
Retrospective Natural History Study of RPGR-Related Cone- and Cone-Rod Dystrophies While Expanding the Mutation Spectrum of the Disease

 , , and
, , and
Abstract
:1. Introduction
2. Results
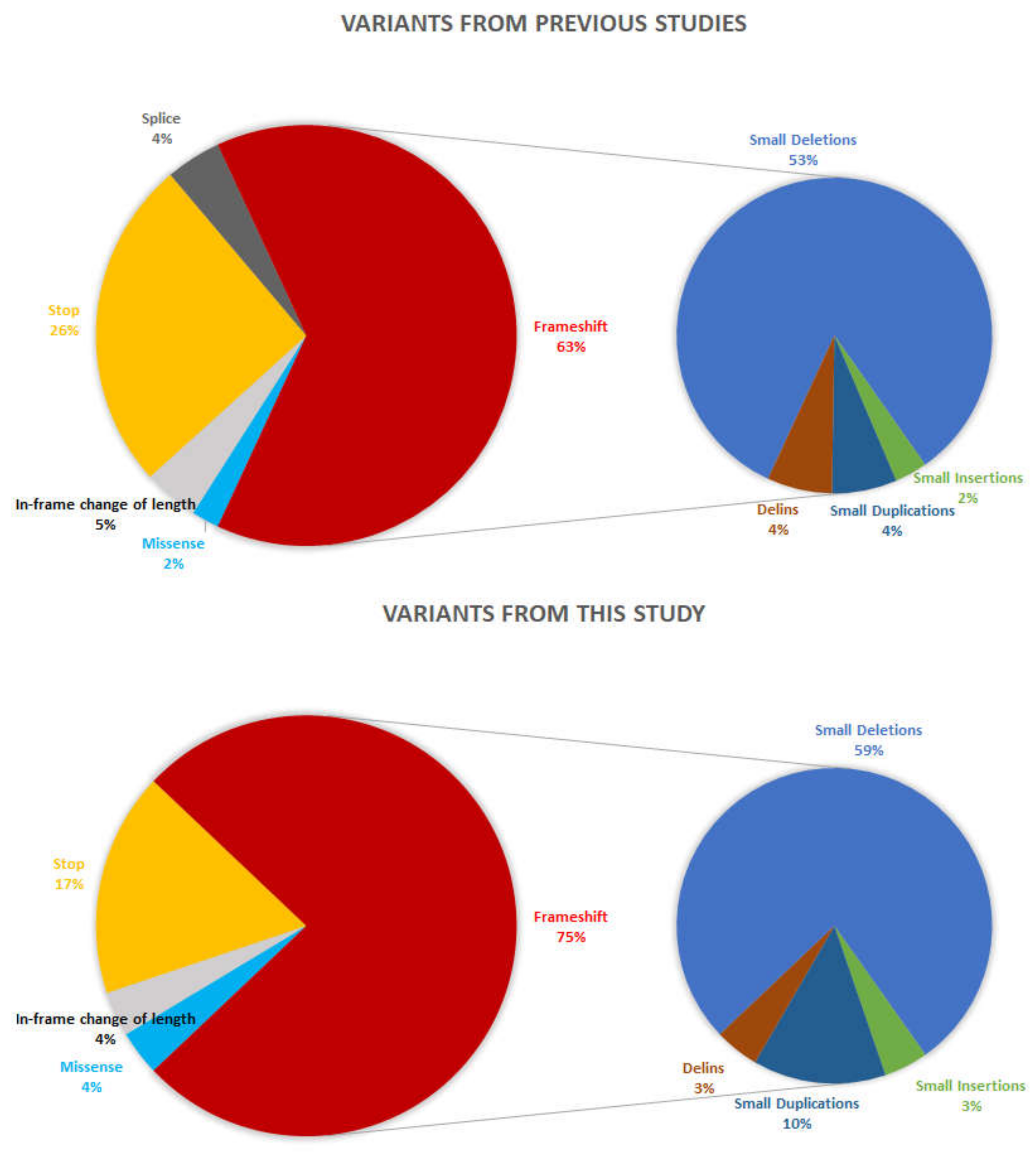
2.1. Genetic Screening
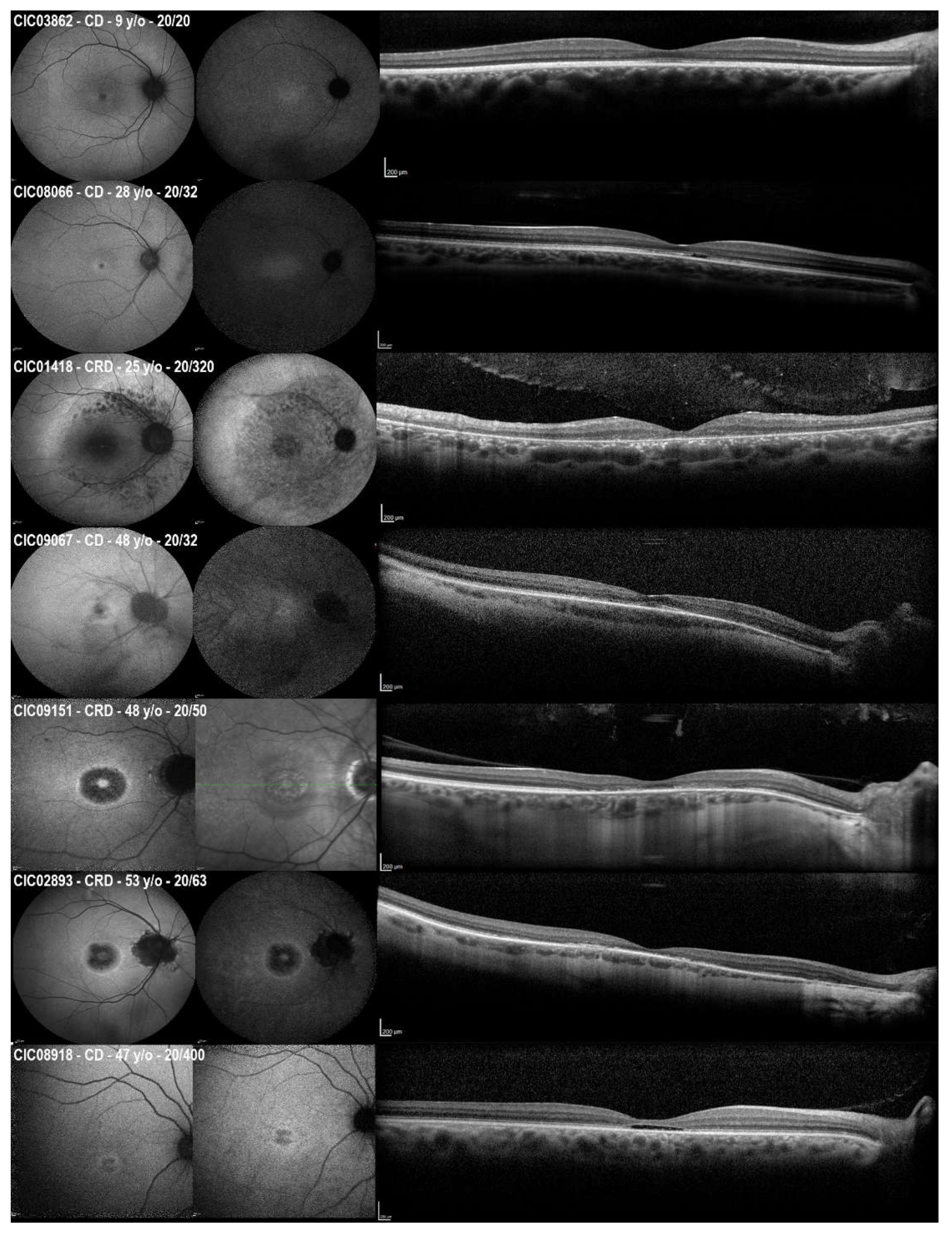
2.2. Clinical Cross-Sectional Data
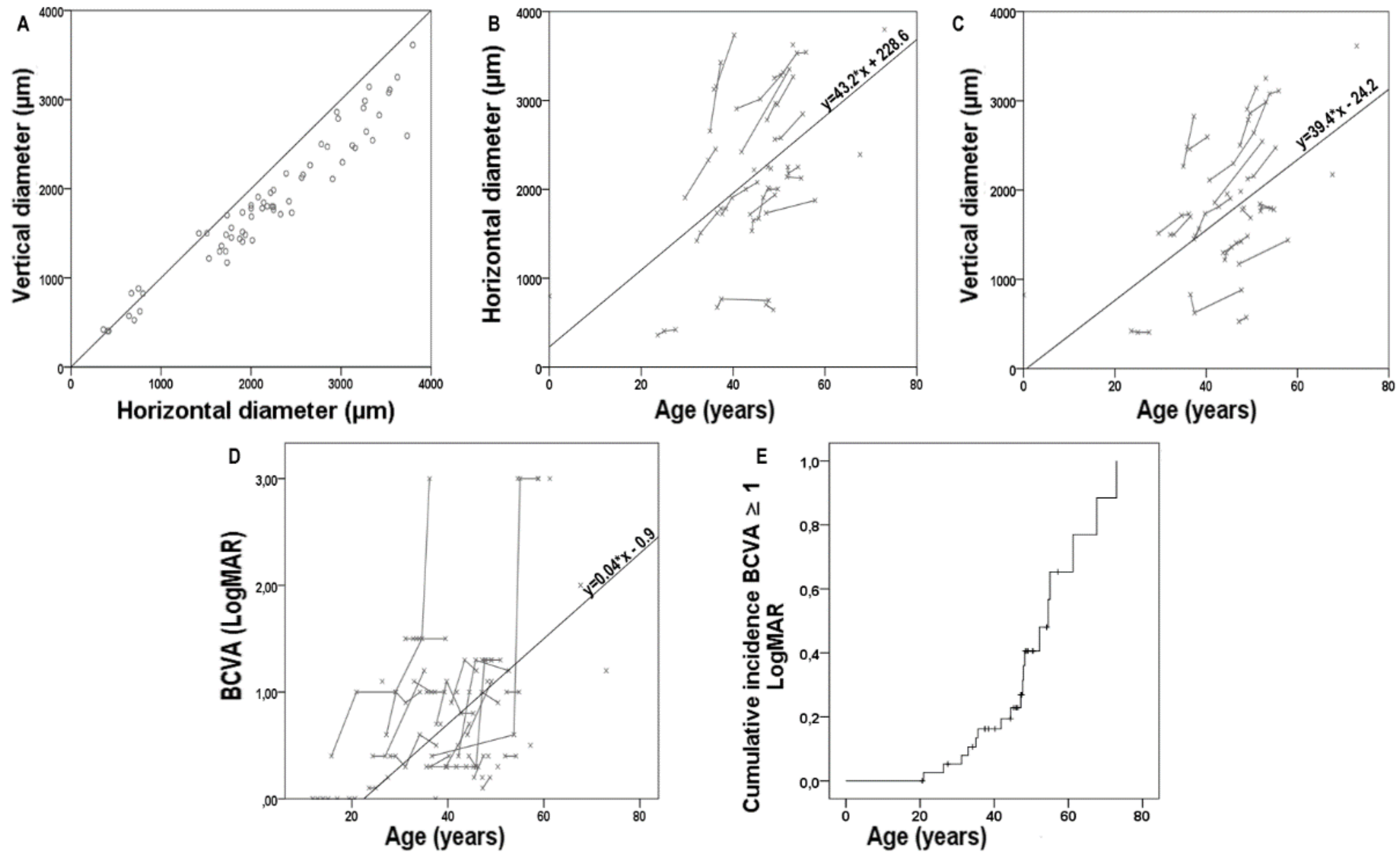
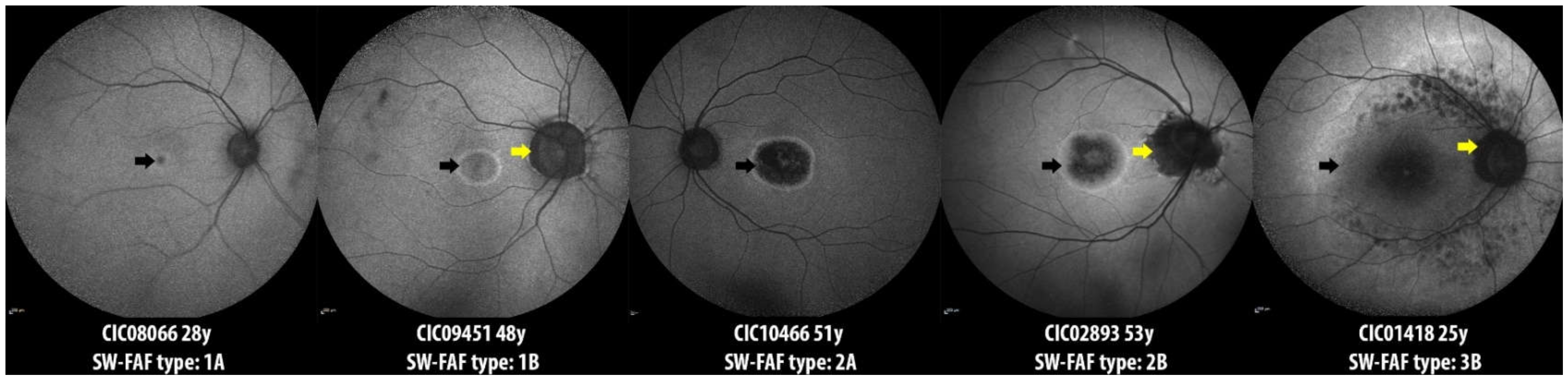
2.3. Longitudinal Data on SW-FAF
2.4. Longitudinal Data on Visual Acuity and Correlations
2.5. Genotype–Phenotype Correlation
3. Discussion
4. Materials and Methods
4.1. Mutation Analysis
4.2. Clinical Data Collection
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamel, C.P. Cone Rod Dystrophies. Orphanet. J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Meindl, A.; Dry, K.; Herrmann, K.; Manson, F.; Ciccodicola, A.; Edgar, A.; Carvalho, M.R.; Achatz, H.; Hellebrand, H.; Lennon, A.; et al. A Gene (RPGR) with Homology to the RCC1 Guanine Nucleotide Exchange Factor Is Mutated in X-Linked Retinitis Pigmentosa (RP3). Nat. Genet. 1996, 13, 35–42. [Google Scholar] [CrossRef]
- Sharon, D.; Sandberg, M.A.; Rabe, V.W.; Stillberger, M.; Dryja, T.P.; Berson, E.L. RP2 and RPGR Mutations and Clinical Correlations in Patients with X-Linked Retinitis Pigmentosa. Am. J. Hum. Genet. 2003, 73, 1131–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talib, M.; van Schooneveld, M.J.; Thiadens, A.A.; Fiocco, M.; Wijnholds, J.; Florijn, R.J.; Schalij-Delfos, N.E.; van Genderen, M.M.; Putter, H.; Cremers, F.P.M.; et al. Clinical and Genetic Characteristics of Male Patients with Rpgr-Associated Retinal Dystrophies: A Long-Term Follow-up Study. Retina 2019, 39, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Megaw, R.D.; Soares, D.C.; Wright, A.F. RPGR: Its Role in Photoreceptor Physiology, Human Disease, and Future Therapies. Exp. Eye Res. 2015, 138, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.; Glaus, E.; Cremers, F.P.M.; Kloeckener-Gruissem, B.; Berger, W.; Neidhardt, J. Mutation- and Tissue-Specific Alterations of RPGR Transcripts. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1628–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vössing, C.; Atigbire, P.; Eilers, J.; Markus, F.; Stieger, K.; Song, F.; Neidhardt, J. The Major Ciliary Isoforms of RPGR Build Different Interaction Complexes with INPP5E and RPGRIP1L. Int. J. Mol. Sci. 2021, 22, 3583. [Google Scholar] [CrossRef]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational Hot Spot within a New RPGR Exon in X-Linked Retinitis Pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef]
- Remans, K.; Bürger, M.; Vetter, I.R.; Wittinghofer, A. C2 Domains as Protein-Protein Interaction Modules in the Ciliary Transition Zone. Cell Rep. 2014, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.H.; Pawlyk, B.S.; Shang, J.; Sandberg, M.A.; Berson, E.L.; Li, T. A Retinitis Pigmentosa GTPase Regulator (RPGR)-Deficient Mouse Model for X-Linked Retinitis Pigmentosa (RP3). Proc. Natl. Acad. Sci. USA 2000, 97, 3649–3654. [Google Scholar] [CrossRef]
