
New Developments and Possibilities in Reanalysis and Reinterpretation of Whole Exome Sequencing Datasets for Unsolved Rare Diseases Using Machine Learning Approaches


{kind=link}
Abstract
:1. Introduction
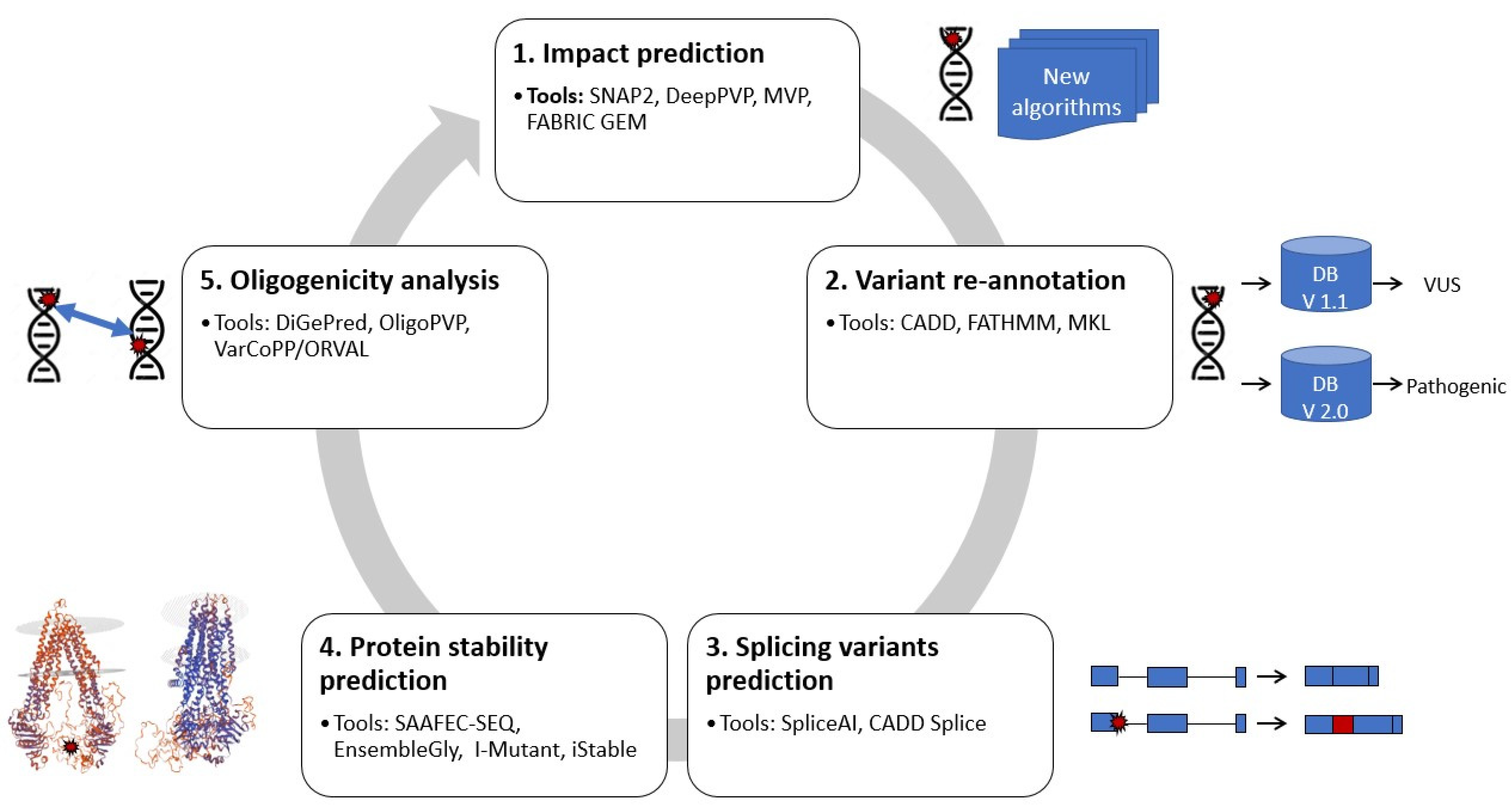
2. Reanalysis Methodologies Using Machine Learning
2.1. Predicting the Impact of Sequence Alterations/Mutations
2.2. Variant Re-Annotation
2.3. Predicting Splicing Variants
2.4. Predicting Protein Stability
2.5. Oligogenicity Analysis
3. Emerging Technologies and Methodologies for Reanalyzing Rare Diseases
3.1. Whole Genome Sequencing and New Sequencing Technologies for Rare Diseases Diagnostics
3.2. Structural Variants Analysis
3.3. Multi-Omics Analysis and Integration
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- About Cord|Canadian Organization for Rare Disorders. Available online: https://www.raredisorders.ca/about-cord/ (accessed on 17 June 2022).
