
Chronic Immune Platelet Activation Is Followed by Platelet Refractoriness and Impaired Contractility

 ,
,
Abstract
:1. Introduction
2. Results
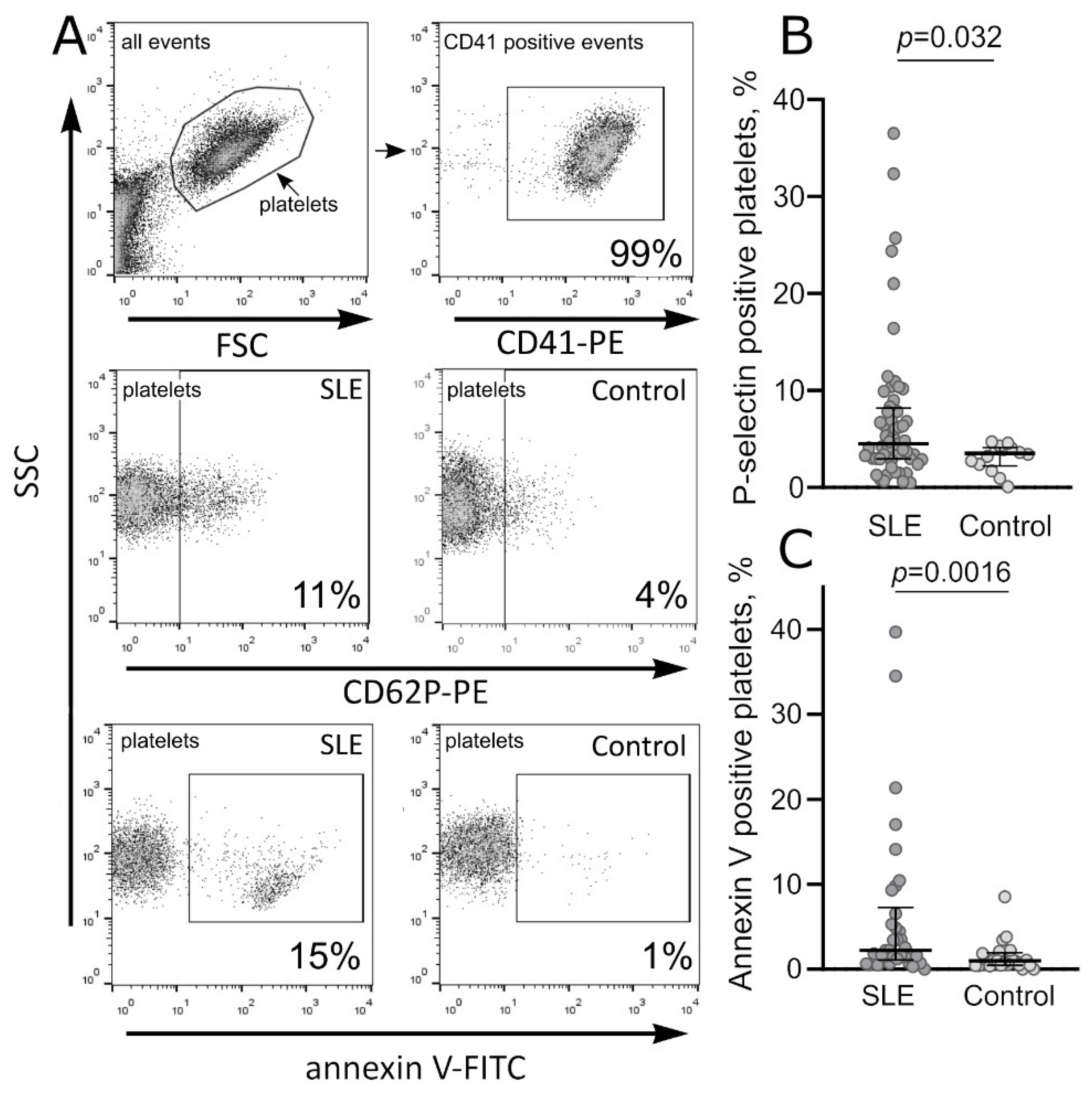
2.1. Continuous Background Activation of Platelets in the Blood of SLE Patients
2.1.1. Molecular Markers of Platelet Activation in SLE
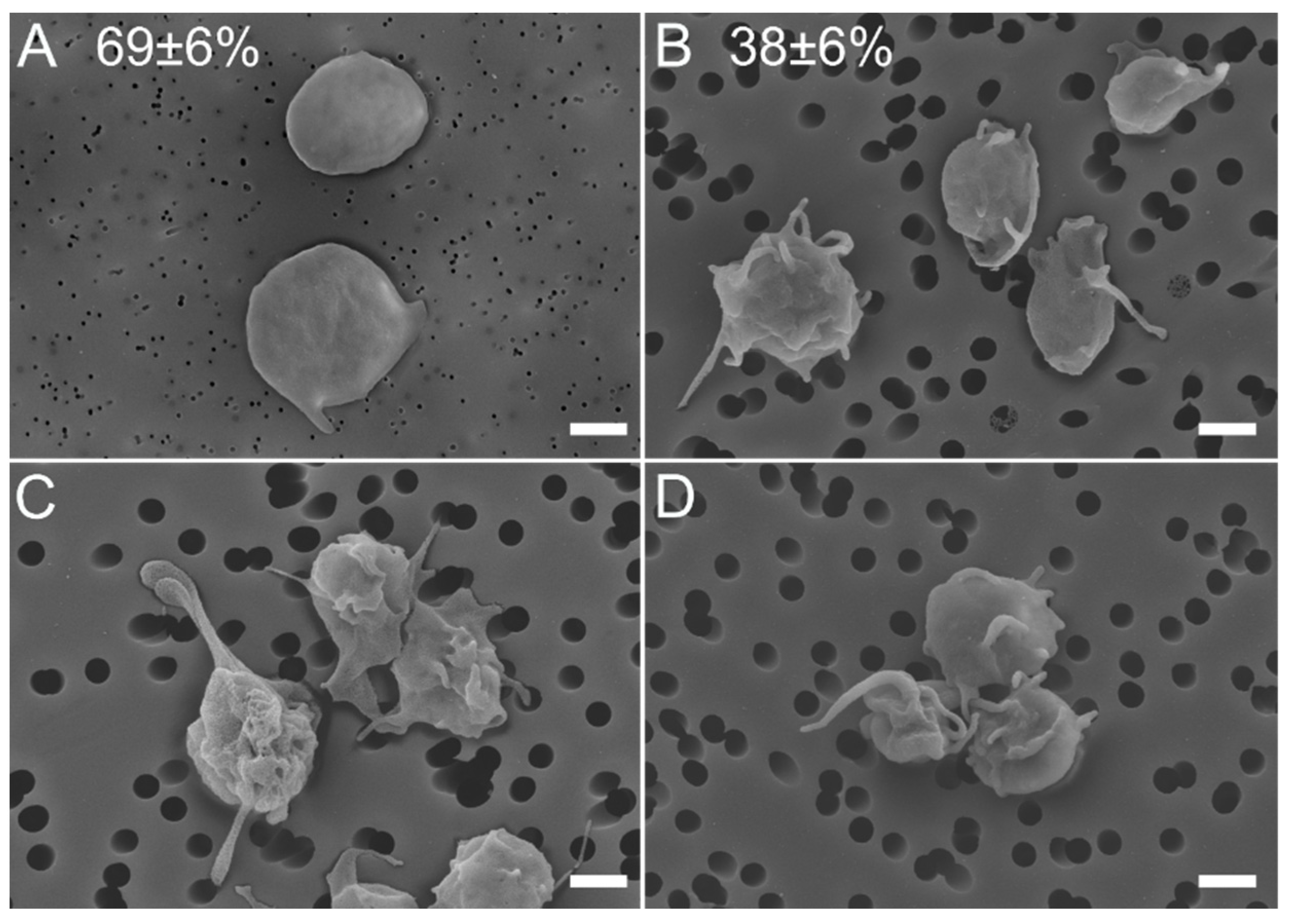
2.1.2. Morphological Signs of Platelet Activation in SLE
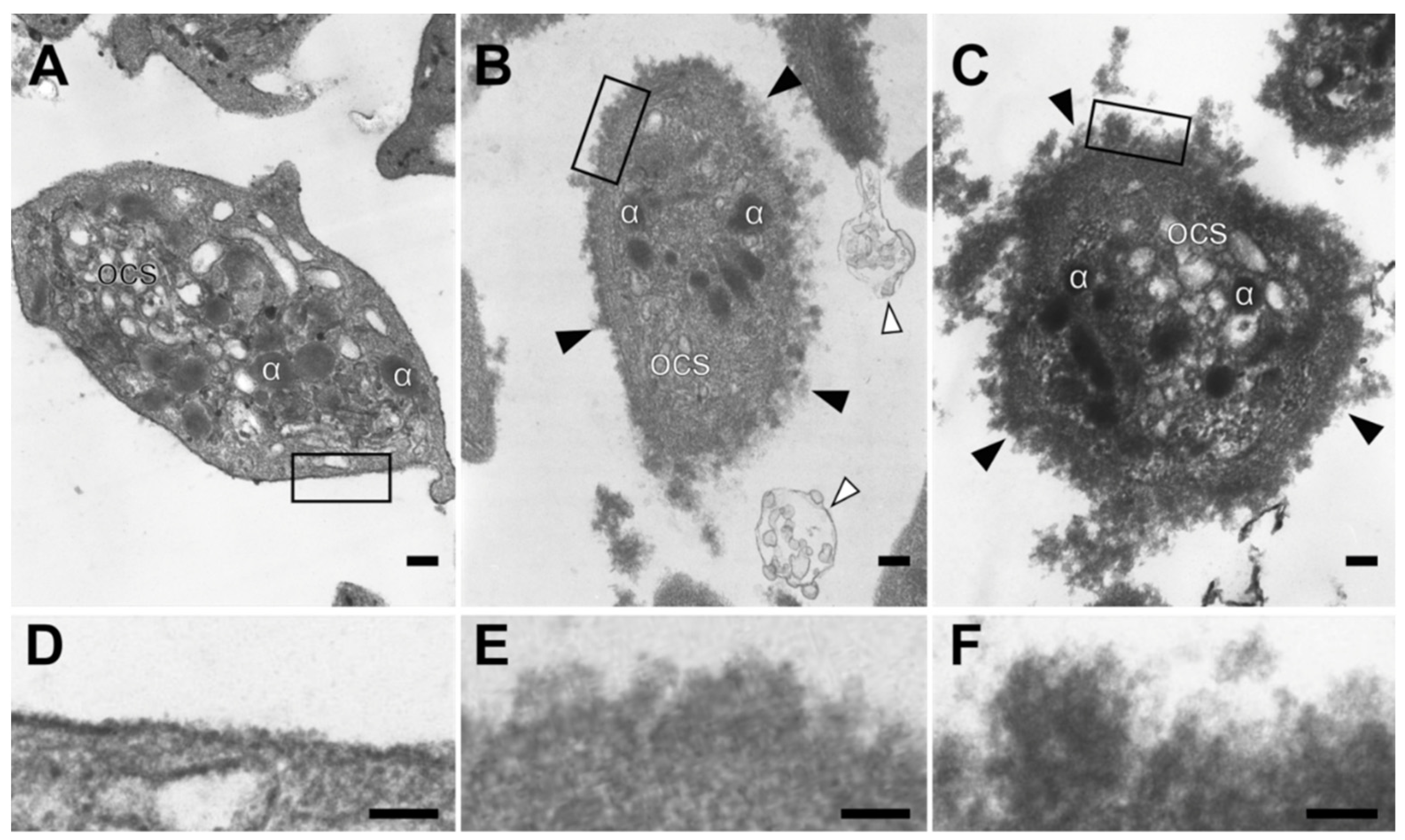
2.1.3. Ultrastructural Alteration of the Platelet Plasma Membrane in SLE
2.2. Impaired Functionality of Platelets in the Blood of SLE Patients
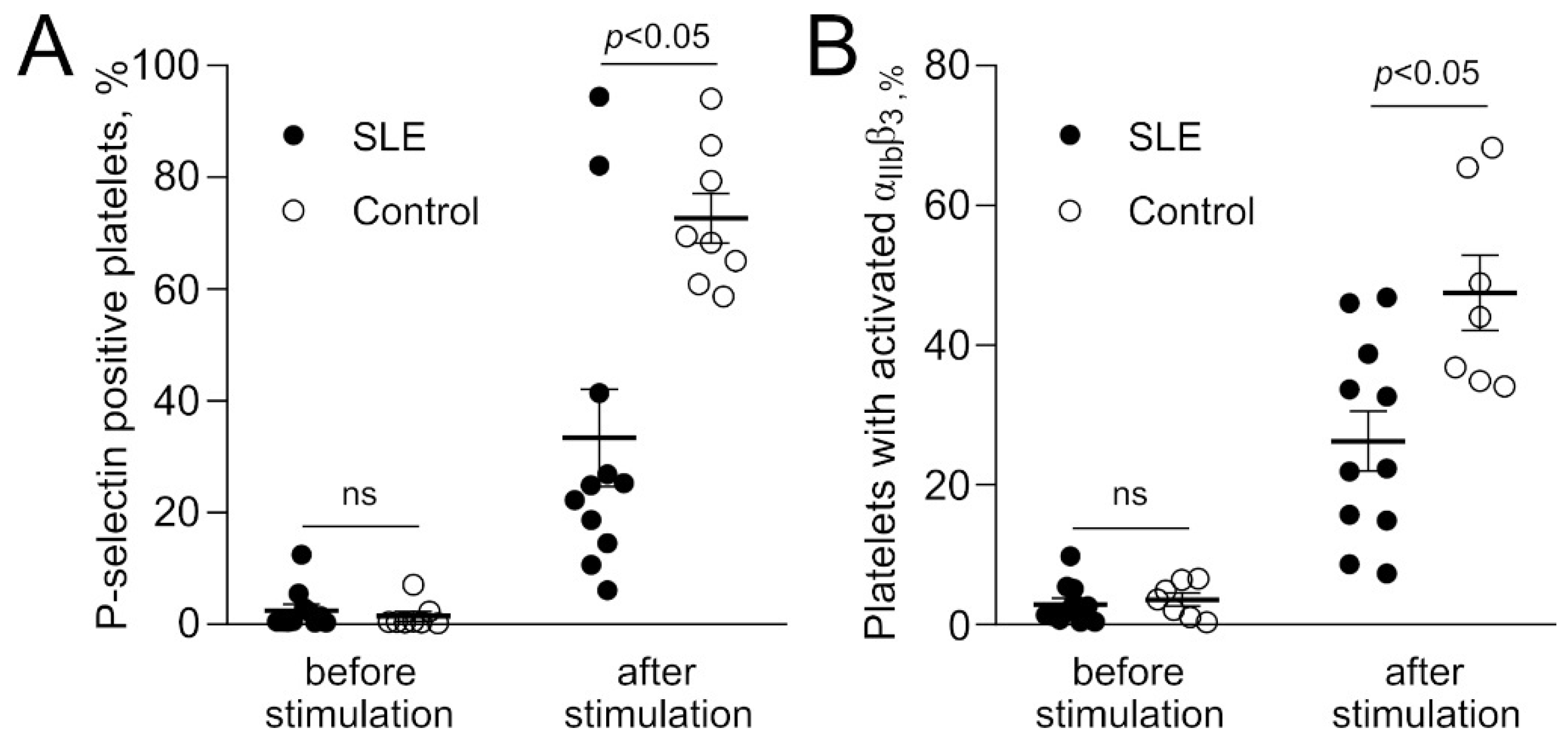
2.2.1. Poor Reactivity of Platelets in Response to Stimulation in SLE
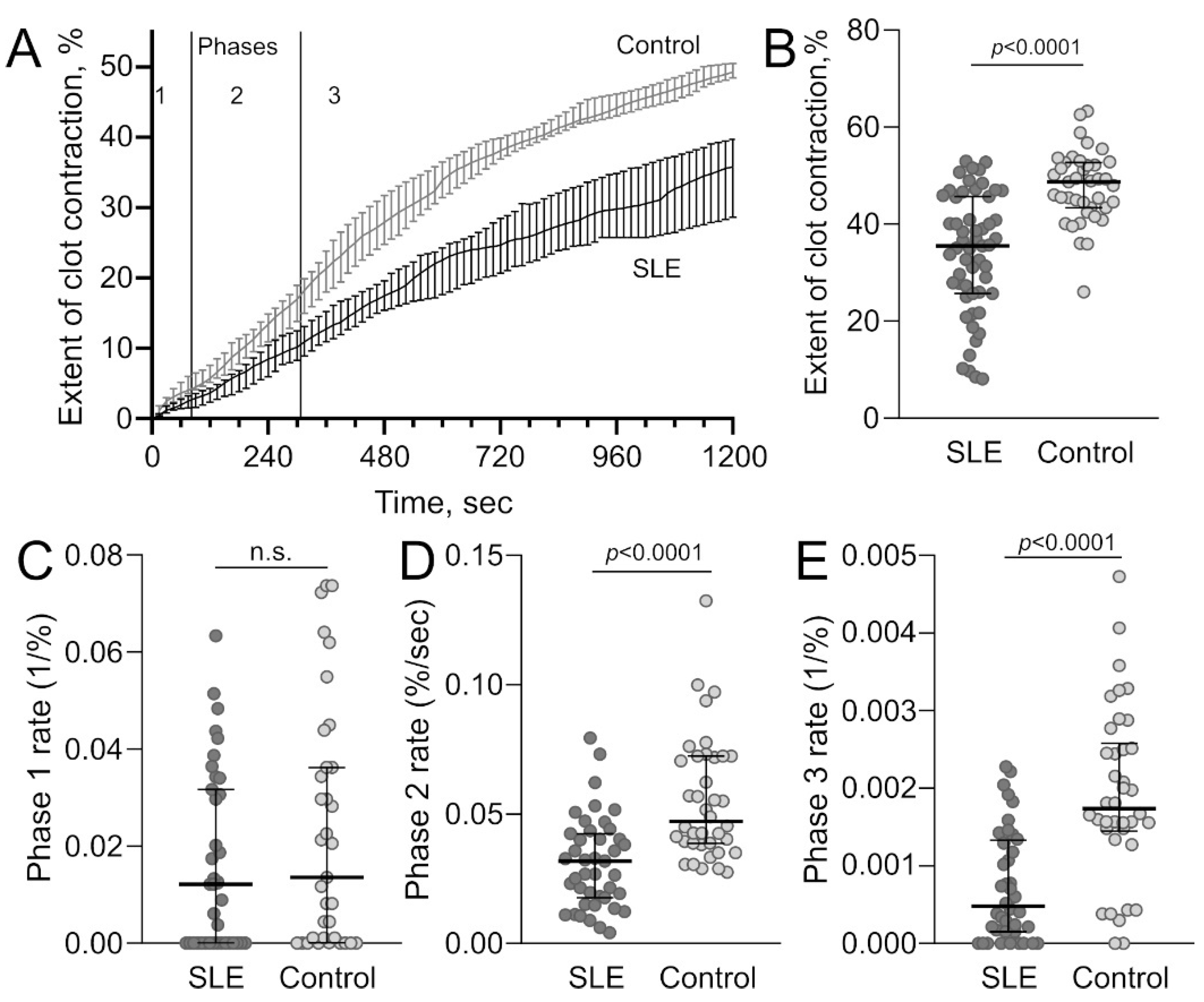
2.2.2. Decreased Platelet Contractility in SLE
2.2.3. Relationship between the Parameters of Platelet Activation, Contractility, and Immune Inflammation in SLE
3. Discussion
4. Materials and Methods
4.1. Clinical Characteristics of the SLE Patients Enrolled in the Study
4.2. Blood Collection and Processing
4.3. Expression Levels of Membrane-Associated P-Selectin and Phosphatidylserines on Platelets
4.4. Concentrations of Soluble P-Selectin in PPP
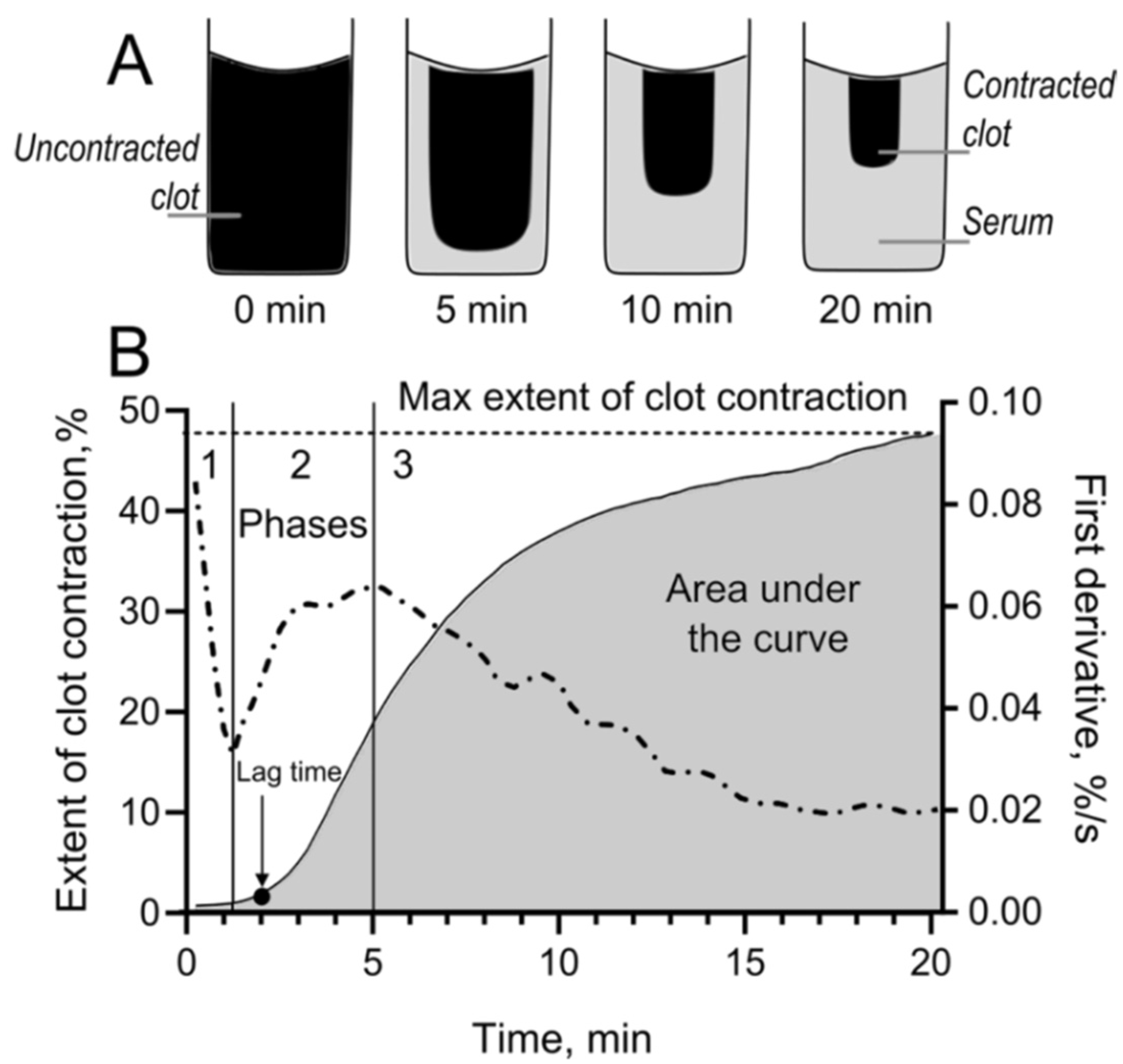
4.5. Blood Clot Contraction Assay
4.6. Isolation of Platelets
4.7. Measuring Platelet Reactivity in Response to Stimulation
4.8. Scanning Electron Microscopy of Platelets
4.9. Transmission Electron Microscopy of Platelets
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APS | antiphospholipid syndrome |
| ALT | alanine transaminase |
| AST | aspartate transaminase |
| FITC | fluorescein isothiocyanate |
| IL | interleukin |
| INR | international normalized ratio |
| PE | phycoerythrin |
| PPP | platelet-poor plasma |
| PRP | platelet rich plasma |
| PSGL-1 | P-selectin glycoprotein ligand-1 |
| SLE | systemic lupus erythematosus |
| SLEDAI | Systemic Lupus Erythematosus Disease Activity Index |
| sP-selectin | soluble P-selectin |
| TRAP | thrombin receptor-activating peptide-6 |
References
- Cervera, R.; Khamashta, M.A.; Font, J.; Sebastiani, G.D.; Gil, A.; Lavilla, P.; Mejía, J.C.; Aydintug, A.O.; Chwalinska-Sadowska, H.; de Ramón, E.; et al. Morbidity and mortality in systemic lupus erythematosus during a 10-year period. Medicine 2003, 82, 299–308. [Google Scholar] [CrossRef]
- Sallai, K.K.; Nagy, E.; Bodó, I.; Mohl, A.; Gergely, P. Thrombosis risk in systemic lupus erythematosus: The role of thrombophilic risk factors. Scand. J. Rheumatol. 2007, 36, 198–205. [Google Scholar] [CrossRef]
- Badsha, H.; Cheng, L.T.; Kok, O.K.; Tsui, Y.L.; Hiok, H.C. Pulmonary hemorrhage in systemic lupus erythematosus. Semin. Arthritis Rheum. 2004, 33, 414–421. [Google Scholar] [CrossRef]
- Al-Homood, I.A. Thrombosis in systemic lupus erythematosus: A review article. ISRN Rheumatol. 2012, 2012, 428269. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Kim, S.T.; Craft, J. The pathogenesis of systemic lupus erythematosus—An update. Curr. Opin. Immunol. 2012, 24, 651–657. [Google Scholar] [CrossRef] [Green Version]
