
Mitochondrial Oxidative Stress Induces Cardiac Fibrosis in Obese Rats through Modulation of Transthyretin

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
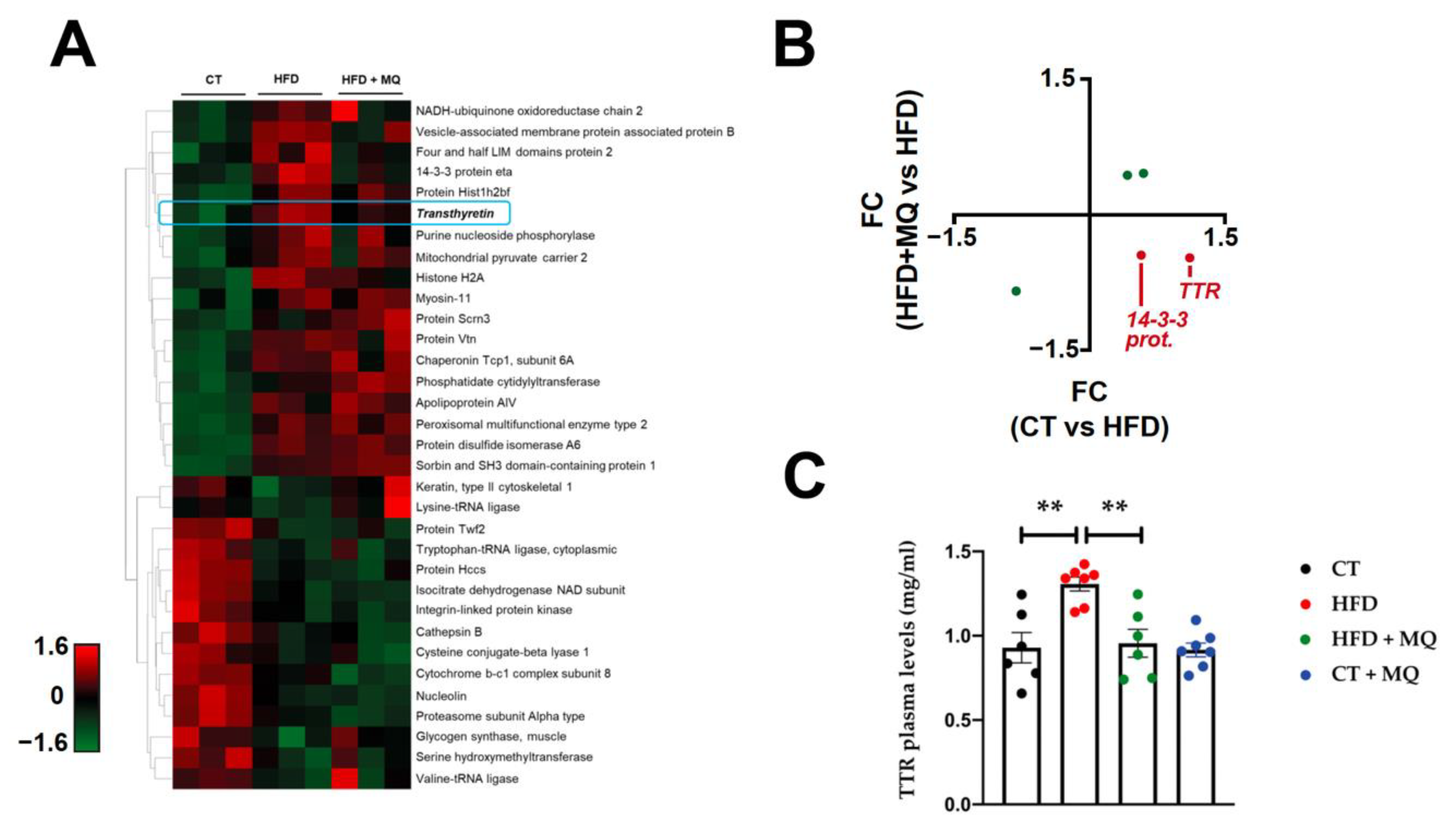
2.1. Proteome-Wide Exploration of Obesity at the Cardiac Level
2.2. Protein Interactome Network Modulated by TTR
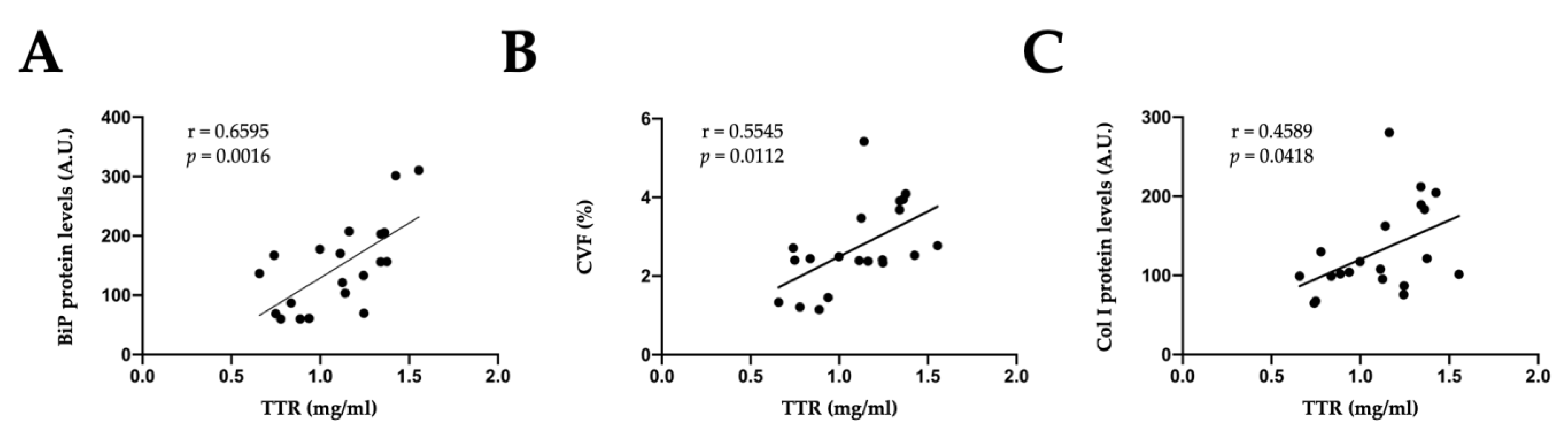
2.3. Effects of TTR on Cardiac Fibroblasts
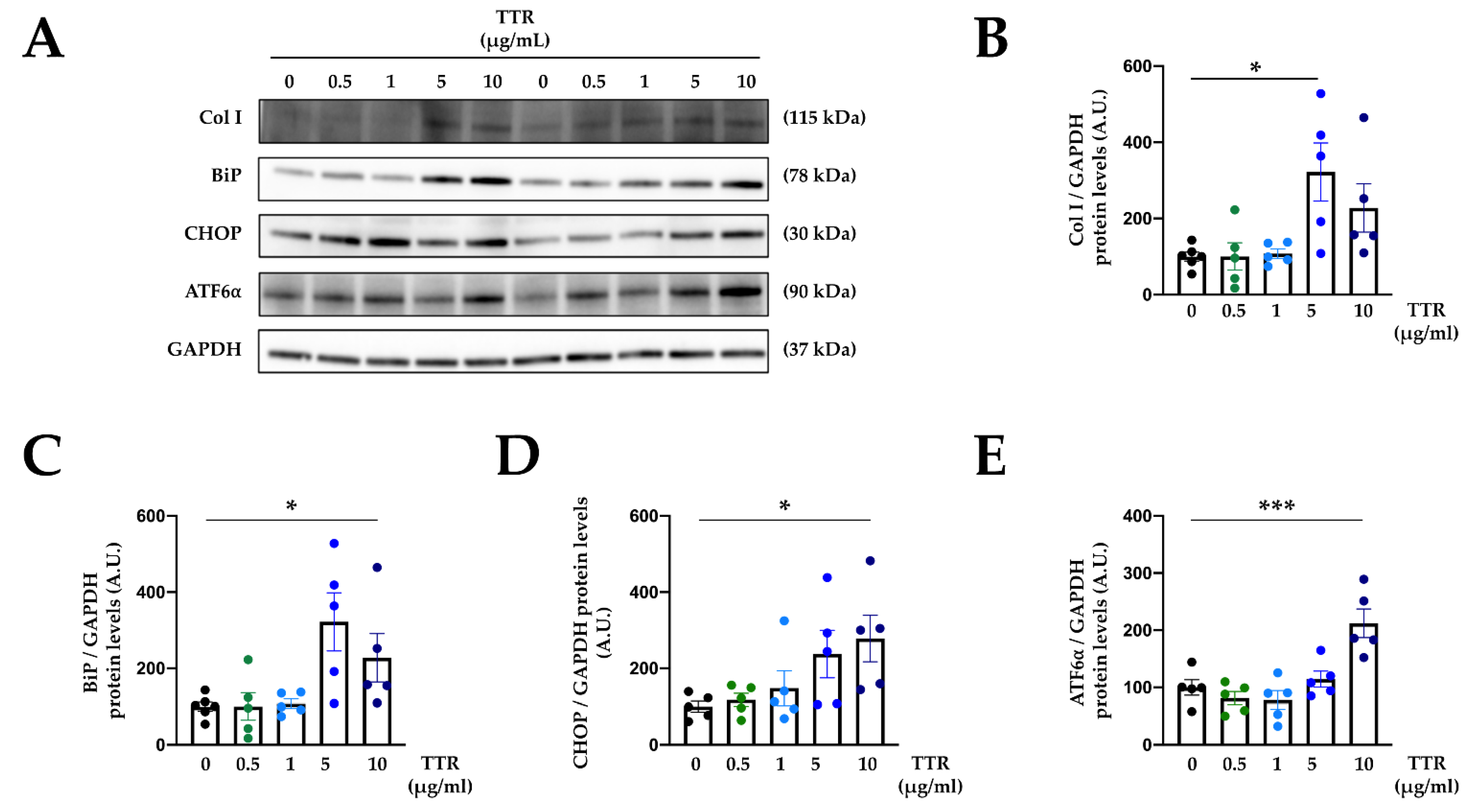
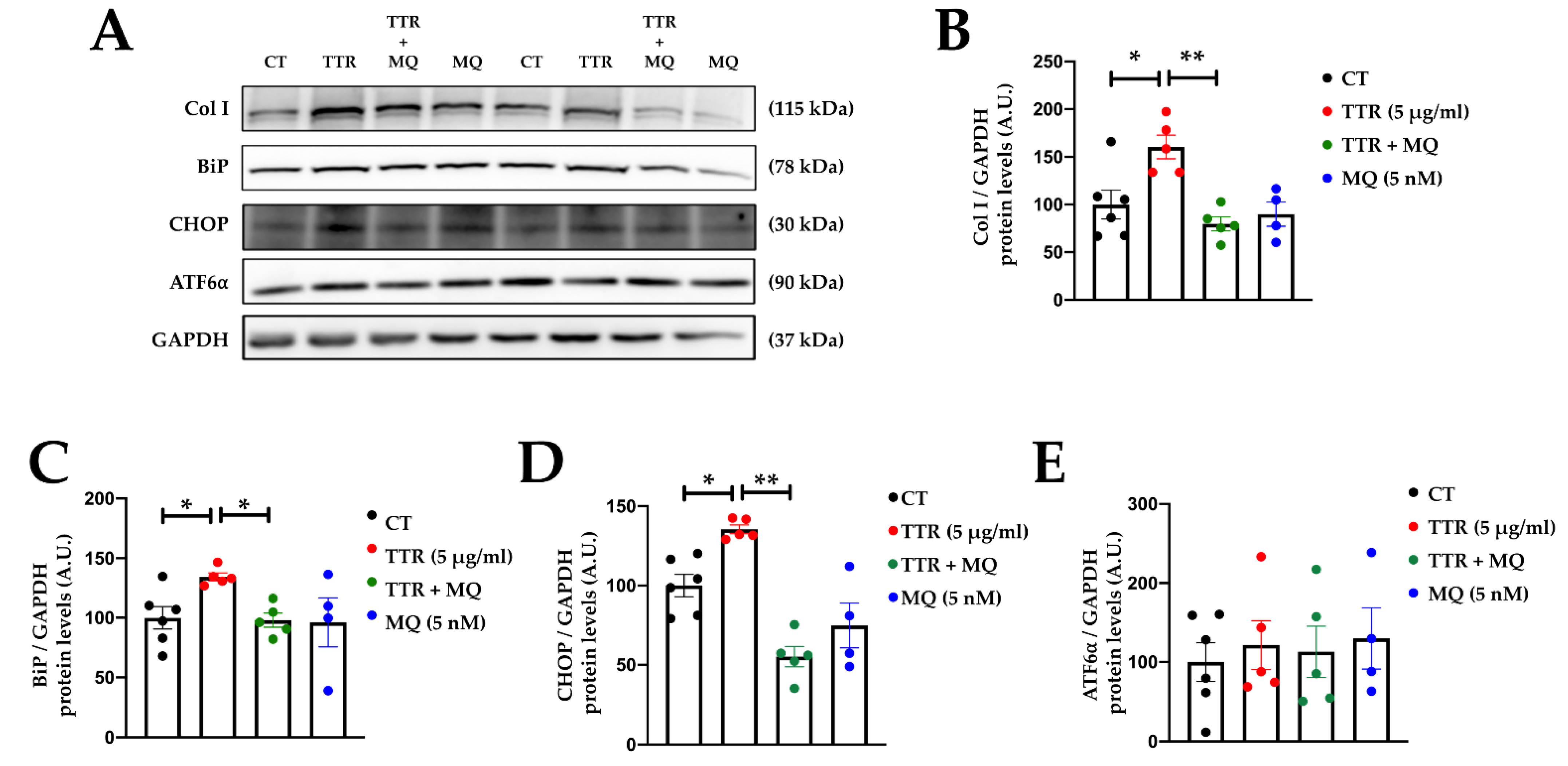
2.4. Mitochondrial Oxidative Stress Mediates the Effects of TTR on Cardiac Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Cell Culture Studies
4.3. Proteomic Analyses
4.3.1. Sample Preparation
4.3.2. Mass Spectrometry Analysis
4.3.3. Data Analysis
4.4. Western Blot
4.5. Circulating Plasma Levels of TTR
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Datta, T.; Lee, A.J.; Cain, R.; McCarey, M.; Whellan, D.J. Weighing in on heart failure: The potential impact of bariatric surgery. Heart Fail. Rev. 2022, 27, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, N.; Ghodsi, A.; Rostami, R.; Torshizian, A.; Jamialahmadi, T.; Jangjoo, A.; Nematy, M.; Bahari, A.; Ebrahimzadeh, F.; Mahmoudabadi, E.; et al. Impact of Bariatric Surgery on Carotid Intima-Media Thickness in Patients with Morbid Obesity: A Prospective Study and Review of the Literature. Obes. Surg. 2022, 32, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.Z.; Lu, W.; Zong, X.F.; Ruan, H.Y.; Liu, Y. Obesity and hypertension. Exp. Ther. Med. 2016, 12, 2395–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wondmkun, Y.T. Obesity, Insulin Resistance, and Type 2 Diabetes: Associations and Therapeutic Implications. Diabetes Metab. Syndr. Obes. 2020, 13, 3611–3616. [Google Scholar] [CrossRef]