- Groft, S.C.; Posada, M.; Taruscio, D. Progress, challenges and global approaches to rare diseases. Acta Paediatr. 2021, 110, 2711–2716. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Hartley, T.; Dyment, D.A.; Beaulieu, C.L.; Schwartzentruber, J.; Smith, A.; Bedford, H.M.; Bernard, G.; Bernier, F.P.; Brais, B.; et al. Boycott, FORGE Canada Consortium, and Care4Rare Canada Consortium. Utility of Whole-Exome Sequencing for Those near the End of the Diagnostic Odyssey: Time to Address Gaps in Care. Clin. Genet. 2016, 89, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. Omim.Org: Online Mendelian Inheritance in Man (Omim®), an Online Catalog of Human Genes and Genetic Disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posey, J.E. Genome Sequencing and Implications for Rare Disorders. Orphanet. J. Rare Dis. 2019, 14, 153. [Google Scholar] [CrossRef] [Green Version]
- Smedley, D.; Smith, K.R.; Martin, A.; Thomas, E.A.; McDonagh, E.M.; Cipriani, V.; Ellingford, J.M.; Arno, G.; Tucci, A.; Vandrovcova, J.; et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care—Preliminary Report. N. Engl. J. Med. 2021, 385, 1868–1880. [Google Scholar]
- McInerney-Leo, A.M.; Duncan, E.L. Massively Parallel Sequencing for Rare Genetic Disorders: Potential and Pitfalls. Front. Endocrinol. 2021, 11, 628946. [Google Scholar] [CrossRef]
- Poon, K.-S.; Tan, K.M.-L. Reclassification of Whole Exome Sequencing-derived Genetic Variants in Pendred Syndrome with ACMG/AMP Standards. Glob. Med Genet. 2021, 8, 129–131. [Google Scholar] [CrossRef]
- De La Vega, F.M.; Chowdhury, S.; Moore, B.; Frise, E.; McCarthy, J.; Hernandez, E.J.; Wong, T.; James, K.; Guidugli, L.; Agrawal, P.B.; et al. Artificial Intelligence Enables Comprehensive Genome Interpretation and Nomination of Candidate Diagnoses for Rare Genetic Diseases. Genome. Med. 2021, 13, 153. [Google Scholar] [CrossRef]
- Matalonga, L.; Hernández-Ferrer, C.; Piscia, D.; Schüle, R.; Synofzik, M.; Töpf, A.; Vissers, L.E.L.M.; de Voer, R.; Tonda, R.; Laurie, S.; et al. Solving Patients with Rare Diseases through Programmatic Reanalysis of Genome-Phenome Data. Eur. J. Hum. Genet. 2021, 29, 1337–1347. [Google Scholar] [CrossRef]
- Salfati, E.L.; Spencer, E.; Topol, S.E.; Muse, E.D.; Rueda, M.; Lucas, J.R.; Wagner, G.N.; Campman, S.; Topol, E.J.; Torkamani, A. Re-analysis of whole-exome sequencing data uncovers novel diagnostic variants and improves molecular diagnostic yields for sudden death and idiopathic diseases. Genome Med. 2019, 11, 83. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using Polyphen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. Cadd: Predicting the Deleteriousness of Variants Throughout the Human Genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Nicora, G.; Zucca, S.; Limongelli, I.; Bellazzi, R.; Magni, P. A Machine Learning Approach Based on Acmg/Amp Guidelines for Genomic Variant Classification and Prioritization. Sci. Rep. 2022, 12, 2517. [Google Scholar] [CrossRef]
