
Discovery of Long Non-Coding RNA MALAT1 Amplification in Precancerous Colorectal Lesions

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
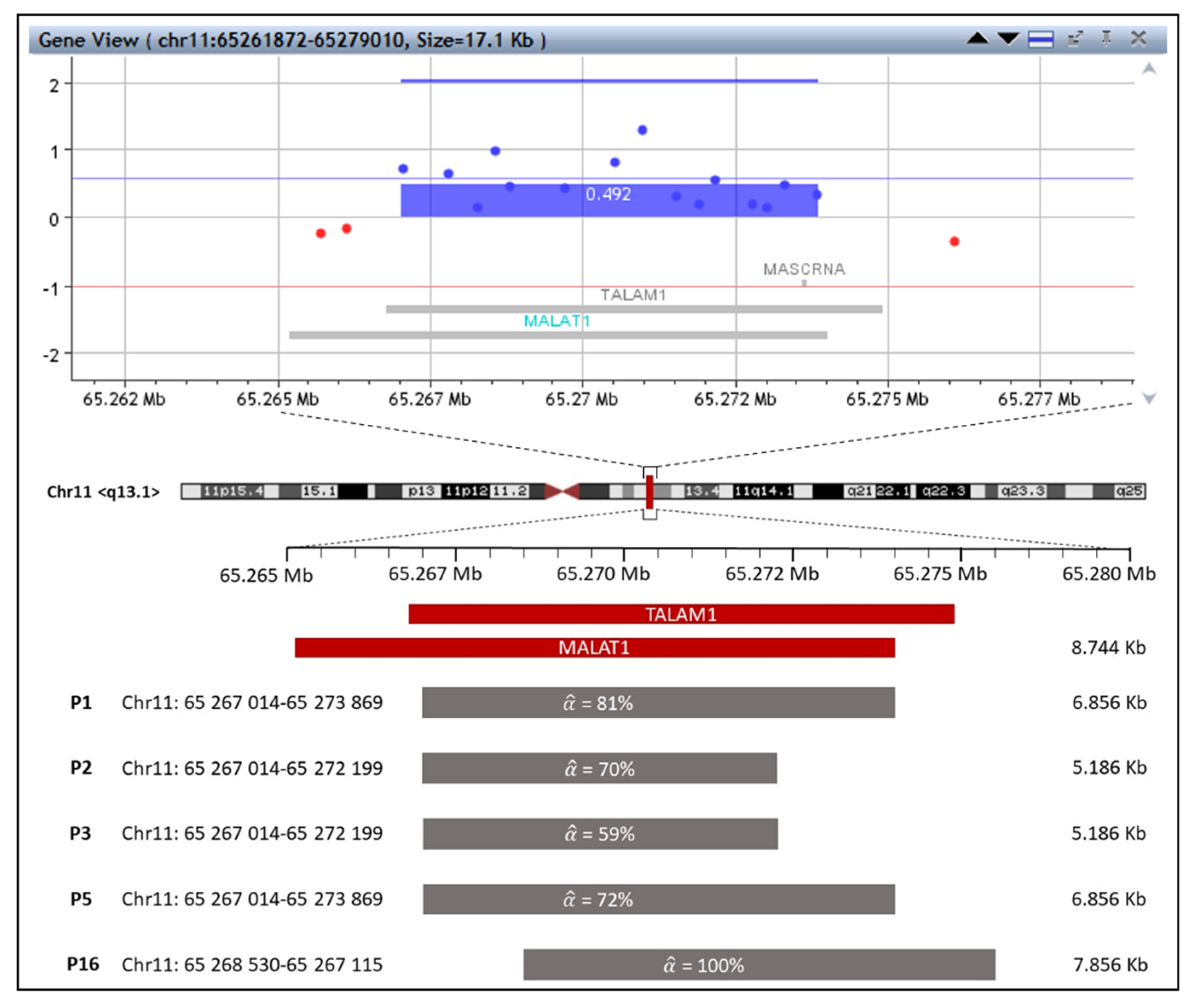
2.1. The First Group with MALAT1 and TALAM1 Gain
2.2. The Second Group with Microdeletions
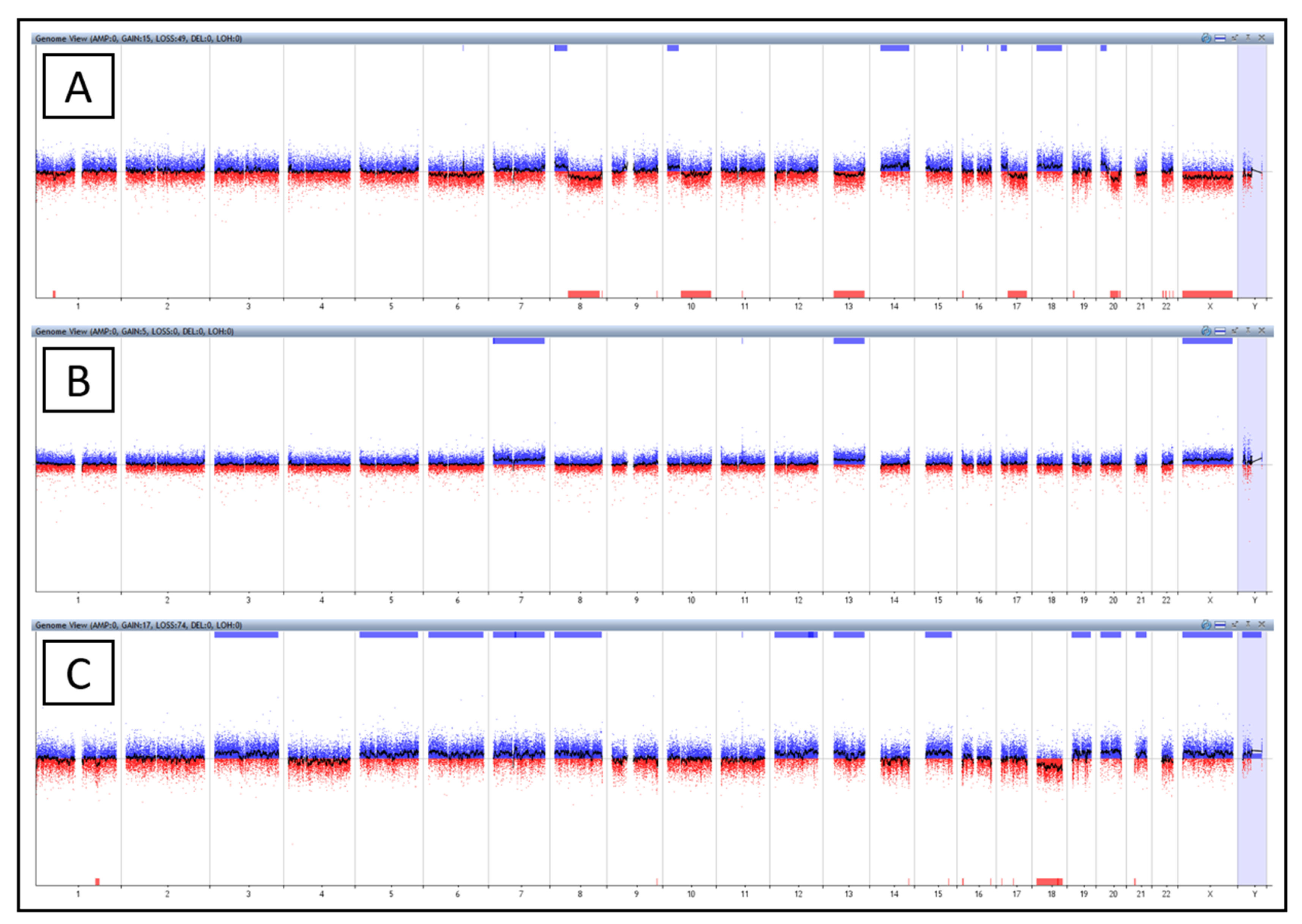
2.3. The Third Group with Affected Karyotype
2.3.1. Patient No. 4
2.3.2. Patient No. 5
2.3.3. Patient No. 16
2.4. The Fourth Group with a Negative Finding
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. DNA Extraction
4.3. Comparative Genomic Hybridization Array Design
4.4. Array Processing and Bioinformatics Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cernat, L.; Blaj, C.; Jackstadt, R.; Brandl, L.; Engel, J.; Hermeking, H.; Jung, A.; Kirchner, T.; Horst, D. Colorectal Cancers Mimic Structural Organization of Normal Colonic Crypts. PLoS ONE 2014, 9, e104284. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Loeve, F.; Boer, R.; Zauber, A.G.; Van Ballegooijen, M.; Van Oortmarssen, G.J.; Winawer, S.J.; Habbema, J.D. National Polyp Study data: Evidence for regression of adenomas. Int. J. Cancer 2004, 111, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, B.; Sillars-Hardebol, A.H.; Postma, C.; Mongera, S.; Terhaar Sive Droste, J.; Obulkasim, A.; van de Wiel, M.; van Criekinge, W.; Ylstra, B.; Fijneman, R.J.; et al. Colorectal adenoma to carcinoma progression is accompanied by changes in gene expression associated with ageing, chromosomal instability, and fatty acid metabolism. Cell. Oncol. 2012, 35, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, M.F.; Ibrahim, A.E.; Arends, M.J. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016, 469, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Kwong, L.N.; Dove, W.F. APC and its modifiers in colon cancer. Adv. Exp. Med. Biol. 2009, 656, 85–106. [Google Scholar] [CrossRef] [Green Version]
- Cross, W.; Kovac, M.; Mustonen, V.; Temko, D.; Davis, H.; Baker, A.M.; Biswas, S.; Arnold, R.; Chegwidden, L.; Gatenbee, C.; et al. The evolutionary landscape of colorectal tumorigenesis. Nat. Ecol. Evol. 2018, 2, 1661–1672. [Google Scholar] [CrossRef] [Green Version]
- Heald, B.; Mester, J.; Rybicki, L.; Orloff, M.S.; Burke, C.A.; Eng, C. Frequent Gastrointestinal Polyps and Colorectal Adenocarcinomas in a Prospective Series of PTEN Mutation Carriers. Gastroenterology 2010, 139, 1927–1933. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, A.; Sharma, A.K.; Damodaran, C. A Review on Notch Signaling and Colorectal Cancer. Cells 2020, 9, 1549. [Google Scholar] [CrossRef]
- Jungwirth, J.; Urbanova, M.; Boot, A.; Hosek, P.; Bendova, P.; Siskova, A.; Svec, J.; Kment, M.; Tumova, D.; Summerova, S.; et al. Mutational analysis of driver genes defines the colorectal adenoma: In situ carcinoma transition. Sci. Rep. 2022, 12, 2570. [Google Scholar] [CrossRef] [PubMed]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siskova, A.; Cervena, K.; Kral, J.; Hucl, T.; Vodicka, P.; Vymetalkova, V. Colorectal Adenomas-Genetics and Searching for New Molecular Screening Biomarkers. Int. J. Mol. Sci. 2020, 21, 3260. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.C.; Torres, M.; Real, F.X. Somatic mosaicism: On the road to cancer. Nat. Rev. Cancer 2016, 16, 43–55. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [Green Version]