- Melki, I.; Allaeys, I.; Tessandier, N.; Mailhot, B.; Cloutier, N.; Campbell, R.A.; Rowley, J.W.; Salem, D.; Zufferey, A.; Laroche, A.; et al. FcγRIIA expression accelerates nephritis and increases platelet activation in systemic lupus erythematosus. Blood 2020, 136, 2933–2945. [Google Scholar] [CrossRef]
- Andrianova, I.A.; Ponomareva, A.A.; Mordakhanova, E.R.; Le Minh, G.; Daminova, A.G.; Nevzorova, T.A.; Rauova, L.; Litvinov, R.I.; Weisel, J.W. In systemic lupus erythematosus anti-dsDNA antibodies can promote thrombosis through direct platelet activation. J. Autoimmun. 2020, 107, 102355. [Google Scholar] [CrossRef]
- Kazzaz, N.M.; Coit, P.; Lewis, E.E.; Mccune, J.; Sawalha, A.H.; Knight, J.S. Systemic lupus erythematosus complicated by diffuse alveolar haemorrhage: Risk factors, therapy and survival. Lupus Sci. Med. 2015, 2, 117. [Google Scholar] [CrossRef] [Green Version]
- Al-Adhoubi, N.K.; Bystrom, J. Systemic lupus erythematosus and diffuse alveolar hemorrhage, etiology and novel treatment strategies. Lupus 2020, 29, 355–363. [Google Scholar] [CrossRef]
- Martínez-Martínez, M.U.; Abud-Mendoza, C. Diffuse alveolar hemorrhage in patients with systemic lupus erythematosus. Clinical Manifestations, Treatment, and Prognosis. Reumatol. Clínica 2014, 10, 248–253. [Google Scholar] [CrossRef]
- Wang, J.; Yu, C.; Zhuang, J.; Qi, W.; Jiang, J.; Liu, X.; Zhao, W.; Cao, Y.; Wu, H.; Qi, J.; et al. The role of phosphatidylserine on the membrane in immunity and blood coagulation. Biomark. Res. 2022, 10, 1–23. [Google Scholar] [CrossRef]
- Bennett, J.S. Structure and function of the platelet integrin αIIbβ3. J. Clin. Investig. 2005, 115, 3363–3369. [Google Scholar] [CrossRef] [Green Version]
- Hantgan, R.R.; Mousa, S.A. Inhibition of platelet-mediated clot retraction by integrin antagonists. Thromb. Res. 1998, 89, 271–279. [Google Scholar] [CrossRef]
- Amin, H.M.; Ahmad, S.; Walenga, J.M.; Hoppensteadt, D.A.; Leitz, H.; Fareed, J. Soluble P-selectin in human plasma: Effect of anticoagulant matrix and its levels in patients with cardiovascular disorders. Clin. Appl. Thromb. 2000, 6, 71–76. [Google Scholar] [CrossRef]
- Blann, A.D.; Lip, G.Y.H. Hypothesis: Is soluble P-selectin a new marker of platelet activation? Atherosclerosis 1997, 128, 135–138. [Google Scholar] [CrossRef]
- Panicker, S.R.; Mehta-D’Souza, P.; Zhang, N.; Klopocki, A.G.; Shao, B.; McEver, R.P. Circulating soluble P-selectin must dimerize to promote inflammation and coagulation in mice. Blood 2017, 130, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Au, A.E.; Josefsson, E.C. Regulation of platelet membrane protein shedding in health and disease. Platelets 2017, 28, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, V.; Smyth, S.S. Interactions between platelets, leukocytes and the endothelium. In Platelets; Michelson, A.D., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 295–312. [Google Scholar]
