
Innate Immunity Crosstalk with Helicobacter pylori: Pattern Recognition Receptors and Cellular Responses


{kind=link}
{kind=link}
Abstract
:1. Introduction
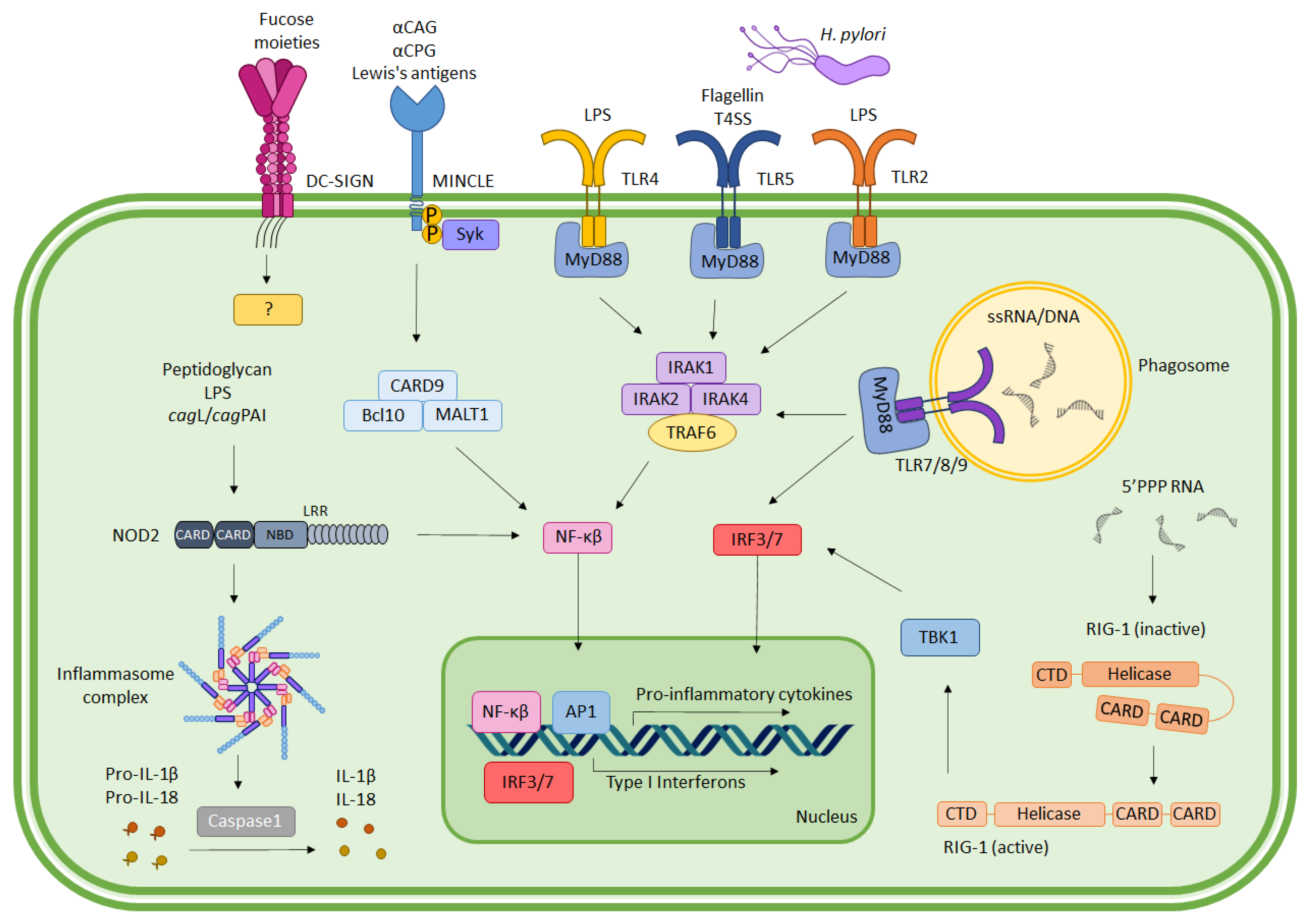
2. Immune Recognition of H. pylori
2.1. Toll-Like Receptors (TLRs)
2.2. C-Type Lectin Receptors (CLRs)
2.3. NOD-Like Receptors (NLRs)
2.4. RIG-I-Like Receptors (RLRs)
3. Innate Immune Cell Activation and Suppression by H. pylori
3.1. Neutrophils
3.2. Monocytes/Macrophages
3.3. Dendritic Cells
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; World Health Organization: Lyon, France, 1994; Volume 61, pp. 1–241. [Google Scholar]
- Peleteiro, B.; Bastos, A.; Ferro, A.; Lunet, N. Prevalence of Helicobacter pylori infection worldwide: A systematic review of studies with national coverage. Dig. Dis. Sci. 2014, 59, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Falkeis-Veits, C.; Vieth, M. Non-malignant Helicobacter pylori-Associated Diseases. Adv. Exp. Med. Biol. 2019, 1149, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Malignant Helicobacter pylori-Associated Diseases: Gastric Cancer and MALT Lymphoma. Adv. Exp. Med. Biol. 2019, 1149, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Reyes, V.E.; Peniche, A.G. Helicobacter pylori Deregulates T and B Cell Signaling to Trigger Immune Evasion. Curr. Top. Microbiol. Immunol. 2019, 421, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Arnold, I.C.; Müller, A. Mechanisms of persistence, innate immune activation and immunomodulation by the gastric pathogen Helicobacter pylori. Curr. Opin. Microbiol. 2020, 54, 1–10. [Google Scholar] [CrossRef]
- Nagashima, H.; Yamaoka, Y. Importance of Toll-like Receptors in Pro-inflammatory and Anti-inflammatory Responses by Helicobacter pylori Infection. Curr. Top. Microbiol. Immunol. 2019, 421, 139–158. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P.Y. The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Castaño-Rodríguez, N.; Kaakoush, N.O.; Mitchell, H.M. Pattern-Recognition Receptors and Gastric Cancer. Front. Immunol. 2014, 5, 336. [Google Scholar] [CrossRef]
- Smith, S.M. Role of Toll-like receptors in Helicobacter pylori infection and immunity. World J. Gastrointest. Pathophysiol. 2014, 5, 133–146. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Toll-like receptors; their physiological role and signal transduction system. Int. Immunopharmacol. 2001, 1, 625–635. [Google Scholar] [CrossRef]
- Smith, M.F.; Mitchell, A.; Li, G.; Ding, S.; Fitzmaurice, A.M.; Ryan, K.; Crowe, S.; Goldberg, J.B. Toll-like Receptor (TLR) 2 and TLR5, but Not TLR4, Are Required for Helicobacter pylori-induced NF-κB Activation and Chemokine Expression by Epithelial Cells. J. Biol. Chem. 2003, 278, 32552–32560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäckhed, F.; Rokbi, B.; Torstensson, E.; Zhao, Y.; Nilsson, C.; Seguin, D.; Normark, S.; Buchan, A.M.J.; Richter-Dahlfors, A. Gastric Mucosal Recognition of Helicobacter pylori Is Independent of Toll-Like Receptor 4. J. Infect. Dis. 2003, 187, 829–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, S.