- Goyal, B.; Yadav, S.R.M.; Awasthee, N.; Gupta, S.; Kunnumakkara, A.B.; Gupta, S.C. Diagnostic, prognostic, and therapeutic significance of long non-coding RNA MALAT1 in cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188502. [Google Scholar] [CrossRef]
- Shi, Z.Z.; Zhang, Y.M.; Shang, L.; Hao, J.J.; Zhang, T.T.; Wang, B.S.; Liang, J.W.; Chen, X.; Zhang, Y.; Wang, G.Q.; et al. Genomic profiling of rectal adenoma and carcinoma by array-based comparative genomic hybridization. BMC Med. Genom. 2012, 5, 52. [Google Scholar] [CrossRef] [Green Version]
- Douglas, E.J.; Fiegler, H.; Rowan, A.; Halford, S.; Bicknell, D.C.; Bodmer, W.; Tomlinson, I.P.; Carter, N.P. Array comparative genomic hybridization analysis of colorectal cancer cell lines and primary carcinomas. Cancer Res. 2004, 64, 4817–4825. [Google Scholar] [CrossRef] [Green Version]
- Rosner, M.; Hanneder, M.; Siegel, N.; Valli, A.; Hengstschlager, M. The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partners. Mutat. Res. 2008, 658, 234–246. [Google Scholar] [CrossRef]
- Slattery, M.L.; Herrick, J.S.; Lundgreen, A.; Fitzpatrick, F.A.; Curtin, K.; Wolff, R.K. Genetic variation in a metabolic signaling pathway and colon and rectal cancer risk: mTOR, PTEN, STK11, RPKAA1, PRKAG2, TSC1, TSC2, PI3K and Akt1. Carcinogenesis 2010, 31, 1604–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, K.; Liu, H.; Xie, D.; Xiao, Q. Differentially expressed genes ASPN, COL1A1, FN1, VCAN and MUC5AC are potential prognostic biomarkers for gastric cancer. Oncol. Lett. 2019, 17, 3191–3202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.; Tian, P.; Pang, J.; Lai, L.; He, G.; Song, Y.; Zheng, Y. Clinical significance of COL1A1 and COL1A2 expression levels in hypopharyngeal squamous cell carcinoma. Oncol. Lett. 2020, 20, 803–809. [Google Scholar] [CrossRef]
- Zou, X.; Feng, B.; Dong, T.; Yan, G.; Tan, B.; Shen, H.; Huang, A.; Zhang, X.; Zhang, M.; Yang, P.; et al. Up-regulation of type I collagen during tumorigenesis of colorectal cancer revealed by quantitative proteomic analysis. J. Proteom. 2013, 94, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Zhou, Y.; Cheng, A.S.; Yu, J.; To, K.F.; Kang, W. NOTCH receptors in gastric and other gastrointestinal cancers: Oncogenes or tumor suppressors? Mol. Cancer 2016, 15, 80. [Google Scholar] [CrossRef] [Green Version]
- Arcaroli, J.J.; Tai, W.M.; McWilliams, R.; Bagby, S.; Blatchford, P.J.; Varella-Garcia, M.; Purkey, A.; Quackenbush, K.S.; Song, E.-K.; Pitts, T.M.; et al. A NOTCH1 gene copy number gain is a prognostic indicator of worse survival and a predictive biomarker to a Notch1 targeting antibody in colorectal cancer. Int. J. Cancer 2016, 138, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dokumcu, K.; Simonian, M.; Farahani, R.M. miR4673 improves fitness profile of neoplastic cells by induction of autophagy. Cell Death Dis. 2018, 9, 1068. [Google Scholar] [CrossRef]
- Thomis, D.C.; Berg, L.J. The role of Jak3 in lymphoid development, activation, and signaling. Curr. Opin. Immunol. 1997, 9, 541–547. [Google Scholar] [CrossRef]
- Bastepe, M.; Juppner, H. GNAS locus and pseudohypoparathyroidism. Horm. Res. 2005, 63, 65–74. [Google Scholar] [CrossRef]