- Suzuki, J.; Hamada, E.; Shodai, T.; Kamoshida, G.; Kudo, S.; Itoh, S.; Koike, J.; Nagata, K.; Irimura, T.; Tsuji, T. Cytokine secretion from human monocytes potentiated by P-selectin-mediated cell adhesion. Int. Arch. Allergy Immunol. 2013, 160, 152–160. [Google Scholar] [CrossRef]
- Del Conde, I.; Nabi, F.; Tonda, R.; Thiagarajan, P.; López, J.A.; Kleiman, N.S. Effect of P-selectin on phosphatidylserine exposure and surface-dependent thrombin generation on monocytes. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1065–1070. [Google Scholar] [CrossRef]
- Aleman, M.M.; Gardiner, C.; Harrison, P.; Wolberg, A.S. Differential contributions of monocyte- and platelet-derived microparticles towards thrombin generation and fibrin formation and stability. J. Thromb. Haemost. 2011, 9, 2251–2261. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Bi, Y.; Kou, J.; Shi, J.; Piao, D. Phosphatidylserine exposing-platelets and microparticles promote procoagulant activity in colon cancer patients. J. Exp. Clin. Cancer Res. 2016, 35, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ay, C.; Jungbauer, L.V.; Sailer, T.; Tengler, T.; Koder, S.; Kaider, A.; Panzer, S.; Quehenberger, P.; Pabinger, I.; Mannhalter, C. High concentrations of soluble P-selectin are associated with risk of venous thromboembolism and the P-selectin Thr715 variant. Clin. Chem. 2007, 53, 1235–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, I.; Choudhury, A.; Patel, J.; Lip, G.Y.H. Soluble, platelet-bound, and total P-selectin as indices of platelet activation in congestive heart failure. Ann. Med. 2009, 41, 45–51. [Google Scholar] [CrossRef]
- Duffau, P.; Seneschal, J.; Nicco, C.; Richez, C.; Lazaro, E.; Douchet, I.; Bordes, C.; Viallard, J.-F.; Goulvestre, C.; Pellegrin, J.-L.; et al. Platelet CD154 potentiates interferon- secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci. Transl. Med. 2010, 2, 47ra63. [Google Scholar] [CrossRef] [PubMed]
- Monzón Manzano, E.; Fernández-Bello, I.; Justo Sanz, R.; Robles Marhuenda, Á.; López-Longo, F.J.; Acuña, P.; Álvarez Román, M.T.; Jiménez Yuste, V.; Butta, N.V. Insights into the procoagulant profile of patients with systemic lupus erythematosus without antiphospholipid antibodies. J. Clin. Med. 2020, 9, 3297. [Google Scholar] [CrossRef] [PubMed]
- Berlacher, M.D.; Vieth, J.A.; Heflin, B.C.; Gay, S.R.; Antczak, A.J.; Tasma, B.E.; Boardman, H.J.; Singh, N.; Montel, A.H.; Kahaleh, M.B.; et al. FcγRIIa Ligation induces platelet hypersensitivity to thrombotic stimuli. Am. J. Pathol. 2013, 182, 244–254. [Google Scholar] [CrossRef]
- Shimomura, H.; Ogawa, H.; Arai, H.; Moriyama, Y.; Takazoe, K.; Hirai, N.; Kaikita, K.; Hirashima, O.; Misumi, K.; Soejima, H.; et al. Serial changes in plasma levels of soluble P-selectin in patients with acute myocardial infarction. Am. J. Cardiol. 1998, 81, 397–400. [Google Scholar] [CrossRef]
