
Molecular Mechanisms of Diabetic Kidney Disease

 , , , ,
, , , ,
Abstract
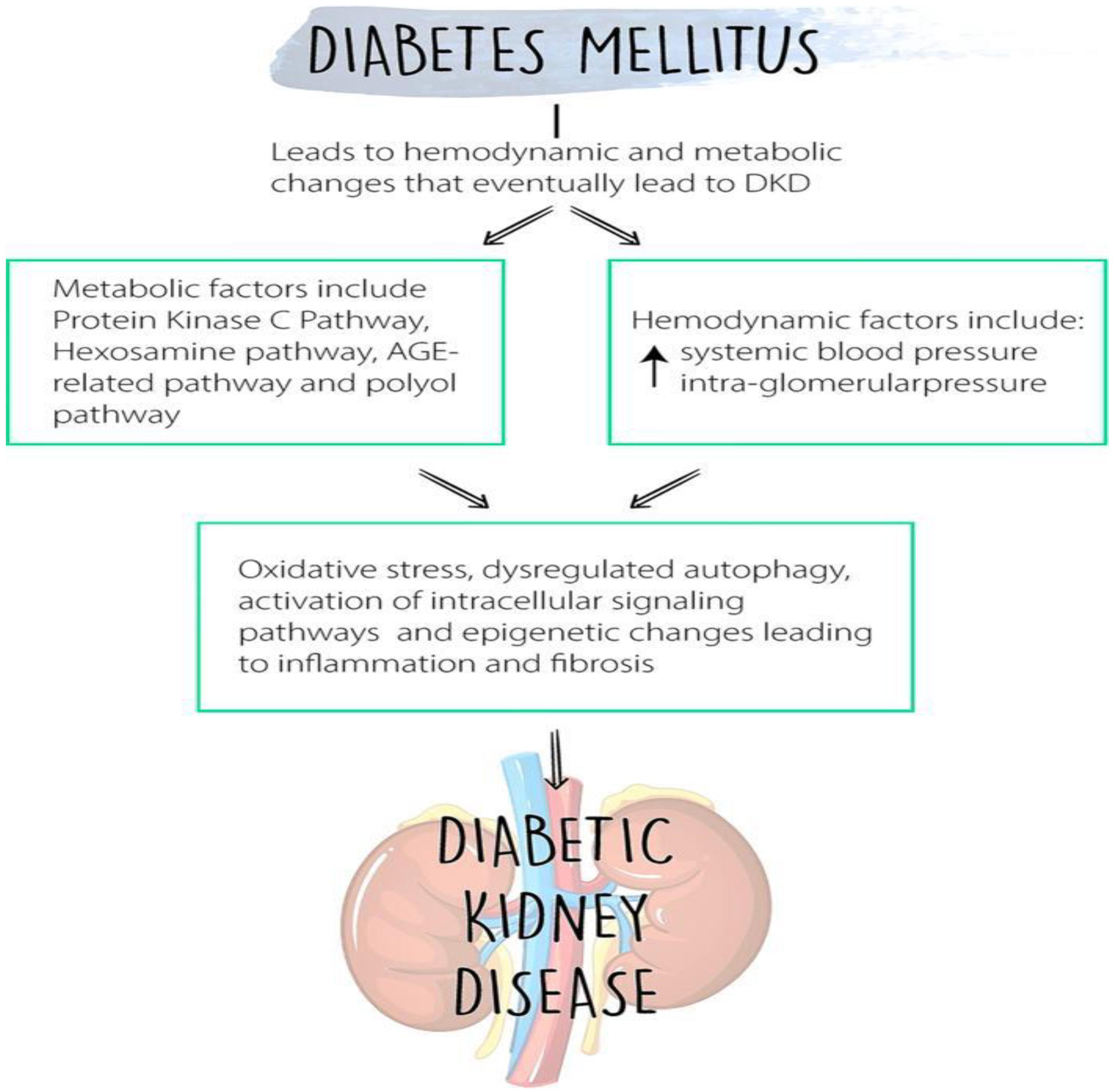
:1. Introduction
2. Genetics and Epigenetics of DKD
3. Fundamentals of Metabolomics and Proteomics in Kidney Disease
4. Toll-like Receptors and Nod-like Receptors
5. The Kallikrein Kinin System (KKS) and DKD
6. Innate Immune Cells
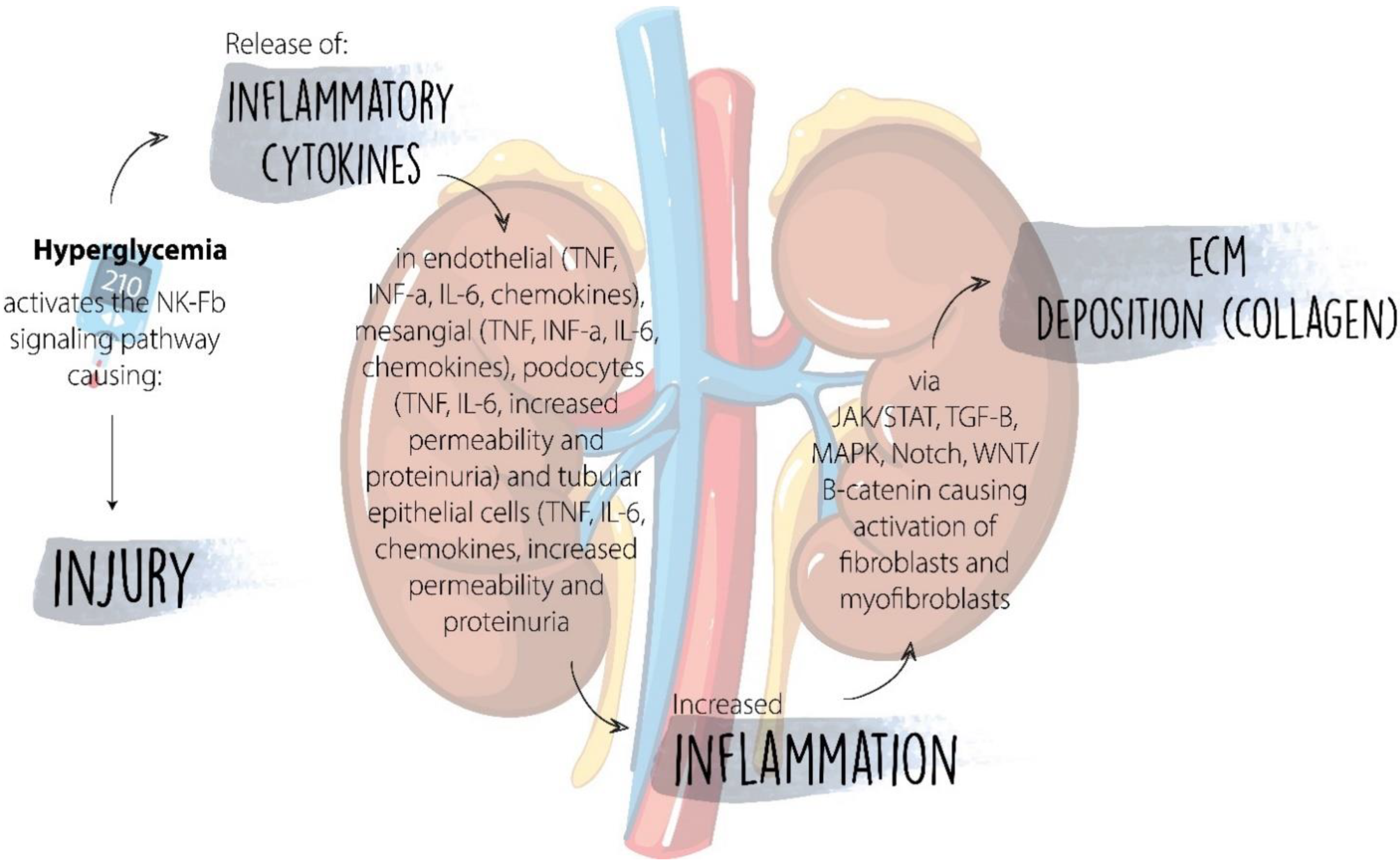
7. Inflammatory Cytokines
8. Chemokines and Their Receptors
9. Complement System
10. Cells of the Adaptive Immune System
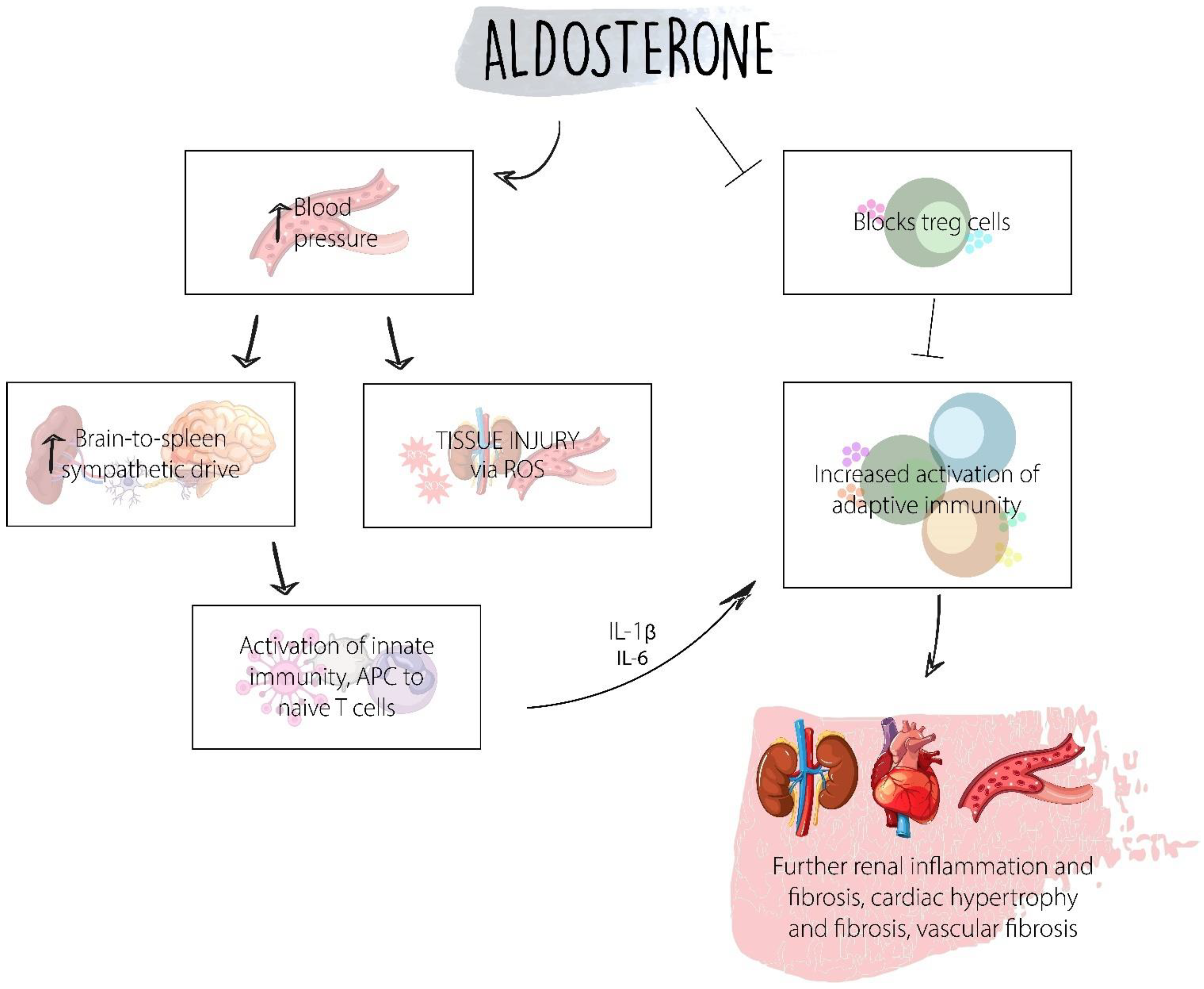
11. Aldosterone
12. Signaling Pathways in DKD
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Turkmen, K. Inflammation, oxidative stress, apoptosis, and autophagy in diabetes mellitus and diabetic kidney disease: The Four Horsemen of the Apocalypse. Int. Urol. Nephrol. 2017, 49, 837–844. [Google Scholar] [CrossRef]
- Samsu, N. Diabetic Nephropathy: Challenges in Pathogenesis, Diagnosis, and Treatment. BioMed. Res. Int. 2021, 2021, 1497449. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.W.; Yiu, W.H. Innate immunity in diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.W.; Moon, J.Y. The role of inflammation in diabetic kidney disease. Korean J. Intern. Med. 2021, 36, 753–766. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Park, A.S.D.; Mohtat, D.; Thomas, D.B.; Pullman, J.M.; Susztak, K. Transcriptome analysis of human diabetic kidney disease. Diabetes 2011, 60, 2354–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiritoshi, S.; Nishikawa, T.; Sonoda, K.; Kukidome, D.; Senokuchi, T.; Matsuo, T.; Matsumura, T.; Tokunaga, H.; Brownlee, M.; Araki, E. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells: Potential role in diabetic nephropathy. Diabetes 2003, 52, 2570–2577. [Google Scholar] [CrossRef] [PubMed]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef]
- Fontalvo, J.E.R. Guía de práctica clínica para la enfermedad renal diabética. Rev. Colomb. Nefrol. 2021, 8. [Google Scholar] [CrossRef]
- Dubin, R.F.; Rhee, E.P. Proteomics and Metabolomics in Kidney Disease, including Insights into Etiology, Treatment, and Prevention. Clin. J. Am. Soc. Nephrol. 2020, 15, 404–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, H.F. Genetic and Epigenetic Studies in Diabetic Kidney Disease. Front. Genet. 2019, 10, 507. [Google Scholar] [CrossRef]
- Rico Fontalvo, J. Enfermedad renal diabética: De cara a la prevención, diagnóstico e intervención temprana. Rev. Colomb. Nefrol. 2020, 7, 15–16. [Google Scholar] [CrossRef]
- Shao, B.-Y.; Zhang, S.-F.; Li, H.-D.; Meng, X.-M.; Chen, H.-Y. Epigenetics and Inflammation in Diabetic Nephropathy. Front. Physiol. 2021, 12, 607. [Google Scholar] [CrossRef] [PubMed]
- Van Zuydam, N.R.; Ahlqvist, E.; Sandholm, N.; Deshmukh, H.; Rayner, N.W.; Abdalla, M.; Ladenvall, C.; Ziemek, D.; Fauman, E.; Robertson, N.R.; et al. A Genome-Wide Association Study of Diabetic Kidney Disease in Subjects with Type 2 Diabetes. Diabetes 2018, 67, 1414–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopera Vargas, J.M.; Rico Fontalvo, J.E.; Melgarejo, R.E.; Castillo Barrios, G.A.; Ramírez Rincón, A.; Gomez, A.M.; Martínez Rojas, S.; Ibatá Bernal, L. Effect of pharmacological therapies for glycemic control in patients with type 2 diabetes mellitus on vascular outcomes. Rev. Colomb. Nefrol. 2020, 7, 44–59. [Google Scholar] [CrossRef]