-I.; Ohnishi, T.; Muroi, M.; Tanamoto, K.-I.; Fujii, N.; Amano, K.-I. Highly-purified Helicobacter pylori LPS preparations induce weak inflammatory reactions and utilize Toll-like receptor 2 complex but not Toll-like receptor 4 complex. FEMS Immunol. Med. Microbiol. 2007, 51, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Tongtawee, T.; Simawaranon, T.; Wattanawongdon, W.; Dechsukhum, C.; Leeanansaksiri, W. Toll-like receptor 2 and 4 polymorphisms associated with Helicobacter pylori susceptibility and gastric cancer. Turk. J. Gastroenterol. 2019, 30, 15–20. [Google Scholar] [CrossRef]
- Hashimoto, M.; Tawaratsumida, K.; Kariya, H.; Aoyama, K.; Tamura, T.; Suda, Y. Lipoprotein is a predominant Toll-like receptor 2 ligand in Staphylococcus aureus cell wall components. Int. Immunol. 2006, 18, 355–362. [Google Scholar] [CrossRef]
- Nagashima, H.; Iwatani, S.; Cruz, M.; Jiménez Abreu, J.A.; Uchida, T.; Mahachai, V.; Vilaichone, R.-k.; Graham, D.Y.; Yamaoka, Y. Toll-like Receptor 10 in Helicobacter pylori Infection. J. Infect. Dis. 2015, 212, 1666–1676. [Google Scholar] [CrossRef] [Green Version]
- Yokota, S.-I.; Okabayashi, T.; Rehli, M.; Fujii, N.; Amano, K.-I. Helicobacter pylori Lipopolysaccharides Upregulate Toll-Like Receptor 4 Expression and Proliferation of Gastric Epithelial Cells via the MEK1/2-ERK1/2 Mitogen-Activated Protein Kinase Pathway. Infect. Immun. 2010, 78, 468–476. [Google Scholar] [CrossRef] [Green Version]
- Chochi, K.; Ichikura, T.; Kinoshita, M.; Majima, T.; Shinomiya, N.; Tsujimoto, H.; Kawabata, T.; Sugasawa, H.; Ono, S.; Seki, S.; et al. Helicobacter pylori Augments Growth of Gastric Cancers via the Lipopolysaccharide-Toll-like Receptor 4 Pathway whereas Its Lipopolysaccharide Attenuates Antitumor Activities of Human Mononuclear Cells. Clin. Cancer Res. 2008, 14, 2909–2917. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.; Wu, Z.; Zhong, S.; Li, M.; Shu, Y. Factors influencing the immunogenicity of influenza vaccines. Hum. Vaccines Immunother. 2021, 17, 2706–2718. [Google Scholar] [CrossRef]
- Pachathundikandi, S.K.; Brandt, S.; Madassery, J.; Backert, S. Induction of TLR-2 and TLR-5 Expression by Helicobacter pylori Switches cagPAI-Dependent Signalling Leading to the Secretion of IL-8 and TNF-α. PLoS ONE 2011, 6, e19614. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Neddermann, M.; Lind, J.; Pachathundikandi, S.K.; Sharafutdinov, I.; Gutiérrez-Escobar, A.J.; Brönstrup, M.; Tegge, W.; Hong, M.; Rohde, M.; et al. Toll-like Receptor 5 Activation by the CagY Repeat Domains of Helicobacter pylori. Cell Rep. 2020, 32, 108159. [Google Scholar] [CrossRef] [PubMed]
