
Aryl Hydrocarbon Receptor in Oxidative Stress as a Double Agent and Its Biological and Therapeutic Significance


Federal Research Center of Fundamental and Translational Medicine, Institute of Molecular Biology and Biophysics, Timakova Str. 2, 630117 Novosibirsk, Russia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(12), 6719; https://doi.org/10.3390/ijms23126719
Submission received: 26 May 2022
/
Revised: 14 June 2022
/
Accepted: 14 June 2022
/
Published: 16 June 2022
(This article belongs to the Special Issue Redox Modulation: New Trends, Biological and Therapeutical Implication)
Abstract
:The aryl hydrocarbon receptor (AhR) has long been implicated in the induction of a battery of genes involved in the metabolism of xenobiotics and endogenous compounds. AhR is a ligand-activated transcription factor necessary for the launch of transcriptional responses important in health and disease. In past decades, evidence has accumulated that AhR is associated with the cellular response to oxidative stress, and this property of AhR must be taken into account during investigations into a mechanism of action of xenobiotics that is able to activate AhR or that is susceptible to metabolic activation by enzymes encoded by the genes that are under the control of AhR. In this review, we examine various mechanisms by which AhR takes part in the oxidative-stress response, including antioxidant and prooxidant enzymes and cytochrome P450. We also show that AhR, as a participant in the redox balance and as a modulator of redox signals, is being increasingly studied as a target for a new class of therapeutic compounds and as an explanation for the pathogenesis of some disorders.
1. Introduction
In live cells, reactive oxygen species are continuously generated, for example, by xanthine oxidase to degrade purine nucleotides, by nitric oxide synthase to form nitric oxide, and by other biochemical reactions as a byproduct of the oxidative energy metabolism for the formation of adenosine triphosphate from glucose in mitochondria [1,2,3,4].
Under normal physiological conditions, small amounts of oxygen are constantly converted into superoxide anions, hydrogen peroxide, and hydroxyl radicals. The biological activity of reactive oxygen species at a physiological concentration plays an important role in cell homeostasis and in a wide range of cellular parameters (proliferation, differentiation, cell cycle, and apoptosis) [5,6,7,8].
In the cell, reactive oxygen species arise under the influence of such exogenous pro-oxidant factors as environmental pollutants, ionizing and ultraviolet radiation, xenobiotics, air pollutants, and heavy metals [9,10].
The main endogenous sites of production of cellular redox-reactive compounds include complexes I and III of the mitochondrial electron transport chain, endoplasmic reticulum, peroxisomes, and such enzymes as membrane-bound nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) isoforms 1–5 (NOX1–NOX5), complexes of dual oxidases 1 and 2, xanthine oxidase, polyamine and amine oxidases, enzymes catabolizing lipids, and cytochrome P450 family 1 (CYP1A) [11,12,13,14,15,16].
The high reactivity of oxygen and its active species necessitates a multi-level antioxidant defense system that blocks the formation of highly active free radicals [10].
Free radicals are usually eliminated by the body’s natural antioxidant system. Redox homeostasis in normal cells is maintained by a nonenzymatic system consisting of carotenoids, flavonoids, glutathione, anserine, carnosine, homocarnosine, melatonin, thioredoxin, and vitamins C and E, as well as a network of antioxidant enzymes such as superoxide dismutases, catalases, peroxiredoxins, glutathione peroxidase (GPX), glutaredoxins, and paraoxonases [17,18]. In redox homeostasis, a certain role is played by the enzymes of phase II xenobiotic biotransformation, e.g., NADPH:quinone oxidoreductase 1 (NQO1), glutathione-S-transferase (GST) P1, GSTA1/2, UDP glucuronosyltransferase (UGT) 1A6, GPX4, and heme oxygenase 1 [19].
An imbalance between the formation of oxidative free radicals and the antioxidant defense capacity of the body’s cells is defined as oxidative stress. An important function in the regulation of oxidative stress is performed by the AhR signaling pathway via pro-oxidant and antioxidant mechanisms.
2. AhR Expression, Functions, and Signaling
2.1. AhR Structure
The aryl hydrocarbon receptor (AhR), its partner protein aryl hydrocarbon receptor nuclear translocator (ARNT), and AhR repressor protein (AhRR) are members of a family of structurally related transcription factors (basic helix–loop–helix (bHLH) motif-containing Per–ARNT–Sim (PAS), whose members carry out critical functions in the gene expression networks that underlie many physiological and developmental processes, especially those participating in responses to signals from the environment [20,21].
Structurally, human AhR has a sequence of 848 amino acid residues and includes 3 functional domains: from the amino (N-) to carboxy (C-)terminus, these are bHLH, PAS A, PAS B, and transcription activation domains (TADs) whose activity is mediated by coactivators called CBP/p300 and RIP140 [21,22].
The amino acid sequence of the bHLH and PAS domains is evolutionarily highly conserved. The bHLH domain can be divided into an HLH domain and a basic domain and is involved in AhR binding to DNA and in protein dimerization [23,24]. The PAS region participates in ligand binding and is thought to be the site of protein–protein interactions during dimer formation; PAS B partially overlaps with the heat shock protein 90 (HSP90)-binding site [21,25]. The transcription activation domain serves as a mediator of the transcriptional activation of downstream genes [26].
The AhRR protein is structurally similar to AhR in the bHLH region, and this property allows AhRR to heterodimerize with ARNT and to bind to a xenobiotic-responsive element (XRE) [27]. The repression domain of AhRR contains three sumoylation sites, all of which must be sumoylated for complete repression of AhR target genes [24,28]. Structure of AhR is shown in Figure 1.
2.2. Main Functions of AhR
AhR is a unique and versatile biological sensor of planar chemical compounds of endogenous and exogenous origin [29,30] and is the only member of the PAS family that binds naturally occurring xenobiotics [31]. By functioning as a transcription factor, AhR takes part in many physiological and pathological processes in cells and tissues.
Traditionally, AhR has been known as a mediator of xenobiotic metabolism ever since AhR was reported to bind to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). AhR over-activates the transcription of target genes, resulting in a release of many toxic compounds; for example, AhR is an activator of TCDD as a carcinogen [32,33]. For many years, AhR has been a research subject of toxicologists owing to its involvement in the metabolism of environmental pollutants and food contaminants such as polycyclic aromatic hydrocarbons, polychlorinated biphenyls, and dioxins [33,34].
Later, numerous studies have shown that AhR is activated by many natural and synthetic ligands, which may or may not be planar molecules of the polycyclic aromatic hydrocarbon type [35,36]. In this context, AhR acts as a sensor that connects the external environment and internal environment. AhR participates in processes of development, immune defense, and homeostasis, including cell differentiation and physiological processes in stem cells. In these cases, its ligands are various endogenous compounds.
In particular, the endogenous stimulation of AhR determines its main function (in mammals), which is related to the normal development of an organism and its homeostasis under the conditions of chemically diverse and dynamic internal and external environments [37,38,39,40,41].
AhR exerts this action by regulating fundamental metabolic processes that modulate cell proliferation, cell cycle, cell differentiation and phenotype formation, and cell adhesion and migration [40,42,43,44,45,46].
The involvement of the AhR in cell cycle regulation confirms its important role in the modulation of cellular homeostasis [33,47,48]. One hypothesis postulates that the endogenous stimulation of AhR triggers the recognition of cellular stress, thereby altering gene expression and causing cell cycle arrest [42,49,50].
In several human cell lines, it has been demonstrated that excessive AhR activation results in cell cycle arrest in the G1 phase; this event makes it impossible for the cell to enter the S phase; this blockade is partly due to a direct interaction of AhR with hypophosphorylated retinoblastoma protein (pRb) [51,52,53,54].
The overstimulation of receptor AhR by anthropogenic pollutants leads to a substantial dysregulation of AhR activity and of its downstream cascades [55,56,57]. Apparently, this phenomenon wreaks havoc on the fine regulation of cellular metabolic processes, e.g., owing to the disruption of mitochondrial structure/function and proliferative activity [58,59].
2.3. AhR Ligands and Target Genes
AhR is activated by a wide range of ligands (Table 1), which can be categorized into endogenous ligands and exogenous ones [60,61,62].
Among the exogenous AhR ligands, halogenated aromatic hydrocarbons are typical, including dioxins (such as TCDD) [63], polychlorinated biphenyls, and polycyclic aromatic hydrocarbons such as benzo[a]pyrene (BaP) and 3-methylcholanthrene [33,34]. AhR also binds to a number of drugs such as omeprazole [64] and to compounds present in foods, such as plant polyphenols and flavonoids (e.g., quercetin) [65,66].