- Aref, S.; Sakrana, M.; Hafez, A.A.; Hamdy, M. Soluble p-selectin levels in diabetes mellitus patients with coronary artery disease. Hematology 2005, 10, 183–187. [Google Scholar] [CrossRef]
- Caine, G.J.; Blann, A.D. Soluble p-selectin should be measured in citrated plasma, not in serum. Br. J. Haematol. 2003, 121, 530–532. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Visualizing thrombosis to improve thrombus resolution. Res. Pract. Thromb. Haemost. 2021, 5, 38–50. [Google Scholar] [CrossRef]
- Tutwiler, V.; Litvinov, R.I.; Lozhkin, A.P.; Peshkova, A.D.; Lebedeva, T.; Ataullakhanov, F.I.; Spiller, K.L.; Cines, D.B.; Weisel, J.W. Kinetics and mechanics of clot contraction are governed by the molecular and cellular composition of the blood. Blood 2016, 127, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woollard, K.J.; Kling, D.; Kulkarni, S.; Dart, A.M.; Jackson, S.; Chin-Dusting, J. Raised plasma soluble p-selectin in peripheral arterial occlusive disease enhances leukocyte adhesion. Circ. Res. 2006, 98, 149–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woollard, K.J.; Suhartoyo, A.; Harris, E.E.; Eisenhardt, S.U.; Jackson, S.P.; Peter, K.; Dart, A.M.; Hickey, M.J.; Chin-Dusting, J.P.F. Pathophysiological levels of soluble p-selectin mediate adhesion of leukocytes to the endothelium through Mac-1 activation. Circ. Res. 2008, 103, 1128–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linge, P.; Fortin, P.R.; Lood, C.; Bengtsson, A.A.; Boilard, E. The non-haemostatic role of platelets in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2018, 14, 195–213. [Google Scholar] [CrossRef]
- Kim, H.; Conway, E.M. Platelets and complement cross-talk in early atherogenesis. Front. Cardiovasc. Med. 2019, 6, 131. [Google Scholar] [CrossRef] [Green Version]
- Blann, A.D.; Nadar, S.K.; Lip, G.Y.H. The adhesion molecule P-selectin and cardiovascular disease. Eur. Heart J. 2003, 24, 2166–2179. [Google Scholar] [CrossRef] [Green Version]
- Ponomareva, A.A.; Nevzorova, T.A.; Mordakhanova, E.R.; Andrianova, I.A.; Rauova, L.; Litvinov, R.I.; Weisel, J.W. Intracellular origin and ultrastructure of platelet-derived microparticles. J. Thromb. Haemost. 2017, 15, 1655–1667. [Google Scholar] [CrossRef] [Green Version]
- Agbani, E.O.; Williams, C.M.; Hers, I.; Poole, A.W. Membrane ballooning in aggregated platelets is synchronised and mediates a surge in microvesiculation. Sci. Rep. 2017, 7, 2770. [Google Scholar] [CrossRef] [Green Version]
- Kim, O.V.O.V.; Nevzorova, T.A.T.A.; Mordakhanova, E.R.E.R.; Ponomareva, A.A.; Andrianova, I.A.; Le Minh, G.; Daminova, A.G.A.G.; Peshkova, A.D.A.D.; Alber, M.S.M.S.; Vagin, O.; et al. Fatal dysfunction and disintegration of thrombin-stimulated platelets. Haematologica 2019, 104, 1866–1878. [Google Scholar] [CrossRef]
