
The Therapeutic Potential of the Restoration of the p53 Protein Family Members in the EGFR-Mutated Lung Cancer


Abstract
:1. Introduction
2. Risk Factors
3. Histological Subtypes of Lung Cancer
4. Non-Small Cell Lung Cancer (NSCLC)
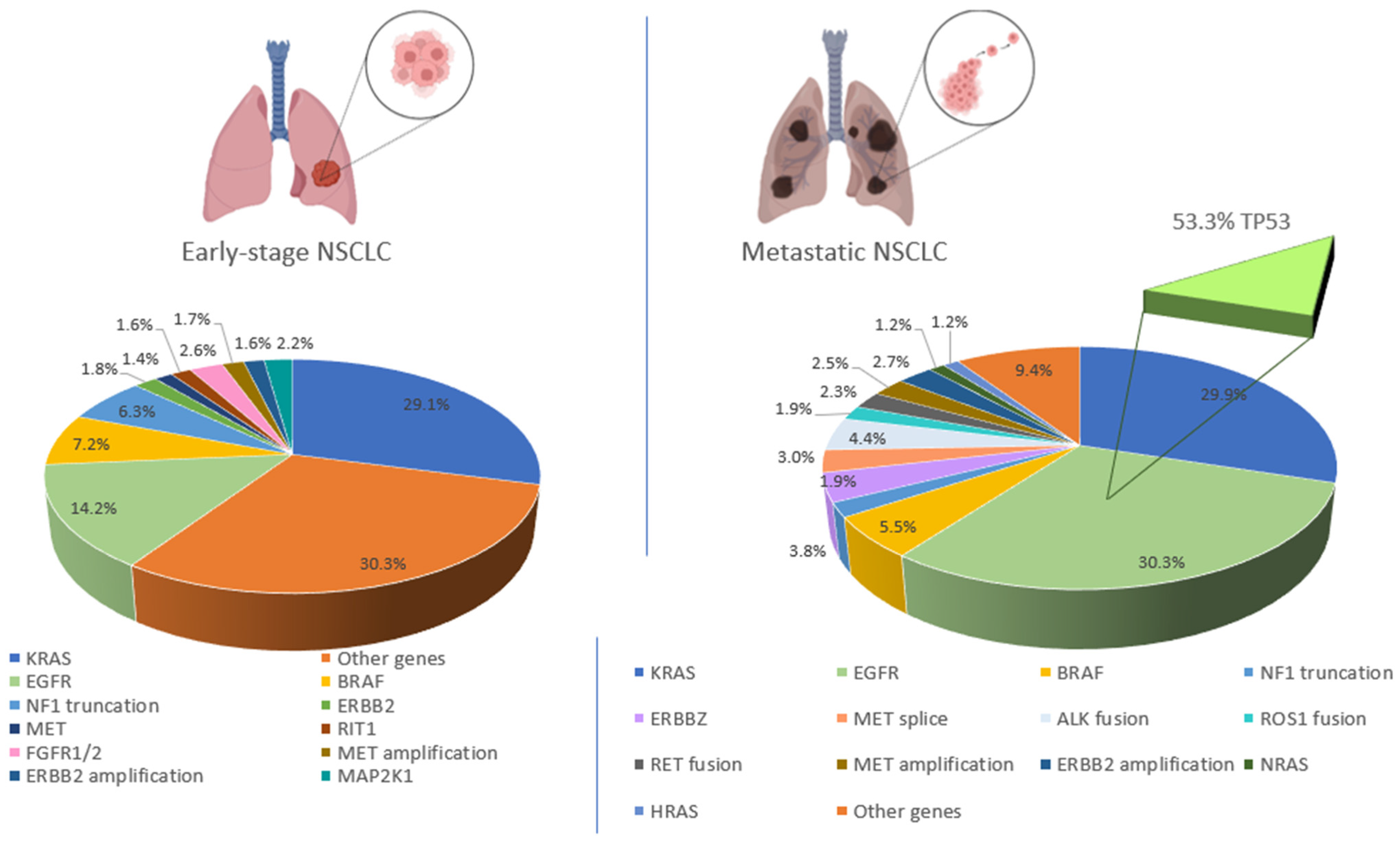
5. Driver Mutations in NSCLC
6. Epidermal Growth Factor Receptor Mutations
7. Targeting Mutated EGFR
8. Tyrosine Kinase Inhibitors
9. p53 Tumor Suppressor
10. TP53 Mutations
11. TP53- and EGFR-Mutated Lung Cancer
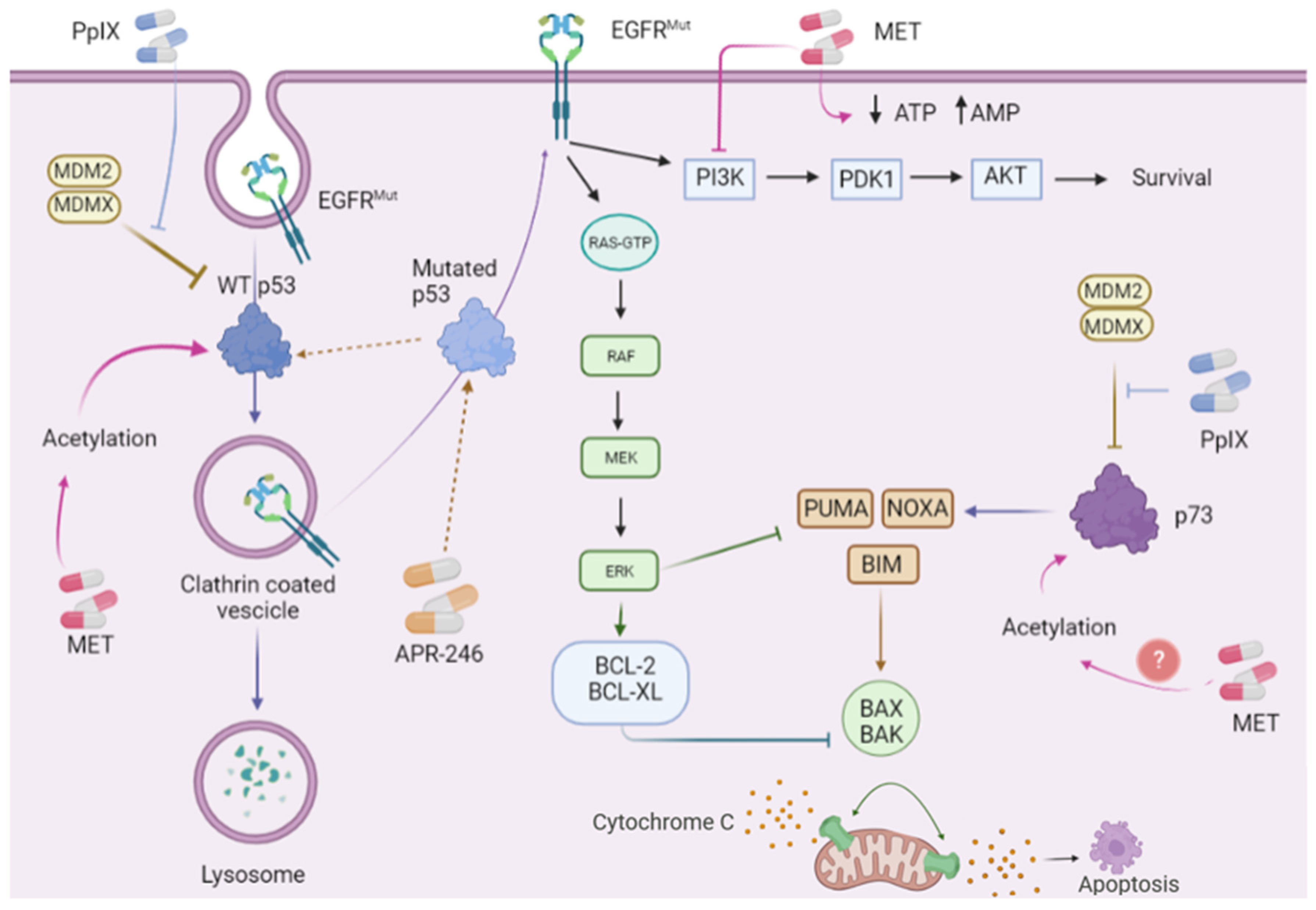
12. Pharmacological Reactivation of p53
13. p53 Isoforms
14. p73 Tumor Suppressor
15. Pharmacological Reactivation of p73
16. p63 Tumor Suppressor
17. p63 in NSCLC
18. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lung Cancer Now a Growing Public Health Threat|MDedge Hematology and Oncology. Available online: https://www.mdedge.com/hematology-oncology/article/252493/lung-cancer/lung-cancer-now-growing-public-health-threat (accessed on 10 March 2022).
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Ryu, W.-S. Adenoviruses. In Molecular Virology of Human Pathogenic Viruses; Elsevier: Amsterdam, The Netherlands; Yonsei University: Seoul, Korea, 2017; pp. 111–124. ISBN 9780128037089. [Google Scholar]
- Lung Cancer Fact Sheet. 2021. Available online: https://www.thoracic.org/about/global-public-health/firs/resources/world-lung-cancer-day-fact-sheet-2021.pdf (accessed on 15 March 2022).
- Wahbah, M.; Boroumand, N.; Castro, C.; El-Zeky, F.; Eltorky, M. Changing trends in the distribution of the histologic types of lung cancer: A review of 4,439 cases. Ann. Diagn. Pathol. 2007, 11, 89–96. [Google Scholar] [CrossRef]
- Chansky, K.; Detterbeck, F.C.; Nicholson, A.G.; Rusch, V.W.; Vallières, E.; Groome, P.; Kennedy, C.; Krasnik, M.; Peake, M.; Shemanski, L.; et al. The IASLC Lung Cancer Staging Project: External Validation of the Revision of the TNM Stage Groupings in the Eighth Edition of the TNM Classification of Lung Cancer. J. Thorac. Oncol. 2017, 12, 1109–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rami-Porta, R.; Bolejack, V.; Giroux, D.J.; Chansky, K.; Crowley, J.; Asamura, H.; Goldstraw, P. The IASLC Lung Cancer Staging Project: The New Database to Inform the Eighth Edition of the TNM Classification of Lung Cancer. J. Thorac. Oncol. 2014, 9, 1618–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, A.G.; Tsao, M.S.; Beasley, M.B.; Borczuk, A.C.; Brambilla, E.; Cooper, W.A.; Dacic, S.; Jain, D.; Kerr, K.M.; Lantuejoul, S.; et al. The 2021 WHO Classification of Lung Tumors: Impact of Advances since 2015. J. Thorac. Oncol. 2021, 17, 362–387. [Google Scholar] [CrossRef]
- Acs, B.; Rantalainen, M.; Hartman, J. Artificial intelligence as the next step towards precision pathology. J. Intern. Med. 2020, 288, 62–81. [Google Scholar] [CrossRef] [Green Version]
- Tunali, I.; Gillies, R.J.; Schabath, M.B. Application of radiomics and artificial intelligence for lung cancer precision medicine. Cold Spring Harb. Perspect. Med. 2021, 11. [Google Scholar] [CrossRef]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.S.; Peled, N.; Pirker, R.; et al. Liquid Biopsy for Advanced Non-Small Cell Lung Cancer (NSCLC): A Statement Paper from the IASLC. J. Thorac. Oncol. 2018, 13, 1248–1268. [Google Scholar] [CrossRef] [Green Version]
