
Sirtuin 3 Deficiency Aggravates Kidney Disease in Response to High-Fat Diet through Lipotoxicity-Induced Mitochondrial Damage

 ,
,
Abstract
:1. Introduction
2. Results
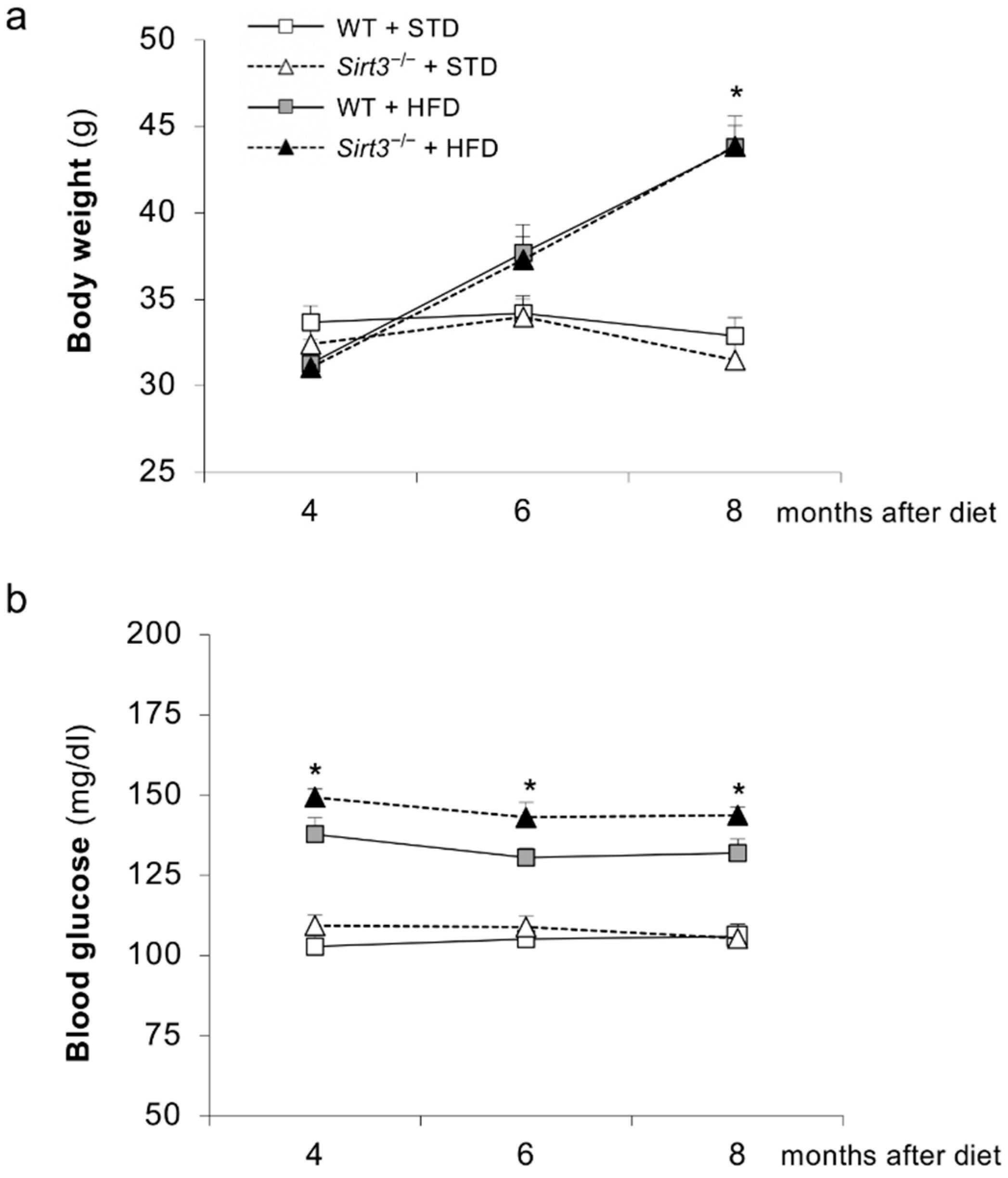
2.1. Laboratory and Systemic Parameters
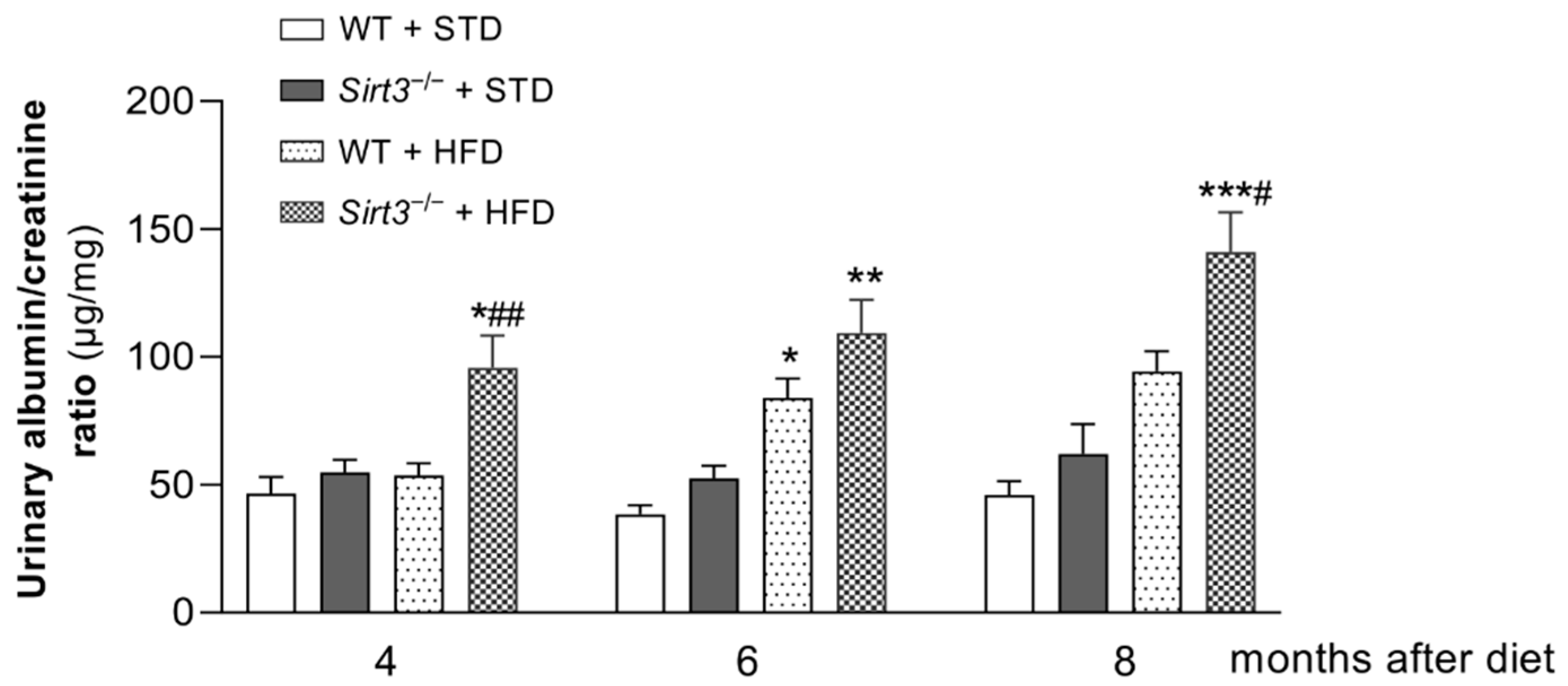
2.2. Sirt3 Deficiency Accelerates and Exacerbates HFD-Induced Albuminuria
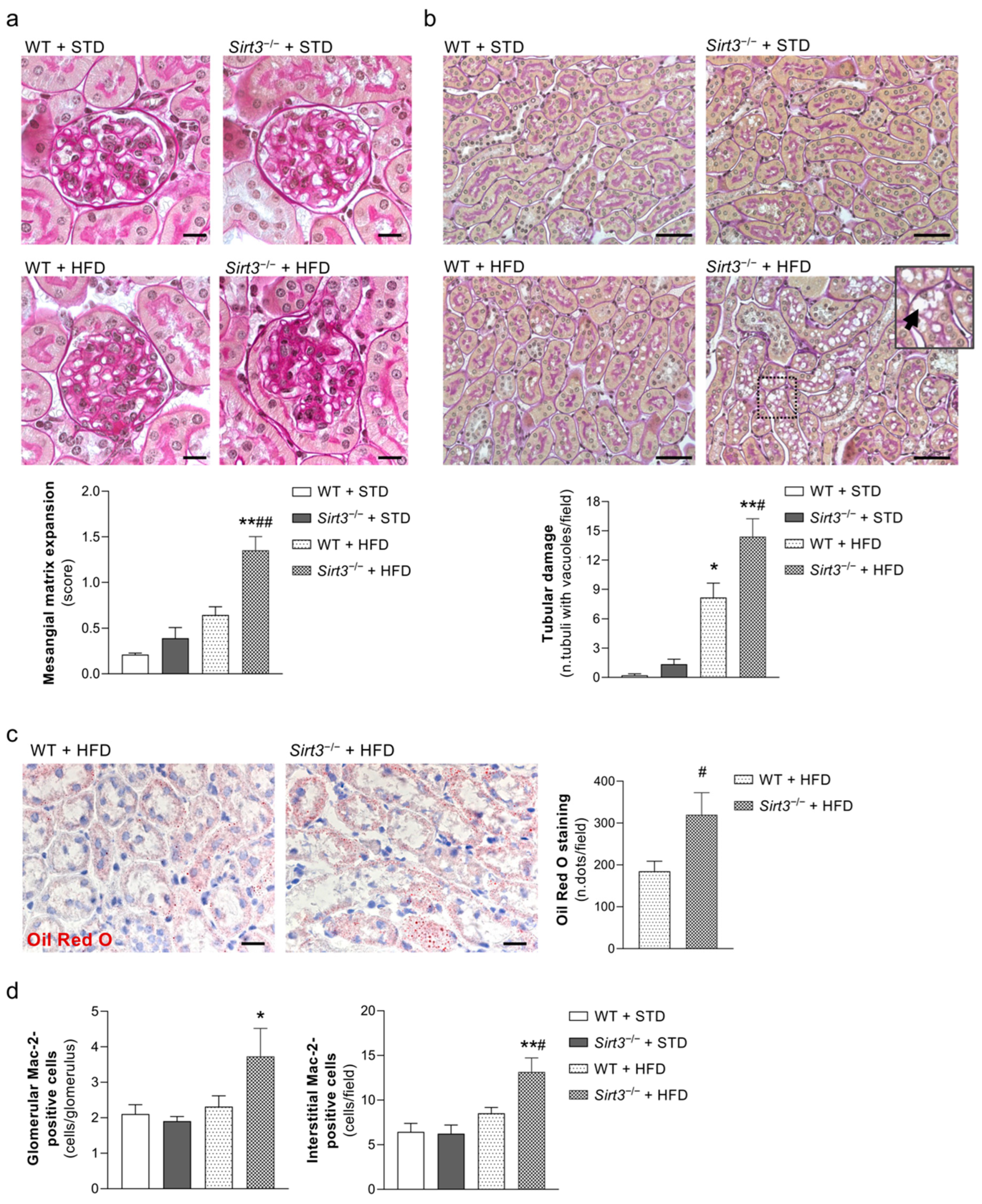
2.3. Sirt3 Deficiency Worsens Glomerular and Tubular Damage and Increases Renal Inflammation in Mice Fed HFD
2.4. Sirt3 Deficiency Alters the Glomerular Filtration Barrier in Mice Fed HFD
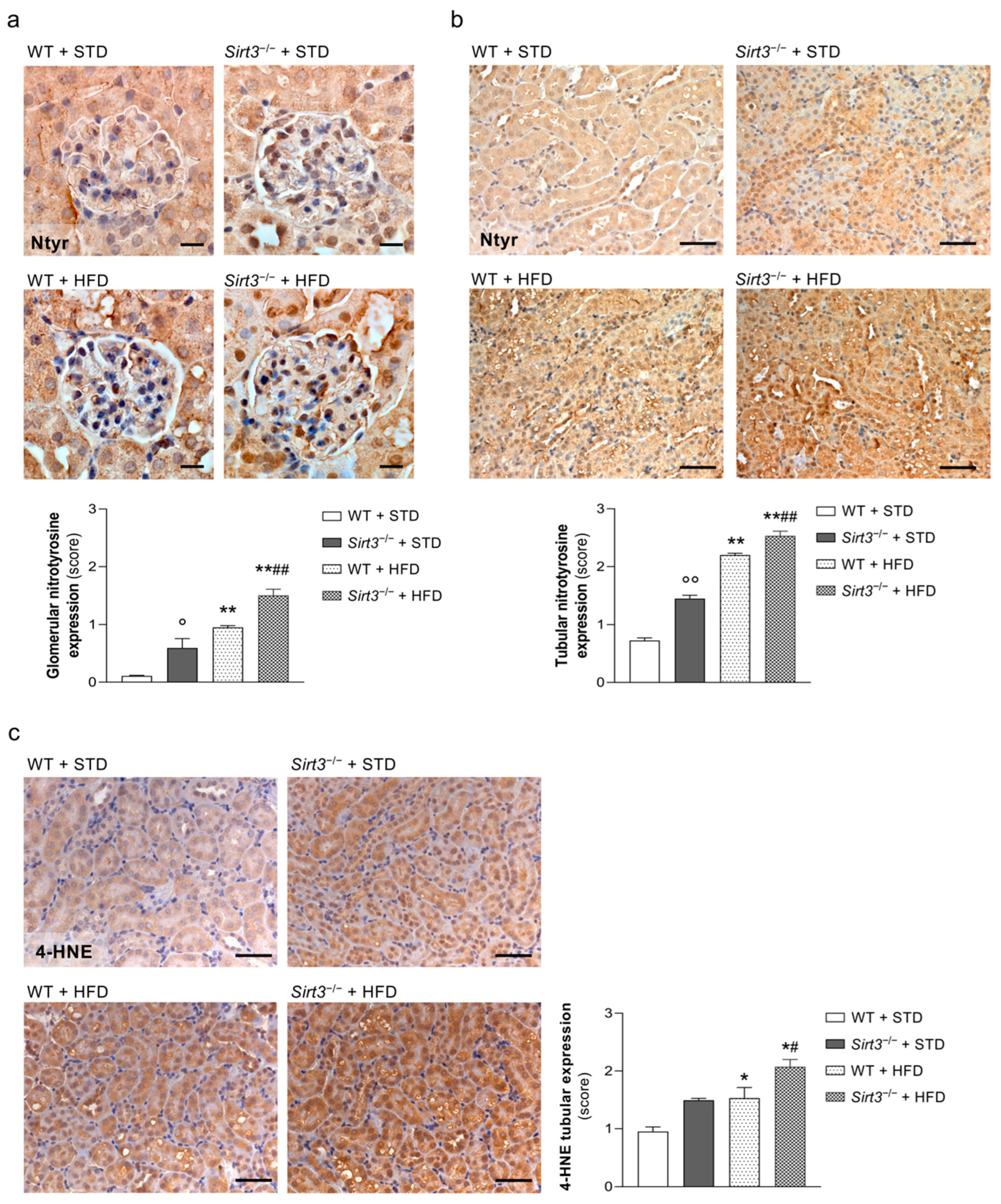
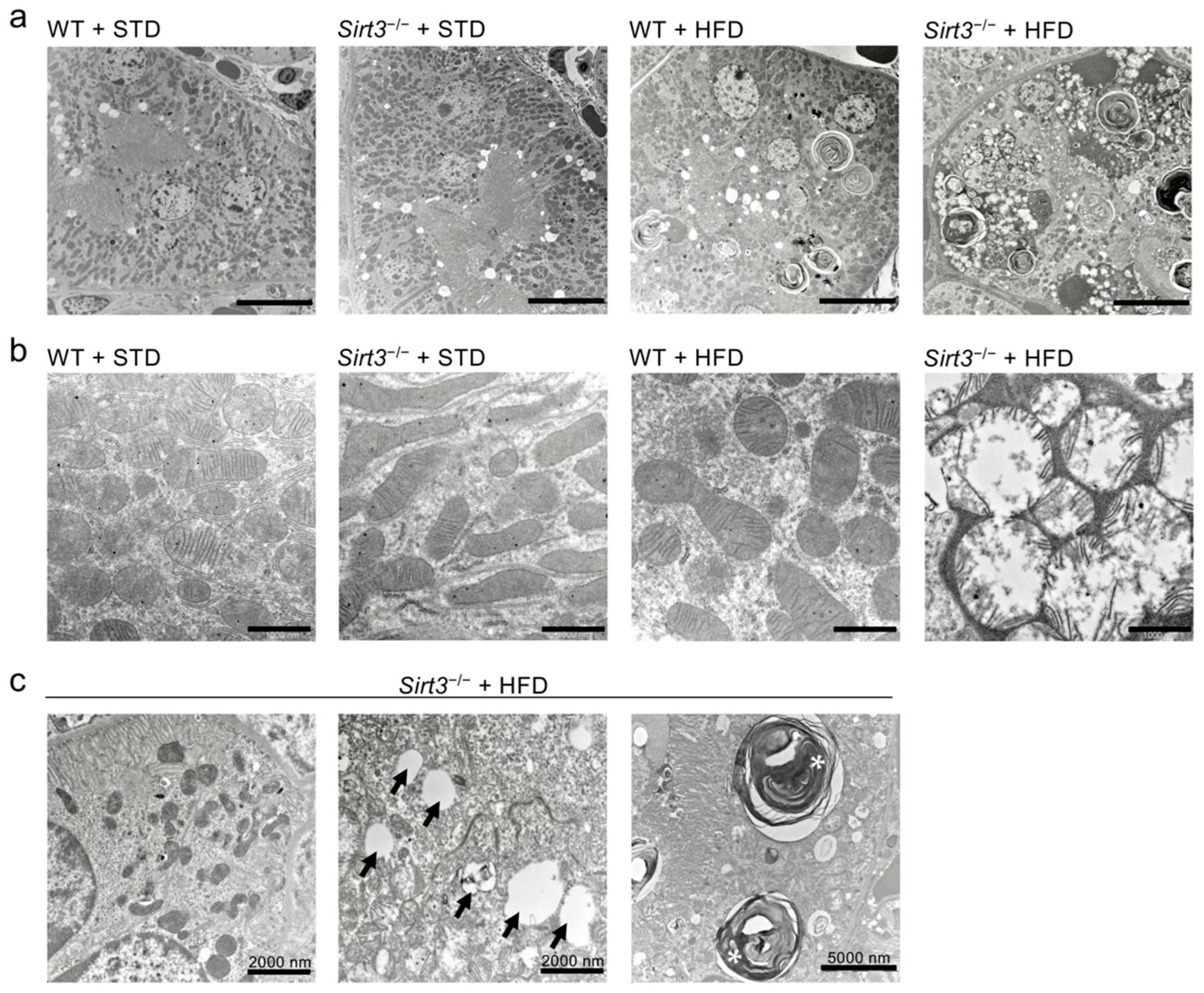
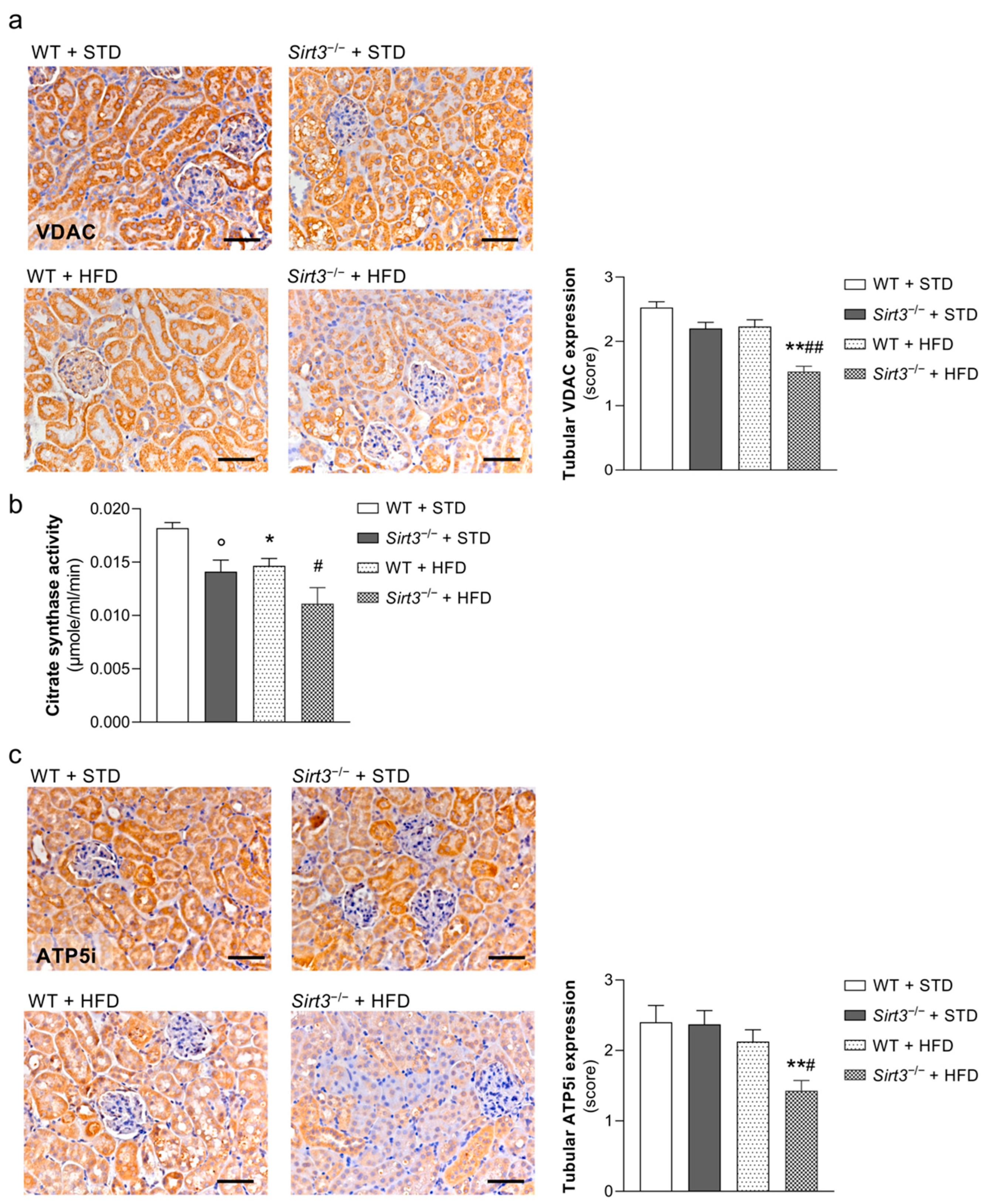
2.5. Sirt3 Deficiency Aggravates HFD-Induced Oxidative Stress and Mitochondrial Damage
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. Blood and Urine Analysis
4.3. Systolic Blood Pressure
4.4. Renal Histology
4.5. Oil Red O Analysis
4.6. Immunohistochemistry
4.7. Transmission Electron Microscopy Analysis
4.8. Citrate Synthase Activity
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [Green Version]
- Fahed, G.; Aoun, L.; Bou Zerdan, M.; Allam, S.; Bou Zerdan, M.; Bouferraa, Y.; Assi, H.I. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int. J. Mol. Sci. 2022, 23, 786. [Google Scholar] [CrossRef] [PubMed]