- Pérez-López, L.; Boronat, M.; Melián, C.; Brito-Casillas, Y.; Wägner, A.M. Animal Models and Renal Biomarkers of Diabetic Nephropathy. In Diabetes: From Research to Clinical Practice; Springer: Berlin/Heidelberg, Germany, 2020; pp. 521–551. [Google Scholar]
- Hall, J.A.; Yerramilli, M.; Obare, E.; Li, J.; Yerramilli, M.; Jewell, D.E. Serum concentrations of symmetric dimethylarginine and creatinine in cats with kidney stones. PLoS ONE 2017, 12, e0174854. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.; Yerramilli, M.; Obare, E.; Yu, S.; Jewell, D. Comparison of serum concentrations of symmetric dimethylarginine and creatinine as kidney function biomarkers in healthy geriatric cats fed reduced protein foods enriched with fish oil, L-carnitine, and medium-chain triglycerides. Veter-J. 2014, 202, 588–596. [Google Scholar] [CrossRef]
- Pelander, L.; Häggström, J.; Larsson, A.; Syme, H.; Elliott, J.; Heiene, R.; Ljungvall, I. Comparison of the diagnostic value of symmetric dimethylarginine, cystatin C, and creatinine for detection of decreased glomerular filtration rate in dogs. J. Veter-Intern. Med. 2018, 33, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Togashi, Y.; Miyamoto, Y. Urinary cystatin C as a biomarker for diabetic nephropathy and its immunohistochemical localization in kidney in Zucker diabetic fatty (ZDF) rats. Exp. Toxicol. Pathol. 2013, 65, 615–622. [Google Scholar] [CrossRef]
- van Hoek, I.; Daminet, S.; Notebaert, S.; Janssens, I.; Meyer, E. Immunoassay of urinary retinol binding protein as a putative renal marker in cats. J. Immunol. Methods 2008, 329, 208–213. [Google Scholar] [CrossRef]
- Steinbach, S.; Weis, J.; Schweighauser, A.; Francey, T.; Neiger, R. Plasma and Urine Neutrophil Gelatinase-Associated Lipocalin (NGAL) in Dogs with Acute Kidney Injury or Chronic Kidney Disease. J. Veter-Intern. Med. 2014, 28, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Hosohata, K.; Ando, H.; Takeshita, Y.; Misu, H.; Takamura, T.; Kaneko, S.; Fujimura, A. Urinary Kim-1 is a sensitive biomarker for the early stage of diabetic nephropathy in Otsuka Long-Evans Tokushima Fatty rats. Diabetes Vasc. Dis. Res. 2014, 11, 243–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colhoun, H.M.; Marcovecchio, M.L. Biomarkers of diabetic kidney disease. Diabetologia 2018, 61, 996–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, A.; Östgren, C.; Länne, T.; Larsson, A.; Nystrom, F.; Ärnlöv, J. The association between endostatin and kidney disease and mortality in patients with type 2 diabetes. Diabetes Metab. 2016, 42, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Dieter, B.P.; McPherson, S.M.; Afkarian, M.; de Boer, I.H.; Mehrotra, R.; Short, R.; Barbosa-Leiker, C.; Alicic, R.Z.; Meek, R.L.; Tuttle, K.R. Serum amyloid a and risk of death and end-stage renal disease in diabetic kidney disease. J. Diabetes Complicat. 2016, 30, 1467–1472. [Google Scholar] [CrossRef] [Green Version]