- Hoffman-Andrews, L. The Known Unknown: The Challenges of Genetic Variants of Uncertain Significance in Clinical Practice. J. Law Biosci. 2018, 4, 648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anna, A.; Monika, G. Splicing Mutations in Human Genetic Disorders: Examples, Detection, and Confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, H.J. Mutation as a Cause of Genetic Disease. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1988, 319, 1194. [Google Scholar]
- de Ligt, J.; Veltman, J.A.; Vissers, L.E. Point Mutations as a Source of De Novo Genetic Disease. Curr. Opin. Genet. Dev. 2013, 23, 257–263. [Google Scholar] [CrossRef]
- Rahit, K.M.; Tarailo-Graovac, M. Genetic Modifiers and Rare Mendelian Disease. Genes 2020, 11, 329. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, J.; Lehne, M.; Schepers, J.; Prasser, F.; Thun, S. The Use of Machine Learning in Rare Diseases: A Scoping Review. Orphanet J. Rare Dis. 2020, 15, 145. [Google Scholar] [CrossRef]
- Sánchez Fernández, I.; Yang, E.; Calvachi, P.; Amengual-Gual, M.; Wu, J.Y.; Krueger, D.; Northrup, H.; Bebin, M.E.; Sahin, M.; Yu, K.H.; et al. Deep Learning in Rare Disease. Detection of Tubers in Tuberous Sclerosis Complex. PLoS ONE 2020, 15, e0232376. [Google Scholar] [CrossRef]
- Ai Driving Breakthroughs on Rare Diseases. Available online: https://nationalpress.org/topic/ai-driving-breakthroughs-on-rare-diseases/ (accessed on 17 June 2022).
- Decherchi, S.; Pedrini, E.; Mordenti, M.; Cavalli, A.; Sangiorgi, L. Opportunities and Challenges for Machine Learning in Rare Diseases. Front. Med. 2021, 8, 747612. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marmiesse, A.; Gouveia, S.; Couce, M.L. Ngs Technologies as a Turning Point in Rare Disease Research, Diagnosis and Treatment. Curr. Med. Chem. 2018, 25, 404–432. [Google Scholar] [CrossRef]
- Ensemble Methods: Bagging, Boosting and Stacking. Available online: https://towardsdatascience.com/ensemble-methods-bagging-boosting-and-stacking-c9214a10a205 (accessed on 17 June 2022).
- Support Vector Machines: A Simple Explanation—Kdnuggets. Available online: https://www.kdnuggets.com/2016/07/support-vector-machines-simple-explanation.html (accessed on 17 June 2022).
- What Are Neural Networks? Available online: https://www.ibm.com/cloud/learn/neural-networks (accessed on 17 June 2022).
- Available online: https://Www.Pharmasug.Org/Proceedings/2019/St/Pharmasug-2019-St-325.Pdf (accessed on 17 June 2022).
- Mitani, A.A.; Haneuse, S. Small Data Challenges of Studying Rare Diseases. JAMA Netw. Open 2020, 3, e201965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Three Rare Disease Diagnostic Opportunities for Ai and Machine Learning. Available online: https://insights.axtria.com/blog/three-rare-disease-diagnoses-opportunities-for-ai/ml-artificial-intelligence-and-machine-learning (accessed on 17 June 2022).