- Le Minh, G.; Peshkova, A.D.; Andrianova, I.A.; Sibgatullin, T.B.; Maksudova, A.N.; Weisel, J.W.; Litvinov, R.I. Impaired contraction of blood clots as a novel prothrombotic mechanism in systemic lupus erythematosus. Clin. Sci. 2018, 132, 243–254. [Google Scholar] [CrossRef] [Green Version]
- George, M.J.; Bynum, J.; Nair, P.; Cap, A.P.; Wade, C.E.; Cox, C.S.; Gill, B.S. Platelet biomechanics, platelet bioenergetics, and applications to clinical practice and translational research. Platelets 2018, 29, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Aibibula, M.; Naseem, K.M.; Sturmey, R.G. Glucose metabolism and metabolic flexibility in blood platelets. J. Thromb. Haemost. 2018, 16, 2300–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, S.; Chacko, B.; Sawada, H.; Kramer, P.A.; Johnson, M.S.; Benavides, G.A.; O’Donnell, V.; Marques, M.B.; Darley-Usmar, V.M. Metabolic plasticity in resting and thrombin activated platelets. PLoS ONE 2015, 10, e0123597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloutier, N.; Allaeys, I.; Marcoux, G.; Machlus, K.R.; Mailhot, B.; Zufferey, A.; Levesque, T.; Becker, Y.; Tessandier, N.; Melki, I.; et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc. Natl. Acad. Sci. USA 2018, 115, E1550–E1559. [Google Scholar] [CrossRef] [Green Version]
- Schrottmaier, W.C.; Pirabe, A.; Pereyra, D.; Heber, S.; Hackl, H.; Schmuckenschlager, A.; Brunnthaler, L.; Santol, J.; Kammerer, K.; Oosterlee, J.; et al. Adverse outcome in COVID-19 Is associated with an aggravating hypo-responsive platelet phenotype. Front. Cardiovasc. Med. 2021, 8, 1–12. [Google Scholar] [CrossRef]
- Pareti, F.; Capitanio, A.; Mannucci, P. Acquired storage pool disease in platelets during disseminated intravascular coagulation. Blood 1976, 48, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Mannucci, P.M.; Cattaneo, M.; Teresa Canciani, M.; Maniezzo, M.; Vaglini, M.; Cascinelli, N. Early presence of activated (‘exhausted’) platelets in malignant tumors (breast adenocarcinoma and malignant melanoma). Eur. J. Cancer Clin. Oncol. 1989, 25, 1413–1417. [Google Scholar] [CrossRef]
- Boneu, B.; Bugat, R.; Boneu, A.; Eche, N.; Sie, P.; Combes, P.F. Exhausted platelets in patients with malignant solid tumors without evidence of active consumption coagulopathy. Eur. J. Cancer Clin. Oncol. 1984, 20, 899–903. [Google Scholar] [CrossRef]
- Pareti, F.I.; Capitanio, A.; Mannucci, L.; Ponticelli, C.; Mannucci, P.M. Acquired dysfunction due to the circulation of “exhausted” platelets. Am. J. Med. 1980, 69, 235–240. [Google Scholar] [CrossRef]
- Michiels, J.J.; Berneman, Z.; Schroyens, W.; Finazzi, G.; Budde, U.; Van Vliet, H.H.D.M. The paradox of platelet activation and impaired function: Platelet-von Willebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestations in essential thrombocythemia and polycythemia vera. Semin. Thromb. Hemost. 2006, 32, 589–604. [Google Scholar] [CrossRef]