- PDQ Adult Treatment Editorial Board. Non-Small cell lung cancer treatment (PDQ®): Patient version. In PDQ Cancer Information Summaries; National Cancer Institute (US): Bethesda, MD, USA, 2002. [Google Scholar]
- Herbst, R.S.; Maddox, A.-M.; Rothenberg, M.L.; Small, E.J.; Rubin, E.H.; Baselga, J.; Rojo, F.; Hong, W.K.; Swaisland, H.; Averbuch, S.D.; et al. Selective Oral Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor ZD1839 Is Generally Well-Tolerated and Has Activity in Non–Small-Cell Lung Cancer and Other Solid Tumors: Results of a Phase I Trial. J. Clin. Oncol. 2002, 20, 3815–3825. [Google Scholar] [CrossRef]
- Herbst, R.S.; Prager, D.; Hermann, R.; Fehrenbacher, L.; Johnson, B.E.; Sandler, A.; Kris, M.; Tran, H.T.; Klein, P.; Li, X.; et al. TRIBUTE: A Phase III Trial of Erlotinib Hydrochloride (OSI-774) Combined with Carboplatin and Paclitaxel Chemotherapy in Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2005, 23, 5892–5899. [Google Scholar] [CrossRef] [PubMed]
- Benesova, L.; Minarik, M.; Jancarikova, D.; Belsanova, B.; Pesek, M. Multiplicity of EGFR and KRAS mutations in non-small cell lung cancer (NSCLC) patients treated with tyrosine kinase inhibitors. Anticancer Res. 2010, 30, 1667–1671. [Google Scholar]
- Shchedrenok, V.V.; Sovakov, A.N.; Sebelev, K.I. Immediate and late results of treating compression forms of lumbar osteochondrosis by the technic of puncture fenestration and decompression of the intervertebral disks. Zhurnal Nevropatol. I Psikhiatrii Im. SS Korsakova 1986, 86, 1158–1161. [Google Scholar]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via Diverse Exon 14 Splicing Alterations Occurs in Multiple Tumor Types and Confers Clinical Sensitivity to MET Inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550, Correction in Nature 2014, 511, 262. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Jordan, E.J.; Kim, H.R.; Arcila, M.E.; Barron, D.; Chakravarty, D.; Gao, J.; Chang, M.T.; Ni, A.; Kundra, R.; Jonsson, P.; et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017, 7, 596–609. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [Green Version]
- Mina, M.; Raynaud, F.; Tavernari, D.; Battistello, E.; Sungalee, S.; Saghafinia, S.; Laessle, T.; Sanchez-Vega, F.; Schultz, N.; Oricchio, E.; et al. Conditional Selection of Genomic Alterations Dictates Cancer Evolution and Oncogenic Dependencies. Cancer Cell 2017, 32, 155–168.e6. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J. Cliques and Schisms of Cancer Genes. Cancer Cell 2017, 32, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [Green Version]
- Helena, A.Y.; Suzawa, K.; Jordan, E.J.; Zehir, A.; Ni, A.; Kim, H.R.; Kris, M.G.; Hellmann, M.D.; Li, B.T.; Somwar, R.; et al. Concurrent Alterations in EGFR-Mutant Lung Cancers Associated with Resistance to EGFR Kinase Inhibitors and Characterization of MTOR as a Mediator of Resistance. Clin. Cancer Res. 2018, 24, 3108–3118. [Google Scholar] [CrossRef] [Green Version]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [Green Version]