- Pachathundikandi, S.K.; Tegtmeyer, N.; Arnold, I.C.; Lind, J.; Neddermann, M.; Falkeis-Veits, C.; Chattopadhyay, S.; Brönstrup, M.; Tegge, W.; Hong, M.; et al. T4SS-dependent TLR5 activation by Helicobacter pylori infection. Nat. Commun. 2019, 10, 5717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gantier, M.P.; Irving, A.T.; Kaparakis-Liaskos, M.; Xu, D.; Evans, V.A.; Cameron, P.U.; Bourne, J.A.; Ferrero, R.L.; John, M.; Behlke, M.A.; et al. Genetic Modulation of TLR8 Response following Bacterial Phagocytosis. Hum. Mutat. 2010, 31, 1069–1079. [Google Scholar] [CrossRef]
- Rad, R.; Ballhorn, W.; Voland, P.; Eisenacher, K.; Mages, J.; Rad, L.; Ferstl, R.; Lang, R.; Wagner, H.; Schmid, R.M.; et al. Extracellular and intracellular pattern recognition receptors cooperate in the recognition of Helicobacter pylori. Gastroenterology 2009, 136, 2247–2257. [Google Scholar] [CrossRef]
- Alvarez-Arellano, L.; Cortés-Reynosa, P.; Sánchez-Zauco, N.; Salazar, E.; Torres, J.; Maldonado-Bernal, C. TLR9 and NF-κB are partially involved in activation of human neutrophils by Helicobacter pylori and its purified DNA. PLoS ONE 2014, 9, e101342. [Google Scholar] [CrossRef] [Green Version]
- Otani, K.; Tanigawa, T.; Watanabe, T.; Nadatani, Y.; Sogawa, M.; Yamagami, H.; Shiba, M.; Watanabe, K.; Tominaga, K.; Fujiwara, Y.; et al. Toll-like receptor 9 signaling has anti-inflammatory effects on the early phase of Helicobacter pylori-induced gastritis. Biochem. Biophys. Res. Commun. 2012, 426, 342–349. [Google Scholar] [CrossRef]
- Varga, M.G.; Piazuelo, M.B.; Romero-Gallo, J.; Delgado, A.G.; Suarez, G.; Whitaker, M.E.; Krishna, U.S.; Patel, R.V.; Skaar, E.P.; Wilson, K.T.; et al. TLR9 activation suppresses inflammation in response to Helicobacter pylori infection. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G852–G858. [Google Scholar] [CrossRef] [Green Version]
- Tone, K.; Stappers, M.H.T.; Willment, J.A.; Brown, G.D. C-type lectin receptors of the Dectin-1 cluster: Physiological roles and involvement in disease. Eur. J. Immunol. 2019, 49, 2127–2133. [Google Scholar] [CrossRef] [Green Version]
- Geijtenbeek, T.B.H.; Gringhuis, S.I. Signalling through C-type lectin receptors: Shaping immune responses. Nat. Rev. Immunol. 2009, 9, 465–479. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; den Dunnen, J.; Litjens, M.; van der Vlist, M.; Geijtenbeek, T.B.H. Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to Mycobacterium tuberculosis, HIV-1 and Helicobacter pylori. Nat. Immunol. 2009, 10, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Bergman, M.P.; Engering, A.; Smits, H.H.; van Vliet, S.J.; van Bodegraven, A.A.; Wirth, H.-P.; Kapsenberg, M.L.; Vandenbroucke-Grauls, C.M.J.E.; van Kooyk, Y.; Appelmelk, B.J. Helicobacter pylori Modulates the T Helper Cell 1/T Helper Cell 2 Balance through Phase-variable Interaction between Lipopolysaccharide and DC-SIGN. J. Exp. Med. 2004, 200, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M.; Toyonaga, K.; Ishikawa, E.; Haji, S.; Okahashi, N.; Takahashi, M.; Izumi, Y.; Imamura, A.; Takato, K.; Ishida, H.; et al. Helicobacter pylori metabolites exacerbate gastritis through C-type lectin receptors. J. Exp. Med. 2020, 218, e20200815. [Google Scholar] [CrossRef]