- Liu, C.; McKeone, D.M.; Walker, N.I.; Bettington, M.L.; Leggett, B.A.; Whitehall, V.L.J. GNAS mutations are present in colorectal traditional serrated adenomas, serrated tubulovillous adenomas and serrated adenocarcinomas with adverse prognostic features. Histopathology 2017, 70, 1079–1088. [Google Scholar] [CrossRef]
- Zauber, P.; Marotta, S.P.; Sabbath-Solitare, M. GNAS gene mutation may be present only transiently during colorectal tumorigenesis. Int. J. Mol. Epidemiol. Genet. 2016, 7, 24–31. [Google Scholar] [PubMed]
- Vashisht, A.A.; Wohlschlegel, J.A. Chapter 8—Role of Human Xeroderma Pigmentosum Group D (XPD) Helicase in Various Cellular Pathways. In Helicases from All Domains of Life; Tuteja, R., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 125–139. [Google Scholar] [CrossRef]
- Zhang, X.; Hamblin, M.H.; Yin, K.J. The long noncoding RNA Malat1: Its physiological and pathophysiological functions. RNA Biol. 2017, 14, 1705–1714. [Google Scholar] [CrossRef]
- Gutschner, T.; Hammerle, M.; Diederichs, S. MALAT1—a paradigm for long noncoding RNA function in cancer. J. Mol. Med. 2013, 91, 791–801. [Google Scholar] [CrossRef]
- Biswas, S.; Thomas, A.A.; Chen, S.; Aref-Eshghi, E.; Feng, B.; Gonder, J.; Sadikovic, B.; Chakrabarti, S. MALAT1: An Epigenetic Regulator of Inflammation in Diabetic Retinopathy. Sci. Rep. 2018, 8, 6526. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hämmerle, M.; Eißmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Groß, M.; et al. The Noncoding RNA MALAT1 Is a Critical Regulator of the Metastasis Phenotype of Lung Cancer Cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meseure, D.; Vacher, S.; Lallemand, F.; Alsibai, K.D.; Hatem, R.; Chemlali, W.; Nicolas, A.; De Koning, L.; Pasmant, E.; Callens, C.; et al. Prognostic value of a newly identified MALAT1 alternatively spliced transcript in breast cancer. Br. J. Cancer 2016, 114, 1395–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Cui, Y.; Liu, L.F.; Ren, W.B.; Li, Q.Q.; Zhou, X.; Li, Y.L.; Li, Y.; Bai, X.Y.; Zu, X.B. High Expression of Long Noncoding RNA MALAT1 Indicates a Poor Prognosis and Promotes Clinical Progression and Metastasis in Bladder Cancer. Clin. Genitourin. Cancer 2017, 15, 570–576. [Google Scholar] [CrossRef]
- Yang, L.; Bai, H.S.; Deng, Y.; Fan, L. High MALAT1 expression predicts a poor prognosis of cervical cancer and promotes cancer cell growth and invasion. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3187–3193. [Google Scholar]
- Konishi, H.; Ichikawa, D.; Yamamoto, Y.; Arita, T.; Shoda, K.; Hiramoto, H.; Hamada, J.; Itoh, H.; Fujita, Y.; Komatsu, S.; et al. Plasma level of metastasis-associated lung adenocarcinoma transcript 1 is associated with liver damage and predicts development of hepatocellular carcinoma. Cancer Sci. 2016, 107, 149–154. [Google Scholar] [CrossRef]
- Ji, Q.; Zhang, L.; Liu, X.; Zhou, L.; Wang, W.; Han, Z.; Sui, H.; Tang, Y.; Wang, Y.; Liu, N.; et al. Long non-coding RNA MALAT1 promotes tumour growth and metastasis in colorectal cancer through binding to SFPQ and releasing oncogene PTBP2 from SFPQ/PTBP2 complex. Br. J. Cancer 2014, 111, 736–748. [Google Scholar] [CrossRef] [Green Version]