- Ellrott, K.; Bailey, M.H.; Saksena, G.; Covington, K.R.; Kandoth, C.; Stewart, C.; Hess, J.; Ma, S.; Chiotti, K.E.; McLellan, M.; et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018, 6, 271–281.e7. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e296. [Google Scholar] [CrossRef] [Green Version]
- Kadara, H.; Choi, M.; Zhang, J.; Parra, E.; Rodriguez-Canales, J.; Gaffney, S.; Zhao, Z.; Behrens, C.; Fujimoto, J.; Chow, C.; et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann. Oncol. 2016, 28, 75–82. [Google Scholar] [CrossRef]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef]
- Gullick, W.J.; Marsden, J.J.; Whittle, N.; Ward, B.; Bobrow, L.; Waterfield, M.D. Expression of epidermal growth factor receptors on human cervical, ovarian, and vulval carcinomas. Cancer Res. 1986, 46, 285–292. [Google Scholar] [PubMed]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2019, 61, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Mitsudomi, T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 2016, 107, 1179–1186. [Google Scholar] [CrossRef]
- Attili, I.; Passaro, A.; Pisapia, P.; Malapelle, U.; de Marinis, F. Uncommon EGFR Compound Mutations in Non-Small Cell Lung Cancer (NSCLC): A Systematic Review of Available Evidence. Curr. Oncol. 2022, 29, 255–266. [Google Scholar] [CrossRef]
- Kannan, S.; Pradhan, M.R.; Tiwari, G.; Tan, W.-C.; Chowbay, B.; Tan, E.H.; Tan, D.S.-W.; Verma, C. Hydration effects on the efficacy of the Epidermal growth factor receptor kinase inhibitor afatinib. Sci. Rep. 2017, 7, 1540. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Landau, M.; Ben-Tal, N. Dynamic equilibrium between multiple active and inactive conformations explains regulation and oncogenic mutations in ErbB receptors. Biochim. Biophys. Acta 2008, 1785, 12–31. [Google Scholar] [CrossRef]
- Eck, M.J.; Yun, C.-H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta 2010, 1804, 559–566. [Google Scholar] [CrossRef] [Green Version]
- Carey, K.D.; Garton, A.J.; Romero, M.S.; Kahler, J.; Thomson, S.; Ross, S.; Park, F.; Haley, J.D.; Gibson, N.; Sliwkowski, M.X. Kinetic Analysis of Epidermal Growth Factor Receptor Somatic Mutant Proteins Shows Increased Sensitivity to the Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor, Erlotinib. Cancer Res. 2006, 66, 8163–8171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung Cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequist, L.V.; Besse, B.; Lynch, T.J.; Miller, V.A.; Wong, K.K.; Gitlitz, B.; Eaton, K.; Zacharchuk, C.; Freyman, A.; Powell, C.; et al. Neratinib, an Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor: Results of a Phase II Trial in Patients with Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2010, 28, 3076–3083. [Google Scholar] [CrossRef]
- Miller, V.A.; Hirsh, V.; Cadranel, J.; Chen, Y.-M.; Park, K.; Kim, S.-W.; Zhou, C.; Su, W.-C.; Wang, M.; Sun, Y.; et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase 2b/3 randomised trial. Lancet Oncol. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Katakami, N.; Atagi, S.; Goto, K.; Hida, T.; Horai, T.; Inoue, A.; Ichinose, Y.; Koboyashi, K.; Takeda, K.; Kiura, K.; et al. LUX-Lung 4: A Phase II Trial of Afatinib in Patients with Advanced Non–Small-Cell Lung Cancer Who Progressed During Prior Treatment with Erlotinib, Gefitinib, or Both. J. Clin. Oncol. 2013, 31, 3335–3341. [Google Scholar] [CrossRef]