Aside from environmental compounds, many small-molecule compounds have been identified that bind to AhR and modulate its activity [33,67].
Growing interest in the physiological functions of AhR has led to the identification of many endogenous ligands of AhR [68]. These include heme metabolites bilirubin and biliverdin [69], tetrapyrroles [70], arachidonic acid metabolites [70,71,72], tryptophan metabolites such as kynurenic acid [73] and kynurenine [68], 6-formylindolo[3,2-b]carbazole (FICZ) (which is a photoproduct of the ultraviolet irradiation of L-tryptophan [74]), indolo[3,2-b]carbazole [68], and estrogen equilenin [72]. Compounds secreted by bacteria can also be AhR ligands [60,75,76].
AhR target genes code for phase I enzymes that metabolize xenobiotics (e.g., CYP1A1, CYP1A2, and CYP1B1) and phase II enzymes including NQO1, GSTA2, aldehyde dehydrogenase 3A1, UGT1A1, and UGT1A6 [21,67,77,78,79,80].
AhR ligands can serve as either agonists or antagonists of the transcription of AhR-controlled genes, depending on various conditions in the cell. In different cell types, there are diverse scenarios of gene activation in response to AhR stimulation. Different AhR ligands can induce dissimilar transcriptome profiles within the same cell type, and the same AhR ligand can give rise to different transcriptome profiles in different cell types [81,82,83].
2.4. Pathways of Transcription Regulation by AhR and Crosstalk with Other Signal Transduction Pathways
The AhR signaling pathway involves both classic (canonical) and non-classic (non-canonical) signal transduction mechanisms (Figure 2) [31,84].
The classic (canonical) pathway of xenobiotic metabolism was the first-studied molecular mechanism of AhR action, and adherence to this paradigm has greatly delayed the understanding of the global biological significance of AhR.
Under physiological conditions, AhR is localized to the cytosol and forms a complex with specific chaperone proteins, such as hepatitis B virus X-associated protein 2 (XAP2, also known as AIP or ARA9), p23, and c-Src [24,84,85,86]. Ligand binding results in a conformational change that causes AhR to disassociate from the above complex, and then the ligand–AhR complex is translocated from the cytosol to the nucleus [87,88].
In the classic mechanism of transcriptional regulation, the complex of AhR with its ligand heterodimerizes with ARNT and binds to xenobiotic-responsive elements in DNA upstream of AhR’s inducible target genes. The AhR–ARNT complex initiates the transcription of several genes, including cytochrome P450 family 1 subfamily A member 1 (CYP1A1) and subfamily B member 1 (CYP1B1), and this action has a wide range of physiological and toxic effects [24,70,89,90,91].
In the non-canonical transcriptional regulatory pathway, the ligand–AhR complex heterodimerizes with partner proteins other than ARNT, for example, Krüppel-like factor 6 (KLF6) and RelB [92,93].
AhR interacts with the signaling pathway of the nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) [94,95,96]. Through interactions of AhR with RelA or RelB, AhR signaling can promote the activation of NF-κB [97,98,99]. AhR and NF-κB form a heterodimer that lead to the inducing of the expression of cytokines and chemokines B-cell-activating factor of the tumor necrosis factor family (BAFF), B-lymphocyte chemoattractant (BLC), CC-chemokine ligand 1 (CCL1), and interferon-responsive factor (IFR3) [100,101].
Additionally, AhR can interact with other signal transduction pathways. There seems to be bidirectional crosstalk between AhR and nuclear factor erythroid 2-related factor 2 (Nrf2) [102,103]. The Nrf2 gene promoter contains at least one functional xenobiotic-responsive element [104], whereas the AhR gene promoter has several antioxidant response elements (AREs) [103]. The crosstalk of the AhR and Nrf2 pathways is discussed in detail below.
AhR signaling is linked with estrogen receptor activity and function [21,67], for which a ligand–AhR complex can serve as a coactivator [92,105]. Additionally, a ligand–AhR complex can function as a coactivator of E2 promoter-binding factor 1 (E2F1): a transcription factor that is crucial for the cell cycle transition from the G1 phase to the S phase [92,106]. The binding of AhR to the hypophosphorylated “active” form of the retinoblastoma tumor suppressor protein (pRb) leads to cell growth arrest in the G1/S phase of the cell cycle [107]. It is reported that two mechanisms contribute to this effect. In the first one, pRb acts as a transcriptional coactivator of classic induction of CYP1A1 by dioxin-like ligands. In the second mechanism, AhR is a component of a repressor complex along with pRb, E2F, and partner protein E2F DP [89,108,109].
Aside from genomic signaling via target genes [33,67,84,110,111], AhR participates in nongenomic signaling [46]. For example, upon the binding of a ligand to a cytosolic complex of AhR with chaperones, kinase c-Src can be released, which relocates to the plasma membrane, thereby activating EGF signaling [66,112].
It has been revealed that certain compounds can directly induce the expression of AhR target gene CYP1A1, suggesting that AhR activation can occur in the absence of direct ligand binding [113]. Indeed, nongenomic effects of AhR have been documented, especially in the context of the induction of inflammatory processes. For instance, TCDD has been reported to increase intracellular calcium concentration, thereby initiating a cascade of reactions ultimately causing cyclooxygenase (COX) 2 activation and an accumulation of inflammatory mediators such as prostaglandins [114,115]. Moreover, AhR is reported to mediate the toxic cellular effects of TCDD through pro-oxidant mechanisms [34].
3. AhR Regulates Enzyme Systems Generating Reactive Oxygen Species
AhR is reported to be responsible for the toxic cellular effects of TCDD via pro-oxidant mechanisms [34,116]. There is convincing evidence that the activation of AhR-dependent detoxification of such environmental stressors as TCDD, polycyclic aromatic hydrocarbons, polychlorinated biphenyls, and effects of ultraviolet radiation gives rise to oxidative stress and to the production of reactive oxygen species, thus inducing oxidative damage to DNA, lipids, and other cellular macromolecules [117,118,119,120]. Several enzyme systems, including CYP1A, NOX, COX, and possibly aldo–keto reductase (AKR) 1, are regulated through the AhR signaling pathway in terms of their ability to generate reactive oxygen species in various cell types and tissues [121,122,123,124].
3.1. CYP1A
The production of reactive oxygen species by cytochromes P450 is associated with a catalytic circle of enzymes, to be precise, with a phenomenon called “uncoupling” [125,126,127]. In the presence of NADPH, CYP monooxygenases reduce molecular oxygen, where one oxygen atom is attached to the substrate, and the second one is reduced to form a water molecule. Stoichiometric analysis of this reaction shows that most CYP enzymes consume more oxygen than necessary to mono-oxygenize their substrate, and hydrogen peroxide can be a byproduct of this reaction [128,129].
When compounds with a stable structure induce the formation of a complex of CYP with oxygen, the absence of an electron acceptor can cause auto-oxidation of CYP and a subsequent release of a superoxide anion radical which dismutates, thereby yielding hydrogen peroxide too. As a result of Fe2+-catalyzed Haber–Weiss and Fenton reactions, both superoxide anions and hydrogen peroxide can be converted into highly reactive hydroxyl radicals [125].
The interaction of TCDD with AhR enhances the expression of such cytochrome P450 family members as CYP1A1, CYP1A2, and CYP1B1. Due to the stable structure of TCDD, these enzymes are unable to metabolize it efficiently. In addition, the formation of reactive oxygen species is caused by excessive CYP1A1 activity resulting from the binding of TCDD to AhR [21,130]. For instance, the AhR-dependent induction of CYP1A is the main source of reactive oxygen species in hepatocytes incubated with TCDD [119]. Similarly, exposure to polycyclic aromatic hydrocarbons such as BaP causes CYP1A1 the overexpression and production of reactive oxygen species [131].
The overproduction of reactive oxygen species under the influence of CYP1A1 may indirectly affect cell metabolism, owing to the direct activation of several signaling pathways. Moreover, the interaction of reactive oxygen species with various biomolecules, such as NF-κB or oncoprotein c-Jun or Rb, can affect the cell cycle [132,133].
3.2. NADPH Oxidases
The metabolic activation of polycyclic aromatic hydrocarbons involves NADPH oxidase in addition to CYP1 isoforms [134]. There is evidence that polycyclic aromatic hydrocarbons can stimulate the production of reactive oxygen species via NADPH oxidases, in particular NOX2 and NOX4 [135,136,137,138,139,140].