- Xiong, M.; Wu, M.; Dan, P.; Huang, W.; Chen, Z.; Ke, H.; Chen, Z.; Song, W.; Zhao, Y.; Xiang, A.P.; et al. LncRNA DANCR represses Doxorubicin-induced apoptosis through stabilizing MALAT1 expression in colorectal cancer cells. Cell Death Dis. 2021, 12, 24. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yang, M.; Tian, J.; Wang, X.; Li, Z. MALAT-1: A long non-coding RNA and its important 3’ end functional motif in colorectal cancer metastasis. Int. J. Oncol. 2011, 39, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cui, Z.; Li, H.; Lv, X.; Gao, M.; Yang, Z.; Bi, Y.; Zhang, Z.; Wang, S.; Zhou, B.; et al. Clinicopathological and prognostic significance of long noncoding RNA MALAT1 in human cancers: A review and meta-analysis. Cancer Cell Int. 2018, 18, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, C.; Zhao, Y.; Li, Y.; Zhang, T.; Ma, Y.; Liu, Y. LncRNA MALAT1 Promotes Lung Cancer Proliferation and Gefitinib Resistance by Acting as a miR-200a Sponge. Arch. De Bronconeumol. 2019, 55, 627–633. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, X.; Hu, X.; Zhou, W.; Zhang, P.; Zhang, J.; Yang, S.; Liu, Y. The effects of lncRNA MALAT1 on proliferation, invasion and migration in colorectal cancer through regulating SOX9. Mol. Med. 2018, 24, 52. [Google Scholar] [CrossRef]
- Su, K.; Wang, N.; Shao, Q.; Liu, H.; Zhao, B.; Ma, S. The role of a ceRNA regulatory network based on lncRNA MALAT1 site in cancer progression. Biomed. Pharmacother. 2021, 137, 111389. [Google Scholar] [CrossRef]
- Ji, Q.; Cai, G.; Liu, X.; Zhang, Y.; Wang, Y.; Zhou, L.; Sui, H.; Li, Q. MALAT1 regulates the transcriptional and translational levels of proto-oncogene RUNX2 in colorectal cancer metastasis. Cell Death Dis. 2019, 10, 378. [Google Scholar] [CrossRef]
- Chaleshi, V.; Irani, S.; Alebouyeh, M.; Mirfakhraie, R.; Aghdaei, H.A. Association of lncRNA-p53 regulatory network (lincRNA-p21, lincRNA-ROR and MALAT1) and p53 with the clinicopathological features of colorectal primary lesions and tumors. Oncol. Lett. 2020, 19, 3937–3949. [Google Scholar] [CrossRef] [Green Version]
- Aisner, D.L.; Nguyen, T.T.; Paskulin, D.D.; Le, A.T.; Haney, J.; Schulte, N.; Chionh, F.; Hardingham, J.; Mariadason, J.; Tebbutt, N.; et al. ROS1 and ALK fusions in colorectal cancer, with evidence of intratumoral heterogeneity for molecular drivers. Mol. Cancer Res. 2014, 12, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Oneyama, C.; Yoshikawa, Y.; Ninomiya, Y.; Iino, T.; Tsukita, S.; Okada, M. Fer tyrosine kinase oligomer mediates and amplifies Src-induced tumor progression. Oncogene 2016, 35, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Medico, E.; Russo, M.; Picco, G.; Cancelliere, C.; Valtorta, E.; Corti, G.; Buscarino, M.; Isella, C.; Lamba, S.; Martinoglio, B.; et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat. Commun. 2015, 6, 7002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, djw332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohonen-Corish, M.R.J.; Sigglekow, N.D.; Susanto, J.; Chapuis, P.H.; Bokey, E.L.; Dent, O.F.; Chan, C.; Lin, B.P.C.; Seng, T.J.; Laird, P.W.; et al. Promoter methylation of the mutated in colorectal cancer gene is a frequent early event in colorectal cancer. Oncogene 2007, 26, 4435–4441. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, G.; Liang, Z.; Mei, Z.; Wu, T.; Cui, A.; Liu, C.; Cui, L. Lysyl oxidase: A colorectal cancer biomarker of lung and hepatic metastasis. Thorac. Cancer 2018, 9, 785–793. [Google Scholar] [CrossRef]