- Garg, V.; Kumar, M.; Mahapatra, H.S.; Chitkara, A.; Gadpayle, A.K.; Sekhar, V. Novel urinary biomarkers in pre-diabetic nephropathy. Clin. Exp. Nephrol. 2015, 19, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Fufaa, G.D.; Weil, E.J.; Nelson, R.G.; Hanson, R.L.; Bonventre, J.V.; Sabbisetti, V.; Waikar, S.S.; Mifflin, T.E.; Zhang, X.; Xie, D.; et al. Association of urinary KIM-1, L-FABP, NAG and NGAL with incident end-stage renal disease and mortality in American Indians with type 2 diabetes mellitus. Diabetologia 2014, 58, 188–198. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Virella, M.F.; Baker, N.L.; Hunt, K.J.; Cleary, P.A.; Klein, R.; Virella, G. The DCCT/EDIC Research Group Baseline Markers of Inflammation Are Associated with Progression to Macroalbuminuria in Type 1 Diabetic Subjects. Diabetes Care 2013, 36, 2317–2323. [Google Scholar] [CrossRef] [Green Version]
- Araki, S.-I.; Haneda, M.; Koya, D.; Sugaya, T.; Isshiki, K.; Kume, S.; Kashiwagi, A.; Uzu, T.; Maegawa, H. Predictive Effects of Urinary Liver-Type Fatty Acid–Binding Protein for Deteriorating Renal Function and Incidence of Cardiovascular Disease in Type 2 Diabetic Patients without Advanced Nephropathy. Diabetes Care 2013, 36, 1248–1253. [Google Scholar] [CrossRef] [Green Version]
- Niewczas, M.A.; Gohda, T.; Skupien, J.; Smiles, A.M.; Walker, W.H.; Rosetti, F.; Cullere, X.; Eckfeldt, J.H.; Doria, A.; Mayadas, T.N.; et al. Circulating TNF Receptors 1 and 2 Predict ESRD in Type 2 Diabetes. J. Am. Soc. Nephrol. 2012, 23, 507–515. [Google Scholar] [CrossRef]
- Fu, W.-J.; Li, B.-L.; Wang, S.-B.; Chen, M.-L.; Deng, R.-T.; Ye, C.-Q.; Liu, L.; Fang, A.-J.; Xiong, S.-L.; Wen, S.; et al. Changes of the tubular markers in type 2 diabetes mellitus with glomerular hyperfiltration. Diabetes Res. Clin. Pr. 2012, 95, 105–109. [Google Scholar] [CrossRef]
- Foster, M.C.; Inker, L.A.; Hsu, C.-Y.; Eckfeldt, J.H.; Levey, A.S.; Pavkov, M.E.; Myers, B.D.; Bennett, P.H.; Kimmel, P.L.; Vasan, R.S.; et al. Filtration Markers as Predictors of ESRD and Mortality in Southwestern American Indians with Type 2 Diabetes. Am. J. Kidney Dis. 2015, 66, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidya, V.S.; Niewczas, M.A.; Ficociello, L.H.; Johnson, A.C.; Collings, F.B.; Warram, J.H.; Krolewski, A.S.; Bonventre, J.V. Regression of microalbuminuria in type 1 diabetes is associated with lower levels of urinary tubular injury biomarkers, kidney injury molecule-1, and N-acetyl-β-D-glucosaminidase. Kidney Int. 2011, 79, 464–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Zhang, Z.; Li, Y. Relevance of the Pyroptosis-Related Inflammasome Pathway in the Pathogenesis of Diabetic Kidney Disease. Front. Immunol. 2021, 12, 603416. [Google Scholar] [CrossRef]
- Pereira, P.R.; Carrageta, D.F.; Oliveira, P.F.; Rodrigues, A.; Alves, M.G.; Monteiro, M.P. Metabolomics as a tool for the early diagnosis and prognosis of diabetic kidney disease. Med. Res. Rev. 2022, 42, 1518–1544. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Dasu, M.R.; Rockwood, J.; Winter, W.; Griffen, S.C.; Jialal, I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: Further evidence of a proinflammatory state. J. Clin. Endocrinol. Metab. 2008, 93, 578–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulay, S.R. Multifactorial functions of the inflammasome component NLRP3 in pathogenesis of chronic kidney diseases. Kidney Int. 2019, 96, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef] [Green Version]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Böttinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef]