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. Revel: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunning, A.C.; Fryer, V.; Fasham, J.; Crosby, A.H.; Ellard, S.; Baple, E.L.; Wright, C.F. Assessing Performance of Pathogenicity Predictors Using Clinically Relevant Variant Datasets. J. Med. Genet. 2021, 58, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Munshani, S.; Ibrahim, E.Y.; Domenicano, I.; Ehrlich, B.E. The Impact of Mutations in Wolframin on Psychiatric Disorders. Front. Pediatrics 2021, 9, 718132. [Google Scholar] [CrossRef] [PubMed]
- Boudellioua, I.; Kulmanov, M.; Schofield, P.N.; Gkoutos, G.V.; Hoehndorf, R. Oligopvp: Phenotype-Driven Analysis of Individual Genomic Information to Prioritize Oligogenic Disease Variants. Sci. Rep. 2018, 8, 14681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, A.; Vg, S.; Joseph, T.; Kotte, S.; Sivadasan, N.; Srinivasan, R. Phenotype-Driven Gene Prioritization for Rare Diseases Using Graph Convolution on Heterogeneous Networks. BMC Med. Genom. 2018, 11, 57. [Google Scholar] [CrossRef]
- Díaz-Santiago, E.; Jabato, F.M.; Rojano, E.; Seoane, P.; Pazos, F.; Perkins, J.R.; Ranea, J.A.G. Phenotype-Genotype Comorbidity Analysis of Patients with Rare Disorders Provides Insight into Their Pathological and Molecular Bases. PLoS Genet. 2020, 16, e1009054. [Google Scholar] [CrossRef]
- Jia, J.; Wang, R.; An, Z.; Guo, Y.; Ni, X.; Shi, T. Rdad: A Machine Learning System to Support Phenotype-Based Rare Disease Diagnosis. Front. Genet. 2018, 9, 587. [Google Scholar] [CrossRef]
- Qi, H.; Zhang, H.; Zhao, Y.; Chen, C.; Long, J.J.; Chung, W.K.; Guan, Y.; Shen, Y. Mvp Predicts the Pathogenicity of Missense Variants by Deep Learning. Nat. Commun. 2021, 12, 510. [Google Scholar] [CrossRef] [PubMed]
- Yandell, M.; Huff, C.; Hu, H.; Singleton, M.; Moore, B.; Xing, J.; Jorde, L.B.; Reese, M.G. A Probabilistic Disease-Gene Finder for Personal Genomes. Genome Res. 2011, 21, 1529–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, M.V.; Guthery, S.L.; Voelkerding, K.V.; Chen, K.; Kennedy, B.; Margraf, R.L.; Durtschi, J.; Eilbeck, K.; Reese, M.G.; Jorde, L.B.; et al. Phevor Combines Multiple Biomedical Ontologies for Accurate Identification of Disease-Causing Alleles in Single Individuals and Small Nuclear Families. Am. J. Hum. Genet. 2014, 94, 599–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, P.N.; Köhler, S.; Oellrich, A.; Wang, K.; Mungall, C.J.; Lewis, S.E.; Washington, N.; Bauer, S.; Seelow, D.; Krawitz, P.; et al. Improved Exome Prioritization of Disease Genes through Cross-Species Phenotype Comparison. Genome Res. 2014, 24, 340–348. [Google Scholar] [CrossRef] [Green Version]
- Available online: Https://Fabricgenomics.Com/Wp-Content/Uploads/2021/09/202011-Fabric-Gem-Data-Sheet-Final.Pdf (accessed on 17 June 2022).
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Hoskinson, D.C.; Dubuc, A.M.; Mason-Suares, H. The Current State of Clinical Interpretation of Sequence Variants. Curr. Opin. Genet. Dev. 2017, 42, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Federici, G.; Soddu, S. Variants of Uncertain Significance in the Era of High-Throughput Genome Sequencing: A Lesson from Breast and Ovary Cancers. J. Exp. Clin. Cancer Res. 2020, 39, 46. [Google Scholar] [CrossRef] [Green Version]
- Schubach, M.; Re, M.; Robinson, P.N.; Valentini, G. Imbalance-Aware Machine Learning for Predicting Rare and Common Disease-Associated Non-Coding Variants. Sci. Rep. 2017, 7, 2959. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Zaucha, J.; Heinzinger, M.; Tarnovskaya, S.; Rost, B.; Frishman, D. Family-Specific Analysis of Variant Pathogenicity Prediction Tools. NAR Genom. Bioinform. 2020, 2, lqaa014. [Google Scholar] [CrossRef] [Green Version]