- Devi, S.; Rajakumara, E.; Ahmed, N. Induction of Mincle by Helicobacter pylori and consequent anti-inflammatory signaling denote a bacterial survival strategy. Sci. Rep. 2015, 5, 15049. [Google Scholar] [CrossRef] [PubMed]
- Platnich, J.M.; Muruve, D.A. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch. Biochem. Biophys. 2019, 670, 4–14. [Google Scholar] [CrossRef]
- Velloso, F.J.; Trombetta-Lima, M.; Anschau, V.; Sogayar, M.C.; Correa, R.G. NOD-like receptors: Major players (and targets) in the interface between innate immunity and cancer. Biosci. Rep. 2019, 39, BSR20181709. [Google Scholar] [CrossRef] [Green Version]
- Castaño-Rodríguez, N.; Kaakoush, N.O.; Goh, K.-L.; Fock, K.M.; Mitchell, H.M. The NOD-Like Receptor Signalling Pathway in Helicobacter pylori Infection and Related Gastric Cancer: A Case-Control Study and Gene Expression Analyses. PLoS ONE 2014, 9, e98899. [Google Scholar] [CrossRef]
- Kim, D.-J.; Park, J.-H.; Franchi, L.; Backert, S.; Núñez, G. The Cag pathogenicity island and interaction between TLR2/NOD2 and NLRP3 regulate IL-1β production in Helicobacter pylori infected dendritic cells. Eur. J. Immunol. 2013, 43, 2650–2658. [Google Scholar] [CrossRef] [Green Version]
- Pachathundikandi, S.K.; Blaser, N.; Bruns, H.; Backert, S. Helicobacter pylori Avoids the Critical Activation of NLRP3 Inflammasome-Mediated Production of Oncogenic Mature IL-1β in Human Immune Cells. Cancers 2020, 12, 803. [Google Scholar] [CrossRef] [Green Version]
- Basak, C.; Pathak, S.K.; Bhattacharyya, A.; Mandal, D.; Pathak, S.; Kundu, M. NF-κB- and C/EBPβ-driven Interleukin-1β Gene Expression and PAK1-mediated Caspase-1 Activation Play Essential Roles in Interleukin-1β Release from Helicobacter pylori Lipopolysaccharide-stimulated Macrophages. J. Biol. Chem. 2005, 280, 4279–4288. [Google Scholar] [CrossRef] [Green Version]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Chen, L.; Feng, J.; Wu, S.; Xu, B.; Zhou, Y.; Wu, C.; Jiang, J. Decreased RIG-I expression is associated with poor prognosis and promotes cell invasion in human gastric cancer. Cancer Cell Int. 2018, 18, 144. [Google Scholar] [CrossRef]
- Tatsuta, T.; Imaizumi, T.; Shimoyama, T.; Sawaya, M.; Kunikazu, T.; Matsumiya, T.; Yoshida, H.; Satoh, K.; Fukuda, S. Expression of melanoma differentiation associated gene 5 is increased in human gastric mucosa infected with Helicobacter pylori. J. Clin. Pathol. 2012, 65, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Cheok, Y.Y.; Lee, C.Y.Q.; Cheong, H.C.; Vadivelu, J.; Looi, C.Y.; Abdullah, S.; Wong, W.F. An Overview of Helicobacter pylori Survival Tactics in the Hostile Human Stomach Environment. Microorganisms 2021, 9, 2502. [Google Scholar] [CrossRef]