- Bhat, Z.F.; Morton, J.D.; Mason, S.; Bekhit, A.E.A.; Bhat, H.F. Obesity and neurological disorders: Dietary perspective of a global menace. Crit. Rev. Food Sci. Nutr. 2019, 59, 1294–1310. [Google Scholar] [CrossRef]
- Jamaly, S.; Carlsson, L.; Peltonen, M.; Andersson-Assarsson, J.C.; Karason, K. Heart failure development in obesity: Underlying risk factors and mechanistic pathways. ESC Heart Fail. 2021, 8, 356–367. [Google Scholar] [CrossRef]
- Turer, A.T.; Hill, J.A.; Elmquist, J.K.; Scherer, P.E. Adipose tissue biology and cardiomyopathy: Translational implications. Circ. Res. 2012, 111, 1565–1577. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Wu, N.N.; Wang, S.; Sowers, J.R.; Zhang, Y. Obesity cardiomyopathy: Evidence, mechanisms, and therapeutic implications. Physiol. Rev. 2021, 101, 1745–1807. [Google Scholar] [CrossRef]
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis—A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef]
- Kruszewska, J.; Cudnoch-Jedrzejewska, A.; Czarzasta, K. Remodeling and Fibrosis of the Cardiac Muscle in the Course of Obesity-Pathogenesis and Involvement of the Extracellular Matrix. Int. J. Mol. Sci. 2022, 23, 4195. [Google Scholar] [CrossRef]
- Aoki, T.; Fukumoto, Y.; Sugimura, K.; Oikawa, M.; Satoh, K.; Nakano, M.; Nakayama, M.; Shimokawa, H. Prognostic impact of myocardial interstitial fibrosis in non-ischemic heart failure—Comparison between preserved and reduced ejection fraction heart failure. Circ. J. 2011, 75, 2605–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyongyosi, M.; Winkler, J.; Ramos, I.; Do, Q.T.; Firat, H.; McDonald, K.; Gonzalez, A.; Thum, T.; Diez, J.; Jaisser, F.; et al. Myocardial fibrosis: Biomedical research from bench to bedside. Eur. J. Heart Fail. 2017, 19, 177–191. [Google Scholar] [CrossRef]