- Iancu, I.F.; Avila-Fernandez, A.; Arteche, A.; Trujillo-Tiebas, M.J.; Riveiro-Alvarez, R.; Almoguera, B.; Martin-Merida, I.; Del Pozo-Valero, M.; Perea-Romero, I.; Ayuso, C. Prioritizing Variants of Uncertain Significance for Reclassification Using a Rule-Based Algorithm in Inherited Retinal Dystrophies. NPJ Genom. Med. 2021, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jhong, J.H.; Lee, J.; Koo, J.Y. Meta-Analytic Support Vector Machine for Integrating Multiple Omics Data. BioData Min. 2017, 10, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Z.; Bromberg, Y. Predicting Functional Effects of Synonymous Variants: A Systematic Review and Perspectives. Front. Genet. 2019, 10, 914. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [Green Version]
- Lord, J.; Baralle, D. Splicing in the Diagnosis of Rare Disease: Advances and Challenges. Front. Genet. 2021, 12, 1146. [Google Scholar] [CrossRef]
- Cheng, J.; Nguyen, T.Y.D.; Cygan, K.J.; Çelik, M.H.; Fairbrother, W.G.; Avsec, Ž.; Gagneur, J. Mmsplice: Modular Modeling Improves the Predictions of Genetic Variant Effects on Splicing. Genome Biol. 2019, 20, 48. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. Cadd-Splice-Improving Genome-Wide Variant Effect Prediction Using Deep Learning-Derived Splice Scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Darling, A.L.; Uversky, V.N. Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front. Genet. 2018, 9, 158. [Google Scholar] [CrossRef]
- Brooks, P.J.; Tagle, D.A.; Groft, S. Expanding Rare Disease Drug Trials Based on Shared Molecular Etiology. Nat. Biotechnol. 2014, 32, 515–518. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Panday, S.K.; Alexov, E. Saafec-Seq: A Sequence-Based Method for Predicting the Effect of Single Point Mutations on Protein Thermodynamic Stability. Int. J. Mol. Sci. 2021, 22, 606. [Google Scholar] [CrossRef]
- Caragea, C.; Sinapov, J.; Silvescu, A.; Dobbs, D.; Honavar, V. Glycosylation Site Prediction Using Ensembles of Support Vector Machine Classifiers. BMC Bioinform. 2007, 8, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting Stability Changes Upon Mutation from the Protein Sequence or Structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.W.; Lin, J.; Chu, Y.W. Istable: Off-the-Shelf Predictor Integration for Predicting Protein Stability Changes. BMC Bioinform. 2013, 14 (Suppl. 2), S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, C.; Timson, D.J. In Silico Prediction of the Effects of Mutations in the Human Mevalonate Kinase Gene: Towards a Predictive Framework for Mevalonate Kinase Deficiency. Ann. Hum. Genet. 2015, 79, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Brasil, S.; Pascoal, C.; Francisco, R.; Dos Reis Ferreira, V.; Videira, P.A.; Valadão, A.G. Artificial Intelligence (Ai) in Rare Diseases: Is the Future Brighter? Genes 2019, 10, 978. [Google Scholar] [CrossRef] [Green Version]
- Kousi, M.; Katsanis, N. Genetic Modifiers and Oligogenic Inheritance. Cold Spring Harb. Perspect. Med. 2015, 5, a017145. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Cogan, J.D.; Newman, J.H.; Phillips, J.A.; Hamid, R.; Meiler, J.; Capra, J.A. Identifying Digenic Disease Genes Via Machine Learning in the Undiagnosed Diseases Network. Am. J. Hum. Genet. 2021, 108, 1946–1963. [Google Scholar] [CrossRef]
- Gazzo, A.M.; Daneels, D.; Cilia, E.; Bonduelle, M.; Abramowicz, M.; Van Dooren, S.; Smits, G.; Lenaerts, T. Dida: A Curated and Annotated Digenic Diseases Database. Nucleic Acids Res. 2016, 44, D900–D907. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, S.; Gazzo, A.; Versbraegen, N.; Nachtegael, C.; Aerts, J.; Moreau, Y.; Van Dooren, S.; Nowé, A.; Smits, G.; Lenaerts, T. Predicting Disease-Causing Variant Combinations. Proc. Natl. Acad. Sci. USA 2019, 116, 11878–11887. [Google Scholar] [CrossRef] [Green Version]
- Dallali, H.; Kheriji, N.; Kammoun, W.; Mrad, M.; Soltani, M.; Trabelsi, H.; Hamdi, W.; Bahlous, A.; Ben Ahmed, M.; Mahjoub, F.; et al. Multiallelic Rare Variants in Bbs Genes Support an Oligogenic Ciliopathy in a Non-Obese Juvenile-Onset Syndromic Diabetic Patient: A Case Report. Front. Genet. 2021, 12, 664963. [Google Scholar] [CrossRef]
- 100,000 Genomes Project 2021 Update: Rare Disease—Genomics Education Programme. Available online: https://www.genomicseducation.hee.nhs.uk/blog/100000-genomes-project-2021-update-rare-disease/ (accessed on 17 June 2022).