- Siriviriyakul, P.; Werawatganon, D.; Phetnoo, N.; Somanawat, K.; Chatsuwan, T.; Klaikeaw, N.; Chayanupatkul, M. Genistein attenuated gastric inflammation and apoptosis in Helicobacter pylori-induced gastropathy in rats. BMC Gastroenterol. 2020, 20, 410. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, O.; Nishizawa, T.; Arita, M.; Kataoka, Y.; Sakitani, K.; Yoshida, S.; Yamashita, H.; Hata, K.; Watanabe, H.; Suzuki, H. Helicobacter pylori infection in subjects negative for high titer serum antibody. World J. Gastroenterol. 2018, 24, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Guclu, M.; Faruq Agan, A. Association of Severity of Helicobacter pylori Infection with Peripheral Blood Neutrophil to Lymphocyte Ratio and Mean Platelet Volume. Euroasian J. Hepatogastroenterol. 2017, 7, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.J.; Evans, D.G.; Takemura, T.; Nakano, H.; Lampert, H.C.; Graham, D.Y.; Granger, D.N.; Kvietys, P.R. Characterization of a Helicobacter pylori neutrophil-activating protein. Infect. Immun. 1995, 63, 2213–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, S.-H.; Hong, Z.-W.; Chen, C.-C.; Chang, H.-W.; Fu, H.-W. Helicobacter pylori Neutrophil-Activating Protein Directly Interacts with and Activates Toll-like Receptor 2 to Induce the Secretion of Interleukin-8 from Neutrophils and ATRA-Induced Differentiated HL-60 Cells. Int. J. Mol. Sci. 2021, 22, 1560. [Google Scholar] [CrossRef]
- Alvarez-Arellano, L.; Camorlinga-Ponce, M.; Maldonado-Bernal, C.; Torres, J. Activation of human neutrophils with Helicobacter pylori and the role of Toll-like receptors 2 and 4 in the response. FEMS Immunol. Med. Microbiol. 2007, 51, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Chu, T.-H.; Huang, S.-T.; Yang, S.-F.; Li, C.-J.; Lin, H.-W.; Weng, B.-C.; Yang, S.-M.; Huang, S.-C.; Wu, J.-C.; Chang, Y.-C.; et al. Hepatoma-derived growth factor participates in Helicobacter Pylori-induced neutrophils recruitment, gastritis and gastric carcinogenesis. Oncogene 2019, 38, 6461–6477. [Google Scholar] [CrossRef]
- Jang, A.-R.; Kang, M.-J.; Shin, J.-I.; Kwon, S.-W.; Park, J.-Y.; Ahn, J.-H.; Lee, T.-S.; Kim, D.-Y.; Choi, B.-G.; Seo, M.-W.; et al. Unveiling the Crucial Role of Type IV Secretion System and Motility of Helicobacter pylori in IL-1β Production via NLRP3 Inflammasome Activation in Neutrophils. Front. Immunol. 2020, 11, 1121. [Google Scholar] [CrossRef] [PubMed]
- Gang Liu, Y.; Teng, Y.S.; Cheng, P.; Kong, H.; Lv, P.Y.; Mao, F.Y.; Wu, X.L.; Hao, C.J.; Chen, W.; Yang, S.M.; et al. Abrogation of cathepsin C by Helicobacter pylori impairs neutrophil activation to promote gastric infection. FASEB J. 2019, 33, 5018–5033. [Google Scholar] [CrossRef] [PubMed]