- Mottillo, S.; Filion, K.B.; Genest, J.; Joseph, L.; Pilote, L.; Poirier, P.; Rinfret, S.; Schiffrin, E.L.; Eisenberg, M.J. The Metabolic Syndrome and Cardiovascular Risk a Systematic Review and Meta-Analysis. J. Am. Coll. Cardiol. 2010, 56, 1113–1132. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Kari, J.A. Metabolic Syndrome and Chronic Kidney Disease. Curr. Opin. Nephrol. Hypertens. 2013, 22, 198–203. [Google Scholar] [CrossRef]
- Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; McCubbin, J.A.; Feinglos, M.N. Diet-Induced Type II Diabetes in C57BL/6J Mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- West, D.B.; Boozer, C.N.; Moody, D.L.; Atkinson, R.L. Dietary Obesity in Nine Inbred Mouse Strains. Am. J. Physiol. 1992, 262, R1025–R1032. [Google Scholar] [CrossRef]
- Deji, N.; Kume, S.; Araki, S.-I.; Soumura, M.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Koya, D.; Haneda, M.; et al. Structural and Functional Changes in the Kidneys of High-Fat Diet-Induced Obese Mice. Am. J. Physiol. Renal Physiol. 2009, 296, F118–F126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial Dysfunction and Oxidative Stress in Metabolic Disorders—A Step towards Mitochondria Based Therapeutic Strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of Cellular Metabolism by Protein Lysine Acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as Regulators of Metabolism and Healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard, D.B.; Alt, F.W.; Cheng, H.-L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 Homolog SIRT3 Regulates Global Mitochondrial Lysine Acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [Green Version]