- Hong, D.-H.; Pawlyk, B.; Sokolov, M.; Strissel, K.J.; Yang, J.; Tulloch, B.; Wright, A.F.; Arshavsky, V.Y.; Li, T. RPGR Isoforms in Photoreceptor Connecting Cilia and the Transitional Zone of Motile Cilia. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2413–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavlyutov, T.A.; Zhao, H.; Ferreira, P.A. Species-Specific Subcellular Localization of RPGR and RPGRIP Isoforms: Implications for the Phenotypic Variability of Congenital Retinopathies among Species. Hum. Mol. Genet. 2002, 11, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Ruddle, J.B.; Ebenezer, N.D.; Kearns, L.S.; Mulhall, L.E.; Mackey, D.A.; Hardcastle, A.J. RPGR ORF15 Genotype and Clinical Variability of Retinal Degeneration in an Australian Population. Br. J. Ophthalmol. 2009, 93, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.R.; Arno, G.; Robson, A.G.; Fakin, A.; Pontikos, N.; Mohamed, M.D.; Bird, A.C.; Moore, A.T.; Michaelides, M.; Webster, A.R.; et al. The X-Linked Retinopathies: Physiological Insights, Pathogenic Mechanisms, Phenotypic Features and Novel Therapies. Prog. Retin. Eye Res. 2021, 82, 100898. [Google Scholar] [CrossRef]
- Thiadens, A.A.H.J.; Soerjoesing, G.G.; Florijn, R.J.; Tjiam, A.G.; den Hollander, A.I.; van den Born, L.I.; Riemslag, F.C.; Bergen, A.A.B.; Klaver, C.C.W. Clinical Course of Cone Dystrophy Caused by Mutations in the RPGR Gene. Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 1527–1535. [Google Scholar] [CrossRef] [Green Version]
- Zahid, S.; Khan, N.; Branham, K.; Othman, M.; Karoukis, A.J.; Sharma, N.; Moncrief, A.; Mahmood, M.N.; Sieving, P.A.; Swaroop, A.; et al. Phenotypic Conservation in Patients with X-Linked Retinitis Pigmentosa Caused by RPGR Mutations. JAMA Ophthalmol. 2013, 131, 1016–1025. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- García-Hoyos, M.; Garcia-Sandoval, B.; Cantalapiedra, D.; Riveiro, R.; Lorda-Sánchez, I.; Trujillo-Tiebas, M.J.; Rodriguez de Alba, M.; Millan, J.M.; Baiget, M.; Ramos, C.; et al. Mutational Screening of the RP2 and RPGR Genes in Spanish Families with X-Linked Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3777–3782. [Google Scholar] [CrossRef] [Green Version]
- Mawatari, G.; Fujinami, K.; Liu, X.; Yang, L.; Yokokawa, Y.-F.; Komori, S.; Ueno, S.; Terasaki, H.; Katagiri, S.; Hayashi, T.; et al. Clinical and Genetic Characteristics of 14 Patients from 13 Japanese Families with RPGR-Associated Retinal Disorder: Report of Eight Novel Variants. Hum. Genome. Var. 2019, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Demirci, F.Y.K.; Gupta, N.; Radak, A.L.; Rigatti, B.W.; Mah, T.S.; Milam, A.H.; Gorin, M.B. Histopathologic Study of X-Linked Cone-Rod Dystrophy (CORDX1) Caused by a Mutation in the RPGR Exon ORF15. Am. J. Ophthalmol. 2005, 139, 386–388. [Google Scholar] [CrossRef] [Green Version]