- Packer, M. Leptin-Aldosterone-Neprilysin Axis: Identification of Its Distinctive Role in the Pathogenesis of the Three Phenotypes of Heart Failure in People With Obesity. Circulation 2018, 137, 1614–1631. [Google Scholar] [CrossRef] [PubMed]
- Wenzl, F.A.; Ambrosini, S.; Mohammed, S.A.; Kraler, S.; Luscher, T.F.; Costantino, S.; Paneni, F. Inflammation in Metabolic Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 742178. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, E.; Jurado-Lopez, R.; Cervantes-Escalera, P.; Cachofeiro, V.; Miana, M. Leptin, a mediator of cardiac damage associated with obesity. Horm. Mol. Biol. Clin. Investig. 2014, 18, 3–14. [Google Scholar] [CrossRef]
- Jimenez-Gonzalez, S.; Marin-Royo, G.; Jurado-Lopez, R.; Bartolome, M.V.; Romero-Miranda, A.; Luaces, M.; Islas, F.; Nieto, M.L.; Martinez-Martinez, E.; Cachofeiro, V. The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells 2020, 9, 451. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef]
- Daverkausen-Fischer, L.; Prols, F. Regulation of calcium homeostasis and flux between the endoplasmic reticulum and the cytosol. J. Biol. Chem. 2022, 298, 102061. [Google Scholar] [CrossRef]
- Souza-Neto, F.V.; Jimenez-Gonzalez, S.; Delgado-Valero, B.; Jurado-Lopez, R.; Genty, M.; Romero-Miranda, A.; Rodriguez, C.; Nieto, M.L.; Martinez-Martinez, E.; Cachofeiro, V. The Interplay of Mitochondrial Oxidative Stress and Endoplasmic Reticulum Stress in Cardiovascular Fibrosis in Obese Rats. Antioxidants 2021, 10, 1274. [Google Scholar] [CrossRef]
- Marin-Royo, G.; Rodriguez, C.; Le Pape, A.; Jurado-Lopez, R.; Luaces, M.; Antequera, A.; Martinez-Gonzalez, J.; Souza-Neto, F.V.; Nieto, M.L.; Martinez-Martinez, E.; et al. The role of mitochondrial oxidative stress in the metabolic alterations in diet-induced obesity in rats. FASEB J. 2019, 33, 12060–12072. [Google Scholar] [CrossRef] [Green Version]