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. 2018, 29, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-H.; Wei, Y.-H. Roles of Mitochondrial Sirtuins in Mitochondrial Function, Redox Homeostasis, Insulin Resistance and Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 5266. [Google Scholar] [CrossRef] [PubMed]
- Bause, A.S.; Haigis, M.C. SIRT3 Regulation of Mitochondrial Oxidative Stress. Exp. Gerontol. 2013, 48, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Hallows, W.C.; Yu, W.; Smith, B.C.; Devries, M.K.; Devires, M.K.; Ellinger, J.J.; Someya, S.; Shortreed, M.R.; Prolla, T.; Markley, J.L.; et al. Sirt3 Promotes the Urea Cycle and Fatty Acid Oxidation during Dietary Restriction. Mol. Cell 2011, 41, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie Restriction Reduces Oxidative Stress by SIRT3-Mediated SOD2 Activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Jing, E.; Emanuelli, B.; Hirschey, M.D.; Boucher, J.; Lee, K.Y.; Lombard, D.; Verdin, E.M.; Kahn, C.R. Sirtuin-3 (Sirt3) Regulates Skeletal Muscle Metabolism and Insulin Signaling via Altered Mitochondrial Oxidation and Reactive Oxygen Species Production. Proc. Natl. Acad. Sci. USA 2011, 108, 14608–14613. [Google Scholar] [CrossRef] [Green Version]
- Lantier, L.; Williams, A.S.; Williams, I.M.; Yang, K.K.; Bracy, D.P.; Goelzer, M.; James, F.D.; Gius, D.; Wasserman, D.H. SIRT3 Is Crucial for Maintaining Skeletal Muscle Insulin Action and Protects Against Severe Insulin Resistance in High-Fat-Fed Mice. Diabetes 2015, 64, 3081–3092. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stančáková, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 Deficiency and Mitochondrial Protein Hyperacetylation Accelerate the Development of the Metabolic Syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, M.; Zoja, C.; Zanchi, C.; Corna, D.; Villa, S.; Bolognini, S.; Novelli, R.; Perico, L.; Remuzzi, G.; Benigni, A.; et al. Manipulating Sirtuin 3 Pathway Ameliorates Renal Damage in Experimental Diabetes. Sci. Rep. 2020, 10, 8418. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Cassis, P.; Conti, S.; Perico, L.; Corna, D.; Cerullo, D.; Zentilin, L.; Zoja, C.; Perna, A.; Lionetti, V.; et al. Sirt3 Deficiency Shortens Life Span and Impairs Cardiac Mitochondrial Function Rescued by Opa1 Gene Transfer. Antioxid. Redox Signal. 2019, 31, 1255–1271. [Google Scholar] [CrossRef] [Green Version]
- Brinkkoetter, P.T.; Ising, C.; Benzing, T. The Role of the Podocyte in Albumin Filtration. Nat. Rev. Nephrol. 2013, 9, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Ballermann, B.J.; Nyström, J.; Haraldsson, B. The Glomerular Endothelium Restricts Albumin Filtration. Front. Med. 2021, 8, 766689. [Google Scholar] [CrossRef]
- Su, W.; Chen, J.; Yang, H.; You, L.; Xu, L.; Wang, X.; Li, R.; Gao, L.; Gu, Y.; Lin, S.; et al. Expression of Nestin in the Podocytes of Normal and Diseased Human Kidneys. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1761–R1767. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ge, X.; Li, X.; He, J.; Wei, X.; Du, J.; Sun, J.; Li, X.; Xun, Z.; Liu, W.; et al. High-Fat Diet Promotes Renal Injury by Inducing Oxidative Stress and Mitochondrial Dysfunction. Cell Death Dis. 2020, 11, 914. [Google Scholar] [CrossRef]