- Har, R.; Scholey, J.W.; Daneman, D.; Mahmud, F.H.; Dekker, R.; Lai, V.; Elia, Y.; Fritzler, M.L.; Sochett, E.B.; Reich, H.N.; et al. The effect of renal hyperfiltration on urinary inflammatory cytokines/chemokines in patients with uncomplicated type 1 diabetes mellitus. Diabetologia 2013, 56, 1166–1173. [Google Scholar] [CrossRef] [Green Version]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ma, F.Y.; Ozols, E.; Rollins, B.J.; Tesch, G.H. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia 2007, 50, 471–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ozols, E.; Atkins, R.C.; Rollin, B.J.; Tesch, G.H. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006, 69, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-González, J.F.; Mora-Fernández, C.; Muros de Fuentes, M.; García-Pérez, J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 327–340. [Google Scholar] [CrossRef]
- Pickup, J.C.; Chusney, G.D.; Thomas, S.M.; Burt, D. Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci. 2000, 67, 291–300. [Google Scholar] [CrossRef]
- Sangoi, M.B.; de Carvalho, J.A.M.; Tatsch, E.; Hausen, B.S.; Bollick, Y.S.; Londero, S.W.K.; Duarte, T.; Scolari, R.; Duarte, M.M.M.F.; Premaor, M.O.; et al. Urinary inflammatory cytokines as indicators of kidney damage in type 2 diabetic patients. Clin. Chim. Acta Int. J. Clin. Chem. 2016, 460, 178–183. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Agarwal, R.; Alpers, C.E.; Bakris, G.L.; Brosius, F.C.; Kolkhof, P.; Uribarri, J. Molecular mechanisms and therapeutic targets for diabetic kidney disease. Kidney Int. 2022, 102, 248–260. [Google Scholar] [CrossRef]
- Park, J.; Guan, Y.; Sheng, X.; Gluck, C.; Seasock, M.J.; Hakimi, A.A.; Qiu, C.; Pullman, J.; Verma, A.; Li, H.; et al. Functional methylome analysis of human diabetic kidney disease. JCI Insight 2019, 4, e128886. [Google Scholar] [CrossRef] [PubMed]
- Niewczas, M.A.; Ficociello, L.H.; Johnson, A.C.; Walker, W.; Rosolowsky, E.T.; Roshan, B.; Warram, J.H.; Krolewski, A.S. Serum concentrations of markers of TNFalpha and Fas-mediated pathways and renal function in nonproteinuric patients with type 1 diabetes. Clin. J. Am. Soc. Nephrol. 2009, 4, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coca, S.G.; Nadkarni, G.N.; Huang, Y.; Moledina, D.G.; Rao, V.; Zhang, J.; Ferket, B.; Crowley, S.T.; Fried, L.F.; Parikh, C.R. Plasma Biomarkers and Kidney Function Decline in Early and Established Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2017, 28, 2786–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruster, C.; Wolf, G. The role of chemokines and chemokine receptors in diabetic nephropathy. Front. Biosci. J. Virtual Libr. 2008, 13, 944–955. [Google Scholar] [CrossRef] [Green Version]
- Tesch, G.H. MCP-1/CCL2: A new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2008, 294, F697–F701. [Google Scholar] [CrossRef] [PubMed]
- Herder, C.; Peltonen, M.; Koenig, W.; Kräft, I.; Müller-Scholze, S.; Martin, S.; Lakka, T.; Ilanne-Parikka, P.; Eriksson, J.G.; Hämäläinen, H.; et al. Systemic immune mediators and lifestyle changes in the prevention of type 2 diabetes: Results from the Finnish Diabetes Prevention Study. Diabetes 2006, 55, 2340–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flyvbjerg, A. The role of the complement system in diabetic nephropathy. Nat. Rev. Nephrol. 2017, 13, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Novel mechanisms and functions of complement. Nat. Immunol. 2017, 18, 1288–1298. [Google Scholar] [CrossRef]