- Behrens, I.-K.; Busch, B.; Ishikawa-Ankerhold, H.; Palamides, P.; Shively John, E.; Stanners, C.; Chan, C.; Leung, N.; Gray-Owen, S.; Haas, R.; et al. The HopQ-CEACAM Interaction Controls CagA Translocation, Phosphorylation, and Phagocytosis of Helicobacter pylori in Neutrophils. mBio 2020, 11, e03256-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peek, R.M., Jr.; Fiske, C.; Wilson, K.T. Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol. Rev. 2010, 90, 831–858. [Google Scholar] [CrossRef] [Green Version]
- Cheok, Y.Y.; Tan, G.M.Y.; Fernandez, K.C.; Chan, Y.T.; Lee, C.Y.Q.; Cheong, H.C.; Looi, C.Y.; Vadivelu, J.; Abdullah, S.; Wong, W.F. Podoplanin Drives Motility of Active Macrophage via Regulating Filamin C During Helicobacter pylori Infection. Front. Immunol. 2021, 12, 702156. [Google Scholar] [CrossRef]
- Algood, H.M.; Gallo-Romero, J.; Wilson, K.T.; Peek, R.M., Jr.; Cover, T.L. Host response to Helicobacter pylori infection before initiation of the adaptive immune response. FEMS Immunol. Med. Microbiol. 2007, 51, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Kaparakis, M.; Walduck, A.K.; Price, J.D.; Pedersen, J.S.; van Rooijen, N.; Pearse, M.J.; Wijburg, O.L.C.; Strugnell, R.A. Macrophages Are Mediators of Gastritis in Acute Helicobacter pylori Infection in C57BL/6 Mice. Infect. Immun. 2008, 76, 2235–2239. [Google Scholar] [CrossRef] [Green Version]
- Faass, L.; Stein, S.C.; Hauke, M.; Gapp, M.; Albanese, M.; Josenhans, C. Contribution of Heptose Metabolites and the cag Pathogenicity Island to the Activation of Monocytes/Macrophages by Helicobacter pylori. Front. Immunol. 2021, 12, 632154. [Google Scholar] [CrossRef]
- Wen, J.; Chen, C.; Luo, M.; Liu, X.; Guo, J.; Wei, T.; Gu, X.; Gu, S.; Ning, Y.; Li, Y. Notch Signaling Ligand Jagged1 Enhances Macrophage-Mediated Response to Helicobacter pylori. Front. Microbiol. 2021, 12, 1741. [Google Scholar] [CrossRef]
- Quiding-Järbrink, M.; Raghavan, S.; Sundquist, M. Enhanced M1 macrophage polarization in human helicobacter pylori-associated atrophic gastritis and in vaccinated mice. PLoS ONE 2010, 5, e15018. [Google Scholar] [CrossRef] [PubMed]
- Fehlings, M.; Drobbe, L.; Moos, V.; Renner Viveros, P.; Hagen, J.; Beigier-Bompadre, M.; Pang, E.; Belogolova, E.; Churin, Y.; Schneider, T.; et al. Comparative Analysis of the Interaction of Helicobacter pylori with Human Dendritic Cells, Macrophages, and Monocytes. Infect. Immun. 2012, 80, 2724–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beceiro, S.; Radin, J.N.; Chatuvedi, R.; Piazuelo, M.B.; Horvarth, D.J.; Cortado, H.; Gu, Y.; Dixon, B.; Gu, C.; Lange, I.; et al. TRPM2 ion channels regulate macrophage polarization and gastric inflammation during Helicobacter pylori infection. Mucosal Immunol. 2017, 10, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Krakowiak, M.S.; Noto, J.M.; Piazuelo, M.B.; Hardbower, D.M.; Romero-Gallo, J.; Delgado, A.; Chaturvedi, R.; Correa, P.; Wilson, K.T.; Peek, R.M. Matrix metalloproteinase 7 restrains Helicobacter pylori-induced gastric inflammation and premalignant lesions in the stomach by altering macrophage polarization. Oncogene 2015, 34, 1865–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobert, A.P.; Verriere, T.; Asim, M.; Barry, D.P.; Piazuelo, M.B.; de Sablet, T.; Delgado, A.G.; Bravo, L.E.; Correa, P.; Peek, R.M.; et al. Heme oxygenase-1 dysregulates macrophage polarization and the immune response to Helicobacter pylori. J. Immunol. 2014, 193, 3013–3022. [Google Scholar] [CrossRef] [Green Version]