- Schaub, J.A.; Venkatachalam, M.A.; Weinberg, J.M. Proximal Tubular Oxidative Metabolism in Acute Kidney Injury and the Transition to CKD. Kidney360 2021, 2, 355–364. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Sivan, S.; Shoshan-Barmatz, V. The Expression Level of the Voltage-Dependent Anion Channel Controls Life and Death of the Cell. Proc. Natl. Acad. Sci. USA 2006, 103, 5787–5792. [Google Scholar] [CrossRef] [Green Version]
- Reisch, A.S.; Elpeleg, O. Biochemical Assays for Mitochondrial Activity: Assays of TCA Cycle Enzymes and PDHc. Methods Cell Biol. 2007, 80, 199–222. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Muneyuki, E.; Hisabori, T. ATP Synthase--a Marvellous Rotary Engine of the Cell. Nat. Rev. Mol. Cell Biol. 2001, 2, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Uzu, T.; Araki, S.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Kubota, N.; Terauchi, Y.; Kadowaki, T.; et al. Role of Altered Renal Lipid Metabolism in the Development of Renal Injury Induced by a High-Fat Diet. J. Am. Soc. Nephrol. JASN 2007, 18, 2715–2723. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Wang, Z.; Proctor, G.; Moskowitz, S.; Liebman, S.E.; Rogers, T.; Lucia, M.S.; Li, J.; Levi, M. Diet-Induced Obesity in C57BL/6J Mice Causes Increased Renal Lipid Accumulation and Glomerulosclerosis via a Sterol Regulatory Element-Binding Protein-1c-Dependent Pathway. J. Biol. Chem. 2005, 280, 32317–32325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, A.P.J.