These membrane-bound enzyme complexes are detectable in the plasma membrane of various cell types, such as phagocytes and endothelial and epithelial cells [141]. In the inactive state, NADPH oxidase subunits—three cytoplasmic (Rac1, p47phox, and p67phox) and two intramembrane ones (p22phox and gp91phox)—are not assembled [141]. After activation by cytokines, by opsonized bacteria, by bacterial lipopolysaccharides, or by other stimuli, the complex assembles and the catalytic subunit, i.e., the heterodimeric flavocytochrome composed of gp91phox and p22phox, and transfers one electron from NADPH to molecular oxygen, thus yielding superoxide anions, which are next dismuted into hydrogen peroxide [141,142,143].
Additional proteins, such as p40phox (one of NADPH oxidase subunits), play an important part in the regulation of NADPH oxidase activity and in the subsequent production of reactive oxygen species [140,141].
According to the literature, there are several mechanisms of NADPH oxidase activation through the AhR signaling pathway. For example, the 0NOX2-mediated formation of reactive oxygen species in epidermal keratinocytes under the action of a polycyclic aromatic hydrocarbon is mediated in an AhR-dependent way by the stimulation of the phosphorylation of p47phox (neutrophil cytosolic factor 1), which is necessary for the assembly of the NOX2 complex on the plasma membrane [137]. In a study on the liver of C57BL/6J mice treated with 3-MX, induction of the NADPH oxidase subunit p40phox was observed, which was not the case in the liver tissue of mice with a conditional AhR b knockout in the liver [140]. In an analysis of Hepa1c1c7 cells, a functional xenobiotic-responsive element was detected in the promoter of the murine p40phox gene [140].
Another mode of NADPH oxidase activation in human and rat macrophages involves the increased transcription of p47phox because of the direct binding to XRE in the promoter region of this gene after treatment with BaP. In addition, BaP promotes the translocation of the p47phox protein to the macrophage plasma membrane and strengthens the production of superoxide anion under the influence of phorbol myristate acetate [136].
Reactive oxygen species that are generated in epidermal keratinocytes during exposure to a polycyclic aromatic hydrocarbon initiate mitogen-activated protein kinase (MAPK) signaling, which drives the activation of transcription factors AP-1 and NF-κB and the subsequent initiation of proinflammatory processes [137].
It has also been shown that AhR ligands, such as TCDD and dioxin-like planar polychlorinated biphenyls, or endogenous substances (e.g., indoxyl sulfate or arachidonic acid) activate NADPH oxidase and thus stimulate the production of reactive oxygen species, thereby leading to damage to vascular endothelial cells [144,145]. During the incubation of human umbilical vein endothelial cells with the endogenous AhR ligand indoxyl sulfate, the production of reactive oxygen species increases through the overexpression of NOX4, thus damaging these cells [146].
NOX4 activation by thiol-reactive agents such as cadmium, arsenic, nickel, and mercury interferes with AhR signaling [147]. For example, the treatment of human HaCaT keratinocytes with arsenic results in NOX4-dependent oxidative stress. The subsequent inhibition of the catalytic activity of CYP1A1 by reactive oxygen species induces an accumulation of the endogenous AhR ligand 6-formylindolo[3,2-b]carbazole and to an AhR-dependent increase in CYP1A1 transcription [130,147]. The same effect is observed in arsenic-treated murine cells [148,149].
The influence of reactive oxygen species on the metabolic degradation of AhR ligands may also explain the high transcriptional activity of AhR that is observed in glutathione-depleted normal and malignant breast cells [150].
3.3. Cyclooxygenase
In the biosynthesis of prostaglandin E2, cyclooxygenase is a key rate-limiting enzyme that catalyzes the conversion of arachidonic acid to prostaglandins [151,152]. Furthermore, there is an alternative enzyme for chemical oxidation: prostaglandin endoperoxide synthase 2, also known as COX2. The latter is an example of an alternative enzyme for xenobiotic metabolism in extrahepatic tissues [153,154].
The activation of AhR by TCDD has been found to induce the expression and activity of COX2 [155,156]. Unlike COX1 expression, the expression of COX2 can be induced by various stimuli, such as growth factors and cytokines [157]. The upregulation of COX2 has been implicated in chronic inflammation and carcinogenesis [158,159,160].
COX2 converts arachidonic acid to prostaglandin (PG) G2, which undergoes peroxidation to PGH2. In this two-step enzymatic process that generates reactive oxygen species, the cyclooxygenase is the rate-limiting enzyme for the formation of prostaglandins [152,161].
Although TCDD and other AhR ligands drive CYP1A1 and CYP1A2 expression via the canonical AhR–ARNT pathway, TCDD-induced expression of COX2 involves non-canonical AhR signaling pathways such as c-Src activation and the subsequent binding of CCAAT/enhancer-binding protein β [162] and MAPK signal transduction [137,163].
The overexpression of COX2 may enhance the production of reactive oxygen species. Elevated levels of COX2 and reactive oxygen species can cause vasoconstriction and renal endothelial dysfunction [164]. In another study, it was hypothesized that lipopolysaccharide inhibits the endothelium-dependent vasodilatory response in middle cerebral arteries of normotensive rats [165]. The effect of lipopolysaccharide in that work was mediated by a release of the superoxide anion that was generated, at least in part, via lipopolysaccharide-induced expression of COX2.
3.4. Aldo–Keto Reductases
The metabolic activation of polycyclic aromatic hydrocarbons involves aldo–keto reductases in addition to CYP1 isoforms, and aldo–keto reductases participate in the formation of reactive oxygen species. These enzymes are cytosolic NADPH-dependent oxidoreductases that convert carbonyl groups to primary and secondary alcohols [166,167].
Aldo–keto reductases, in particular human AKR1A1 and AKR1C1–AKR1C4, can oxidize trans-dihydrodiols (which are intermediates of CYP1-mediated oxidation of polycyclic aromatic hydrocarbons) to the corresponding catechols [3,168]. For example, BaP is oxidized by CYP1A1 to BaP-7,8-epoxide [169]. In a dihydroxylation reaction, microsomal epoxide hydrolase 1 transforms this epoxide into BaP-7,8-trans-dihydrodiol, which is detoxified by phase II enzymes, re-oxidized by CYP1 isoforms, or converted by aldo–keto reductases into BaP-7,8-catechol [166,169]. In the presence of oxygen, BaP-7,8-catechin undergoes one-electron oxidation, giving rise to the o-semiquinone anion radical and resulting in a release of hydrogen peroxide [170]. If the o-semiquinone anion radical is not detoxified by catechol-O-methyltransferases or phase II conjugating enzymes, another one-electron oxidation gives BaP-7,8-dione (o-quinone) and superoxide anion radicals [170]. BaP-7,8-dione is highly reactive, and either forms DNA adducts [171] or undergoes a redox cycle, i.e., is reduced back to BaP-7,8-catechin, in the presence of NADPH [172].
Consequently, the aldo–keto reductase-mediated metabolism of polycyclic aromatic hydrocarbons contributes to oxidative damage and their genotoxicity.
The role of AhR in the regulation of AKR1 gene expression is not yet clear. High expression of AKR1C enzymes is observed in BaP-exposed cell lines, including human hepatoma, colon carcinoma, and breast cancer cells [122,123,124]. Moreover, an AhR knockdown in breast cancer MDA-MB-231 cells drastically lowers both basal and 3-methylcholanthrene-induced AKR1C3 expression [124]. On the other hand, the AhR ligand prototype TCDD is known to be ineffective in terms of AKR1C induction, and promoter sequences of the gene encoding human AKR1C do not contain sensitive xenobiotic-responsive elements [122,123,124]. suggesting that the regulation of these enzymes is not mediated by the canonical AhR–ARNT signaling pathway.
4. Participation of AhR in Antioxidant Defense
Aside from the AhR-dependent production of intracellular reactive oxygen species, the AhR signaling pathway modulates the expression of genes of the antioxidant system and thereby regulates cell functions that ensure protection from oxidative stress. Numerous studies indicate that the protective action of antioxidants against oxidative stress is mediated by AhR through a response to such AhR ligands as flavonoids, phytochemicals, and azoles [173,174,175,176,177,178,179,180].
When this type of ligand binds to AhR, the production of reactive oxygen species does not occur because of the induction of the nuclear translocation of AhR; instead, Nrf2 is activated. Nrf2 is a key biomolecule that provides cell protection against the oxidative damage caused by reactive oxygen species: Nrf2 is a transcription factor that regulates the genes encoding enzymes of the antioxidant system [102,181].