- Aboumsallem, J.P.; Muthuramu, I.; Mishra, M.; Kempen, H.; De Geest, B. Effective Treatment of Diabetic Cardiomyopathy and Heart Failure with Reconstituted HDL (Milano) in Mice. Int. J. Mol. Sci. 2019, 20, 1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manrique, C.; DeMarco, V.G.; Aroor, A.R.; Mugerfeld, I.; Garro, M.; Habibi, J.; Hayden, M.R.; Sowers, J.R. Obesity and insulin resistance induce early development of diastolic dysfunction in young female mice fed a Western diet. Endocrinology 2013, 154, 3632–3642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Chandrasekera, P.C.; Pippin, J.J. Leptin- and leptin receptor-deficient rodent models: Relevance for human type 2 diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, M.; Saraiva, M.J. Transthyretin: A multifaceted protein. Biomol. Concepts 2014, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Khan, S.; Rahman, S.; Singh, L.R. The Extracellular Protein, Transthyretin Is an Oxidative Stress Biomarker. Front. Physiol. 2019, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.; Saraiva, M.J. Transthyretin regulates hippocampal 14-3-3zeta protein levels. FEBS Lett. 2013, 587, 1482–1488. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Choi, J.W.; Yun, J.W.; Chung, I.S.; Cho, H.C.; Song, S.E.; Im, S.S.; Song, D.K. Proteomics approach to identify serum biomarkers associated with the progression of diabetes in Korean patients with abdominal obesity. PLoS ONE 2019, 14, e0222032. [Google Scholar] [CrossRef]
- Saito, S.; Ando, Y.; Nakamura, M.; Ueda, M.; Kim, J.; Ishima, Y.; Akaike, T.; Otagiri, M. Effect of nitric oxide in amyloid fibril formation on transthyretin-related amyloidosis. Biochemistry 2005, 44, 11122–11129. [Google Scholar] [CrossRef]