- Khost, D.E.; Eickbush, D.G.; Larracuente, A.M. Single-Molecule Sequencing Resolves the Detailed Structure of Complex Satellite DNA Loci in Drosophila Melanogaster. Genome Res. 2017, 27, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameur, A.; Kloosterman, W.P.; Hestand, M.S. Single-Molecule Sequencing: Towards Clinical Applications. Trends Biotechnol. 2019, 37, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Sedlazeck, F.J.; Lam, T.W.; Schatz, M.C. A Multi-Task Convolutional Deep Neural Network for Variant Calling in Single Molecule Sequencing. Nat. Commun. 2019, 10, 998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Q.; Wang, Y.; Guan, J.; Ji, G. Sciae: An Integrative Autoencoder-Based Ensemble Classification Framework for Single-Cell Rna-Seq Data. Brief. Bioinform. 2022, 23, bbab508. [Google Scholar] [CrossRef]
- Li, H.; Brouwer, C.R.; Luo, W. A Universal Deep Neural Network for in-Depth Cleaning of Single-Cell Rna-Seq Data. Nat. Commun. 2022, 13, 1–11. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H. Non-Linear Archetypal Analysis of Single-Cell Rna-Seq Data by Deep Autoencoders. PLoS Comput. Biol. 2022, 18, e1010025. [Google Scholar] [CrossRef]
- Pratella, D.; Ait-El-Mkadem Saadi, S.; Bannwarth, S.; Paquis-Fluckinger, V.; Bottini, S. A Survey of Autoencoder Algorithms to Pave the Diagnosis of Rare Diseases. Int. J. Mol. Sci. 2021, 22, 10891. [Google Scholar] [CrossRef]
- Ergin, S.; Kherad, N.; Alagoz, M. RNA sequencing and its applications in cancer and rare diseases. Mol. Biol. Rep. 2022, 49, 2325–2333. [Google Scholar] [CrossRef]
- Komlósi, K.; Gyenesei, A.; Bene, J. Editorial: Copy Number Variation in Rare Disorders. Front. Genet. 2022, 13, 898059. [Google Scholar] [CrossRef]
- Requena, F.; Abdallah, H.H.; García, A.; Nitschké, P.; Romana, S.; Malan, V.; Rausell, A. Cnvxplorer: A Web Tool to Assist Clinical Interpretation of Cnvs in Rare Disease Patients. Nucleic Acids Res. 2021, 49, W93–W103. [Google Scholar] [CrossRef]
- Gabrielaite, M.; Torp, M.H.; Rasmussen, M.S.; Andreu-Sánchez, S.; Vieira, F.G.; Pedersen, C.B.; Kinalis, S.; Madsen, M.B.; Kodama, M.; Demircan, G.S.; et al. A Comparison of Tools for Copy-Number Variation Detection in Germline Whole Exome and Whole Genome Sequencing Data. Cancers 2021, 13, 6283. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; Glessner, J.T.; Coe, B.P.; Li, J.; Mohebnasab, M.; Chang, X.; Connolly, J.; Kao, C.; Wei, Z.; Bradfield, J.; et al. Rare Copy Number Variants in over 100,000 European Ancestry Subjects Reveal Multiple Disease Associations. Nat. Commun. 2020, 11, 255. [Google Scholar] [CrossRef] [PubMed]
- Sharo, A.G.; Hu, Z.; Sunyaev, S.R.; Brenner, S.E. Strvctvre: A Supervised Learning Method to Predict the Pathogenicity of Human Genome Structural Variants. Am. J. Hum. Genet. 2022, 109, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Barseghyan, H.; Délot, E.C.; Vilain, E. Nanotator: A Tool for Enhanced Annotation of Genomic Structural Variants. BMC Genom. 2021, 22, 10. [Google Scholar] [CrossRef]