- Olm, M.A.K.; Marson, F.A.L.; Athanazio, R.A.; Nakagawa, N.K.; Macchione, M.; Loges, N.T.; Omran, H.; Rached, S.Z.; Bertuzzo, C.S.; Stelmach, R.; et al. Severe Pulmonary Disease in an Adult Primary Ciliary Dyskinesia Population in Brazil. Sci. Rep. 2019, 9, 8693. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Black, G.C.; Rice, J.M.; Hart-Holden, N.; Jones, A.; O’Grady, A.; Ramsden, S.; Wright, A.F. RPGR Mutation Analysis and Disease: An Update. Hum. Mutat. 2007, 28, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Bader, I.; Brandau, O.; Achatz, H.; Apfelstedt-Sylla, E.; Hergersberg, M.; Lorenz, B.; Wissinger, B.; Wittwer, B.; Rudolph, G.; Meindl, A.; et al. X-Linked Retinitis Pigmentosa: RPGR Mutations in Most Families with Definite X Linkage and Clustering of Mutations in a Short Sequence Stretch of Exon ORF15. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1458–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holladay, J.T. Proper Method for Calculating Average Visual Acuity. J. Refract. Surg. 1997, 13, 388–391. [Google Scholar] [CrossRef]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Müller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and Genetic Characteristics of 251 Consecutive Patients with Macular and Cone/Cone-Rod Dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive Cone and Cone-Rod Dystrophies: Clinical Features, Molecular Genetics and Prospects for Therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 Update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef]
- Sharon, D.; Bruns, G.A.; McGee, T.L.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. X-Linked Retinitis Pigmentosa: Mutation Spectrum of the RPGR and RP2 Genes and Correlation with Visual Function. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2712–2721. [Google Scholar]
- Ayyagari, R.; Demirci, F.Y.; Liu, J.; Bingham, E.L.; Stringham, H.; Kakuk, L.E.; Boehnke, M.; Gorin, M.B.; Richards, J.E.; Sieving, P.A. X-Linked Recessive Atrophic Macular Degeneration from RPGR Mutation. Genomics 2002, 80, 166–171. [Google Scholar] [CrossRef] [Green Version]
- Ebenezer, N.D.; Michaelides, M.; Jenkins, S.A.; Audo, I.; Webster, A.R.; Cheetham, M.E.; Stockman, A.; Maher, E.R.; Ainsworth, J.R.; Yates, J.R.; et al. Identification of Novel RPGR ORF15 Mutations in X-Linked Progressive Cone-Rod Dystrophy (XLCORD) Families. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1891–1898. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.P.-W.; Lamey, T.; McLaren, T.; Thompson, J.A.; Montgomery, H.; De Roach, J. Progress and Prospects of Next-Generation Sequencing Testing for Inherited Retinal Dystrophy. Expert. Rev. Mol. Diagn. 2015, 15, 1269–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.P.W.; Lamey, T.M.; Wang, N.K.; Duan, J.; Zhou, W.; McLaren, T.L.; Thompson, J.A.; Ruddle, J.; De Roach, J.N. Development of High-Throughput Clinical Testing of RPGR ORF15 Using a Large Inherited Retinal Dystrophy Cohort. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4434–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linari, M.; Ueffing, M.; Manson, F.; Wright, A.; Meitinger, T.; Becker, J. The Retinitis Pigmentosa GTPase Regulator, RPGR, Interacts with the Delta Subunit of Rod Cyclic GMP Phosphodiesterase. Proc. Natl. Acad. Sci. USA 1999, 96, 1315–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, M.; Khanna, H. Ciliary Transition Zone (TZ) Proteins RPGR and CEP290: Role in Photoreceptor Cilia and Degenerative Diseases. Expert. Opin. Ther. Targets 2012, 16, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Park, J.H.; Gumerson, J.; Wu, Z.; Swaroop, A.; Qian, H.; Roll-Mecak, A.; Li, T. Loss of RPGR Glutamylation Underlies the Pathogenic Mechanism of Retinal Dystrophy Caused by TTLL5 Mutations. Proc. Natl. Acad. Sci. USA 2016, 113, E2925–E2934. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.-H.; Pawlyk, B.S.; Adamian, M.; Li, T. Dominant, Gain-of-Function Mutant Produced by Truncation of RPGR. Investig. Ophthalmol. Vis. Sci. 2004, 45, 36–41. [Google Scholar] [CrossRef] [Green Version]
- Fahim, A.T.; Bowne, S.J.; Sullivan, L.S.; Webb, K.D.; Williams, J.T.; Wheaton, D.K.; Birch, D.G.; Daiger, S.P. Allelic Heterogeneity and Genetic Modifier Loci Contribute to Clinical Variation in Males with X-Linked Retinitis Pigmentosa Due to RPGR Mutations. PLoS ONE 2011, 6, e23021. [Google Scholar] [CrossRef] [Green Version]
- Körschen, H.G.; Illing, M.; Seifert, R.; Sesti, F.; Williams, A.; Gotzes, S.; Colville, C.; Müller, F.; Dosé, A.; Godde, M. A 240 KDa Protein Represents the Complete Beta Subunit of the Cyclic Nucleotide-Gated Channel from Rod Photoreceptor. Neuron 1995, 15, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Pearring, J.N.; Martínez-Márquez, J.; Willer, J.R.; Lieu, E.C.; Salinas, R.Y.; Arshavsky, V.Y. The GARP Domain of the Rod CNG Channel’s B1-Subunit Contains Distinct Sites for Outer Segment Targeting and Connecting to the Photoreceptor Disk Rim. J. Neurosci. 2021, 41, 3094–3104. [Google Scholar] [CrossRef]
- Nassisi, M.; Smirnov, V.M.; Solis Hernandez, C.; Mohand-Saïd, S.; Condroyer, C.; Antonio, A.; Kühlewein, L.; Kempf, M.; Kohl, S.; Wissinger, B.; et al. CNGB1-Related Rod-Cone Dystrophy: A Mutation Review and Update. Hum. Mutat. 2021, 42, 641–666. [Google Scholar] [CrossRef]
- Rinaldi, C.; Donato, L.; Alibrandi, S.; Scimone, C.; D’Angelo, R.; Sidoti, A. Oxidative Stress and the Neurovascular Unit. Life 2021, 11, 767. [Google Scholar] [CrossRef] [PubMed]
- Scimone, C.; Donato, L.; Alibrandi, S.; Vadalà, M.; Giglia, G.; Sidoti, A.; D’Angelo, R. N-Retinylidene-N-Retinylethanolamine Adduct induces Expression of Chronic Inflammation Cytokines in Retinal Pigment Epithelium Cells. Exp. Eye Res. 2021, 209, 108641. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Abdalla, E.M.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Nabil, K.M.; D’Angelo, R.; Sidoti, A. Impairments of Photoreceptor Outer Segments Renewal and Phototransduction Due to a Peripherin Rare Haplotype Variant: Insights from Molecular Modeling. Int. J. Mol. Sci. 2021, 22, 3484. [Google Scholar] [CrossRef] [PubMed]