4.1. Nrf2 Expression, Functions and Signaling
In a normal physiological state, Nrf2 is located in the cytoplasm and binds to Kelch-like ECH-associated protein 1 (Keap1) [182,183]. In response to stress signals, Keap1 is inactivated, resulting in Nrf2 stabilization. Nrf2 is translocated to the nucleus where it binds to members of the Maf protein family (e.g., MafK, MafF, and MafG). In a sequence-specific manner, the Nrf2–sMaf complex binds to an ARE, 5′-TGACXXXGC-3′, in a promoter region and activates the transcription of target genes of Nrf2 [19,181,184,185,186].
Nrf2 regulates the transcription of genes encoding components of the antioxidant systems based on glutathione and thioredoxin as well as genes coding for enzymes involved in the phase II detoxification of exogenous and endogenous compounds or in NADPH regeneration and heme metabolism (heme oxygenase 1): GPX4, superoxide dismutase, sulfiredoxin, paraoxonases, NQO1, GSTP1, GSTA1/2, UGT1A6, and various other enzymes that remain to be identified [187,188,189,190,191,192].
In addition to ensuring redox homeostasis, Nrf2 functions encompass multiple cellular processes, including the regulation of cell survival, metabolic and protein homeostasis, inflammation, and cell proliferation and differentiation [193,194,195,196,197].
Nrf2 is at the center of a complicated regulatory network. Its activity is modulated at several levels, including transcriptional regulation (by NF-κB, AhR, ATF4, and other transcription factors and cofactors), post-transcriptional regulation (by microRNA, RBPs, or alternative splicing), post-translational regulation (by ERK, JNK, PKC, CK2, PERK, GSK3, or p38), and the regulation of Nrf2 protein stability (by KEAP1, βTrCP, HRD1, WDR23, or CRIF1) [181,198].
4.2. Participation of AhR in Mechanisms of Nrf2 Activation
At present, there is some understanding of the mechanisms underlying Nrf2 activation by AhR. (Figure 3) One of them is the transcriptional activation of Nrf2 as a target gene of AhR, and the other is the indirect activation of Nrf2 via CYP1A1-generated reactive oxygen species [199].
4.2.1. Nrf2 as a Target Gene of AhR
AhR is one of the transcription factors regulating Nrf2 [199,200]. Research on the Nrf2 promoter indicates that Nrf2 is a target gene of AhR. That Nrf2 gene transcription is directly modulated by AhR activation has been demonstrated by DNA sequence analyses of the mouse Nrf2 promoter; this work revealed one xenobiotic-responsive element-like element (XREL1) located at position –712 and two additional xenobiotic-responsive element-like elements located at positions +755 (XREL2) and +850 (XREL3). In those studies, functional analysis by a luciferase assay revealed that XREL1, XREL2, and XREL3 are all inducible by TCDD treatment, with XREL2 being the most potent [104,200].
There is also confirmation of the functionality of these xenobiotic-responsive element-like elements, and a direct binding of AhR to the Nrf2 promoter has been proven [200]. It has been reported that Nrf2 expression is at least partly regulated by AhR inducers through the activation of multiple xenobiotic-responsive elements in the Nrf2 promoter. This molecular event represents a direct connection between AhR and Nrf2 and places the Nrf2–ARE pathway downstream of AhR–XRE activation in certain scenarios [104].
There is fine-tuned crosstalk between AhR and Nrf2, which mutually enhance or weaken their activation states. The antioxidant response resulting from AhR activation and mediated by Nrf2 depends on the type of AhR ligand. Such AhR ligands as dioxins, BaP, and other polycyclic aromatic hydrocarbons bind to AhR with high affinity and induce extremely high CYP1A1 expression along with reactive oxygen species production [131,201]. Although Nrf2 is also activated in this case [102], the oxidative stress suppresses antioxidant defense [131,177].
Other AhR ligands, such as phytochemicals and flavonoids, bind to AhR less strongly [202,203,204], and among antioxidant phytochemicals, there are those that activate the AhR signaling pathway without the production of reactive oxygen species. These include ketoconazole and cynaropicrin [173,174].
Antioxidant phytochemicals capable of modulating Nrf2, AhR, and CYP1A1 have been described. By means of epidermal keratinocytes, as an example, it has been revealed that phytochemicals exerting their antioxidant actions through AhR and Nrf2 signaling can be categorized into three groups depending on their ability to increase and decrease AhR and CYP1A1 activities [10]. Group 1 contains Nrf2 agonists with AhR-agonistic activity. This group includes soybean tar, Opuntia Ficus-Indica extract, Houttuynia cordata extract, Bidens pilosa extract, and cynaropicrin. Group 2 contains Nrf2 agonists with AhR-antagonistic activity. This group includes cinnamaldehyde and epigallocatechin gallate. Group 3 contains Nrf2 agonists with CYP1A1-inhibitory activity. This group includes Z-ligustilide, quercetin, kaempferol, pterostilbene, and resveratrol.
4.2.2. Indirect Activation of Nrf2 via CYP1A1-Generated Reactive Oxygen Species
4.2.3. Direct Crosstalk between AhR–XRE and Nrf2–ARE Signaling Pathways
AhR and Nrf2 signaling pathways coordinate the expression regulation of genes of phase II xenobiotic metabolism, e.g., GSTA2, UGT1A6, and NQO1 [191,199,200,207,208,209]. The mechanism of direct crosstalk between the AhR–XRE and Nrf2–ARE signaling cascades has been described for the NQO1 enzyme and involves the close proximity of a xenobiotic-responsive element and ARE in the regulatory region of the NQO1 gene [199,210].
5. AhR in the Pathogenesis of Diseases Related to Oxidative Stress
Although initial studies on AhR were focused on its function as a signaling molecule of a chemical sensor responsive to environmental pollutants, lately, the range of subject areas has widened significantly. Our understanding has expanded regarding the role of the AhR signaling pathway in the regulation of a variety of physiological and pathological phenomena. AhR’s functions cover many cellular processes, including the regulation of cell survival, metabolic and protein homeostasis, inflammation, cell proliferation and differentiation, apoptosis, and cellular adhesion and migration. Reactive oxygen species-induced activation of transcription factors and proinflammatory genes increases inflammation. Accordingly, research on various diseases in which AhR induces an oxidative stress response—by switching on inflammation and antioxidant, prooxidant, and cytochrome P450 enzymes—is now within the scope of the interest of investigators.
It is known that oxidative stress causes inflammation and toxicity, and these problems can lead to such pathologies as cardiovascular, liver, kidney, lung, brain, eye, skin, and joint diseases, as well as aging and cancer [7,211,212,213,214,215]. In recent decades, AhR has been increasingly recognized as an important modulator of disease because of AhR’s role in the regulation of the redox system and of immune and inflammatory responses [120,216].
Below are examples of AhR involvement in the pathogenesis of many human diseases associated with oxidative stress.
5.1. Neurological Diseases
Oxidative stress has been studied in neurological diseases, including Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and amyotrophic lateral sclerosis, and in some psychiatric disorders such as depression [213,215,217,218,219,220]. It is now proven that AhR is involved in the initiation of oxidative stress in the brain because AhR activation by some ligands shifts the cellular redox balance toward oxidative stress [120,221,222]. In most areas of the brain and in the pituitary gland, AhR activation induces CYP1A1 and CYP1B1 expression [223]. This event can result in the mitochondrial production of reactive oxygen species [124,224] and in the higher production of reactive oxygen species through the activation of the arachidonic acid pathway by CYP enzymes and other intracellular signaling processes [70,225].
AhR plays an important part in the initiation of benign and malignant brain tumors [226,227]. In glioblastoma cells, neuroactive hormone dopamine has been identified as an inducer of enzymes CYP1A1, CYP1B1, and UGT1A1 [228].
It has been proven that AhR signaling pathways (especially after activation by such endogenous AhR ligands as tryptophan metabolites) are implicated in neurodegenerative diseases, in particular in well-known age-related brain diseases (Parkinson’s disease, Alzheimer’s disease, multiple sclerosis, and amyotrophic lateral sclerosis) and other diseases of the central nervous system [229,230,231,232,233]. The hypothesis that AhR is involved in the neurodegenerative processes in Parkinson’s disease and Alzheimer’s disease derives from both human and in vitro studies. An important question addressed in these studies is the contribution of environmental factors to the risk of neurodegenerative diseases [67,233,234].