- Schuler, M.; Yang, J.C.-H.; Park, K.; Kim, J.-H.; Bennouna, J.; Chen, Y.-M.; Chouaid, C.; De Marinis, F.; Feng, J.-F.; Grossi, F.; et al. Afatinib beyond progression in patients with non-small-cell lung cancer following chemotherapy, erlotinib/gefitinib and afatinib: Phase III randomized LUX-Lung 5 trial. Ann. Oncol. 2015, 27, 417–423. [Google Scholar] [CrossRef]
- Yosaatmadja, Y.; Silva, S.; Dickson, J.M.; Patterson, A.V.; Smaill, J.B.; Flanagan, J.U.; McKeage, M.J.; Squire, C.J. Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J. Struct. Biol. 2015, 192, 539–544. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Tagrisso Significantly Improves Overall Survival in the Phase III FLAURA Trial for 1st-Line egfr-Mutated Non-Small Cell Lung Cancer. Available online: https://www.astrazeneca.com/media-centre/press-releases/2019/tagrisso-significantly-improves-overall-survival-in-the-phase-iii-flaura-trial-for-1st-line-egfr-mutated-non-small-cell-lung-cancer-09082019.html#! (accessed on 9 March 2022).
- Final FLAURA Results Demonstrate Overall Survival Benefit. Available online: https://www.esmo.org/oncology-news/final-flaura-results-demonstrate-overall-survival-benefit-with-osimertinib-over-tkis-in-advanced-nsclc (accessed on 9 March 2022).
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Zawacka-Pankau, J.E. The Role of p53 Family in Cancer. Cancers 2022, 14, 823. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The many faces of p53: Something for everyone. J. Mol. Cell Biol. 2019, 11, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Janic, A.; Valente, L.J.; Wakefield, M.J.; Di Stefano, L.; Milla, L.; Wilcox, S.; Yang, H.; Tai, L.; Vandenberg, C.J.; Kueh, A.J.; et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat. Med. 2018, 24, 947–953. [Google Scholar] [CrossRef]
- Zawacka-Pankau, J. The Undervalued Avenue to Reinstate Tumor Suppressor Functionality of the p53 Protein Family for Improved Cancer Therapy-Drug Repurposing. Cancers 2020, 12, 2717. [Google Scholar] [CrossRef]
- Joseph, T.W.; Zaika, A.; Moll, U.M. Nuclear and cytoplasmic degradation of endogenous p53 and HDM2 occurs during down-regulation of the p53 response after multiple types of DNA damage. FASEB J. 2003, 17, 1622–1630. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.-P.; Gu, W. Modes of p53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Tournillon, A.-S.; López, I.; Malbert-Colas, L.; Findakly, S.; Naski, N.; Olivares-Illana, V.; Karakostis, K.; Vojtesek, B.; Nylander, K.; Fåhraeus, R. p53 binds the mdmx mRNA and controls its translation. Oncogene 2016, 36, 723–730. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [Green Version]
- de Andrade, K.C.; Khincha, P.P.; Hatton, J.N.; Frone, M.N.; Wegman-Ostrosky, T.; Mai, P.L.; Best, A.F.; Savage, S.A. Cancer incidence, patterns, and genotype–phenotype associations in individuals with pathogenic or likely pathogenic germline TP53 variants: An observational cohort study. Lancet Oncol. 2021, 22, 1787–1798. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant p53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. IARC TP53 Database. Available online: https://p53.fr (accessed on 15 May 2022).