- Vrigkou, E.; Tsangaris, I.; Bonovas, S.; Kopterides, P.; Kyriakou, E.; Konstantonis, D.; Pappas, A.; Anthi, A.; Gialeraki, A.; Orfanos, S.E.; et al. Platelet and coagulation disorders in newly diagnosed patients with pulmonary arterial hypertension. Platelets 2019, 30, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Urbanus, R.T.; ten Cate, H.; de Groot, P.G.; de Laat, B.; Heemskerk, J.W.M.; Roest, M. Platelet activation mechanisms and consequences of immune thrombocytopenia. Cells 2021, 10, 3386. [Google Scholar] [CrossRef] [PubMed]
- Mohan, G.; Malayala, S.V.; Mehta, P.; Balla, M. A Comprehensive review of congenital platelet disorders, thrombocytopenias and thrombocytopathies. Cureus 2020, 12, e11275. [Google Scholar] [CrossRef] [PubMed]
- Evtugina, N.G.; Peshkova, A.D.; Pichugin, A.A.; Weisel, J.W.; Litvinov, R.I. Impaired contraction of blood clots precedes and predicts postoperative venous thromboembolism. Sci. Rep. 2020, 10, 18261. [Google Scholar] [CrossRef]
- Tutwiler, V.; Peshkova, A.D.; Andrianova, I.A.; Khasanova, D.R.; Weisel, J.W.; Litvinov, R.I. Contraction of blood clots is impaired in acute ischemic stroke. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Peshkova, A.; Malyasyov, D.; Bredikhin, R.; Le Minh, G.; Andrianova, I.; Tutwiler, V.; Nagaswami, C.; Weisel, J.; Litvinov, R. Reduced contraction of blood clots in venous thromboembolism is a potential thrombogenic and embologenic mechanism. TH Open 2018, 2, e104–e115. [Google Scholar] [CrossRef] [Green Version]
- Khismatullin, R.R.; Nagaswami, C.; Shakirova, A.Z.; Vrtková, A.; Procházka, V.; Gumulec, J.; Mačák, J.; Litvinov, R.I.; Weisel, J.W. Quantitative morphology of cerebral thrombi related to intravital contraction and clinical features of ischemic stroke. Stroke 2020, 51, 3640–3650. [Google Scholar] [CrossRef]
- Peshkova, A.D.; Evdokimova, T.A.; Sibgatullin, T.B.; Ataullakhanov, F.I.; Litvinov, R.I.; Weisel, J.W. Accelerated spatial fibrin growth and impaired contraction of blood clots in patients with rheumatoid arthritis. Int. J. Mol. Sci. 2020, 21, 9434. [Google Scholar] [CrossRef]
- Tutwiler, V.; Peshkova, A.D.; Le Minh, G.; Zaitsev, S.; Litvinov, R.I.; Cines, D.B.; Weisel, J.W. Blood clot contraction differentially modulates internal and external fibrinolysis. J. Thromb. Haemost. 2019, 17, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Khismatullin, R.R.; Abdullayeva, S.; Peshkova, A.D.; Sounbuli, K.; Evtugina, N.G.; Litvinov, R.I.; Weisel, J.W. Extent of intravital contraction of arterial and venous thrombi and pulmonary emboli. Blood Adv. 2021, 6, 1708–1718. [Google Scholar] [CrossRef]
- Bhoria, P.; Sharma, S.; Varma, N.; Malhotra, P.; Varma, S.; Luthra-Guptasarma, M. Effect of steroids on the activation status of platelets in patients with Immune thrombocytopenia (ITP). Platelets 2015, 26, 119–126. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SLE Patients | Control | ||||
|---|---|---|---|---|---|
| n | Median (IQR) | n | Median (IQR) | p Value 1 | |