- Hirsch, D.; Camps, J.; Varma, S.; Kemmerling, R.; Stapleton, M.; Ried, T.; Gaiser, T. A new whole genome amplification method for studying clonal evolution patterns in malignant colorectal polyps. Genes. Chromosomes Cancer 2012, 51, 490–500. [Google Scholar] [CrossRef] [Green Version]
- Hermsen, M.; Postma, C.; Baak, J.; Weiss, M.; Rapallo, A.; Sciutto, A.; Roemen, G.; Arends, J.W.; Williams, R.; Giaretti, W.; et al. Colorectal adenoma to carcinoma progression follows multiple pathways of chromosomal instability. Gastroenterology 2002, 123, 1109–1119. [Google Scholar] [CrossRef] [Green Version]
- Paraskeva, C.; Buckle, B.G.; Sheer, D.; Wigley, C.B. The isolation and characterization of colorectal epithelial cell lines at different stages in malignant transformation from familial polyposis coli patients. Int. J. Cancer 1984, 34, 49–56. [Google Scholar] [CrossRef]
- Longy, M.; Saura, R.; Schouler, L.; Mauhin, C.; Goussot, J.F.; Grison, O.; Couzigou, P. Chromosomal analysis of colonic adenomatous polyps. Cancer Genet. Cytogenet. 1990, 49, 249–257. [Google Scholar] [CrossRef]
- Bomme, L.; Bardi, G.; Pandis, N.; Fenger, C.; Kronborg, O.; Heim, S. Cytogenetic analysis of colorectal adenomas: Karyotypic comparisons of synchronous tumors. Cancer Genet. Cytogenet. 1998, 106, 66–71. [Google Scholar] [CrossRef]
- Ahnen, D.J.; Wade, S.W.; Jones, W.F.; Sifri, R.; Mendoza Silveiras, J.; Greenamyer, J.; Guiffre, S.; Axilbund, J.; Spiegel, A.; You, Y.N. The increasing incidence of young-onset colorectal cancer: A call to action. Mayo. Clin. Proc. 2014, 89, 216–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loomans-Kropp, H.A.; Umar, A. Increasing Incidence of Colorectal Cancer in Young Adults. J. Cancer Epidemiol. 2019, 2019, 9841295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mieulet, V.; Lamb, R.F. Tuberous sclerosis complex: Linking cancer to metabolism. Trends Mol. Med. 2010, 16, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.E.; Dunlop, E.A.; Seifan, S.; McCann, H.D.; Hay, T.; Parfitt, G.J.; Jones, A.T.; Giles, P.J.; Shen, M.H.; Sampson, J.R.; et al. Loss of tuberous sclerosis complex 2 sensitizes tumors to nelfinavir-bortezomib therapy to intensify endoplasmic reticulum stress-induced cell death. Oncogene 2018, 37, 5913–5925. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Li, F.; Hu, H.; Fang, Z.; Gao, Z.; Xia, G.; Ng, W.L.; Khodadadi-Jamayran, A.; Chen, T.; Deng, J.; et al. Loss of TSC1/TSC2 sensitizes immune checkpoint blockade in non-small cell lung cancer. Sci. Adv. 2022, 8, eabi9533. [Google Scholar] [CrossRef]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Cheung, S.; Shaw, C.; Scott, D.; Patel, A.; Sahoo, T.; Bacino, C.; Pursley, A.; Li, J.; Erickson, R.; Gropman, A.; et al. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am. J. Med. Genetics. Part A 2007, 143A, 1679–1686. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Patient | Type | Chromosome | Location | Cytoband | Size | Gene Name | |
|---|---|---|---|---|---|---|---|
| P1 | loss | 16 | 2,110,696–2,136,380 | p13.3 | 25.685 kb | 38% | TSC2 |
| gain | 1 | 2,260,756–3,080,894 | p36.33–p36.32 | 820.139 kb | 39% | PEX10,PLCH2, PANK4, HES5, TNFRSF14, MMEL1, ACTRT2, PRDM16, MORN1, LOC100129534, RER1, TNFRSF14-AS1, LOC100996583, PRXL2B, TTC34, PRDM16-DT, MIR4251 | |
