Vasc. Biol. 2013, 33, 201–205. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, L.; Lin, K.; Zhang, Q.; Chen, Y.; Li, R. Micrornas in Acute Myocardial Infarction: Evident Value as Novel Biomarkers? Anatol. J. Cardiol. 2018, 19, 140–147. [Google Scholar] [CrossRef]
- Bavelloni, A.; Ramazzotti, G.; Poli, A.; Piazzi, M.; Focaccia, E.; Blalock, W.; Faenza, I. MiRNA-210: A Current Overview. Anticancer Res. 2017, 37, 6511–6521. [Google Scholar]
- Chan, S.Y.; Loscalzo, J. MicroRNA-210: A unique and pleiotropic hypoxamir. Cell Cycle 2010, 9, 1072–1083. [Google Scholar] [CrossRef]
- Chan, Y.C.; Banerjee, J.; Choi, S.Y.; Sen, C.K. miR-210: The Master Hypoxamir. Microcirculation 2012, 19, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Song, X.; Sun, W.; Wang, Y.; Liu, B. Effect of Hypoxia-Induced MicroRNA-210 Expression on Cardiovascular Disease and the Underlying Mechanism. Oxidative Med. Cell. Longev. 2019, 2019, 4727283. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ding, L.; Bennewith, K.L.; Tong, R.T.; Welford, S.; Ang, K.K.; Story, M.; Le, Q.-T.; Giaccia, A.J. Hypoxia-Inducible mir-210 Regulates Normoxic Gene Expression Involved in Tumor Initiation. Mol. Cell 2009, 35, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Huang, X. miR-210: Fine-Tuning the Hypoxic Response. Adv. Exp. Med. Biol. 2014, 772, 205–227. [Google Scholar] [PubMed]
- Chan, S.Y.; Zhang, Y.-Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. Microrna-210 Controls Mitochondrial Metabolism During Hypoxia by Repressing the Iron-Sulfur Cluster Assembly Proteins Iscu1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef]
- Kim, H.W.; Haider, H.K.; Jiang, S.; Ashraf, M. Ischemic Preconditioning Augments Survival of Stem Cells via miR-210 Expression by Targeting Caspase-8-associated Protein 2. J. Biol. Chem. 2009, 284, 33161–33168. [Google Scholar] [CrossRef]
- Gou, D.; Ramchandran, R.; Peng, X.; Yao, L.; Kang, K.; Sarkar, J.; Wang, Z.; Zhou, G.; Raj, J.U. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L682–L691. [Google Scholar] [CrossRef]
- Hu, S.; Huang, M.; Li, Z.; Jia, F.; Ghosh, Z.; Lijkwan, M.A.; Fasanaro, P.; Sun, N.; Wang, X.; Martelli, F.; et al. MicroRNA-210 as a Novel Therapy for Treatment of Ischemic Heart Disease. Circulation 2010, 122, S124–S131. [Google Scholar] [CrossRef]
- Chio, C.-C.; Lin, J.-W.; Cheng, H.-A.; Chiu, W.-T.; Wang, Y.-H.; Wang, J.-J.; Hsing, C.-H.; Chen, R.-M. MicroRNA-210 targets antiapoptotic Bcl-2 expression and mediates hypoxia-induced apoptosis of neuroblastoma cells. Arch. Toxicol. 2012, 87, 459–468. [Google Scholar] [CrossRef]
- Qiu, J.; Zhou, X.-Y.; Cheng, R.; Liu, H.-Y.; Li, Y. Neuroprotective effects of microRNA-210 against oxygen-glucose deprivation through inhibition of apoptosis in PC12 cells. Mol. Med. Rep. 2013, 7, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xiong, L.; Huang, X.; Zhao, T.; Wu, L.-Y.; Liu, Z.-H.; Ding, X.; Liu, S.; Wu, Y.; Zhao, Y.; et al. miR-210 suppresses BNIP3 to protect against the apoptosis of neural progenitor cells. Stem Cell Res. 2013, 11, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Grosso, S.; Doyen, J.; Parks, S.K.; Bertero, T.; Paye, A.; Cardinaud, B.; Gounon, P.; Lacas-Gervais, S.; Noel, A.; Pouysségur, J.; et al. MiR-210 promotes a hypoxic phenotype and increases radioresistance in human lung cancer cell lines. Cell Death Dis. 2013, 4, e544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, H.; Dai, H.; Walsh, R.M.; Imakura, M.; Schelter, J.; Burchard, J.; Dai, X.; Chang, A.N.; Diaz, R.L.; et al. MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle 2009, 8, 2756–2768. [Google Scholar] [CrossRef]