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.D.; Lamb, H.J.; Pongrac Barlovic, D.; Hojs, R.; et al. Fatty Kidney: Emerging Role of Ectopic Lipid in Obesity-Related Renal Disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef]
- Opazo-Ríos, L.; Mas, S.; Marín-Royo, G.; Mezzano, S.; Gómez-Guerrero, C.; Moreno, J.A.; Egido, J. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 2632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered Renal Lipid Metabolism and Renal Lipid Accumulation in Human Diabetic Nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Gewin, L.S. Sugar or Fat? Renal Tubular Metabolism Reviewed in Health and Disease. Nutrients 2021, 13, 1580. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 Regulates Mitochondrial Fatty-Acid Oxidation by Reversible Enzyme Deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Li, C.-M.; Ye, Z.-C.; Huang, J.; Li, Y.; Lai, W.; Peng, H.; Lou, T.-Q. Sirt3 Modulates Fatty Acid Oxidation and Attenuates Cisplatin-Induced AKI in Mice. J. Cell. Mol. Med. 2020, 24, 5109–5121. [Google Scholar] [CrossRef] [Green Version]
- Castro, B.B.A.; Foresto-Neto, O.; Saraiva-Camara, N.O.; Sanders-Pinheiro, H. Renal Lipotoxicity: Insights from Experimental Models. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Kume, S.; Koya, D.; Araki, S.; Isshiki, K.; Chin-Kanasaki, M.; Sugimoto, T.; Haneda, M.; Sugaya, T.; Kashiwagi, A.; et al. SIRT3 Attenuates Palmitate-Induced ROS Production and Inflammation in Proximal Tubular Cells. Free Radic. Biol. Med. 2011, 51, 1258–1267. [Google Scholar] [CrossRef]