| 11 | 65,267,014–65,273,869 | q13.1 | 6.856 kb | 81% | MALAT1, TALAM1 | ||
| P2 | loss | 6 | 107,338,10–127,407,686 | q21–q22.33 | 20,069.584 kb | 65% | MTRES1, BEND3, PDSS2, SOBP, SEC63, OSTM1, NR2E1, SNX3, AFG1L, FOXO3, ARMC2, SESN1, CD164, SMPD2, MICAL1, ZBTB24, AK9, FIG4, GPR6, WASF1, CDC40, DDO, SLC22A16, CDK19, AMD1, GTF3C6, RPF2, SLC16A10, MFSD4B, REV3L, TRAF3IP2, FYN, CCN6, TUBE1, LAMA4, MARCKS, HDAC2, HS3ST5, FRK, COL10A1, DSE, TSPYL1, CALHM6, TRAPPC3L, RSPH4A, KPNA5, GPRC6A, RFX6, VGLL2, ROS1, GOPC, NUS1, PLN, MCM9, ASF1A, MAN1A1, TBC1D32, GJA1, HSF2, SERINC1, PKIB, FABP7, SMPDL3A, CLVS2, TRDN, NKAIN2, RNF217-AS1, RNF217, TPD52L1, HEY2, NCOA7, HINT3, CENPW, SCML4, OSTM1-AS1, LINC00222, ARMC2-AS1, CEP57L1, CCDC162P, PPIL6, METTL24, SNORA40C, GSTM2P1, SNORD166, TRAF3IP2-AS1, LINC02527, FAM229B, LOC101927640, RFPL4B, LINC02518, LINC02541, MROCKI, FLJ34503, HDAC2-AS2, LINC02534, TPI1P3, NT5DC1, TSPYL4, LOC100287467, CALHM5, CALHM4, RWDD1, ZUP1, FAM162B, DCBLD1, LOC101927919, SLC35F1, LOC105377967, CEP85L, BRD7P3, SELENOKP3, FAM184A, MIR548B, LOC285762, LOC105377975, MIR3144, TRDN-AS1, LOC100126584, HDDC2, LINC02523, NCOA7-AS1, TRMT11, MIR588 |
| gain | 11 | 65,267,014–65,272,199 | q13.1 | 5.186 kb | 70% | MALAT1, TALAM1 | |
| P3 | loss | 5 | 102,026,08–127,375,136 | q21.1–q23.3 | 25,349.057 kb | 59% | PAM, PPIP5K2, C5orf30, NUDT12, EFNA5, FBXL17, FER, MAN2A1, SLC25A46, TSLP, WDR36, CAMK4, STARD4, NREP, EPB41L4A, APC, SRP19, REEP5, DCP2, MCC, TSSK1B, YTHDC2, KCNN2, TRIM36, PGGT1B, FEM1C, TICAM2, CDO1, ATG12, AP3S1, LVRN, COMMD10, SEMA6A, DMXL1, TNFAIP8, HSD17B4, FAM170A, PRR16, FTMT, SRFBP1, LOX, SNCAIP, SNX2, PPIC, PRDM6, CEP120, CSNK1G3, ALDH7A1, PHAX, LMNB1, MARCHF3, MEGF10, GIN1, LINC02115, RAB9BP1, LINC01950, LINC01023, LOC285638, PJA2, LINC01848, TMEM232, MIR548F3, STARD4-AS1, NREP-AS1, EPB41L4A-AS1, SNORA13, LOC101927023, EPB41L4A-DT, LINC02200, LOC102467216, LOC101927078, LINC01957, CCDC112, TMED7-TICAM2, LOC101927100, TMED7, LOC102467217, LINCADL, ARL14EPL, MIR12130, LOC101927190, SEMA6A-AS1, SEMA6A-AS2, LINC02214, LINC00992, LINC02147, LINC02148, LINC02208, LINC02215, DTWD2, MIR1244-1, MIR1244-2, MIR1244-3, MIR1244-4, LOC105379143, MIR5706, LOC102467226, ZNF474, LOC100505841, MGC32805, LOC101927357, LINC02201, SNX24, LOC105379152, LINC01170, ZNF608, LOC101927421, LINC02240, LINC02039, LOC101927488, GRAMD2B, TEX43, LMNB1-DT, C5orf63, PRRC1, CTXN3, CCDC192, LINC01184 |
| gain | 11 | 65,267,014–65,272,199 | q13.1 | 5.186 kb | 70% | MALAT1, TALAM1 |
| Patient | Type | Chromosome | Location | Cytoband | Size | Gene Name | |
|---|---|---|---|---|---|---|---|
| P6 | loss | 16 | 2,103,321–2,138,073 | p13.3 | 34.753 kb | 51% | TSC2 |
| 17 | 48,263,792–48,273,321 | q21.33 | 9.530 kb | 34% | COL1A1 | ||
| P7 | loss | 7 | 73,442,449–73,481,111 | q11.23 | 38.663 kb | 35% | ELN |
| 16 | 2,105,434–2,138,073 | p13.3 | 32.640 kb | 58% | TSC2 | ||
| 17 | 48,263,792–48,273,777 | q21.33 | 9.986 kb | 40% | COL1A1 | ||
| 20 | 57,407,840–57,495,925 | q13.32 | 88.086 kb | 38% | GNAS-AS1, GNAS, LOC1019 | ||
| P10 | loss | 9 | 21,549,338–23,792,459 | p21.3 | 2243.122 kb | 46% | MIR31HG, MTAP, CDKN2A, CDKN2B-AS1, CDKN2B, DMRTA1, ELAVL2, CDKN2A-DT, LINC01239, LOC101929563 |
| Patient | Type | Chromosome | Location | Cytoband | Size | Gene Name | |