- Marwarha, G.; Røsand, Ø.; Scrimgeour, N.; Slagsvold, K.H.; Høydal, M.A. miR-210 Regulates Apoptotic Cell Death during Cellular Hypoxia and Reoxygenation in a Diametrically Opposite Manner. Biomedicines 2021, 10, 42. [Google Scholar] [CrossRef]
- Beurel, E.; Jope, R.S. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006, 79, 173–189. [Google Scholar] [CrossRef]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Juhaszova, M.; Zorov, D.B.; Kim, S.-H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen Synthase Kinase-3beta Mediates Convergence of Protection Signaling to Inhibit the Mitochondrial Permeability Transition Pore. J. Clin. Investig. 2004, 113, 1535–1549. [Google Scholar] [CrossRef]
- Zhu, J.; Rebecchi, M.J.; Glass, P.S.; Brink, P.R.; Liu, L. Interactions of Gsk-3β with Mitochondrial Permeability Transition Pore Modulators During Preconditioning: Age-Associated Differences. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 395–403. [Google Scholar] [CrossRef]
- Gomez, L.; Paillard, M.; Thibault, H.; Derumeaux, G.; Ovize, M. Inhibition of Gsk3beta by Postconditioning Is Required to Prevent Opening of the Mitochondrial Permeability Transition Pore During Reperfusion. Circulation 2008, 117, 2761–2768. [Google Scholar] [CrossRef]
- Tanno, M.; Kuno, A.; Ishikawa, S.; Miki, T.; Kouzu, H.; Yano, T.; Murase, H.; Tobisawa, T.; Ogasawara, M.; Horio, Y.; et al. Translocation of Glycogen Synthase Kinase-3β (GSK-3β), a Trigger of Permeability Transition, Is Kinase Activity-dependent and Mediated by Interaction with Voltage-dependent Anion Channel 2 (VDAC2). J. Biol. Chem. 2014, 289, 29285–29296. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Cassina, A.; Lim, F.; Cerrato, T.; Palomo, G.M.; Diaz-Nido, J. Mitochondrial Hexokinase II Promotes Neuronal Survival and Acts Downstream of Glycogen Synthase Kinase-3. J. Biol. Chem. 2009, 284, 3001–3011. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3β by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Lal, H.; Ahmad, F.; Woodgett, J.; Force, T. The GSK-3 Family as Therapeutic Target for Myocardial Diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef]
- Tong, H.; Imahashi, K.; Steenbergen, C.; Murphy, E. Phosphorylation of Glycogen Synthase Kinase-3β During Preconditioning Through a Phosphatidylinositol-3-Kinase–Dependent Pathway Is Cardioprotective. Circ. Res. 2002, 90, 377–379. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Does Inhibition of Glycogen Synthase Kinase Protect in Mice? Circ. Res. 2008, 103, 226–228. [Google Scholar] [CrossRef]
- Yin, Z.; Gao, H.; Wang, H.; Li, L.; Di, C.; Luan, R.; Tao, L. Ischaemic Post-Conditioning Protects Both Adult and Aged Sprague-Dawley Rat Heart from Ischaemia-Reperfusion Injury through the Phosphatidylinositol 3-Kinase-Akt and Glycogen Synthase Kinase-3beta Pathways. Clin. Exp. Pharmacol. Physiol. 2009, 36, 756–763. [Google Scholar] [CrossRef]