- Uchida, K. 4-Hydroxy-2-Nonenal: A Product and Mediator of Oxidative Stress. Prog. Lipid Res. 2003, 42, 318–343. [Google Scholar] [CrossRef]
- Roede, J.R.; Jones, D.P. Reactive Species and Mitochondrial Dysfunction: Mechanistic Significance of 4-Hydroxynonenal. Environ. Mol. Mutagen. 2010, 51, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Wani, W.Y.; Redmann, M.; Benavides, G.A.; Johnson, M.S.; Ouyang, X.; Cofield, S.S.; Mitra, K.; Darley-Usmar, V.; Zhang, J. Regulation of Autophagy, Mitochondrial Dynamics, and Cellular Bioenergetics by 4-Hydroxynonenal in Primary Neurons. Autophagy 2017, 13, 1828–1840. [Google Scholar] [CrossRef] [Green Version]
- Fritz, K.S.; Galligan, J.J.; Smathers, R.L.; Roede, J.R.; Shearn, C.T.; Reigan, P.; Petersen, D.R. 4-Hydroxynonenal Inhibits SIRT3 via Thiol-Specific Modification. Chem. Res. Toxicol. 2011, 24, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brèthes, D.; di Rago, J.-P.; Velours, J. The ATP Synthase Is Involved in Generating Mitochondrial Cristae Morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S.V. Protection of Mitochondria Prevents High-Fat Diet-Induced Glomerulopathy and Proximal Tubular Injury. Kidney Int. 2016, 90, 997–1011. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Plasma Cholesterol (mg/dL) | Plasma Triglycerides (mg/dL) | SBP (mmHg) | Heart Rate (beats/min) | BUN (mg/dL) |
|---|---|---|---|---|---|
| WT + STD | 102.3 ± 1.8 | 123.9 ± 9.2 | 103.4 ± 2.3 | 561.1 ± 16.3 | 22.4 ± 1.1 |