Parkinson’s disease is an extrapyramidal disorder characterized by decreased motor function due to the loss of dopaminergic neurons. Toxic exogenous ligands such as TCDD enhance the degeneration of dopaminergic neurons in the midbrain owing to enhanced oxidative stress, thereby inducing experimental Parkinson’s disease; in contrast, several phytochemicals such as a flavonoid called tangeretin as well as natural compounds from the plant Withaferin sominifera act via AhR to protect against Parkinson’s symptoms in several models of this disease [235,236]. An experimental murine model of Parkinson’s disease has revealed that AhR activation by BaP may have a protective effect against this pathology [237]. It should be noted that AhR is activated by carbidopa, which is used to treat Parkinson’s disease [238].
AhR is associated with Alzheimer’s disease too, which is a neurodegenerative illness featuring aggregation of β-amyloid plaques, which cause neuroinflammation and promote neuronal loss [239]. In a mouse model of Alzheimer’s disease, AhR activation alleviates cognitive deficits through the upregulation of neprilysin: the main endogenous enzyme of β-amyloid catabolism [239]. Those authors showed that neprilysin expression and enzymatic activity are higher when AhR is activated by endogenous ligands L-kynurenine or 6-formylindolo[3,2-b]carbazole or by exogenous ligands diosmin or indole-3-carbinol; they also found that AhR is a direct transcription factor of the neprilysin gene [239]. Substantial amounts of AhR and indolamine-2,3-dioxygenase 1 (IDO1) are detectable in glial cells in postmortem brain samples from Alzheimer’s patients and in postmortem hippocampus and serum samples from such patients; these amounts are elevated as compared with young people and elderly patients without dementia [230].
It has been established that β-amyloid neurotoxicity depends on AhR activation through the IDO1–kynurenine–AhR cascade, where—via the increased activity of IDO1 (an enzyme responsible for tryptophan degradation)—the production of tryptophan metabolites is accelerated, which can act as AhR ligands and may be neurotoxic [240]. On the contrary, inhibitors of IDO1 attenuate the neurotoxic activity of AhR. Because of the evidence of the neuroprotective effects of AhR against neurodegenerative diseases of the brain, research on non-toxic AhR agonists will be necessary and may help to alleviate the symptoms as a new therapeutic strategy against these diseases.
In multiple sclerosis and amyotrophic lateral sclerosis, the enhancement of inflammatory and neurodegenerative processes in the central nervous system is caused by environmental factors, among other reasons. In particular, the risk of multiple sclerosis correlates with smoking, which drives AhR gene demethylation and inhibits AhR signaling pathways, followed by the enhancement of inflammatory processes in the central nervous system in multiple sclerosis [241,242,243]. Patients with multiple sclerosis have lower levels of circulating AhR than healthy controls do, suggesting that AhR is involved in the pathogenesis of this disorder [244]. AhR may limit the central nervous system-inflammation characteristic of multiple sclerosis by suppressing astrocyte activation [245,246]. In multiple sclerosis, the gut microbiome is altered, which represents an interesting opportunity for investigating the role of the kynurenine–AhR pathway in this pathology [247]. In an animal model of multiple sclerosis (autoimmune encephalomyelitis), it has been shown that AhR may be a therapeutic target in multiple sclerosis too: a knockdown of AhR worsens disease signs, whereas AhR activation by such AhR agonists as TCDD, indole-3-carbinol, or diindolylmethane slows the progression of experimental allergic encephalomyelitis, owing to the overexpression of Forkhead Box P3, greater numbers of anti-inflammatory regulatory T cells, and the attenuation of proinflammatory expansion of T helper 17 (Th17) cells [246,248,249].
6-Formylindolo[3,2-b]carbazole, another AhR agonist, also alleviates disease progression when systemically administered in mouse models [250]. On the other hand, topical administration of 6-formylindolo[3,2-b]carbazole promotes Th17 cell expansion, thus worsening disease signs [249]. Furthermore, in an animal model of multiple sclerosis (autoimmune encephalomyelitis), treatment with laquinimod, which crosses the blood–brain barrier, reduces astrogliosis and prevents the production of downstream proinflammatory cytokines in an AhR-dependent manner [251]. These results suggest that laquinimod is a first-in-class drug that targets AhR for the treatment of multiple sclerosis and other neurodegenerative diseases.
In amyotrophic lateral sclerosis, TDP-43 has been identified as the major pathological protein [252]. Drugs targeting this protein have become a therapeutic approach to this disease. One work on a cell line (induced pluripotent stem cells differentiated into motor neurons) on a mouse brain and on human neuronal cell lines (BE-M17 cells) has revealed that AhR activation by an exogenous ligand (TCDD) or an endogenous ligand (6-formylindolo[3,2-b]carbazole) raises the TDP-43 protein level in human neuronal cell lines and motor neurons [243]. The observed effects were abrogated by AhR antagonists, implying that exposure to the environmental toxic substances that activate AhR may be a risk factor for the onset or progression of amyotrophic lateral sclerosis [243].
Available data suggest that AhR activation may have a bidirectional effect on diseases of the brain. At the same time, only the IDO1-kynurenine-AhR cascade has been clearly shown to be involved in the development of Alzheimer’s disease. AhR function may depend on additional stress events, or cell types. Effects on the AhR signaling pathway may depend on interaction with various coactivators or ligand-selective binding of the AhR complex to nontraditional sequences of XRE [226,253]. Apparently, in different cases, AhR antagonists or agonists can promote or hinder the development of neurodegenerative disorders, as well as exert either pro- or antitumor influence.
5.2. Ocular Diseases
Oxidative stress participates in age-related macular degeneration and cataracts by altering various types of eye cells photochemically or nonphotochemically [254]. Under the action of free radicals, crystalline proteins in the lens can become crosslinked and aggregate, causing the formation of cataracts [255]. In the retina, prolonged exposure to radiation can inhibit mitosis in the retinal pigment epithelium and choroid, may damage outer segments of photoreceptors, and can be implicated in lipid peroxidation [256].
Because environmental factors and metabolites have been shown to affect the central nervous system, researchers have demonstrated a role of AhR in some diseases of the central nervous system and a functional contribution of AhR to the regulation of the behavior of astrocytes, other microglial cells, and neurons; given that the retina is an extension of the brain, a growing area of research deals with the involvement of AhR in ocular diseases [233].
The importance of AhR and AhR signaling in eye development, toxicology, and diseases is currently being uncovered. AhR is expressed in many ocular tissues, including the retina, choroid, cornea, and orbit. A considerable role of AhR in age-related macular degeneration, glaucoma, and other eye diseases has been identified [257].
Glaucoma is caused by an increase in intraocular pressure, which damages the optic nerve and induces vision loss and blindness. CYP1B1, an AhR-inducible gene, is associated with various types of glaucoma, including two main ones: primary open-angle glaucoma and primary congenital glaucoma [258,259]. Mutations correlating with various types of glaucoma have been identified in CYP1B1 and cause a low CYP1B1 enzymatic activity or its absence [258,260].
Additionally, TCDD can induce CYP1B1 expression in non-pigmented human ciliary epithelial cells that constitute the ciliary body, whose main function is the production of aqueous humor in the eye [261]. CYP1B1 takes part in the metabolism of steroids, retinol, retinal, arachidonate, and melatonin. Consequently, CYP1B1 expression, which is elevated during AhR activation, alters the biosynthesis of critical metabolites as well as metabolic pathways that may lead to the initiation and/or progression of glaucoma [258].
Age-related macular degeneration is a complex multifactorial disease of the elderly and has unclear pathogenesis. In age-related macular degeneration, one of the destructive processes is oxidative stress, which gives an imbalance among the processes responsible for the production and detoxification of reactive oxygen species. In the dry type of age-related macular degeneration, there is a thinning and degradation of choroid capillaries, expanding atrophy of the outer retinal part, and irreversible damage to photoreceptors. In wet age-related macular degeneration, the presence of proinflammatory cytokines, in particular vascular endothelial growth factor (VEGF), promotes angiogenesis and vascular permeability.
During an investigation into the AhR function in retinal homeostasis, a loss of AhR signaling was found to increase retinal susceptibility to environmental stressors such as intense light [262]. In the retina of Ahr−/− mice, there is a subretinal microglia accumulation concurrent with changes in autofluorescence, degeneration of the retinal pigment epithelium, and immune activation [262]. That study implies that AhR plays a protective part in the retina as a sensor of environmental stress, whereas altered AhR function may contribute to the progression of age-related macular degeneration in humans.