- Miura, T.; Tanno, M. Mitochondria and GSK-3β in Cardioprotection Against Ischemia/Reperfusion Injury. Cardiovasc. Drugs Ther. 2010, 24, 255–263. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Wang, L.; Song, Y.; Wang, H.; Liu, K.; Shao, Z.; Shang, Z. Mir-210-3p-Ephrina3-Pi3k/Akt Axis Regulates the Progression of Oral Cancer. J. Cell. Mol. Med. 2020, 24, 4011–4022. [Google Scholar] [CrossRef]
- Lee, D.W.; Futami, M.; Carroll, M.; Feng, Y.; Wang, Z.; Fernandez, M.; Whichard, Z.; Chen, Y.; Kornblau, S.; Shpall, E.J.; et al. Loss of SHIP-1 protein expression in high-risk myelodysplastic syndromes is associated with miR-210 and miR-155. Oncogene 2012, 31, 4085–4094. [Google Scholar] [CrossRef] [PubMed]
- Pap, M.; Cooper, G.M. Role of Glycogen Synthase Kinase-3 in the Phosphatidylinositol 3-Kinase/Akt Cell Survival Pathway. J. Biol. Chem. 1998, 273, 19929–19932. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Johnson, G.V. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 2004, 29, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Welsh, G.I.; Proud, C. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem. J. 1993, 294, 625–629. [Google Scholar] [CrossRef]
- Hughes, K.; Nikolakaki, E.; Plyte, S.; Totty, N.F.; Woodgett, J.R. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 1993, 12, 803–808. [Google Scholar] [CrossRef]
- Ryves, W.J.; Dajani, R.; Pearl, L.; Harwood, A.J. Glycogen Synthase Kinase-3 Inhibition by Lithium and Beryllium Suggests the Presence of Two Magnesium Binding Sites. Biochem. Biophys. Res. Commun. 2002, 290, 967–972. [Google Scholar] [CrossRef]
- Ryves, W.; Fryer, L.; Dale, T.; Harwood, A. An Assay for Glycogen Synthase Kinase 3 (GSK-3) for Use in Crude Cell Extracts. Anal. Biochem. 1998, 264, 124–127. [Google Scholar] [CrossRef]
- Stambolic, V.; Woodgett, J.R. Mitogen Inactivation of Glycogen Synthase Kinase-3 Beta in Intact Cells Via Serine 9 Phosphorylation. Biochem. J. 1994, 303, 701–704. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Lazebnik, Y.A.; Kaufmann, S.; Desnoyers, S.; Poirier, G.G.; Earnshaw, W. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 1994, 371, 346–347. [Google Scholar] [CrossRef]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Soldani, C.; Scovassi, A.I. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: An update. Apoptosis 2002, 7, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.R.; Little, J.B.; Shipley, W.U. Association of mammalian cell death with a specific endonucleolytic degradation of DNA. Nature 1974, 252, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, A.H.; Morris, R.G.; Smith, A.L.; Dunlop, D. Chromatin cleavage in apoptosis: Association with condensed chromatin morphology and dependence on macromolecular synthesis. J. Pathol. 1984, 142, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Gorczyca, W.; Bruno, S.; Darzynkiewicz, R.; Gong, J. DNA Strand breaks occurring during apoptosis—Their early insitu detection by the terminal deoxynucleotidyl transferase and nick translation assays and prevention by serine protease inhibitors. Int. J. Oncol. 1992, 1, 639–648. [Google Scholar] [CrossRef]