- Khateb, S.; Mohand-Saïd, S.; Nassisi, M.; Bonnet, C.; Roux, A.-F.; Andrieu, C.; Antonio, A.; Condroyer, C.; Zeitz, C.; Devisme, C.; et al. Phenotypic Characteristics of Rod-Cone Dystrophy Associated with Myo7a Mutations in A Large French Cohort. Retina 2020, 40, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Khateb, S.; Nassisi, M.; Bujakowska, K.M.; Méjécase, C.; Condroyer, C.; Antonio, A.; Foussard, M.; Démontant, V.; Mohand-Saïd, S.; Sahel, J.-A.; et al. Longitudinal Clinical Follow-up and Genetic Spectrum of Patients With Rod-Cone Dystrophy Associated With Mutations in PDE6A and PDE6B. JAMA Ophthalmol. 2019, 137, 669–679. [Google Scholar] [CrossRef]
- Greenstein, V.C.; Duncker, T.; Holopigian, K.; Carr, R.E.; Greenberg, J.P.; Tsang, S.H.; Hood, D.C. Structural and Functional Changes Associated with Normal and Abnormal Fundus Autofluorescence in Patients with Retinitis Pigmentosa. Retina 2012, 32, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Packer, O.; Hendrickson, A.E.; Curcio, C.A. Photoreceptor Topography of the Retina in the Adult Pigtail Macaque (Macaca Nemestrina). J. Comp. Neurol. 1989, 288, 165–183. [Google Scholar] [CrossRef]
- Vrabec, F. The Temporal Raphe of the Human Retina. Am. J. Ophthalmol. 1966, 62, 926–938. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Herrmann, P.; MacLaren, R.E.; Bolz, H.J.; Charbel Issa, P. Peripapillary Sparing in Autosomal Recessive Bestrophinopathy. Ophthalmol. Retina 2020, 4, 523–529. [Google Scholar] [CrossRef]
- Burke, T.R.; Rhee, D.W.; Smith, R.T.; Tsang, S.H.; Allikmets, R.; Chang, S.; Lazow, M.A.; Hood, D.C.; Greenstein, V.C. Quantification of Peripapillary Sparing and Macular Involvement in Stargardt Disease (STGD1). Investig. Ophthalmol. Vis. Sci. 2011, 52, 8006–8015. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Lee, W.; Sengillo, J.D.; Allikmets, R.; Garg, K.; Tsang, S.H. Peripapillary Sparing in RDH12-Associated Leber Congenital Amaurosis. Ophthalmic. Genet. 2017, 38, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Nassisi, M.; Mohand-Saïd, S.; Andrieu, C.; Antonio, A.; Condroyer, C.; Méjécase, C.; Dhaenens, C.-M.; Sahel, J.-A.; Zeitz, C.; Audo, I. Peripapillary Sparing with Near Infrared Autofluorescence Correlates with Electroretinographic Findings in Patients With Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4951–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Sumaroka, A.; Schwartz, S.B.; Roman, M.I.; Milam, A.H.; Bennett, J.; Stone, E.M.; Jacobson, S.G. ABCA4-Associated Retinal Degenerations Spare Structure and Function of the Human Parapapillary Retina. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4739–4746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neidhardt, J.; Glaus, E.; Lorenz, B.; Netzer, C.; Li, Y.; Schambeck, M.; Wittmer, M.; Feil, S.; Kirschner-Schwabe, R.; Rosenberg, T.; et al. Identification of Novel Mutations in X-Linked Retinitis Pigmentosa Families and Implications for Diagnostic Testing. Mol. Vis. 2008, 14, 1081–1093. [Google Scholar] [PubMed]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next Generation in Gene Variant Databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Casper, J.; Zweig, A.S.; Villarreal, C.