- Zhang, L.; Shi, J.; Ouyang, J.; Zhang, R.; Tao, Y.; Yuan, D.; Lv, C.; Wang, R.; Ning, B.; Roberts, R.; et al. X-Cnv: Genome-Wide Prediction of the Pathogenicity of Copy Number Variations. Genome Med. 2021, 13, 132. [Google Scholar] [CrossRef]
- Schlieben, L.D.; Prokisch, H.; Yépez, V.A. How Machine Learning and Statistical Models Advance Molecular Diagnostics of Rare Disorders Via Analysis of Rna Sequencing Data. Front. Mol. Biosci. 2021, 8, 647277. [Google Scholar] [CrossRef]
- Taroni, J.N.; Grayson, P.C.; Hu, Q.; Eddy, S.; Kretzler, M.; Merkel, P.A.; Greene, C.S. Multiplier: A Transfer Learning Framework for Transcriptomics Reveals Systemic Features of Rare Disease. Cell Syst. 2019, 8, 380–394. [Google Scholar] [CrossRef]
- Kerr, K.; McAneney, H.; Smyth, L.J.; Bailie, C.; McKee, S.; McKnight, A.J. A Scoping Review and Proposed Workflow for Multi-Omic Rare Disease Research. Orphanet J. Rare Dis. 2020, 15, 107. [Google Scholar] [CrossRef]
- Labory, J.; Fierville, M.; Ait-El-Mkadem, S.; Bannwarth, S.; Paquis-Flucklinger, V.; Bottini, S. Multi-Omics Approaches to Improve Mitochondrial Disease Diagnosis: Challenges, Advances, and Perspectives. Front. Mol. Biosci. 2020, 7, 327. [Google Scholar] [CrossRef]
- Liu, X.; Yang, Z.; Lin, H.; Simmons, M.; Lu, Z. Dignifi: Discovering Causative Genes for Orphan Diseases Using Protein-Protein Interaction Networks. BMC Syst. Biol. 2017, 11 (Suppl. 3), 23. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Kushwaha, A.; Berman, K.; Jegga, A.G. A Vertex Similarity-Based Framework to Discover and Rank Orphan Disease-Related Genes. BMC Syst. Biol. 2012, 6 (Suppl. 3), S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodra, Y.; Weinbach, J.; Posada-de-la-Paz, M.; Coi, A.; Lemonnier, S.L.; van Enckevort, D.; Roos, M.; Jacobsen, A.; Cornet, R.; Ahmed, S.F.; et al. Recommendations for Improving the Quality of Rare Disease Registries. Int. J. Environ. Res. Public Health 2018, 15, 1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Setty, S.T.; Scott-Boyer, M.-P.; Cuppens, T.; Droit, A. New Developments and Possibilities in Reanalysis and Reinterpretation of Whole Exome Sequencing Datasets for Unsolved Rare Diseases Using Machine Learning Approaches. Int. J. Mol. Sci. 2022, 23, 6792. https://doi.org/10.3390/ijms23126792
Setty ST, Scott-Boyer M-P, Cuppens T, Droit A. New Developments and Possibilities in Reanalysis and Reinterpretation of Whole Exome Sequencing Datasets for Unsolved Rare Diseases Using Machine Learning Approaches. International Journal of Molecular Sciences. 2022; 23(12):6792. https://doi.org/10.3390/ijms23126792
Chicago/Turabian StyleSetty, Samarth Thonta, Marie-Pier Scott-Boyer, Tania Cuppens, and Arnaud Droit. 2022. "New Developments and Possibilities in Reanalysis and Reinterpretation of Whole Exome Sequencing Datasets for Unsolved Rare Diseases Using Machine Learning Approaches" International Journal of Molecular Sciences 23, no. 12: 6792. https://doi.org/10.3390/ijms23126792