- Jiang, L.; Zawacka-Pankau, J. The p53/MDM2/MDMX-targeted therapies—A clinical synopsis. Cell Death Dis. 2020, 11, 237. [Google Scholar] [CrossRef]
- Li, G.; Tang, L.; Zhou, X.; Tron, V.; Ho, V. Chemotherapy-induced apoptosis in melanoma cells is p53 dependent. Melanoma Res. 1998, 8, 17–18. [Google Scholar] [CrossRef]
- Asada, N.; Tsuchiya, H.; Tomita, K. De novo deletions of p53 gene and wild-type p53 correlate with acquired cisplatin-resistance in human osteosarcoma OST cell line. Anticancer Res. 2000, 19, 5131–5137. [Google Scholar]
- Berns, E.M.; Foekens, J.A.; Vossen, R.; Look, M.P.; Devilee, P.; Henzen-Logmans, S.C.; Van Staveren, I.L.; Van Putten, W.L.; Inganäs, M.; Gelder, M.E.M.-V.; et al. Complete sequencing of TP53 predicts poor response to systemic therapy of advanced breast cancer. Cancer Res. 2000, 60, 2155–2162. [Google Scholar]
- Houldsworth, J.; Xiao, H.; Murty, V.; Chen, W.; Ray, B.; Reuter, V.E.; Bosl, G.J.; Chaganti, R. Human male germ cell tumor resistance to cisplatin is linked to TP53 gene mutation. Oncogene 1998, 16, 2345–2349. [Google Scholar] [CrossRef] [Green Version]
- Righetti, S.C.; Perego, P.; Corna, E.; Pierotti, M.A.; Zunino, F. Emergence of p53 mutant cisplatin-resistant ovarian carcinoma cells following drug exposure: Spontaneously mutant selection. Cell Growth Differ. 1999, 10, 473–478. [Google Scholar]
- Mogi, A.; Kuwano, H. TP53 mutations in nonsmall cell lung cancer. J. Biomed. Biotechnol. 2011, 2011, 583929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Custodio, A.B.; González-Larriba, J.L.; Bobokova, J.; Calles, A.; Álvarez, R.; Cuadrado, E.; Manzano, A.; Díaz-Rubio, E. Prognostic and Predictive Markers of Benefit from Adjuvant Chemotherapy in Early-Stage Non-small Cell Lung Cancer. J. Thorac. Oncol. 2009, 4, 891–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, M.-S.; Aviel-Ronen, S.; Ding, K.; Lau, D.; Liu, N.; Sakurada, A.; Whitehead, M.; Zhu, C.-Q.; Livingston, R.; Johnson, D.H.; et al. Prognostic and Predictive Importance of p53 and RAS for Adjuvant Chemotherapy in Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2007, 25, 5240–5247. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.-C.; Hsu, S.-L.; Tsai, J.-R.; Liang, F.-P.; Lin, S.-Y.; Sheu, G.-T.; Chen, C.-Y. Molecular mechanisms of ZD1839-induced G1-cell cycle arrest and apoptosis in human lung adenocarcinoma A549 cells. Biochem. Pharmacol. 2004, 68, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Labbé, C.; Cabanero, M.; Korpanty, G.J.; Tomasini, P.; Doherty, M.K.; Mascaux, C.; Jao, K.; Pitcher, B.; Wang, R.; Pintilie, M.; et al. Prognostic and predictive effects of TP53 co-mutation in patients with EGFR -mutated non-small cell lung cancer (NSCLC). Lung Cancer 2017, 111, 23–29. [Google Scholar] [CrossRef]
- Molina-Vila, M.A.; Bertran-Alamillo, J.; Gascó, A.; Mayo-De-Las-Casas, C.; Sánchez-Ronco, M.; Pujantell-Pastor, L.; Bonanno, L.; Favaretto, A.G.; Cardona, A.F.; Vergnenègre, A.; et al. Nondisruptive p53 Mutations Are Associated with Shorter Survival in Patients with Advanced Non–Small Cell Lung Cancer. Clin. Cancer Res. 2014, 20, 4647–4659. [Google Scholar] [CrossRef] [Green Version]
- Munsch, D.; Watanabe-Fukunaga, R.; Bourdon, J.-C.; Nagata, S.; May, E.; Yonish-Rouach, E.; Reisdorf, P. Human and Mouse Fas (APO-1/CD95) Death Receptor Genes Each Contain a p53-responsive Element That Is Activated by p53 Mutants Unable to Induce Apoptosis. J. Biol. Chem. 2000, 275, 3867–3872. [Google Scholar] [CrossRef] [Green Version]
- Rho, J.K.; Choi, Y.J.; Ryoo, B.-Y.; Na, I.I.; Yang, S.H.; Kim, C.H.; Lee, J.C. p53 Enhances Gefitinib-Induced Growth Inhibition and Apoptosis by Regulation of Fas in Non–Small Cell Lung Cancer. Cancer Res. 2007, 67, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- VanderLaan, P.; Rangachari, D.; Mockus, S.M.; Spotlow, V.; Reddi, H.V.; Malcolm, J.; Huberman, M.S.; Joseph, L.J.; Kobayashi, S.S.; Costa, D.B. Mutations in TP53, PIK3CA, PTEN and other genes in EGFR mutated lung cancers: Correlation with clinical outcomes. Lung Cancer 2017, 106, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Canale, M.; Petracci, E.; Delmonte, A.; Chiadini, E.; Dazzi, C.; Papi, M.; Capelli, L.; Casanova, C.; De Luigi, N.; Mariotti, M.; et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2017, 23, 2195–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, V.; Roisman, L.; Kian, W.; Daniel, L.; Dudnik, J.; Nechushtan, H.; Goldstein, I.; Dvir, A.; Soussan-Gutman, L.; Grinberg, R.; et al. The impact of osimertinib’ line on clonal evolution in EGFRm NSCLC through NGS-based liquid biopsy and overcoming strategies for resistance. Lung Cancer 2021, 153, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Duncavage, E.J.; Chang, G.S.; Jacoby, M.A.; Miller, C.A.; Shao, J.; Heath, S.; Elliott, K.; Reineck, T.; Fulton, R.S.; et al. Dynamic changes in the clonal structure of MDS and AML in response to epigenetic therapy. Leukemia 2017, 31, 872–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, C.-W.; Lin, C.-H.; Hsiao, T.-H.; Lo, C.-C.; Hsieh, C.-Y.; Huang, C.-C.; Sher, Y.-P. Therapeutic effects of statins against lung adenocarcinoma via p53 mutant-mediated apoptosis. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- AXL Gene-GeneCards|UFO Protein|UFO Antibody. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=AXL (accessed on 9 March 2022).