- Fong, V.H.; Vieira, A. Transthyretin aggregates induce production of reactive nitrogen species. Neurodegener. Dis. 2013, 11, 42–48. [Google Scholar] [CrossRef]
- Chen, J.J.; Genereux, J.C.; Suh, E.H.; Vartabedian, V.F.; Rius, B.; Qu, S.; Dendle, M.T.A.; Kelly, J.W.; Wiseman, R.L. Endoplasmic Reticulum Proteostasis Influences the Oligomeric State of an Amyloidogenic Protein Secreted from Mammalian Cells. Cell Chem. Biol. 2016, 23, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and treatment of cardiac amyloidosis: A position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2021, 42, 1554–1568. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed-Salem, L.; Santos-Mateo, J.J.; Sanchez-Serna, J.; Hernandez-Vicente, A.; Reyes-Marle, R.; Castellon Sanchez, M.I.; Claver-Valderas, M.A.; Gonzalez-Vioque, E.; Haro-Del Moral, F.J.; Garcia-Pavia, P.; et al. Prevalence of wild type ATTR assessed as myocardial uptake in bone scan in the elderly population. Int. J. Cardiol. 2018, 270, 192–196. [Google Scholar] [CrossRef]
- Gonzalez-Lopez, E.; Lopez-Sainz, A.; Garcia-Pavia, P. Diagnosis and Treatment of Transthyretin Cardiac Amyloidosis. Progress and Hope. Rev. Esp. Cardiol. 2017, 70, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Tschope, C.; Elsanhoury, A. Treatment of Transthyretin Amyloid Cardiomyopathy: The Current Options, the Future, and the Challenges. J. Clin. Med. 2022, 11, 2148. [Google Scholar] [CrossRef]
- Nguyen, T.P.; Qu, Z.; Weiss, J.N. Cardiac fibrosis and arrhythmogenesis: The road to repair is paved with perils. J. Mol. Cell. Cardiol. 2014, 70, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Sant’Anna, R.; Gallego, P.; Robinson, L.Z.; Pereira-Henriques, A.; Ferreira, N.; Pinheiro, F.; Esperante, S.; Pallares, I.; Huertas, O.; Almeida, M.R.; et al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat. Commun. 2016, 7, 10787. [Google Scholar] [CrossRef]