- Sakahira, H.; Enari, M.; Nagata, S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998, 391, 96–99. [Google Scholar] [CrossRef]
- Liu, X.; Zou, H.; Slaughter, C.; Wang, X. DFF, a Heterodimeric Protein That Functions Downstream of Caspase-3 to Trigger DNA Fragmentation during Apoptosis. Cell 1997, 89, 175–184. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Scherer, D.C.; van Kaer, L.; Wang, X.; Xu, M. Resistance to DNA fragmentation and chromatin condensation in mice lacking the DNA fragmentation factor 45. Proc. Natl. Acad. Sci. USA 1998, 95, 12480–12485. [Google Scholar] [CrossRef]
- Guihard, G.; Bellot, G.; Moreau, C.; Pradal, G.; Ferry, N.; Thomy, R.; Fichet, P.; Meflah, K.; Vallette, F. The Mitochondrial Apoptosis-induced Channel (MAC) Corresponds to a Late Apoptotic Event. J. Biol. Chem. 2004, 279, 46542–46550. [Google Scholar] [CrossRef]
- Guo, L.; Pietkiewicz, D.; Pavlov, E.V.; Grigoriev, S.M.; Kasianowicz, J.J.; Dejean, L.M.; Korsmeyer, S.J.; Antonsson, B.; Kinnally, K.W. Effects of Cytochrome C on the Mitochondrial Apoptosis-Induced Channel Mac. Am. J. Physiol.-Cell Physiol. 2004, 286, C1109–C1117. [Google Scholar] [CrossRef]
- Pavlov, E.V.; Priault, M.; Pietkiewicz, D.; Cheng, E.H.-Y.; Antonsson, B.; Manon, S.; Korsmeyer, S.J.; Mannella, C.A.; Kinnally, K.W. A Novel, High Conductance Channel of Mitochondria Linked to Apoptosis in Mammalian Cells and Bax Expression in Yeast. J. Cell Biol. 2001, 155, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Dewson, G.; Kluck, R.M. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J. Cell Sci. 2009, 122, 2801–2808. [Google Scholar] [CrossRef] [PubMed]
- Westphal, D.; Kluck, R.M.; Dewson, G. Building blocks of the apoptotic pore: How Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014, 21, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Wolter, K.G.; Hsu, Y.-T.; Smith, C.L.; Nechushtan, A.; Xi, X.-G.; Youle, R.J. Movement of Bax from the Cytosol to Mitochondria during Apoptosis. J. Cell Biol. 1997, 139, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-T.; Wolter, K.G.; Youle, R.J. Cytosol-to-Membrane Redistribution of Bax and Bcl-XL During Apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 3668–3672. [Google Scholar] [CrossRef]
- Westphal, D.; Dewson, G.; Menard, M.; Frederick, P.; Iyer, S.; Bartolo, R.; Gibson, L.; Czabotar, P.E.; Smith, B.J.; Adams, J.M.; et al. Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc. Natl. Acad. Sci. USA 2014, 111, 4076–4085. [Google Scholar] [CrossRef]
- Fujiki, Y.; Fowler, S.; Shio, H.; Hubbard, A.L.; Lazarow, P.B. Polypeptide and phospholipid composition of the membrane of rat liver peroxisomes: Comparison with endoplasmic reticulum and mitochondrial membranes. J. Cell Biol. 1982, 93, 103–110. [Google Scholar] [CrossRef]
- Kim, H.; Botelho, S.C.; Park, K.; Kim, H. Use of carbonate extraction in analyzing moderately hydrophobic transmembrane proteins in the mitochondrial inner membrane. Protein Sci. 2015, 24, 2063–2069. [Google Scholar] [CrossRef]