- Bremnes, R.M.; Veve, R.; Gabrielson, E.; Hirsch, F.R.; Baron, A.; Bemis, L.; Gemmill, R.M.; Drabkin, H.A.; Franklin, W.A. High-Throughput Tissue Microarray Analysis Used to Evaluate Biology and Prognostic Significance of the E-Cadherin Pathway in Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2002, 20, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Deeb, G.; Wang, J.; Ramnath, N.; Slocum, H.K.; Wiseman, S.; Beck, A.; Tan, D. Altered E-cadherin and epidermal growth factor receptor expressions are associated with patient survival in lung cancer: A study utilizing high-density tissue microarray and immunohistochemistry. Mod. Pathol. 2004, 17, 430–439. [Google Scholar] [CrossRef]
- Rho, J.K.; Choi, Y.J.; Lee, J.K.; Ryoo, B.-Y.; Na, I.I.; Yang, S.H.; Kim, C.H.; Lee, J.C. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer 2009, 63, 219–226. [Google Scholar] [CrossRef]
- Ji, W.; Choi, Y.J.; Kang, M.-H.; Sung, K.J.; Kim, D.H.; Jung, S.; Choi, C.-M.; Lee, J.C.; Rho, J.K. Efficacy of the CDK7 Inhibitor on EMT-Associated Resistance to 3rd Generation EGFR-TKIs in Non-Small Cell Lung Cancer Cell Lines. Cells 2020, 9, 2596. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Kim, D.H.; Choi, Y.J.; Kim, S.Y.; Park, H.; Lee, H.; Choi, C.-M.; Sung, Y.H.; Lee, J.C.; Rho, J.K. Contribution of p53 in sensitivity to EGFR tyrosine kinase inhibitors in non-small cell lung cancer. Sci. Rep. 2021, 11, 19667. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 Drives Invasion by Promoting Integrin Recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef]
- Comel, A.; Sorrentino, G.; Capaci, V.; Del Sal, G. The cytoplasmic side of p53’s oncosuppressive activities. FEBS Lett. 2014, 588, 2600–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, Y.; Sugiyama, A.; Li, S.-A.; Ohmori, K.; Ohata, H.; Yoshida, Y.; Shibuya, M.; Takei, K.; Enari, M.; Taya, Y. Regulation of clathrin-mediated endocytosis by p53. Genes Cells 2008, 13, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, M.-Q.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.; Arnér, E.S.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2017, 8, e2751. [Google Scholar] [CrossRef] [Green Version]
- Available online: clinicaltrials.gov (accessed on 7 June 2022).
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Malik, N.; Acedo, P.; Zawacka-Pankau, J. Protoporphyrin IX is a dual inhibitor of p53/MDM2 and p53/MDM4 interactions and induces apoptosis in B-cell chronic lymphocytic leukemia cells. Cell Death Discov. 2019, 5, 77. [Google Scholar] [CrossRef] [Green Version]
- Sznarkowska, A.; Kostecka, A.; Kawiak, A.; Acedo, P.; Lion, M.; Inga, A.; Zawacka-Pankau, J. Reactivation of TAp73 tumor suppressor by protoporphyrin IX, a metabolite of aminolevulinic acid, induces apoptosis in TP53-deficient cancer cells. Cell Div. 2018, 13, 10. [Google Scholar] [CrossRef] [Green Version]
- Prast-Nielsen, S.; Dexheimer, T.S.; Schultz, L.; Stafford, W.C.; Cheng, Q.; Xu, J.; Jadhav, A.; Arnér, E.S.J.; Simeonov, A. Inhibition of thioredoxin reductase 1 by porphyrins and other small molecules identified by a high-throughput screening assay. Free Radic. Biol. Med. 2011, 50, 1114–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acedo, P.; Fernandes, A.; Zawacka-Pankau, J. Activation of TAp73 and inhibition of TrxR by Verteporfin for improved cancer therapy in TP53 mutant pancreatic tumors. Future Sci. OA 2019, 5, FSO366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef] [Green Version]
- Mohell, N.; Alfredsson, J.; Fransson, A.; Uustalu, M.; Bystrom, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.N.; Bjorklund, U.; Wiman, K. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef] [Green Version]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Lambot, S.M.; Oullier, T.; le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wu, J.-L.; Liang, Y.; Tang, Y.-G.; Song, H.-X.; Wu, L.-L.; Xing, Y.-F.; Yan, N.; Li, Y.-T.; Wang, Z.-Y.; et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2020, 39, 225–239.e8. [Google Scholar] [CrossRef]
- Synnott, N.C.; O’Connell, D.; Crown, J.; Duffy, M.J. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat. 2019, 179, 47–56. [Google Scholar] [CrossRef]
- Joseph, T.L.; Madhumalar, A.; Brown, C.J.; Lane, D.; Verma, C.S. Differential binding of p53 and nutlin to MDM2 and MDMX: Computational studies. Cell Cycle 2010, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marine, J.-C.; Francoz, S.; Maetens, M.; Wahl, G.M.; Toledo, F.; Lozano, G. Keeping p53 in check: Essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006, 13, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Patton, J.T.; Mayo, L.D.; Singhi, A.D.; Gudkov, A.V.; Stark, G.R.; Jackson, M.W. Levels of HdmX Expression Dictate the Sensitivity of Normal and Transformed Cells to Nutlin-3. Cancer Res. 2006, 66, 3169–3176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvajal, L.A.; Ben Neriah, D.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.-R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10, eaao3003. [Google Scholar] [CrossRef] [Green Version]