- Tang, S.; Zhou, W.; Sheerin, N.S.; Vaughan, R.W.; Sacks, S.H. Contribution of renal secreted complement C3 to the circulating pool in humans. J Immunol 1999, 162, 4336–4341. [Google Scholar]
- Budge, K.; Dellepiane, S.; Yu, S.M.W.; Cravedi, P. Complement, a Therapeutic Target in Diabetic Kidney Disease. Front. Med. 2020, 7, 599236. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.L.; Nordestgaard, B.G.; Nielsen, S.F. Complement C3 and Risk of Diabetic Microvascular Disease: A Cohort Study of 95202 Individuals from the General Population. Clin. Chem. 2018, 64, 1113–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, T.K.; Gall, M.A.; Tarnow, L.; Thiel, S.; Stehouwer, C.D.; Schalkwijk, C.G.; Parving, H.-H.; Flyvbjerg, A. Mannose-binding lectin and mortality in type 2 diabetes. Arch. Intern. Med. 2006, 166, 2007–2013. [Google Scholar] [CrossRef]
- Yiu, W.H.; Li, R.X.; Wong, D.W.L.; Wu, H.J.; Chan, K.W.; Chan, L.Y.Y.; Leung, J.C.K.; Lai, K.N.; Sacks, S.H.; Zhou, W.; et al. Complement C5a inhibition moderates lipid metabolism and reduces tubulointerstitial fibrosis in diabetic nephropathy. Nephrol. Dial. Transpl. 2018, 33, 1323–1332. [Google Scholar] [CrossRef]
- Anand, G.; Vasanthakumar, R.; Mohan, V.; Babu, S.; Aravindhan, V. Increased IL-12 and decreased IL-33 serum levels are associated with increased Th1 and suppressed Th2 cytokine profile in patients with diabetic nephropathy (CURES-134). Int. J. Clin. Exp. Pathol. 2014, 7, 8008–8015. [Google Scholar]
- Zeng, C.; Shi, X.; Zhang, B.; Liu, H.; Zhang, L.; Ding, W.; Zhao, Y. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: Relationship with metabolic factors and complications. J. Mol. Med. 2012, 90, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.Y.; Jeong, K.H.; Lee, T.W.; Ihm, C.G.; Lim, S.J.; Lee, S.H. Aberrant recruitment and activation of T cells in diabetic nephropathy. Am. J. Nephrol. 2012, 35, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Selye, H. Production of Nephrosclerosis by Overdosage with Desoxycorticosterone Acetate. Can. Med. Assoc. J. 1942, 47, 515–519. [Google Scholar] [PubMed]
- Kang, Y.S.; Cha, D.R. Aldosterone and diabetic kidney disease. Curr. Diab. Rep. 2009, 9, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Brosius, F.C.; Adler, S.G.; Kretzler, M.; Mehta, R.L.; Tumlin, J.A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M.E.; et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: Results from a Phase 2 randomized controlled clinical trial. Nephrol. Dial. Transplant. 2018, 33, 1950–1959. [Google Scholar] [CrossRef] [Green Version]
- Härma, M.-A.; on behalf of the FinnDiane Study Group; Dahlström, E.H.; Sandholm, N.; Forsblom, C.; Groop, P.-H.; Lehto, M. Decreased plasma kallikrein activity is associated with reduced kidney function in individuals with type 1 diabetes. Diabetologia 2020, 63, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Cefalu, W.T.; A Leiter, L.; Yoon, K.-H.; Arias, P.; Niskanen, L.; Xie, J.; A Balis, D.; Canovatchel, W.; Meininger, G. Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet 2013, 382, 941–950. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef]