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Karolchik, D.; et al. The UCSC Genome Browser Database: 2018 Update. Nucleic Acids Res. 2018, 46, D762–D769. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J. BLAT—The BLAST-like Alignment Tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for Full-Field Clinical Electroretinography (2015 Update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sex | cDNA | Protein Change | Reference | |
|---|---|---|---|---|---|
| CIC03862 | M | Index | c.2931_2932ins29 | p.(Glu978Lysfs*121) | This study |
| 782819 | M | Index | c.3276del | p.(Gly1093Aspfs*3) | This study |
| CIC09352 | M | Index | c.3261del | p.(Val1088*) | This study |
| CIC04404 | M | Index | c.3178_3179del | p.(Glu1060Argfs*18) | García-Hoyos, 2006 [18] |
| CIC02893 | M | Index | c.3399del | p.(Pro1134Hisfs*18) | Mawatary, 2019 [19] |
| CIC03560 | M | Index | c.3248_3252del | p.(Glu1083Valfs*17) | This study |
| CIC06538 | M | Index | c.3317dup | p.(Ser1107Valfs*4) | Demirci, 2005 [20] |
| CIC02863 | M | Index | c.2719G>T | p.(Glu907*) | This study |
| CIC07494 | M | Index | c.3039_3040del | p.(Glu1014Glyfs*64) | Zahid, 2013 [16] |
| CIC06631 | M | Index | c.3119_3120del | p.(Glu1040Glyfs*38) | This study |
| c.3074_3085del | p.(Val1025_Glu1028del) | Olm, 2019 [21] | |||
| CIC04447 | M | Index | c.3134_3137del | p.(Glu1045Glyfs*43) | This study |
| CIC04647 | M | Index | c.2872G>T | p.(Glu958*) | Shu, 2007 [22] |
| CIC09451 | M | Index | c.3178_3179del | p.(Glu1060Argfs*18) | García-Hoyos, 2006 [18] |
| CIC07574 | M | Index | c.3039_3040del | p.(Glu1014Glyfs*64) | Zahid, 2013 [16] |
| CIC07658 | M | Index | c.2985_2993delinsAGAAGGGG | p.(Glu997Glyfs*92) | This study |
| CIC07798 | M | Index | c.3146_3147del | p.(Glu1049Glyfs*29) | This study |
| CIC08152 | M | Index | c.3334C>T | p.(Gln1112*) | This study |
| CIC08901 | M | Index | c.3212dup | p.(Thr1072Aspfs*7) | This study |
| CIC09159 | M | Affected brother | c.3212dup | p.(Thr1072Aspfs*7) | This study |
| CIC08918 | M | Index | c.2678G>T | p.(Gly893Val) | This study |
| CIC09067 | M | Index | c.3178_3179del | p.(Glu1060Argfs*18) | García-Hoyos, 2006 [18] |
| CIC09151 | M | Affected brother | c.3178_3179del | p.(Glu1060Argfs*18) | García-Hoyos, 2006 [18] |
| CIC8950 | M | Index | c.3104_3105del | p.(Glu1035Glyfs*43) | Mawatary, 2019 [19] |
| CIC01063 | M | Index | c.3074_3085del | p.(Val1025_Glu1028del) | Olm, 2019 [21] |
| c.3092del | p.(Glu1031Glyfs*58) | Bader, 2003 [23] | |||
| CIC10466 | M | Index | c.3395dup | p.(Asn1132Lysfs*12) | This study |
| CIC09949 | M | Index | c.2966del | p.(Glu989Glyfs*100) | This study |
| CIC10733 | M | Index | c.3134_3137del | p.(Glu1045Glyfs*43) | This study |