- Grinkevich, V.V.; Vema, A.; Fawkner, K.; Issaeva, N.; Andreotti, V.; Dickinson, E.R.; Hedström, E.; Spinnler, C.; Inga, A.; Larsson, L.-G.; et al. Novel Allosteric Mechanism of Dual p53/MDM2 and p53/MDM4 Inhibition by a Small Molecule. Front. Mol. Biosci. 2022, 9, 823195. [Google Scholar] [CrossRef]
- Pierotti, M.A.; Berrino, F.; Gariboldi, M.; Melani, C.; Mogavero, A.; Negri, T.; Pasanisi, P.; Pilotti, S. Targeting metabolism for cancer treatment and prevention: Metformin, an old drug with multi-faceted effects. Oncogene 2012, 32, 1475–1487. [Google Scholar] [CrossRef]
- Bernard, H.; Garmy-Susini, B.; Ainaoui, N.; Van Den Berghe, L.; Peurichard, A.; Javerzat, S.; Bikfalvi, A.; Lane, D.P.; Bourdon, J.-C.; Prats, A.-C. The p53 isoform, Δ133p53α, stimulates angiogenesis and tumour progression. Oncogene 2012, 32, 2150–2160. [Google Scholar] [CrossRef] [Green Version]
- Arsic, N.; Gadea, G.; Lagerqvist, E.L.; Busson, M.; Cahuzac, N.; Brock, C.; Hollande, F.; Gire, V.; Pannequin, J.; Roux, P. The p53 Isoform Δ133p53β Promotes Cancer Stem Cell Potential. Stem Cell Rep. 2015, 4, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. p53 isoforms Δ133p53 and p53β are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef]
- Candeias, M.M.; Hagiwara, M.; Matsuda, M. Cancer-specific mutations in p53 induce the translation of Δ160p53 promoting tumorigenesis. EMBO Rep. 2016, 17, 1542–1551. [Google Scholar] [CrossRef]
- Gadea, G.; Arsic, N.; Fernandes, K.; Diot, A.; Joruiz, S.M.; Abdallah, S.; Meuray, V.; Vinot, S.; Anguille, C.; Remenyi, J.; et al. TP53 drives invasion through expression of its Δ133p53β variant. eLife 2016, 5, e14734. [Google Scholar] [CrossRef] [PubMed]
- Campbell, H.; Fleming, N.; Roth, I.; Mehta, S.; Wiles, A.; Williams, G.; Vennin, C.; Arsic, N.; Parkin, A.; Pajic, M.; et al. ∆133p53 isoform promotes tumour invasion and metastasis via interleukin-6 activation of JAK-STAT and RhoA-ROCK signalling. Nat. Commun. 2018, 9, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazantseva, M.; Mehta, S.; Eiholzer, R.A.; Gimenez, G.; Bowie, S.; Campbell, H.; Reily-Bell, A.L.; Roth, I.; Ray, S.; Drummond, C.J.; et al. The Δ133p53β isoform promotes an immunosuppressive environment leading to aggressive prostate cancer. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Steffens Reinhardt, L.; Zhang, X.; Wawruszak, A.; Groen, K.; De Iuliis, G.N.; Avery-Kiejda, K.A. Good Cop, Bad Cop: Defining the Roles of Δ40p53 in Cancer and Aging. Cancers 2020, 12, 1659. [Google Scholar] [CrossRef] [PubMed]
- Tadijan, A.; Precazzini, F.; Hanžić, N.; Radić, M.; Gavioli, N.; Vlašić, I.; Ozretić, P.; Pinto, L.; Škreblin, L.; Barban, G.; et al. Altered Expression of Shorter p53 Family Isoforms Can Impact Melanoma Aggressiveness. Cancers 2021, 13, 5231. [Google Scholar] [CrossRef]
- Tomasini, R.; Tsuchihara, K.; Wilhelm, M.; Fujitani, M.; Rufini, A.; Cheung, C.C.; Khan, F.; Itie-Youten, A.; Wakeham, A.; Tsao, M.-S.; et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008, 22, 2677–2691. [Google Scholar] [CrossRef] [Green Version]
- Pozniak, C.D.; Radinovic, S.; Yang, A.; McKeon, F.; Kaplan, D.R.; Miller, F.D. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 2000, 289, 304–306. [Google Scholar] [CrossRef]
- Agostini, M.; Annicchiarico-Petruzzelli, M.; Melino, G.; Rufini, A. Metabolic pathways regulated by TAp73 in response to oxidative stress. Oncotarget 2016, 7, 29881–29900. [Google Scholar] [CrossRef] [Green Version]
- Conforti, F.; Sayan, A.E.; Sreekumar, R.; Sayan, B.S. Regulation of p73 activity by post-translational modifications. Cell Death Dis. 2012, 3, e285. [Google Scholar] [CrossRef] [Green Version]
- Daskalos, A.; Logotheti, S.; Markopoulou, S.; Xinarianos, G.; Gosney, J.R.; Kastania, A.N.; Zoumpourlis, V.; Field, J.K.; Liloglou, T. Global DNA hypomethylation-induced ΔNp73 transcriptional activation in non-small cell lung cancer. Cancer Lett. 2011, 300, 79–86. [Google Scholar] [CrossRef]
- Liu, K.; Zhuang, X.; Mai, Z. p73 expression is associated with cellular chemosensitivity in human non-small cell lung cancer cell lines. Oncol. Lett. 2012, 5, 583–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez, G.; García, J.M.; Peña, C.; Silva, J.; García, V.; Martínez, L.; Maximiano, C.; Gómez, M.E.; Rivera, J.A.; García-Andrade, C.; et al. ΔTAp73 Upregulation Correlates with Poor Prognosis in Human Tumors: Putative In Vivo Network Involving p73 Isoforms, p53, and E2F-1. J. Clin. Oncol. 2006, 24, 805–815. [Google Scholar] [CrossRef]
- Hofstetter, G.; Berger, A.; Chamson, M.; Müller-Holzner, E.; Reimer, D.; Ulmer, H.; Uramoto, H.; Marth, C.; Zeimet, A.G.; Zeillinger, R.; et al. Clinical Relevance of TAp73 and ΔNp73 Protein Expression in Ovarian Cancer. Int. J. Gynecol. Pathol. 2011, 30, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Bunch, B.; Krishnan, N.; Greenspan, R.D.; Ramakrishnan, S.; Attwood, K.; Yan, L.; Qi, Q.; Wang, D.; Morrison, C.; Omilian, A.; et al. TAp73 expression and P1 promoter methylation, a potential marker for chemoresponsiveness to cisplatin therapy and survival in muscle-invasive bladder cancer (MIBC). Cell Cycle 2019, 18, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.M.S.; Nugent, J.K.; Zhao, X.; Irwin, M.S. HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene 2008, 27, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.; Rossi, M.; Roperch, J.; Ansell, K.; Simpson, K.; Taylor, D.; Mathon, N.; Knight, R.; Melino, G. Itch inhibition regulates chemosensitivity in vitro. Biochem. Biophys. Res. Commun. 2007, 361, 33–36. [Google Scholar] [CrossRef]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307. [Google Scholar] [CrossRef] [Green Version]