- Zhang, Y.; Jin, D.; Kang, X.; Zhou, R.; Sun, Y.; Lian, F.; Tong, X. Signaling Pathways Involved in Diabetic Renal Fibrosis. Front. Cell Dev. Biol. 2021, 9, 696542. [Google Scholar] [CrossRef] [PubMed]
- Fontalvo, J.E.R.; Anedo, R.D.; Sarabia, M.R.; Galvis, N.P.; Espinosa, A.B.; Gulfo, I.U.; Calvo, C.P.; Lara, A.P.; Almeida, Z.M.; Serpa, O.V.; et al. Proteoma urinario en la enfermedad renal diabética. Rev. Colomb. Nefrol. 2021, 8, e546. [Google Scholar]
- Aghadavoud, E.; Nasri, H.; Amiri, M. Molecular signaling pathways of diabetic kidney disease; new concepts. J. Prev. Epidemiol. 2017, 2, e03. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Biomarker (s) | Reference | Population | Study Design | Findings | Potential Clinical Application |
|---|---|---|---|---|---|
| Serum SDMA (symmetric dymethylarginine) | Hall et al. (2016) [16] | 19 dogs with CKD and 20 control dogs | Retrospective | SDMA detected CKD earlier than creatinine | SDMA as a potential target for evaluation of kidney disease in dogs |
| Serum SDMA (symmetric dymethylarginine) | Hall et al. (2014) [17] | 21 cats with CKD and 21 healthy control cats | Retrospective | SDMA detects CKD earlier than creatinine | SDMA as a potential target for evaluation of kidney disease in cats |
| Serum SDMA and Cystatin C | Pelander et al. (2019) [18] | 30 healthy dogs and 67 dogs with diagnosis or suspicion of CKD | Cross-sectional | Creatinine and SDMA were similar in detecting reduced GFR, whereas cystatin C was inferior | SDMA can be measured together with creatinine for evaluation of kidney function in dogs |
| β2-microglobulin, calbindin, clusterin, EGF, GST-α, GST-μ, KIM-1, NGAL, osteopontin, TIMP-1, and VEGF | Togashi and Miyamoto et al. (2013) [19] | 5 male Zucker diabetic fatty rats (ZDF/CrlCrlj-Leptfa/fa) and 5 male non-diabetic lean rats (ZDF/CrlCrlj-Lept?/+ | Cross-sectional | Urinary levels of cystatin C, β2-microglobulin, clusterin, GST-μ, and KIM-1 were increased before the development of histopathological changes consistent with DN | Cystatin C, β2-microglobulin, clusterin, GST-μ, and KIM-1 could be used as markers of DN in mice models of DN |
| Serum RBP4 (Retinol-binding protein 4) | Van Hoek et al. (2018) [20] | 10 cats with CKD, 10 cats with hyperthyroidism, and 10 healthy cats | Cross-sectional | Cats with CKD and hyperthyroidism had higher concentrations of RBP4 than healthy cats | RBP4 can be used as a marker of kidney dysfunction in cats |
| Plasma NGAL and UNCR | Steinbach et al. (2014) [21] | 17 dogs with CKD, 48 dogs with AKI, and 18 control subjects | Cross-sectional | Plasma NGAL concentration and UNCR were significantly higher in dogs with AKI or CKD compared to healthy dogs. In addition, these markers were higher in dogs with AKI compared with dogs with CKD | NGAL is an established marker of AKI, but can also be used to distinguish dogs with CKD from healthy dogs |
| Urinary KIM-1, NGAL, and vanin-1 | Hosohata et al. (2014) [22] | 8 male spontaneous type 2 diabetic OLETF rats and 8 male non-diabetic Long Evans, Tokushima Otsuka (LETO) rats | Cross-sectional | Urinary KIM-1 was more sensitive than albumin in detecting DN | KIM-1 can detect early tubular damage in rats with DN |