- Martinez-Martinez, E.; Jurado-Lopez, R.; Valero-Munoz, M.; Bartolome, M.V.; Ballesteros, S.; Luaces, M.; Briones, A.M.; Lopez-Andres, N.; Miana, M.; Cachofeiro, V. Leptin induces cardiac fibrosis through galectin-3, mTOR and oxidative stress: Potential role in obesity. J. Hypertens. 2014, 32, 1104–1114, discussion 1114. [Google Scholar] [CrossRef]
- Fu, H.Y.; Okada, K.; Liao, Y.; Tsukamoto, O.; Isomura, T.; Asai, M.; Sawada, T.; Okuda, K.; Asano, Y.; Sanada, S.; et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 2010, 122, 361–369. [Google Scholar] [CrossRef]
- Blackwood, E.A.; Hofmann, C.; Santo Domingo, M.; Bilal, A.S.; Sarakki, A.; Stauffer, W.; Arrieta, A.; Thuerauf, D.J.; Kolkhorst, F.W.; Muller, O.J.; et al. ATF6 Regulates Cardiac Hypertrophy by Transcriptional Induction of the mTORC1 Activator, Rheb. Circ. Res. 2019, 124, 79–93. [Google Scholar] [CrossRef]
- Stauffer, W.T.; Blackwood, E.A.; Azizi, K.; Kaufman, R.J.; Glembotski, C.C. The ER Unfolded Protein Response Effector, ATF6, Reduces Cardiac Fibrosis and Decreases Activation of Cardiac Fibroblasts. Int. J. Mol. Sci. 2020, 21, 1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, Y.C.; Chen, C.L.; Zhang, Y.; Mellor, R.L.; Kanter, E.M.; Fang, Y.; Wang, H.C.; Hung, C.T.; Nong, J.Y.; Chen, H.J.; et al. Endoplasmic Reticulum Protein TXNDC5 Augments Myocardial Fibrosis by Facilitating Extracellular Matrix Protein Folding and Redox-Sensitive Cardiac Fibroblast Activation. Circ. Res. 2018, 122, 1052–1068. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Ding, X.; Zhao, T.; Wu, L.; Perkins, S.; Du, H.; Yan, C. Transthyretin Stimulates Tumor Growth through Regulation of Tumor, Immune, and Endothelial Cells. J. Immunol. 