The effects of AhR have been analyzed in retinal pigment epithelial and choroid cell lines using AhR small interfering RNA [263]. That paper indicates that the expression of the VEGFA gene and of a proinflammatory chemokine (CC motif ligand 2; CCL2) goes up after AhR depletion in a human retinal pigment epithelial cell line, ARPE-19 [263]. Additionally, in that study, AhR depletion increased collagen IV synthesis and secretion in ARPE-19 cells and choroidal RF/6A cells. The depletion of AhR in RF/6A cells also raised the expression of the macrophage chemotactic factor gene, of secreted phosphoprotein 1 (SPP1), and of transforming growth factor (TGF)-β, while reducing the expression of antiangiogenic factor SERPINF1 [263].
Mice treated with TCDD show elevated VEGFA levels and choroidal vascularization: a hallmark of age-related macular degeneration [264]. These findings suggest that either a loss of AhR expression or AhR activation by TCDD (and/or by other toxic substances in cigarette smoke) promotes angiogenesis, inflammation, and alterations in the extracellular matrix, all of which are seen in the wet type of age-related macular degeneration.
In an experimental model of age-related macular degeneration (OXYS rats with signs of age-related macular-degeneration-like retinopathy of varying severity), it has been found that an imbalance between the prooxidant (AhR-dependent) system and antioxidant (Nrf2-dependent) system may be key to the pathogenesis of age-related macular degeneration and its initiation and/or progression [265]. Moreover, mitochondria-targeted antioxidant SkQ1 has been shown to ameliorate clinical signs of retinopathy in OXYS rats (manifesting signs of age-related macular-degeneration-like retinopathy) by influencing the transcriptional activity of AhR and Nrf2 and mRNA expression levels of CYP1A2 and CYP1B1 in the retina of OXYS and Wistar rats. These data are evidence that the enzymes CYP1A2 and CYP1B1 are implicated in the pathogenesis of age-related macular-degeneration-like retinopathy in OXYS rats and are possible therapeutic targets of SkQ1 [266].
Investigation into the impact of melatonin on mRNA expression of genes of AhR and Nrf2 signaling pathways in OXYS rats has shown that melatonin reduces the mRNA level of AhR-dependent genes of cytochromes CYP1A2 and CYP1B1 in the retina but does not affect mRNA expression of Nrf2-dependent genes in OXYS rats. This means that in age-related macular degeneration, the efficacy of melatonin is attributable to its ability to modulate the expression of AhR signaling pathway genes [267].
The discovery of a new synthetic AhR ligand (2,2′-aminophenylindole) should also be mentioned regarding the development of therapeutic strategies promoting cell homeostasis during the degeneration of the retinal pigment epithelium layer [268]. This compound protects retinal pigment epithelium cells from lipid peroxidation cytotoxicity in vitro and the retina from light-induced damage in vivo. In addition, metabolic characterization of this agent by liquid chromatography coupled with mass spectrometry suggests that 2,2′-aminophenylindole alters lipid metabolism in retinal pigment epithelium cells, thereby increasing the intracellular concentration of palmitoleic acid. The latter, as a downstream effector of 2,2′-aminophenylindole-mediated activation of AhR, has also been reported to protect cells of the retinal pigment epithelium from 4HNE-mediated stress and light-induced retinal degeneration in mice [268].
The synthetic AhR agonist 2,2′-aminophenylindole also regulates microglial homeostasis, thus making AhR a potential target for immunomodulatory and antioxidant therapies. This notion has been illustrated in a study on the anti-inflammatory and antioxidant effects of 2,2′-aminophenylindole on microglial reactivity; the results showed that 2,2′-aminophenylindole strongly represses the expression of proinflammatory genes and induces antioxidant genes in activated human and murine microglial cells, in lipopolysaccharide-stimulated explants of the retina, and in stressed human ARPE-19 cells [269].
5.3. Pulmonary Diseases
There is now firm proof that inflammatory lung disorders such as asthma and chronic obstructive pulmonary disease are characterized by systemic and localized chronic inflammation and oxidative stress [270,271,272,273]. Oxidants may contribute to inflammation by activating various kinases and redox transcription factors such as NF-κB and AP-1 [272,273].
The production of an inflammatory mediator called bronchial mucin-containing mucus is usually mediated by a release of cytokines or lipid mediators or by an increase in reactive oxygen species levels [274,275,276,277]. AhR is expressed in many lung cells, including macrophages, club cells, type II alveolar cells, and endothelial cells, and plays an important part in lung function modulation [278]. AhR serves as a regulator of mucosal-barrier function and may influence an immune response in the lungs through changes in gene expression, in intercellular adhesion, in mucin production, and in cytokine expression. AhR is expressed in cells of innate immune responses and in cells crucial for adaptive immunity [278].
By means of gene-deficient mice and the administration of AhR agonists and antagonists, numerous researchers have demonstrated that AhR modulates an immune response in various respiratory diseases and that lungs are sensitive to AhR ligands [279,280,281]. Allergic and inflammatory diseases such as bronchitis, asthma, and chronic obstructive pulmonary disease were recently linked with exposure to environmental toxic compounds. AhR mediates the effects of these substances through the arachidonic acid pathway, cell differentiation, intercellular adhesion interactions, cytokine expression, and mucin production. Human bronchial epithelial cells are reported to express AhR, and AhR activation induces mucin production via reactive oxygen species [277,279,282,283,284].
Chronic obstructive pulmonary disease develops as a result of exposure to risk factors that cause oxidative stress, inflammation, and the aberrant proliferation, death, and aging of lung cells, with the consequent destruction of parenchymal tissue [285,286]. AhR has a ligand-specific impact on the lungs and can either aggravate or ameliorate chronic obstructive pulmonary disease. For example, the toxic effects of dioxins and polycyclic aromatic hydrocarbons from tobacco smoke and from particulate matter on the lungs are mediated by AhR signal transduction. These ligands induce inflammation, increase the expression of mucin 5AC and matrix metalloproteinases (MMPs), and damage ciliated cells, club cells, and alveolar macrophages, thereby contributing to the pathogenesis of chronic obstructive pulmonary disease [287,288,289].
The exact molecular mechanisms behind the pathogenetic effects of endogenous AhR are unclear, but research indicates that the RelB protein (encoded by a target gene of NF-κB) may be partially responsible: AhR is known to interact with RelB and to modulate its expression [290]. Moreover, AhR regulates oxidative stress. When exposed to cigarette smoke, AhR-deficient lung cells show a higher production of reactive oxygen species and a lower expression of antioxidant enzymes (NQO1 and sulfiredoxin) than do lung cells with normal AhR levels, suggesting that cigarette smoke-induced oxidative stress is enhanced in AhR-deficient lungs [291].
AhR activation can influence the inflammatory phase in both asthma and chronic obstructive pulmonary disease through inflammatory and resident cells in the lungs [282,284]. The relation between AhR function and airway inflammation in the initial phase is important not only in chronic obstructive pulmonary disease but also in asthma. Airway Clara cells are sensitive to AhR activation by the ligand TCDD, and these cells are capable of secreting a wide range of glycoproteins such as mucins and SP-D [274,292]. TCDD upregulates inflammatory cytokines, COX2, MUC5AC, and MMPs via AhR signaling in a Clara cell-derived cell line. Research articles about AhR agonists and inhibitors have shown that AhR activation induces the production of such cytokines as TGF-α and tumor necrosis factor and of MMPs through receptors in human hematocytes and epithelial cells [282,292,293,294]. Thus, typical AhR xenobiotic ligands such as TCDD and BaP may contribute to the development of lung diseases.
Nevertheless, there are observations strongly indicating that AhR signaling can be beneficial in lung diseases mediated by inflammation and oxidative damage [295]. A deficiency of AhR signaling affects immune and nonimmune cells such as neutrophils, macrophages, and fibroblasts in the lungs, thereby resulting in greater pulmonary inflammation after exposure to tobacco smoke, lipopolysaccharide, and hyperoxia [296,297]. Conversely, AhR activation is known to reduce airway inflammation in rodent models of asthma by modulating the production and secretion of Th2 cytokines such as interleukin (IL) 4, IL-5, and IL-15 [298,299]. The activation of AhR by omeprazole alleviates lung inflammation in a model of acute hyperoxic lung injury based on adult mice, while neutrophil infiltration and MCP-1 expression are weaker as compared to vehicle-treated animals [300].
Because AhR takes part in the initiation of the aforementioned lung diseases, the development of biologics and small-molecule compounds that target the AhR signaling pathway is a possible approach to the prevention and treatment of these pathologies. For instance, AhR antagonist resveratrol has been shown to diminish mucin production [279]. AhR antagonist CH223191 attenuates BaP-induced allergic lung inflammation via AhR [301]. This AhR antagonist has also been reported to reverse experimental pulmonary hypertension induced by Sugen 5146 in rats [302].