- Ramarao, N.; Gray-Owen, S.D.; Backert, S.; Meyer, T.F. Helicobacter pylori inhibits phagocytosis by professional phagocytes involving type IV secretion components. Mol. Microbiol. 2000, 37, 1389–1404. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.-A.H.; Schlesinger, L.S.; Kang, B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J. Exp. Med. 2000, 191, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.D.; Asim, M.; Barry, D.P.; de Sablet, T.; Singh, K.; Piazuelo, M.B.; Gobert, A.P.; Chaturvedi, R.; Wilson, K.T. Immune evasion by Helicobacter pylori is mediated by induction of macrophage arginase II. J. Immunol. 2011, 186, 3632–3641. [Google Scholar] [CrossRef] [Green Version]
- Gobert, A.P.; Cheng, Y.; Wang, J.-Y.; Boucher, J.-L.; Iyer, R.K.; Cederbaum, S.D.; Casero, R.A.; Newton, J.C.; Wilson, K.T. Helicobacter pylori induces macrophage apoptosis by activation of arginase II. J. Immunol. 2002, 168, 4692–4700. [Google Scholar] [CrossRef] [Green Version]
- Tan, G.M.Y.; Looi, C.Y.; Fernandez, K.C.; Vadivelu, J.; Loke, M.F.; Wong, W.F. Suppression of cell division-associated genes by Helicobacter pylori attenuates proliferation of RAW264.7 monocytic macrophage cells. Sci. Rep. 2015, 5, 11046. [Google Scholar] [CrossRef] [Green Version]
- Asim, M.; Chaturvedi, R.; Hoge, S.; Lewis, N.D.; Singh, K.; Barry, D.P.; Algood, H.S.; de Sablet, T.; Gobert, A.P.; Wilson, K.T. Helicobacter pylori induces ERK-dependent formation of a phospho-c-Fos c-Jun activator protein-1 complex that causes apoptosis in macrophages. J. Biol. Chem. 2010, 285, 20343–20357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, R.; Cheng, Y.; Asim, M.; Bussière, F.I.; Xu, H.; Gobert, A.P.; Hacker, A.; Casero, R.A., Jr.; Wilson, K.T. Induction of polyamine oxidase 1 by Helicobacter pylori causes macrophage apoptosis by hydrogen peroxide release and mitochondrial membrane depolarization. J. Biol. Chem. 2004, 279, 40161–40173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranzer, K.; Eckhardt, A.; Aigner, M.; Knoll, G.; Deml, L.; Speth, C.; Lehn, N.; Rehli, M.; Schneider-Brachert, W. Induction of Maturation and Cytokine Release of Human Dendritic Cells by Helicobacter pylori. Infect. Immun. 2004, 72, 4416–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bimczok, D.; Clements, R.H.; Waites, K.B.; Novak, L.; Eckhoff, D.E.; Mannon, P.J.; Smith, P.D.; Smythies, L.E. Human primary gastric dendritic cells induce a Th1 response to H. pylori. Mucosal Immunol. 