| Biomarker (s) | Reference | Population | Study Design | Findings | Potential Clinical Application |
|---|---|---|---|---|---|
| Plasma endostatin | Carlsson et al. (2016) [24] | 607 patients with type 2 DM | Prospective | Edostatin levels are associated with increased risk of GFR decline and mortality | Potential use as a marker of kidney dysfunction in type 2 diabetics |
| Serum amyloid A | Dieter et al. (2016) [25] | 135 patients with type 2 DM | Prospective | Higher levels of serum amyloid A are associated with higher risk of death and ESRD | Amyloid A is a potential target for evaluating diabetic nephropathy in patients with type 2 diabetes |
| Urinary NGAL and cystatin C | Garg et al. (2015) [26] | 91 patients with type 2 DM, 30 patients with prediabetes. | Cross-sectional | NGAL and cystatin C were significantly higher in participants with vs those without microalbuminuria | Early detection of microalbuminuria in patients with type 2 DM |
| Urinary KIM-1,L-FABP, NAG, and NGAL | Fufaa et al. (2015) [27] | 260 patients with type 2 DM | Prospective | NGAL and L-FABP are independently associated with ESRD and mortality | Prediction of ESRD and mortality in patients with type 2 DM |
| Serum E-selectin, IL-6, PAI-1, sTNFR1, TNFR2 | Lopes-Virella et al. (2013) [28] | 1237 patients with type 1 DM | Prospective | TNFR1 and TNFR2 and E-selectin are the best predictors of progression to macroalbuminuria | Marker of progression to macroalbuminuria in patients with type 1 DM |
| Urinary L-FABP | Araki et al. (2013) [29] | 618 patients with type 2 DM | Prospective | L-FABP is associated with decline in eGFR | Potential use as a marker of progression of GFR in DN |
| Plasma TNF-α, TNFR1, and TNFR2, ICAM-1, VCAM-1, PAI-1, IL-6, and CRP | Niewczas et al. (2012) [30] | 410 patients with type 2 DM, CKD stages 1-3 | Prospective | TNFR1 and TNFR2 were strongly associated with risk of ESRD | Potential use as a marker of progression towards ESRD in patients with CKD stages 1–3 in patients with diabetic nephropathy |
| Urinary NAGL, NAG, and KIM-1 | Fu et al. (2012) [31] | 101 T2DM patients, 28 control subjects | Cross-sectional | Every marker showed increased levels in patients with DM; NGAL and NAG were positively correlated with albuminuria; NGAL showed important differences between micro- and macroalbuminuric patients | KIM-1 and NGAL could be potential early markers of DN |
| β-Trace protein and B2M | Foster et al. (2015) [32] | 250 patients with type 2 DM | Prospective | β-Trace protein is associated with ESRD | β-Trace protein is a potential marker for progression towards ESRD in type 2 diabetics |
| Urinary IL-6, CXCL10/ IP-10, NAG, and KIM-1 | Vaidya et al. (2011) [33] | 659 patients with type 1 DM, 38 controls | Cross-sectional and prospective | KIM-1 and NAG both individually and collectively were significantly associated with regression of microalbuminuria | Both molecules are potential markers for regression of microalbuminuria in patients with type 1 DM |
| Molecular Mechanisms of DKD | Main Findings | Association with Clinical Trials |
|---|---|---|
| Genetics and Epigenetics | DNA methylation, histone post-translational modifications, microRNA (miRNA), lcnRNA, and circRNA are exacerbated by hyperglycemia, promoting renal inflammation and fibrosis. | A cohort by van Zuydam et al associated the GABRR1 gene with microalbuminuria [13]. |