5.4. Rheumatoid Arthritis
This is an autoimmune disease characterized by the chronic inflammation of the joints and the tissues around the joints along with infiltration by macrophages and activated T cells [126,303]. The pathogenesis of this disease is due to the formation of reactive oxygen and nitrogen species at the site of inflammation. The oxidative damage and inflammation have been confirmed in various rheumatic diseases by means of elevated levels of isoprostane and prostaglandins in serum and synovial fluid as compared with a control group [304]. Environmental factors, especially polycyclic aromatic hydrocarbons, are implicated in the pathogenesis of rheumatoid arthritis; hence, the role of AhR in this pathogenesis is of interest. Many papers indicate that polycyclic aromatic hydrocarbons play a critical part in the development of rheumatoid arthritis in various ways [305,306,307], primarily by influencing changes in the diversity of immune cells and the related downstream cytokines, and the main route of these effects goes through the AhR signaling pathway.
AhR, a major immunomodulator, is expressed in various cells (T and B cells, natural killers, macrophages, and dendritic cells) involved in rheumatoid arthritis [308]. Of note, in different cells implicated in rheumatoid arthritis, the effects of AhR activation are opposite. For instance, in Th1 and Th17 cells, B cells, dendritic cells, M1 monocytes, natural killers, and osteoclasts, AhR activation has an exacerbating impact on rheumatoid arthritis [309,310,311,312,313,314]; however, in Th2 cells, regulatory T cells, regulatory dendritic cells, M2 monocytes, and osteoblasts, AhR activation protects against rheumatoid arthritis [311,314,315,316].
AhR ligands such as 6-formylindolo[3,2-b]carbazole, TCDD and BaP relieve experimental arthritis; nonetheless, the long-term administration of these compounds has serious adverse effects such as high embryonic mortality, hepatotoxicity, and carcinogenicity [317,318,319], which limit the use of these compounds as therapeutic agents in animals or humans. AhR agonists with fewer adverse effects may be candidate therapeutics for rheumatoid arthritis [308]. Although much basic research has been conducted on the role of polycyclic aromatic hydrocarbon-activated AhR in rheumatoid arthritis, clinical studies on its effects on the mechanism of AhR signaling in rheumatoid arthritis are still lacking, and further research is needed.
5.5. Skin Diseases
These diseases may also be linked with environmental pollutants having high affinity for AhR [84,320]. Low doses of these compounds cause skin irritation or worsen symptoms of diseases [321]. Exposure to high doses of air pollutants leads to such skin pathologies as chloracne and hyperpigmentation [322,323,324,325]. AhR participates in many pathological processes in the skin through alterations in the signaling pathways controlled by AhR.
Early studies pointed to a partial role of AhR in the pathogenesis of various skin diseases, including inflammatory diseases, skin pigmentation disorders, and skin cancer [131,179,326], as well as a dependence of the outcome of AhR activation on the cell type and ligand [130,327]. Several papers offer evidence of AhR’s involvement in the pathogenesis of chloracne, hyperpigmentation, and vitiligo, as well as inflammatory diseases such as psoriasis and atopic dermatitis [116,328,329,330,331]. Additionally, many different biological responses to AhR stimulation or inhibition are observed in the skin [177].
On the one hand, the activation of AhR by ligands can induce the overexpression of proinflammatory cytokines and the production of reactive oxygen species, yielding an inflammatory disease or carcinogenesis [173]. On the other hand, AhR activity can influence the differentiation of regulatory T cells, thereby promoting immune tolerance [332]. A large class of tryptophan derivatives that are AhR ligands may play a part in the pathogenesis or treatment of many skin diseases [73,333]. Tryptophan derivatives are generated by enzymatic reactions or by exposure to ultraviolet light in various skin cells, and some of these compounds are present in herbs and plant extracts commonly used for skin care and therapies. Their biological activities require further research [100,334].
5.6. Nephropathies
Oxidative stress participates in various renal pathologies such as glomerulonephritis, tubulointerstitial nephritis, proteinuria, uremia, diabetic nephropathy, and chronic renal failure [335]. In chronic kidney disease, especially with uremia, oxidative stress can be explained by high pro-oxidant activity driving the overproduction of reactive oxygen species. The reasons for this phenomenon are complicated and multifactorial, and are mostly linked with elevated NOX activity, markedly upregulated xanthine oxidase, and concomitant mitochondrial dysfunction [336].
The roles of oxidative stress and inflammation in kidney failure lie in their influence on the most common disorders accompanying chronic kidney disease, which progress further via a positive-feedback mechanism, promoting an additional enhancement of oxidative stress and the progression of chronic kidney disease with a full range of complications [336]. Many authors have reported that AhR is associated with chronic kidney disease and its complications. A review of current knowledge on the participation of AhR in chronic kidney disease [337] shows that AhR mediates chronic kidney disease complications, including cardiovascular disorders, anemia, bone disorders, cognitive dysfunction, and malnutrition, and that AhR affects drug metabolism in patients with chronic kidney disease [337].
AhR also helps to reduce the harmful effects of uremic toxins by strengthening intestinal-barrier function [337,338]. In patients with chronic kidney disease, the key AhR ligands are the uremic toxins that arise during the metabolism of tryptophan, which is a precursor of a large number of microbial and host metabolites [338]. Derivatives of kynurenine, of serotonin, and of indole are (by-)products of the three main tryptophan metabolic pathways, which are directly or indirectly modulated by the gut microbiota [338]. The tryptophan metabolites indoxyl sulfate, indole-3-acetic acid, and kynurenine are not only important uremic toxins but are also potent AhR ligands [339,340,341]. Activated AhR is proven to exacerbate kidney damage [342,343,344]. An increase in AhR activity—in the periglomerular region and in proximal and distal renal tubules—under the action of adenine and indoxyl sulfate leads to renal fibrosis [345]. Indoxyl sulfate-activated AhR causes podocyte injury, progressive glomerular damage, and a proinflammatory phenotype [346]. Chronic kidney disease may be aggravated by indoxyl sulfate via the OAT3–AhR–STAT3 cascade in proximal tubular cells owing to the downregulation of a receptor called MAS, the subsequent inhibition of the renin–angiotensin system, and TGF-β activation [347]. Renal fibrosis may be mediated by AhR signaling [338,348].
AhR activity in patients with diabetes mellitus positively correlates with the progression of diabetic nephropathy and kidney failure severity [349]. A paper about the function of AhR in the pathophysiological processes of diabetic nephropathy (on the basis of an AhR knockout mouse model and a pharmacological inhibitor, α-naphthoflavone) has revealed that AhR mediates renal oxidative stress in a diabetic mouse model, thereby inducing infiltration by macrophages, extracellular matrix accumulation, and mesangial cell activation [350].
5.7. Cardiovascular Diseases
These diseases have a multifactorial etiology related to various risk factors, including but not limited to hypercholesterolemia, hypertension, tobacco smoking, diabetes mellitus, poor diet, stress, and lack of physical activity. Evidence has been published supporting the participation of oxidative stress in a number of cardiovascular disorders such as atherosclerosis, ischemia, hypertension, cardiomyopathy, cardiac hypertrophy, and congestive heart failure [351,352,353,354].
AhR is closely connected with cardiovascular diseases in terms of cardiac function, vascular development, and blood pressure regulation. In some diseases associated with atherosclerosis, AhR can serve as a transmitter of an oxidative stress signal [62,355,356].
There is a strong association between the accumulation of uremic toxic compounds and cardiovascular complications of chronic kidney disease [357]. AhR activation directly correlates with cardiovascular risk, even in the absence of uremia [358,359]. People exposed to AhR agonists are at an increased cardiovascular risk. To give an example, people who were exposed to TCDD as part of Agent Orange during the Vietnam War were more likely to experience relevant cardiovascular complications such as coronary heart disease and stroke [360].