|---|---|---|---|---|---|---|---|
| P4 | loss | 1 | 55,108,604–61,921,519 | p32.3–p31.3 | 6812.92 kb | 33% | TTC4, PARS2, LEXM, DHCR24, BSND, PCSK9, USP24, PLPP3, PRKAA2, FYB2, C8A, C8B, DAB1, OMA1, TACSTD2, MYSM1, JUN, FGGY, HOOK1, CYP2J2, NFIA, MROH7, MROH7-TTC4, TTC22, TMEM61, LOC100507634, MIR4422HG, MIR4422, LINC01753, LINC01755, LINC01767, LOC101929935, DAB1-AS1, LINC01135, LINC01358, HSD52, MIR4711, LOC101926944, C1orf87, LINC01748, LOC101926964, NFIA-AS2, NFIA-AS1 |
| 4 | 1,802,707–1,809,469 | p16.3 | 6.763 kb | 65% | FGFR3 | ||
| 9 | 139,394,991–139,418,283 | q34.3 | 23.293 kb | 50% | NOTCH1, MIR4673 | ||
| 11 | 65,265,673–65,273,325 | q13.1 | 7.653 kb | 100% | MALAT1, TALAM1 | ||
| 16 | 2,105,434–2,138,073 | p13.3 | 32.640 kb | 50% | TSC2 | ||
| gain | 6 | 107,068,675–109,166,111 | q21 | 2097.437 kb | 47% | RTN4IP1, QRSL1, MTRES1, BEND3, PDSS2, SOBP, SEC63, OSTM1, NR2E1, SNX3, AFG1L, FOXO3, LINC02526, LINC02532, MIR587, SCML4, OSTM1-AS1, LINC00222 | |
| 7 | 83,325–2,737,748 | p22.3 | 2654.42 kb | 40% | FAM20C, PDGFA, PRKAR1B, DNAAF5, SUN1, GET4, ADAP1, COX19, CYP2W1, MIR339, GPER1, ZFAND2A, INTS1, MAFK, PSMG3, ELFN1, MAD1L1, MRM2, NUDT1, SNX8, EIF3B, CHST12, LFNG, BRAT1, IQCE, TTYH3, AMZ1, LOC102723672, LOC100507642, LOC105375115, LOC442497, HRAT92, LOC101927000, LOC101926963, C7orf50, GPR146, LOC101927021, UNCX, MICALL2, LOC100128653, TMEM184A, PSMG3-AS1, TFAMP1, ELFN1-AS1, MIR4655, SNORA114, MIR6836, GRIFIN, MIR4648 | ||
| P4 | monosomy | 8q | - | 25% | - | ||
| 13 | 14% | ||||||
| 17q | 20% | ||||||
| 20q | 31% | ||||||
| X | 12% | ||||||
| trisomy | 8p | - | 23% | - | |||
| 10p | 19% | ||||||
| 14 | 23% | ||||||
| 17p | 20% | ||||||
| 18 | 23% | ||||||
| 20p | 31% | ||||||
| P5 | gain | 11 | 65,267,014–65,273,869 | q13.1 | 6.856 kb | 100% | MALAT1, TALAM1 |
| trisomy | 7 | - | 23% | - | |||
| 13 | 22% | ||||||
| X | 11% | ||||||
| P16 | loss | 1 | 185,274268–199,118,773 | q25.3–q32.1 | 13,828 kb | 24% | HMCN1, PRG4, TPR, ODR4, OCLM, PDC, PTGS2, PACERR, PLA2G4A, BRINP3, RGS18, RGS21, RGS1, RGS13, RGS2, UCHL5, RO60, GLRX2, CDC73, B3GALT2, KCNT2, CFH, CFHR3, CFHR1, CFHR4, CFHR2, CFHR5, F13B, ASPM, CRB1, DENND1B, LHX9, NEK7, ATP6V1G3, PTPRC, MIR181B1, MIR181A1, LOC102724919, LINC01036, LINC01037, LINC01351, LINC01720, LINC01680, MIR4426, LINC01032, SCARNA18B, MIR1278, LINC01031, LINC01724, MIR4735, ZBTB41, C1orf53, MIR181A1HG, LINC01222, LINC01221, LINC02789 |
| 1 | 890,945–3,729,090 | p36.33–p36.32 | 2838.146 kb | 40% | NOC2L, PERM1, HES4, ISG15, AGRN, MIR200B, MIR200A, MIR429, TNFRSF18, TNFRSF4, SDF4, B3GALT6, C1QTNF12, SCNN1D, INTS11, CPTP, TAS1R3, DVL1, MXRA8, AURKAIP1, CCNL2, MRPL20, VWA1, ATAD3C, ATAD3B, ATAD3A, TMEM240, SSU72, MIB2, MMP23B, MMP23A, CDK11B, CDK11A, NADK, GNB1, CALML6, GABRD, PRKCZ, FAAP20, SKI, PEX10, PLCH2, PANK4, HES5, TNFRSF14, MMEL1, ACTRT2, PRDM16, MEGF6, MIR551A, TPRG1L, WRAP73, TP73, SMIM1, CEP104, KLHL17, PLEKHN1, LOC100288175, RNF223, C1orf159, LINC01342, TTLL10, UBE2J2, ACAP3, MIR6726, SNORD167, PUSL1, MIR6727, MIR6808, MRPL20-AS1, ANKRD65, TMEM88B, LINC01770, FNDC10, LOC105378586, SLC35E2B, SLC35E2A, TMEM52, CFAP74, LOC105378591, PRKCZ-AS1, MORN1, LOC100129534, RER1, TNFRSF14-AS1, LOC100996583, PRXL2B, TTC34, PRDM16-DT, MIR4251, ARHGEF16, TP73-AS1, CCDC27, LRRC47 | ||