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| Sirt3−/− + STD | 106.4 ± 3.1 | 129.8 ± 3.8 | 111.9 ± 2.2 | 564.3 ± 22.2 | 20.3 ± 1.1 |
| (n = 12) | (n = 12) | (n = 12) | (n = 12) | (n = 12) | |
| WT + HFD | 128.7 ± 5.1 * | 154.5 ± 5.4 * | 113.4 ± 4.7 | 590.1 ± 11.8 | 19.8 ± 1.1 |
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| Sirt3−/− + HFD | 139.3 ± 6.2 ** | 144.4 ± 4.2 | 109.6 ± 3.4 | 651.3 ± 15.6 *# | 20.7 ± 0.9 |
| (n = 14) | (n = 14) | (n = 14) | (n = 14) | (n = 14) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Locatelli, M.; Macconi, D.; Corna, D.; Cerullo, D.; Rottoli, D.; Remuzzi, G.; Benigni, A.; Zoja, C. Sirtuin 3 Deficiency Aggravates Kidney Disease in Response to High-Fat Diet through Lipotoxicity-Induced Mitochondrial Damage. Int. J. Mol. Sci. 2022, 23, 8345. https://doi.org/10.3390/ijms23158345
Locatelli M, Macconi D, Corna D, Cerullo D, Rottoli D, Remuzzi G, Benigni A, Zoja C. Sirtuin 3 Deficiency Aggravates Kidney Disease in Response to High-Fat Diet through Lipotoxicity-Induced Mitochondrial Damage. International Journal of Molecular Sciences. 2022; 23(15):8345. https://doi.org/10.3390/ijms23158345
Chicago/Turabian StyleLocatelli, Monica, Daniela Macconi, Daniela Corna, Domenico Cerullo, Daniela Rottoli, Giuseppe Remuzzi, Ariela Benigni, and Carlamaria Zoja. 2022. "Sirtuin 3 Deficiency Aggravates Kidney Disease in Response to High-Fat Diet through Lipotoxicity-Induced Mitochondrial Damage" International Journal of Molecular Sciences 23, no. 15: 8345. https://doi.org/10.3390/ijms23158345