2019, 202, 991–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilov, I.V.; Seymour, S.L.; Patel, A.A.; Loboda, A.; Tang, W.H.; Keating, S.P.; Hunter, C.L.; Nuwaysir, L.M.; Schaeffer, D.A. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteom. 2007, 6, 1638–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.; Shilov, I.V.; Seymour, S.L. Nonlinear fitting method for determining local false discovery rates from decoy database searches. J. Proteome Res. 2008, 7, 3661–3667. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [Green Version]
- Okuda, S.; Watanabe, Y.; Moriya, Y.; Kawano, S.; Yamamoto, T.; Matsumoto, M.; Takami, T.; Kobayashi, D.; Araki, N.; Yoshizawa, A.C.; et al. jPOSTrepo: An international standard data repository for proteomes. Nucleic Acids Res. 2017, 45, D1107–D1111. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Martínez, E.; Fernández-Irigoyen, J.; Santamaría, E.; Nieto, M.L.; Bravo-San Pedro, J.M.; Cachofeiro, V. Mitochondrial Oxidative Stress Induces Cardiac Fibrosis in Obese Rats through Modulation of Transthyretin. Int. J. Mol. Sci. 2022, 23, 8080. https://doi.org/10.3390/ijms23158080
Martínez-Martínez E, Fernández-Irigoyen J, Santamaría E, Nieto ML, Bravo-San Pedro JM, Cachofeiro V. Mitochondrial Oxidative Stress Induces Cardiac Fibrosis in Obese Rats through Modulation of Transthyretin. International Journal of Molecular Sciences. 2022; 23(15):8080. https://doi.org/10.3390/ijms23158080
Chicago/Turabian StyleMartínez-Martínez, Ernesto, Joaquín Fernández-Irigoyen, Enrique Santamaría, María Luisa Nieto, José Manuel Bravo-San Pedro, and Victoria Cachofeiro. 2022. "Mitochondrial Oxidative Stress Induces Cardiac Fibrosis in Obese Rats through Modulation of Transthyretin" International Journal of Molecular Sciences 23, no. 15: 8080. https://doi.org/10.3390/ijms23158080