| Innate Immunity | Macrophages and dendritic cells are activated by the interaction of TLRs and NLRs by hyperglycemia, leading to inflammation and renal injury. These cells require the presence of CSF-1. | A phase 2 randomized controlled clinical trial showed that baricitinib, a JAK1/2 inhibitor, reduces albuminuria in patients with type 2 DM. The JAK-STAT pathway is important in the initiation and regulation of innate and adaptive immunity [66]. |
| Adaptive Immunity | Seen in lower proportion than innate immune cells; predominantly Th1 and Th17 cells. Increased expression of CD4+, CD8+, and CD20+ cells in DKD. | A phase 2 randomized controlled clinical trial showed that baricitinib, a JAK1/2 inhibitor, reduces albuminuria in patients with type 2 DM. The JAK-STAT pathway is important in the initiation and regulation of innate and adaptive immunity [66]. |
| KKS | In hyperglycemia, the binding of bradykinin and kallidin metabolites to B1R induces an NK-Fβ-dependent pro-inflammatory response. B2R stimulation leads to a pro-inflammatory response via MAPK pathway. | A cross-sectional study by Härma et al measured plasma kallikrein activity in 295 individuals with type 1 DM, showing that lower levels of plasma kallikrein were associated with higher GFRs [67]. |
| Cytokines and Chemokines | Cytokines including IL-1, IL-6, IL-18, (TNF-α), and IL-17 have a strong pro-inflammatory effect in the pathogenesis of DKD; chemokines include CCL2 (MCP-1), CCL5 (RANTES), and C-X3-C motif chemokine 1 (CX3CL1, fractalkine). | Patients with DKD from the CANTATA-SU trial showed that treatment with canagliflozin decreased circulating levels of IL-6, TNF receptor-1, matrix metalloproteinase-7, and fibronectin-1 [68]. |
| Complement System | Activation of lectin and glycation of the complement regulatory protein CD 59 lead to activation of MAC and endothelial cell damage. | |
| Aldosterone | Induces increased expression of TGF-β1 and type IV collagen in hyperglycemic conditions. | As shown in the FIDELIO-DKD trial and the FIGARO-DKD, finerenone, minelarocorticoid receptor blocker, has positive results in reducing kidney failure/progression and leads to a reduction in cardiovascular morbidity and mortality in patients with DKD [69,70]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rico-Fontalvo, J.; Aroca, G.; Cabrales, J.; Daza-Arnedo, R.; Yánez-Rodríguez, T.; Martínez-Ávila, M.C.; Uparella-Gulfo, I.; Raad-Sarabia, M. Molecular Mechanisms of Diabetic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 8668. https://doi.org/10.3390/ijms23158668
Rico-Fontalvo J, Aroca G, Cabrales J, Daza-Arnedo R, Yánez-Rodríguez T, Martínez-Ávila MC, Uparella-Gulfo I, Raad-Sarabia M. Molecular Mechanisms of Diabetic Kidney Disease. International Journal of Molecular Sciences. 2022; 23(15):8668. https://doi.org/10.3390/ijms23158668
Chicago/Turabian StyleRico-Fontalvo, Jorge, Gustavo Aroca, Jose Cabrales, Rodrigo Daza-Arnedo, Tomas Yánez-Rodríguez, María Cristina Martínez-Ávila, Isabella Uparella-Gulfo, and María Raad-Sarabia. 2022. "Molecular Mechanisms of Diabetic Kidney Disease" International Journal of Molecular Sciences 23, no. 15: 8668. https://doi.org/10.3390/ijms23158668