| CIC08066 | M | Index | c.3178_3179del | p.(Glu1060Argfs*18) | García-Hoyos, 2006 [18] |
| CIC04835 | M | Index | c.2956G>T | p.(Gly986*) | This study |
| CIC03515 | M | Index | c.3178_3179del | p.(Glu1060Argfs*18) | García-Hoyos, 2006 [18] |
| CIC07400 | M | Index | c.3077_3081del | p.(Glu1026Glyfs*51) | This study |
| CIC09653 | M | Index | c.2236_2237del | p.(Glu746Argfs*23) | Vervoort, 2000 [8] |
| CIC04850 | M | Index | c.3071_3080del | p.(Glu1024Glyfs*62) | This study |
| CIC01503 | M | Index | c.2820_2840dup | p.(Asp943_Glu949dup) | Vervoort, 2000 [8] |
| c.3134_3138del | p.(Glu1045Glyfs*32) | This study | |||
| 1659193 | M | Index | c.2731G>T | p.(Glu911*) | This study |
| CIC01418 | M | Index | c.3146_3149del | p.(Glu1049Glyfs*39) | This study |
| Genomic Start Position (hg19) | cDNA RPGRORF15: NM_001034853.2 | Protein Change | ACMG Classification (Criteria) |
|---|---|---|---|
| chrX-38145574 | c.2678G>T | p.(Gly893Val) | Uncertain significance (PM2, PP3, BP1) |
| chrX-38145533 | c.2719G>T | p.(Glu907*) | Likely Pathogenic (PVS1, PM2) |
| chrX-38145521 | c.2731del | p.(Glu911Argfs*178) | Pathogenic (PVS1, PM2, PP1) |
| chrX-38145321 | c.2931_2932ins AAGGAAAAGGGGAGAAGGGGAAGGGGAGGAAGGA | p.(Glu978Lysfs*121) | Likely Pathogenic (PVS1, PM2) |
| chrX-38145296 | c.2956G>T | p.(Gly986*) | Likely Pathogenic (PVS1, PM2) |
| chrX-38145286 | c.2966del | p.(Glu989Glyfs*100) | Likely Pathogenic (PVS1, PM2) |
| chrX-38145259 | c.2985_2993delinsAGAAGGGG | p.(Glu997Glyfs*92) | Likely Pathogenic (PVS1, PM2) |
| chrX-38145172 | c.3071_3080del | p.(Glu1024Glyfs*62) | Likely Pathogenic (PVS1, PM2) |
| chrX-38145171 | c.3077_3081del | p.(Glu1026Glyfs*51) | Likely Pathogenic (PVS1, PM2) |
| chrX-38145132 | c.3119_3120del | p.(Glu1040Glyfs*38) | Likely Pathogenic (PVS1, PM2) |
| chrX-38145115 | c.3134_3137del | p.(Glu1045Glyfs*43) | Likely Pathogenic (PVS1, PM2) |
| chrX-38145115 | c.3134_3138del | p.(Glu1045Glyfs*32) | Likely Pathogenic (PVS1, PM2) |
| chrX-38145105 | c.3146_3147del | p.(Glu1049Glyfs*29) | Likely Pathogenic (PVS1, PM2) |
| chrX-38145105 | c.3146_3149del | p.(Glu1049Glyfs*39) | Likely Pathogenic (PVS1, PM2) |
| chrX-38145040 | c.3212dup | p.(Thr1072Aspfs*7) | Pathogenic (PVS1, PM2, PP1) |
| chrX-38145000 | c.3248_3252del | p.(Glu1083Valfs*17) | Likely Pathogenic (PVS1, PM2) |
| chrX-38144991 | c.3261del | p.(Val1088*) | Likely Pathogenic (PVS1, PM2) |
| chrX-38144976 | c.3276del | p.(Gly1093Aspfs*3) | Likely Pathogenic (PVS1, PM2) |
| chrX-38144918 | c.3334C>T | p.(Gln1112*) | Likely Pathogenic (PVS1, PM2) |
| chrX-38144857 | c.3395dup | p.(Asn1132Lysfs*12) | Likely Pathogenic (PVS1, PM2) |
| RPGRORF15 | |
|---|---|
| Male, No./Total No. (%) | 36/36 (100) |
| Age of Onset | n = 18 |
| 1st decade | 4 |
| 2nd decade | 9 |
| 3rd decade | 4 |
| ≥4th decade | 1 |
| High Myopia, No./Total No. (%) | 14/33 (42.42) |
| Cataract and/or previous cataract surgery in at least one eye, No./Total No. (%) | 4/33 (12.12) |