| (a) Phosphatidylserine and P-selectin expression | |||||
| Phosphatidylserine-positive unstimulated platelets, % | 34 | 2.2 (1.1–7.2) | 22 | 1.0 (0.5–1.9) | 0.002 |
| P-selectin-positive unstimulated platelets, % | 56 | 4.5 (2.9; 8.2) | 14 | 3.5 (2.2; 4.1) | 0.032 |
| (b) soluble P-selectin secretion | |||||
| sP-selectin in blood plasma, ng/mL | 53 | 51 (38; 75) | 15 | 34 (29; 39) | <0.001 |
| (c) Platelet contractility, parameters of blood clot contraction | |||||
| Extent of blood clot contraction, % | 55 | 35.3 (25.8; 45.8) | 40 | 48 (43; 52) | <0.001 |
| Lag time of blood clot contraction, sec | 55 | 150 (108; 236) | 40 | 75 (45; 120) | <0.001 |
| Area over the kinetic curve of blood clot contraction, a.u. | 55 | 259 (176; 317) | 40 | 380 (316; 447) | <0.001 |
| Average velocity of blood clot contraction, (%/sec) × 10−3 | 55 | 29 (20; 37) | 40 | 38(35; 42) | <0.001 |
| (d) Kinetic phase analysis of clot contraction | |||||
| Rate constant for phase 1, s−1 | 55 | 0.012 | 40 | 0.014 | n.s. |
| Rate constant for phase 2, %/s | 55 | 0.032 | 40 | 0.047 | <0.0001 |
| Rate constant for phase 3, s−1 | 55 | 0.0005 | 40 | 0.0017 | <0.0001 |
| (a) Phosphatidylserine-expressing platelets vs. the markers of immune inflammation and clot contraction | ||||
| SLEDAI | Leukocyte Count | Lag Time of Clot Contraction | ||
| Phosphatidylserine-positive platelets in all SLE patients | r = 0.48 p = 0.012 | r = −0.35 p = 0.043 | r = −0.39 p = 0.029 | |
| Phosphatidylserine-positive platelets in SLE patients with APS | r = 0.84 p = 0.029 | n.s. | n.s. | |
| Phosphatidylserine-positive platelets in SLE patients without APS | n.s. | n.s. | n.s. | |
| (b) P-selectin-expressing platelets vs. the markers of immune inflammation and clot contraction | ||||
| IgG | IgA | Anti-dsDNA Abs | Extent of Clot Contraction | |
| Platelet-associated P-selectin in all SLE patients | r = 0.33 p = 0.031 | n.s. | r = 0.46 p = 0.013 | r = 0.31 p = 0.029 |
| Platelet-associated P-selectin in SLE patients with APS | r = 0.67 p = 0.039 | r = 0.69 p = 0.032 | n.s. | n.s. |
| Platelet-associated P-selectin in SLE patients without APS | n.s. | n.s. | n.s. | r = 0.35 p = 0.02 |
| (c) sP-selectin vs. the markers of immune inflammation | ||||
| IgM | IgA | Anti-dsDNA Abs | ||
| sP-selectin levels in all SLE patients | r = 0.47 p = 0.003 | r = 0.35 p = 0.031 | r = 0.43 p = 0.030 | |
| sP-selectin levels in SLE patients with APS | r = 0.74 p = 0.045 | r = 0.67 p = 0.039 | r = 0.82 p = 0.034 | |
| sP-selectin levels in SLE patients without APS | n.s. | n.s. | n.s. | |
| Demographic and Clinical Characteristics | SLE Patients (n = 61) | |
|---|---|---|
| Sex, male/female, n | 3 (5%)/58 (95%) | |
| Age (years), median; min-max | 40; 19–64 | |
| Duration of SLE * (years), median; IQR | 4; 1–8.5 | |
| Patients receiving immunosuppressive treatment *, n | 47 (77%) | |
| SLEDAI *, median; IQR | 4; 2–10 | |
| Antiphospholipid syndrome, n | 12 (20%) | |