| P16 | loss | 9 | 139,389,744–139,440,753 | q34.3 | 51.010 kb | 70% | NOTCH1, MIR4673, MIR4674 |
| 16 | 2,044,093–2,263,638 | p13.3 | 219.546 kb | 52% | SYNGR3, ZNF598, NPW, SLC9A3R2, NTHL1, TSC2, PKD1, MIR1225, RAB26, TRAF7, CASKIN1, MLST8, PGP, LOC105371049, MIR6511B1, MIR6511B2, MIR4516, MIR3180-5, SNHG19, SNORD60, BRICD5 | ||
| 16 | 88,365,786–89,383,369 | q24.2–q24.3 | 1017.584 kb | 43% | ZNF469, ZFPM1, IL17C, CYBA, MVD, SNAI3, RNF166, CTU2, PIEZO1, CDT1, APRT, GALNS, TRAPPC2L, CBFA2T3, ACSF3, CDH15, ANKRD11, MIR5189, LOC100128882, ZC3H18-AS1, ZC3H18, SNAI3-AS1, MIR4722, LOC100289580, LOC339059, PABPN1L, LOC101927793, LOC100129697, LINC00304, LINC02138, SLC22A31, ZNF778, LOC105371414 | ||
| loss | 17 | 36,861,875–36,896,355 | q12 | 34.481 kb | 54% | MLLT6, CISD3, PCGF2, MIR4726 | |
| 22 | 19,702,774–19,851,138 | q11.21 | 148.365 kb | 47% | SEPTIN5, SEPT5-GP1BB, GP1BB, TBX1, GNB1L, RTL10 | ||
| gain | 11 | 65,268,530–65,276,115 | q13.1 | 7.586 kb | 100% | MALAT1, TALAM1, MASCRNA | |
| monosomy | 18 | - | 31% | - | |||
| trisomy | 3 | - | 23% | - | |||
| 5 | 20% | ||||||
| 6 | 20% | ||||||
| 7 | 20% | ||||||
| 8 | 20% | ||||||
| 12 | 20% | ||||||
| 13 | 25% | ||||||
| 15 | 26% | ||||||
| 19 | 30% | ||||||
| 20 | 32% | ||||||
| 21 | 20% | ||||||
| X | 11% | ||||||
| Y | 10% |
| Sample ID | Gender | Age | Histology Type of Adenoma | Size of Adenoma (mm) | Vienna Classification | Grade | Localization |
|---|---|---|---|---|---|---|---|
| P1 | F a | 61 | T c | 15 × 8 × 10 | 3 | LG e | colon |
| P2 | M b | 60 | TL d | 7 × 4 × 4 | 4.1. | HG f | colon |
| P3 | F | 56 | T | 24 × 12 × 8 | 3 | LG | rectum |
| P4 | F | 29 | T | 8 × 8 g | 3 | LG | colon |
| P5 | F | 43 | T | 7 × 5 g | 3 | LG | rectum |
| P6 | M | 64 | T | 4 × 2 g | 3 | LG | colon |
| P7 | M | 63 | T | 3 × 4 g | 3 | LG | colon |
| P8 | M | 67 | T | 4 × 2 × 2 | 3 | LG | colon |
| P9 | M | 61 | T | 5 × 5 g | 4.1. | HG | rectum |
| P10 | M | 68 | T | 12 × 8 × 10 | 3 | LG | rectum |
| P11 | F | 44 | TL | 10 × 10 × 4 | 3 | LG | rectum |
| P12 | M | 53 | T | 3 × 10 g | 3 | LG | colon |
| P13 | M | 54 | T | 9 × 3 × 3 | 3 | LG | colon |
| P14 | M | 49 | TL | 10 × 3 × 2 | 3 | LG | colon |
| P15 | F | 57 | T | 2 × 2 g | 3 | LG | colon |
| P16 | M | 43 | TL | 18 × 13 × 11 | 3 | LG | rectum |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siskova, A.; Kral, J.; Drabova, J.; Cervena, K.; Tomasova, K.; Jungwirth, J.; Hucl, T.; Kohout, P.; Summerova, S.; Vodickova, L.; et al. Discovery of Long Non-Coding RNA MALAT1 Amplification in Precancerous Colorectal Lesions. Int. J. Mol. Sci. 2022, 23, 7656. https://doi.org/10.3390/ijms23147656
Siskova A, Kral J, Drabova J, Cervena K, Tomasova K, Jungwirth J, Hucl T, Kohout P, Summerova S, Vodickova L, et al. Discovery of Long Non-Coding RNA MALAT1 Amplification in Precancerous Colorectal Lesions. International Journal of Molecular Sciences. 2022; 23(14):7656. https://doi.org/10.3390/ijms23147656
Chicago/Turabian StyleSiskova, Anna, Jan Kral, Jana Drabova, Klara Cervena, Kristyna Tomasova, Jiri Jungwirth, Tomas Hucl, Pavel Kohout, Sandra Summerova, Ludmila Vodickova, and et al. 2022. "Discovery of Long Non-Coding RNA MALAT1 Amplification in Precancerous Colorectal Lesions" International Journal of Molecular Sciences 23, no. 14: 7656. https://doi.org/10.3390/ijms23147656