2010, 3, 260–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khamri, W.; Walker Marjorie, M.; Clark, P.; Atherton John, C.; Thursz Mark, R.; Bamford Kathleen, B.; Lechler Robert, I.; Lombardi, G. Helicobacter pylori Stimulates Dendritic Cells To Induce Interleukin-17 Expression from CD4+ T Lymphocytes. Infect. Immun. 2010, 78, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Guiney Donald, G.; Hasegawa, P.; Cole Sheri, P. Helicobacter pylori Preferentially Induces Interleukin 12 (IL-12) Rather than IL-6 or IL-10 in Human Dendritic Cells. Infect. Immun. 2003, 71, 4163–4166. [Google Scholar] [CrossRef] [Green Version]
- León, M.A.; Palma, C.; Hernández, C.; Sandoval, M.; Cofre, C.; Perez-Mateluna, G.; Borzutzky, A.; Harris, P.R.; Serrano, C.A. Helicobacter pylori pediatric infection changes FcεRI expression in dendritic cells and Treg profile in vivo and in vitro. Microbes Infect. 2019, 21, 449–455. [Google Scholar] [CrossRef]
- Russler-Germain, E.V.; Yi, J.; Young, S.; Nutsch, K.; Wong, H.S.; Ai, T.L.; Chai, J.N.; Durai, V.; Kaplan, D.H.; Germain, R.N.; et al. Gut Helicobacter presentation by multiple dendritic cell subsets enables context-specific regulatory T cell generation. Elife 2021, 10, e54792. [Google Scholar] [CrossRef]
- Go, D.-M.; Lee, S.H.; Lee, S.-H.; Woo, S.-H.; Kim, K.; Kim, K.; Park, K.S.; Park, J.-H.; Ha, S.-J.; Kim, W.H.; et al. Programmed Death Ligand 1-Expressing Classical Dendritic Cells Mitigate Helicobacter-Induced Gastritis. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 715–739. [Google Scholar] [CrossRef]
- Mitchell, P.; Germain, C.; Fiori, P.L.; Khamri, W.; Foster, G.R.; Ghosh, S.; Lechler, R.I.; Bamford, K.B.; Lombardi, G. Chronic exposure to Helicobacter pylori impairs dendritic cell function and inhibits Th1 development. Infect. Immun. 2007, 75, 810–819. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Lu, L.; Tang, Y.; Luo, W.; Li, J.; Tang, M. Association between Toll-like receptor gene polymorphisms and risk of Helicobacter pylori infection: A protocol for systematic review and meta-analysis. Medicine 2021, 100, e25729. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheok, Y.Y.; Tan, G.M.Y.; Lee, C.Y.Q.; Abdullah, S.; Looi, C.Y.; Wong, W.F. Innate Immunity Crosstalk with Helicobacter pylori: Pattern Recognition Receptors and Cellular Responses. Int. J. Mol. Sci. 2022, 23, 7561. https://doi.org/10.3390/ijms23147561
Cheok YY, Tan GMY, Lee CYQ, Abdullah S, Looi CY, Wong WF. Innate Immunity Crosstalk with Helicobacter pylori: Pattern Recognition Receptors and Cellular Responses. International Journal of Molecular Sciences. 2022; 23(14):7561. https://doi.org/10.3390/ijms23147561
Chicago/Turabian StyleCheok, Yi Ying, Grace Min Yi Tan, Chalystha Yie Qin Lee, Suhailah Abdullah, Chung Yeng Looi, and Won Fen Wong. 2022. "Innate Immunity Crosstalk with Helicobacter pylori: Pattern Recognition Receptors and Cellular Responses" International Journal of Molecular Sciences 23, no. 14: 7561. https://doi.org/10.3390/ijms23147561