- Fujiki, Y.; Hubbard, A.L.; Fowler, S.; Lazarow, P.B. Isolation of intracellular membranes by means of sodium carbonate treatment: Application to endoplasmic reticulum. J. Cell Biol. 1982, 93, 97–102. [Google Scholar] [CrossRef]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen Synthase Kinase-3 (Gsk3): Regulation, Actions, and Diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Zaccagnini, G.; Maimone, B.; Fuschi, P.; Maselli, D.; Spinetti, G.; Gaetano, C.; Martelli, F. Overexpression of miR-210 and its significance in ischemic tissue damage. Sci. Rep. 2017, 7, 9563. [Google Scholar] [CrossRef] [PubMed]
- Arif, M.; Pandey, R.; Alam, P.; Jiang, S.; Sadayappan, S.; Paul, A.; Ahmed, R.P.H. MicroRNA-210-mediated proliferation, survival, and angiogenesis promote cardiac repair post myocardial infarction in rodents. Klin. Wochenschr. 2017, 95, 1369–1385. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-N.; Wang, S.-H.; Su, X.-L.; Komal, S.; Fan, H.-K.; Xia, L.; Zhang, L.-R.; Han, S.-N. Targeting Glycogen Synthase Kinase 3 Beta Regulates Cd47 Expression after Myocardial Infarction in Rats Via the Nf-Κb Signaling Pathway. Front. Pharmacol. 2021, 12, 662726. [Google Scholar] [CrossRef]
- Jacobs, K.M.; Bhave, S.R.; Ferraro, D.J.; Jaboin, J.J.; Hallahan, D.E.; Thotala, D. Gsk-3β: A Bifunctional Role in Cell Death Pathways. Int. J. Cell Biol. 2012, 2012, 930710. [Google Scholar] [CrossRef]
- Yoshino, Y.; Ishioka, C. Inhibition of glycogen synthase kinase-3 beta induces apoptosis and mitotic catastrophe by disrupting centrosome regulation in cancer cells. Sci. Rep. 2015, 5, 13249. [Google Scholar] [CrossRef]
- Dennison, C. A Guide to Protein Isolation; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T.; Irwin, N. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Deverre, J.-R.; Boutet, V.; Boquet, D.; Ezan, E.; Grassi, J.; Grognet, J.-M. A competitive enzyme hybridization assay for plasma determination of phosphodiester and phosphorothioate antisense oligonucleotides. Nucleic Acids Res. 1997, 25, 3584–3589. [Google Scholar] [CrossRef]
- Tremblay, G.; Khalafaghian, G.; Legault, J.; Nielsen, P.; Bartlett, A. Dual ligation hybridization assay for the specific determination of oligonucleotide therapeutics. Bioanalysis 2011, 3, 499–508. [Google Scholar] [CrossRef]
- Chaiet, L.; Wolf, F.J. The properties of streptavidin, a biotin-binding protein produced by Streptomycetes. Arch. Biochem. Biophys. 1964, 106, 1–5. [Google Scholar] [CrossRef]
- Dundas, C.M.; Demonte, D.; Park, S. Streptavidin–biotin technology: Improvements and innovations in chemical and biological applications. Appl. Microbiol. Biotechnol. 2013, 97, 9343–9353. [Google Scholar] [CrossRef] [PubMed]
- Diamandis, E.P.; Christopoulos, T.K. The biotin-(strept)avidin system: Principles and applications in biotechnology. Clin. Chem. 1991, 37, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Sélo, I.; Négroni, L.; Créminon, C.; Grassi, J.; Wal, J.M. Preferential Labeling of Alpha-Amino N-Terminal Groups in Peptides by Biotin: Application to the Detection of Specific Anti-Peptide Antibodies by Enzyme Immunoassays. J. Immunol. Methods 1996, 199, 127–138. [Google Scholar] [CrossRef]
- Coghlan, M.P.; Culbert, A.A.; Cross, D.A.; Corcoran, S.L.; Yates, J.W.; Pearce, N.J.; Rausch, O.L.; Murphy, G.J.; Carter, P.S.; Cox, L.R.; et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7, 793–803. [Google Scholar] [CrossRef]