| ǂ CD/CRD, No/No | 15/21 |
| Age at last visit, mean ± SD, y | n = 36; 43.97 ± 11.24 |
| Light perception or no light perception in at least one eye, No./total No. (%) | 1/35 (2.86) |
| § BCVA at last visit, LogMar, Mean ± SD [Snellen Equivalent] | n = 35; 0.97 ± 0.71 [20/200] |
| Follow-up BCVA, No | n = 35 [range: 0–19 y] |
| 0 y | 11 |
| 1–5 y | 14 |
| 6–10 y | 9 |
| >10 y | 1 |
| Estimation of BCVA decline | n = 24 |
| Annual rate, LogMAR/year ± SD | 0.04 ± 0.06 |
| Mean regression slope ± SD | 0.04 ± 0.08 |
| Mean regression intercept ± SD | −0.90 ± 3.34 |
| Binocular normal color vision, No./Total No. (%) | 1/32 (3.12) |
| Bilateral undetectable ff-ERG, No./Total No. (%) | 5/36 (13.89) |
| SW-FAF Phenotype type | n = 35 |
| Group 1 | 6 |
| Group 2 | 21 |
| Group 3 | 8 |
| Peripapillary sparing, No./Total No. (%) | 10/35 (28.57) |
| Estimation of central hyperAF ring enlargement | n = 19 |
| Horizontal diameter, µm/y | 42.88 ± 48.98 |
| Slope | 43.16 ± 47.81 |
| Intercept | 228.60 ± 1689.24 |
| Vertical diameter, µm/y | 39.44 ± 40.90 |
| Slope | 39.36 ± 40.57 |
| Intercept | −24.16 ± 1658.86 |
| CRT, µm, mean ± SD | n = 35; 148.97 ± 27.17 |
| Unilateral or bilateral ERM, No./Total No. (%) | 2/35 (2.78) |
| Unilateral or bilateral iHRF, No./Total No. (%) | 15/35 (42.86) |
| Univariate | Multivariate | |||
|---|---|---|---|---|
| β Coefficient | p Value | β Coefficient | p Value | |
| Age | 0.306 | 0.078 | - | - |
| Decade of onset | −0.241 | 0.315 | - | - |
| High Myopia | −0.094 | 0.609 | - | - |
| ff-ERG | 0.140 | 0.438 | - | - |
| iHRF | −0.266 | 0.135 | - | - |
| SW-FAF phenotype | 0.353 | 0.044 | 0.092 | 0.573 |
| Peripapillary sparing | −0.376 | 0.031 | −0.385 | 0.019 |
| CRT | −0.523 | 0.003 | −0.456 | 0.006 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nassisi, M.; De Bartolo, G.; Mohand-Said, S.; Condroyer, C.; Antonio, A.; Lancelot, M.-E.; Bujakowska, K.; Smirnov, V.; Pugliese, T.; Neidhardt, J.; et al. Retrospective Natural History Study of RPGR-Related Cone- and Cone-Rod Dystrophies While Expanding the Mutation Spectrum of the Disease. Int. J. Mol. Sci. 2022, 23, 7189. https://doi.org/10.3390/ijms23137189
Nassisi M, De Bartolo G, Mohand-Said S, Condroyer C, Antonio A, Lancelot M-E, Bujakowska K, Smirnov V, Pugliese T, Neidhardt J, et al. Retrospective Natural History Study of RPGR-Related Cone- and Cone-Rod Dystrophies While Expanding the Mutation Spectrum of the Disease. International Journal of Molecular Sciences. 2022; 23(13):7189. https://doi.org/10.3390/ijms23137189
Chicago/Turabian StyleNassisi, Marco, Giuseppe De Bartolo, Saddek Mohand-Said, Christel Condroyer, Aline Antonio, Marie-Elise Lancelot, Kinga Bujakowska, Vasily Smirnov, Thomas Pugliese, John Neidhardt, and et al. 2022. "Retrospective Natural History Study of RPGR-Related Cone- and Cone-Rod Dystrophies While Expanding the Mutation Spectrum of the Disease" International Journal of Molecular Sciences 23, no. 13: 7189. https://doi.org/10.3390/ijms23137189