| Renal features, n | Lupus nephritis | 40 (66%) |
| Hematuria | 6 (10%) | |
| Proteinuria | 11 (18%) | |
| Musculoskeletal features, n | 24 (39%) | |
| Mucocutaneous features, n | 29 (48%) | |
| Cardiovascular features, n | 17 (28%) | |
| Neurological manifestations, n | 5 (8%) | |
| Pulmonary manifestations, n | 7 (11%) | |
| History of thrombosis, n | 8 (13%) | |
| Obstetric manifestations (miscarriages/stillbirths), n | 5 (8%) | |
| History of hematologic features, n | Leukopenia | 20 (33%) |
| Anemia | 29 (48%) | |
| Thrombocytopenia | 8 (13%) | |
| Blood Tests (Normal Ranges Are Shown in Parentheses) | SLE Patients (n = 61) |
|---|---|
| Immunologic parameters | Median (IQR) |
| Anti-dsDNA antibodies (<10), IU/mL | 22 (7–200) |
| Anti-cardiolipin antibodies (<10), IU/mL | 1.8 (1.4–5.1) |
| IgA (1.1–3.5), mg/mL | 2.0 (1.5–3.8) |
| IgM (0.7–2.5), mg/mL | 1.3 (0.9–2.6) |
| IgG (6.7–16.5), mg/mL | 11.3 (7.5–17.7) |
| Total complement activity (31–60) U/mL | 31 (28–36) |
| Circulating immune complexes (<50), U/mL | 102 (62–128) |
| Prothrombin time (9.2–12.2), s | 11.0 (10.3–11.8) |
| Fibrinogen (2–4), g/L | 3.9 (3.5–4.4) |
| INR (0.9–1.5) | 0.9 (0.88–1) |
| Hematologic parameters | |
| Platelets (140–380), × 109/L | 230 (188–286) |
| Red blood cells (3.9–4.7), × 1012/L | 4.2 (3.8–4.4) |
| Leukocytes (4–9), × 109/L | 6.2 (4.8–7.9) |
| Neutrophils (40–70), % | 63 (53–71) |
| Lymphocytes (19–37), % | 28 (21–35) |
| Monocytes (3–9), % | 7 (5.1–9.8) |
| Hemoglobin (12–14), g/dL | 11.3 (9–12.5) |
| Erythrocyte sedimentation rate (3–16), mm/h | 17 (8.5–27.5) |
| Biochemical parameters | |
| Glucose (3.8–6.1), Mmol/L | 4.6 (4.1–4.9) |
| ALT (<45), U/L | 15.5 (11.3–19.1) |
| AST (<35), U/L | 13.6 (11–17) |
| Creatinine (45–100), μmol/L | 60 (50–79) |
| Cholesterol (2.2–5.7), mmol/L | 5.5 (4.5–6.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrianova, I.A.; Khabirova, A.I.; Ponomareva, A.A.; Peshkova, A.D.; Evtugina, N.G.; Le Minh, G.; Sibgatullin, T.B.; Weisel, J.W.; Litvinov, R.I. Chronic Immune Platelet Activation Is Followed by Platelet Refractoriness and Impaired Contractility. Int. J. Mol. Sci. 2022, 23, 7336. https://doi.org/10.3390/ijms23137336
Andrianova IA, Khabirova AI, Ponomareva AA, Peshkova AD, Evtugina NG, Le Minh G, Sibgatullin TB, Weisel JW, Litvinov RI. Chronic Immune Platelet Activation Is Followed by Platelet Refractoriness and Impaired Contractility. International Journal of Molecular Sciences. 2022; 23(13):7336. https://doi.org/10.3390/ijms23137336
Chicago/Turabian StyleAndrianova, Izabella A., Alina I. Khabirova, Anastasia A. Ponomareva, Alina D. Peshkova, Natalia G. Evtugina, Giang Le Minh, Timur B. Sibgatullin, John W. Weisel, and Rustem I. Litvinov. 2022. "Chronic Immune Platelet Activation Is Followed by Platelet Refractoriness and Impaired Contractility" International Journal of Molecular Sciences 23, no. 13: 7336. https://doi.org/10.3390/ijms23137336