- Butler, J.; Ni, L.; Nessler, R.; Joshi, K.; Suter, M.; Rosenberg, B.; Chang, J.; Brown, W.; Cantarero, L. The physical and functional behavior of capture antibodies adsorbed on polystyrene. J. Immunol. Methods 1992, 150, 77–90. [Google Scholar] [CrossRef]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef]
- Liu, Z.-Q.; Du, J.-J.; Ren, J.-J.; Zhang, Z.-Y.; Guo, X.-B.; Yan, Y.-E.; Jia, X.-T.; Gu, N.-B.; Di, Z.-L.; Li, S.-Z. miR-183-96-182 clusters alleviated ox-LDL-induced vascular endothelial cell apoptosis in vitro by targeting FOXO1. RSC Adv. 2018, 8, 35031–35041. [Google Scholar] [CrossRef]
- Riccio, G.; Esposito, G.; Leoncini, E.; Contu, R.; Condorelli, G.; Chiariello, M.; Laccetti, P.; Hrelia, S.; D’Alessio, G.; De Lorenzo, C. Cardiotoxic Effects, or Lack Thereof, of Anti-Erbb2 Immunoagents. FASEB J. 2009, 23, 3171–3178. [Google Scholar] [CrossRef]
- Bollo, E.; Guglielmino, R.; Sant, S.; Pregel, P.; Riondato, F.; Miniscalco, B.; Cornaglia, E.; Nebbia, C.; Dacasto, M. Biochemical, ultrastructural and molecular characterization of the triphenyltin acetate (TPTA)-induced apoptosis in primary cultures of mouse thymocytes. Cell Biol. Toxicol. 2006, 22, 275–284. [Google Scholar] [CrossRef]
- Wang, G.; Wu, Y.; Zhu, Y. Mechanism of Malat1 Preventing Apoptosis of Vascular Endothelial Cells Induced by Oxygen–Glucose Deficiency and Reoxidation. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. S1), 798–805. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Stetler, R.A.; Cao, G.; Pei, W.; O’Horo, C.; Yin, X.M.; Chen, J. Characterization of the Rat DNA Fragmentation Factor 35/Inhibitor of Caspase-Activated Dnase (Short Form). The Endogenous Inhibitor of Caspase-Dependent DNA Fragmentation in Neuronal Apoptosis. J. Biol. Chem. 2000, 275, 38508–38517. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Pei, W.; Lan, J.; Stetler, R.A.; Luo, Y.; Nagayama, T.; Graham, S.H.; Yin, X.-M.; Simon, R.P.; Chen, J. Caspase-Activated DNase/DNA Fragmentation Factor 40 Mediates Apoptotic DNA Fragmentation in Transient Cerebral Ischemia and in Neuronal Cultures. J. Neurosci. 2001, 21, 4678–4690. [Google Scholar] [CrossRef]
- Link, A.J.; LaBaer, J. Trichloroacetic acid (TCA) precipitation of proteins. Cold Spring Harb. Protoc. 2011, 2011, 993–994. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Empty Vector (pEZX-MR04-Scrambled) | miR-210 Overexpression (OE) Vector (pEZX-MR04-miR-210) | Control Empty Vector (pcDNA3) | HA-Tagged GSK3 Beta S9A (pcDNA3-HA-GSK3β S9A) | |
|---|---|---|---|---|
| Normoxia, 18 h | n = 4 | n = 4 | n = 4 | n = 4 |
| Hypoxia, 18 h | n = 4 | n = 4 | n = 4 | n = 4 |
| Antibody | Application | Amount | Host | Manufacturer | Catalogue # | Resource Identifier ID (RRID) |
|---|---|---|---|---|---|---|
| β-Actin | WB 1:5000 | 1 µg | Mouse | Santa Cruz Biotechnology | sc-47778 | AB_2714189 |
| β-Actin | ELISA capture | 20 ng/well | Mouse | Santa Cruz Biotechnology | sc-47778 | AB_2714189 |
| β-Actin | ELISA detection | 20 ng/well | Rabbit | Cell Signaling Technology | 4970 | AB_2223172 |
| β-Actin antibody blocking peptide | ELISA detection | N/A | N/A | Cell Signaling Technology | 1025 | N/A |
| BAK | ELISA capture | 20 ng/well | Mouse | Thermo Fisher Scientific | MA5-36225 | AB_2884059 |