- Luh, L.M.; Kehrloesser, S.; Deutsch, G.B.; Gebel, J.; Coutandin, D.; Schäfer, B.; Agostini, M.; Melino, G.; Dotsch, V. Analysis of the oligomeric state and transactivation potential of TAp73α. Cell Death Differ. 2013, 20, 1008–1016. [Google Scholar] [CrossRef]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef]
- Celli, J.; Duijf, P.; Hamel, B.C.; Bamshad, M.; Kramer, B.; Smits, A.P.; Newbury-Ecob, R.; Hennekam, R.C.; Van Buggenhout, G.; van Haeringen, A.; et al. Heterozygous Germline Mutations in the p53 Homolog p63 Are the Cause of EEC Syndrome. Cell 1999, 99, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Keyes, W.M.; Papazoglu, C.; Zuber, J.; Li, W.; Lowe, S.W.; Vogel, H.; Mills, A.A. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat. Cell Biol. 2009, 11, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Montagner, M.; Enzo, E.; Forcato, M.; Zanconato, F.; Parenti, A.; Rampazzo, E.; Basso, G.; Leo, G.; Rosato, A.; Bicciato, S.; et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature 2012, 487, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad Complex Opposes p63 to Empower TGFβ-Induced Metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Chakravarti, D.; Cho, M.S.; Liu, L.-Z.; Gi, Y.J.; Lin, Y.-L.; Leung, M.L.; El-Naggar, A.; Creighton, C.J.; Suraokar, M.B.; et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010, 467, 986–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyes, W.M.; Pecoraro, M.; Aranda, V.; Vernersson-Lindahl, E.; Li, W.; Vogel, H.; Guo, X.; Garcia, E.L.; Michurina, T.V.; Enikolopov, G.; et al. ΔNp63α Is an Oncogene that Targets Chromatin Remodeler Lsh to Drive Skin Stem Cell Proliferation and Tumorigenesis. Cell Stem Cell 2011, 8, 164–176. [Google Scholar] [CrossRef] [Green Version]
- Bid, H.K.; Roberts, R.D.; Cam, M.; Audino, A.; Kurmasheva, R.T.; Lin, J.; Houghton, P.J.; Cam, H. ΔNp63 Promotes Pediatric Neuroblastoma and Osteosarcoma by Regulating Tumor Angiogenesis. Cancer Res. 2014, 74, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Rocco, J.W.; Leong, C.-O.; Kuperwasser, N.; DeYoung, M.P.; Ellisen, L.W. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 2006, 9, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Venkatanarayan, A.; Raulji, P.; Norton, W.; Chakravarti, D.; Coarfa, C.; Su, X.; Sandur, S.K.; Ramirez, M.S.; Lee, J.; Kingsley, C.V.; et al. IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature 2014, 517, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Conde, E.; Angulo, B.; Redondo, P.; Toldos, O.; García-García, E.; Suarez-Gauthier, A.; Rubio-Viqueira, B.; Marrón, C.; García-Luján, R.; Sánchez-Céspedes, M.; et al. The Use of P63 Immunohistochemistry for the Identification of Squamous Cell Carcinoma of the Lung. PLoS ONE 2010, 5, e12209. [Google Scholar] [CrossRef] [Green Version]
- Osmani, L.; Askin, F.; Gabrielson, E.; Li, Q.K. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): Moving from targeted therapy to immunotherapy. Semin. Cancer Biol. 2018, 52, 103–109. [Google Scholar] [CrossRef]
- Napoli, M.; Wu, S.J.; Gore, B.L.; Abbas, H.A.; Lee, K.; Checker, R.; Dhar, S.; Rajapakshe, K.; Tan, A.C.; Lee, M.G.; et al. ΔNp63 regulates a common landscape of enhancer associated genes in non-small cell lung cancer. Nat. Commun. 2022, 13, 614. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug | Mechanism of Action | FDA Approvals/Clinical Trials |
|---|---|---|
| APR-246 (eprenetapopt) | Binding to and refolding mutant p53 [104,105] Inhibition of thioredoxin reductase and glutaredoxin [106,107] | 13 clinical trials registered in cancer [108] |
| Metformin (MET) | Inhibition of mitochondrial complex I) [109] Activation of AMP-dependent kinase [110] | 400 clinical trials registered in cancer [108] |
| Protoporphyrin IX (PpIX) | Inhibition of p53/MDM2/MDM4 interactions [111] Inhibition of TAp73/MDM2/MDM4 interactions [112] Inhibition of thioredoxin reductase [113,114] | 29 clinical trials registered in cancer [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fregni, M.; Ciribilli, Y.; Zawacka-Pankau, J.E. The Therapeutic Potential of the Restoration of the p53 Protein Family Members in the EGFR-Mutated Lung Cancer. Int. J. Mol. Sci. 2022, 23, 7213. https://doi.org/10.3390/ijms23137213
Fregni M, Ciribilli Y, Zawacka-Pankau JE. The Therapeutic Potential of the Restoration of the p53 Protein Family Members in the EGFR-Mutated Lung Cancer. International Journal of Molecular Sciences. 2022; 23(13):7213. https://doi.org/10.3390/ijms23137213
Chicago/Turabian StyleFregni, Matilde, Yari Ciribilli, and Joanna E. Zawacka-Pankau. 2022. "The Therapeutic Potential of the Restoration of the p53 Protein Family Members in the EGFR-Mutated Lung Cancer" International Journal of Molecular Sciences 23, no. 13: 7213. https://doi.org/10.3390/ijms23137213