| BAK | ELISA detection | 20 ng/well | Rabbit | Novus Biologicals | NBP1-77152 | AB_11014847 |
| BAK antibody blocking peptide | ELISA detection | N/A | N/A | Novus Biologicals | NBP1-77152PEP | N/A |
| BAX | ELISA capture | 20 ng/well | Mouse | Thermo Fisher Scientific | 33-6600 | AB_2533133 |
| BAX | ELISA detection | 20 ng/well | Rabbit | Novus Biologicals | NBP1-88682 | AB_11014342 |
| BAX antibody blocking peptide | ELISA detection | N/A | N/A | Novus Biologicals | NBP1-88682PEP | N/A |
| BCL2 | ELISA capture | 20 ng/well | Mouse | Thermo Fisher Scientific | BMS1028 | AB_10597451 |
| BCL2 | ELISA detection | 20 ng/well | Rabbit | Thermo Fisher Scientific | PA5-20068 | AB_11152761 |
| BCL2 antibody blocking peptide | ELISA detection | N/A | N/A | Thermo Fisher Scientific | PEP-0187 | N/A |
| Caspase-3 | WB 1:1000 | 5 µg | Rabbit | Cell Signaling Technology | 14220 | AB_2798429 |
| cleaved-caspase-3 (Asp175) | ELISA capture | 20 ng/well | Rabbit | Cell Signaling Technology | 9579 | AB_10897512 |
| cleaved-caspase-3 (Asp175) (biotinylated) | ELISA detection | 20 ng/well | Rabbit | Cell Signaling Technology | 9654 | AB_10694088 |
| cleaved-caspase-3 (Asp175) antibody blocking peptide | ELISA detection | N/A | N/A | Cell Signaling Technology | 1050 | N/A |
| COX4 | WB 1:1000 | 5 µg | Rabbit | Cell Signaling Technology | 4844 | AB_2085427 |
| COX4 | ELISA capture | 20 ng/well | Mouse | Thermo Fisher Scientific | MA5-15686 | AB_10977841 |
| COX4 | ELISA detection | 20 ng/well | Rabbit | Cell Signaling Technology | 4844 | AB_2085427 |
| COX4 antibody blocking peptide | ELISA detection | N/A | N/A | Cell Signaling Technology | 1034 | N/A |
| DFF45 | WB 1:500 | 5 µg | Rabbit | Cell Signaling Technology | 9732 | AB_329956 |
| cleaved-DFF45 (Asp224) | WB 1:500 | 5 µg | Rabbit | Cell Signaling Technology | 9731 | AB_329954 |
| cleaved-DFF45 (Asp224) | ELISA capture | 20 ng/well | Rabbit | Cell Signaling Technology | 9731 | AB_329954 |
| cleaved-DFF45 (Asp224) (biotinylated) | ELISA detection | 20 ng/well | Rabbit | Cell Signaling Technology | 9731 | AB_329954 |
| Cytochrome C | ELISA capture | 20 ng/well | Mouse | Thermo Fisher Scientific | BMS1037 | AB_10598651 |
| Cytochrome C | ELISA detection | 20 ng/well | Rabbit | Cell Signaling Technology | 4280 | AB_10695410 |
| Goat Anti-Mouse IgG (H + L)-HRP Conjugate | 1:5000 | 1 µg | Goat | Bio-Rad | 1706516 | AB_11125547 |
| Goat Anti-Mouse IgG-AP Conjugate | 1:5000 | N/A € | Goat | Bio-Rad | 1706520 | AB_11125348 |
| Goat Anti-Rabbit IgG (H + L)-HRP Conjugate | 1:5000 | 1 µg | Goat | Bio-Rad | 1706515 | AB_11125142 |
| Goat Anti-Rabbit IgG-AP Conjugate | 1:20,000 | N/A € | Goat | Sigma Aldrich/Merck Life Science | A3687 | AB_258103 |
| GSK3β | WB 1:1000 | 5 µg | Rabbit | Cell Signaling Technology | 9315 | AB_490890 |
| p-Ser9 GSK3β | WB 1:1000 | 5 µg | Rabbit | Cell Signaling Technology | 9322 | AB_2115196 |
| p-Tyr279/Tyr216 GSK3α/β | WB 1:1000 | 5 µg | Rabbit | Thermo Fisher Scientific | PA5-36646 | AB_2553634 |
| HA tag | WB 1:1000 | 5 µg | Mouse | Thermo Fisher Scientific | 26183 | AB_10978021 |
| HA tag | ELISA capture | 30 ng/well | Mouse | Thermo Fisher Scientific | 26183 | AB_10978021 |
| HA tag | ELISA detection | 30 ng/well | Rabbit | Abcam | ab13834 | AB_443010 |
| HA tag antibody blocking peptide | ELISA detection | N/A | N/A | Abcam | ab13835 | N/A |
| HK2 | ELISA capture | 30 ng/well | Rabbit | Thermo Fisher Scientific | PA5-97828 | AB_2812442 |
| HK2 | ELISA detection | 30 ng/well | Mouse | Thermo Fisher Scientific | MA5-15679 | AB_10986812 |
| LDH | ELISA capture | 30 ng/well | Mouse | Santa Cruz Biotechnology | sc-133123 | AB_2134964 |
| LDH-A | ELISA detection | 30 ng/well | Rabbit | Novus Biologicals | NBP1-48336 | AB_10011099 |
| LDH-A antibody blocking peptide | ELISA detection | N/A | N/A | Novus Biologicals | NBP1-48336PEP | N/A |
| LDH-B | ELISA detection | 30 ng/well | Rabbit | Novus Biologicals | NBP2-38131 | N/A |
| LDH-A antibody blocking peptide | ELISA detection | N/A | N/A | Novus Biologicals | NBP2-38131PEP | N/A |
| MCL1 | ELISA capture | 20 ng/well | Mouse | Thermo Fisher Scientific | MA5-15236 | AB_10986161 |
| MCL1 | ELISA detection | 20 ng/well | Rabbit | Thermo Fisher Scientific | PA5-20121 | AB_11152825 |
| MCL1 antibody blocking peptide | ELISA detection | N/A | N/A | Thermo Fisher Scientific | PEP-0239 | N/A |
| PARP | WB 1:1000 | 5 µg | Rabbit | Cell Signaling Technology | 9542 | AB_2160739 |
| cleaved-PARP (Asp214) | WB 1:1000 | 5 µg | Mouse | Cell Signaling Technology | 9546 | AB_2160593 |
| cleaved-PARP (Asp214) | ELISA capture | 20 ng/well | Mouse | Cell Signaling Technology | 9546 | AB_2160593 |
| cleaved-PARP (Asp214) (biotinylated) | ELISA detection | 20 ng/well | Rabbit | Cell Signaling Technology | 9185 | AB_10858875 |
| p-Ser | GSK3β activity | 100 ng/well | Mouse | Santa Cruz Biotechnology | sc-81516 | AB_1128626 |
| SDHA (SDH2) | ELISA capture | 30 ng/well | Mouse | Thermo Fisher Scientific | 459200 | AB_2532231 |
| SDHA (SDH2) | ELISA detection | 30 ng/well | Rabbit | Cell Signaling Technology | 11998 | AB_2750900 |
| TOM40 | ELISA capture | 30 ng/well | Mouse | Santa Cruz Biotechnology | sc-365467 | AB_10847086 |
| TOM40 | ELISA detection | 30 ng/well | Rabbit | Thermo Fisher Scientific | 18409-1-AP | AB_2303725 |
| TIM22 | ELISA capture | 30 ng/well | Mouse | Sigma Aldrich/Merck Life Science | SAB1400520- | AB_1858016 |
| TIM22 | ELISA detection | 30 ng/well | Rabbit | Thermo Fisher Scientific | 14927-1-AP | AB_11183050 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marwarha, G.; Røsand, Ø.; Slagsvold, K.H.; Høydal, M.A. GSK3β Inhibition Is the Molecular Pivot That Underlies the Mir-210-Induced Attenuation of Intrinsic Apoptosis Cascade during Hypoxia. Int. J. Mol. Sci. 2022, 23, 9375. https://doi.org/10.3390/ijms23169375
Marwarha G, Røsand Ø, Slagsvold KH, Høydal MA. GSK3β Inhibition Is the Molecular Pivot That Underlies the Mir-210-Induced Attenuation of Intrinsic Apoptosis Cascade during Hypoxia. International Journal of Molecular Sciences. 2022; 23(16):9375. https://doi.org/10.3390/ijms23169375
Chicago/Turabian StyleMarwarha, Gurdeep, Øystein Røsand, Katrine Hordnes Slagsvold, and Morten Andre Høydal. 2022. "GSK3β Inhibition Is the Molecular Pivot That Underlies the Mir-210-Induced Attenuation of Intrinsic Apoptosis Cascade during Hypoxia" International Journal of Molecular Sciences 23, no. 16: 9375. https://doi.org/10.3390/ijms23169375