Environmental pollutants that are AhR ligands promote the onset and progression of atherosclerosis, indicating that AhR may partake in the regulation of atherosclerosis [361,362,363]. Inflammatory responses contribute to AhR-regulated atherosclerosis. There are three hypotheses based on the AhR signaling pathways that mediate inflammation and promote atherosclerosis. The first hypothesis involves downstream inflammatory signaling factors such as VCAM-1 acting through the AhR–NF-κB signaling pathway, resulting in monocyte chemotaxis. Macrophages and monocytes are targets of polycyclic aromatic hydrocarbons, which are implicated in the physiological and pathological processes of atherosclerosis [364,365]. The second hypothesis postulates that AhR promotes absorption of oxidized low-density lipoprotein by macrophages giving rise to foam cells with the help of endogenous and exogenous ligands such as oxidized low-density lipoprotein, lipopolysaccharides, and TCDD. In vitro experiments have revealed that cholesterol accumulation in foam cells under the influence of particulate matter-induced inflammation is an early sign of cardiovascular diseases. Nonetheless, the suppressive action of AhR inhibitors on foam cells and on inflammation has not been investigated. It is believed that these mechanisms will be clarified by extensive studies on AhR [366,367]. The third hypothesis is the accelerated proliferation of vascular smooth muscle cells, which is key to the initiation of vascular complications according to the finding that indoxyl sulfate induces the proliferation of vascular smooth muscle cells through AhR activation, NF-κB signal transduction, and the production of reactive oxygen species [368]. It should be said that the division of the pathological involvement of AhR in the development of atherosclerosis into three hypotheses is largely arbitrary. These processes are related, and this division can only reflect the structuring and emphasis in future studies that should clarify the activity of this complex molecular network.
The regulation of AhR at the AhR transcription level in humans has not been elucidated yet. AhR is connected with other signaling pathways, including Wnt and E2 cascades, and further research is needed to clarify the function of AhR and identify new endogenous ligands in order to elucidate the role, regulation, and possible usefulness of AhR in the treatment of atherosclerosis. AhR is reported to be a major participant in the pathogenesis of such cardiovascular pathologies as myocarditis, hypertension, coronary heart disease, and pulmonary arterial hypertension [62]. The pathogenesis driven by AhR varies among cardiovascular diseases, but includes inflammatory responses, immune responses, oxidative stress, and endothelial dysfunction. The molecular mechanisms behind AhR signaling and behind the crosstalk between AhR signaling and other signal transduction cascades still require further investigation [62].
6. Conclusions
Major breakthroughs were recently made in the biology of redox modulation by AhR. Despite all the gained knowledge, the remaining intriguing questions concern the mechanism underlying the cell- and tissue-specific effects of AhR ligands and the dependence of responses on the type of ligands. The function of AhR is complicated because the outcome of its activation depends on a wide range of endogenous and exogenous ligands (which are characterized by different affinity values and diverse combinatorial effects) and on different AhR functions in many physiological and pathological processes in cells and tissues. The molecular mechanisms of AhR signaling and of the crosstalk between AhR signaling and other signal transduction cascades require further research. It is mostly the inconsistency of scientific findings that makes it difficult to determine the signaling pathways through which AhR can exert its beneficial or detrimental actions. There is growing evidence that AhR activation can have multidirectional effects on many aspects of human physiology and pathology, and that these may depend on cell and tissue types, or on the interaction of the AhR complex with non-traditional XRE sequences, or interaction with various coactivators and corepressors. Depending on many factors, the action of AhR agonists or antagonists can cause positive or negative effects on human health (Figure 4).
The recognition that AhR is implicated in the pathogenesis of many human diseases has arisen in conjunction with numerous examples of diseases in which AhR modulates disease activity through interaction with environmental factors. The pathogenesis driven by AhR often includes oxidative stress and immune and inflammatory responses. The weight of evidence indicates that, in diseases of various organs and tissues, AhR activation can be beneficial or detrimental. The ultimate effect depends both on the context of the disease and on the nature of AhR ligands. In this context, AhR activation aggravates the symptoms of some diseases, but alleviates the symptoms of other diseases.
Currently, in the literature, there are few examples of disorders where the molecular mechanisms of AhR’s involvement in the pathogenesis are clear. More numerous are findings about various biological responses to the stimulation or inhibition of AhR in various diseases. At the current stage of our insight into AhR’s biology and its role in the pathogenesis of diverse diseases, the utility of AhR as a therapeutic target has already been established, and a foundation has been laid for the selection and design of effective AhR ligands as new treatments of various diseases. Although much basic research has been conducted on the functions of AhR in pathological processes, clinical studies about the effects on the mechanism of the AhR signaling pathway in different pathologies are still scarce, and further investigation is necessary.
Author Contributions
Conceptualization, A.Y.G.; writing—original draft preparation, A.Y.G. and M.L.P.; writing—review and editing, A.Y.G. and M.L.P.; project administration, A.Y.G.; funding acquisition, A.Y.G.; All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by budgetary financing project FGMU-2022-0004, N1021050601082-2-1.6.4;3.1.6.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Shevchuk N.A. for language help and for proofreading the article.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AhR | aryl hydrocarbon receptor |
| AhRR | aryl hydrocarbon receptor repressor |
| AKR | aldo–keto reductase |
| ARE | antioxidant response element |
| ARNT | aryl hydrocarbon receptor nuclear translocator |
| BAFF | B-cell-activating factor of the tumor necrosis factor family |
| BaP | benzo[a]pyrene |
| bHLH | basic helix–loop–helix |
| BLC | B-lymphocyte chemoattractant |
| CCL1 | CC-chemokine ligand 1 |
| COX | cyclooxygenase |
| CYP | cytochrome P450 |
| E2F1 | E2 promoter binding factor 1 |
| EGF | epidermal growth factor |
| GPx | glutathione peroxidase |
| GST | glutathione S-transferase |
| HSP90 | heat shock protein 90 |
| IDO1 | indolamine-2,3-dioxygenase 1 |
| IFR3 | interferon responsive factor |
| IL | interleukin |
| Keap1 | Kelch-like ECH-associated protein 1 |
| KLF6 | Kruppel-like Factor 6 |
| MAPK | mitogen-activated protein kinase |
| MMP | matrix metalloproteinase |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NF-κB | nuclear factor κB |
| NOX | NADPH oxidase |
| NQO1 | NADPH:quinone oxidoreductase-1 |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PAS | Per–ARNT–Sim |
| pRB | retinoblastoma protein |
| TAD | transactivation domain |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TDP-43 | TAR DNA-binding protein 43 kDa |
| TGF | transforming growth factor |
| UGT | UDP glucuronosyltransferase |
| VEGF | vascular endothelial growth factor |
| XRE | xenobiotic-responsive element |
| XREL1 | XRE-like element |
References
- Pierre, S.; Chevallier, A.; Teixeira-Clerc, F.; Ambolet-Camoit, A.; Bui, L.C.; Bats, A.S.; Fournet, J.C.; Fernandez-Salguero, P.; Aggerbeck, M.; Lotersztajn, S.; et al. Aryl hydrocarbon receptor-dependent induction of liver fibrosis by dioxin. Toxicol. Sci. 2014, 137, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalkhof, S.; Dautel, F.; Loguercio, S.; Baumann, S.; Trump, S.; Jungnickel, H.; Otto, W.; Rudzok, S.; Potratz, S.; Luch, A.; et al. Pathway and time-resolved benzo[a]pyrene toxicity on Hepa1c1c7 cells at toxic and subtoxic exposure. J. Proteome Res. 2015, 14, 164–182. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Bargen, I.; Weighardt, H.; Haarmann-Stemmann, T.; Krutmann, J. Functions of the aryl hydrocarbon receptor in the skin. Semin. Immunopathol. 2013, 35, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Xue, C.H.; Hwang, S.K.; Li, W.H.; Chen, Z.; Zhang, J.Z. Exposure to fine particulate matter associated with senile lentigo in Chinese women: A cross-sectional study. J. Eur. Acad. Dermatol. Venereol 2017, 31, 355–360. [Google Scholar] [CrossRef]
- Guo, Y.L.; Yu, M.L.; Hsu, C.C.; Rogan, W.J. Chloracne, goiter, arthritis, and anemia after polychlorinated biphenyl poisoning: 14-year follow-Up of the Taiwan Yucheng cohort. Environ. Health Perspect. 1999, 107, 715–719. [Google Scholar] [CrossRef]
- Caputo, R.; Monti, M.; Ermacora, E.; Carminati, G.; Gelmetti, C.; Gianotti, R.; Gianni, E.; Puccinelli, V. Cutaneous manifestations of tetrachlorodibenzo-p-dioxin in children and adolescents. Follow-up 10 years after the Seveso, Italy, accident. J. Am. Acad. Dermatol. 1988, 19, 812–819. [Google Scholar] [CrossRef]
- Furue, M.; Uenotsuchi, T.; Urabe, K.; Ishikawa, T.; Kuwabara, M. Overview of Yusho. J. Dermatol. Sci. Suppl. 2005, 1, S3–S10. [Google Scholar] [CrossRef]
- Mitoma, C.; Mine, Y.; Utani, A.; Imafuku, S.; Muto, M.; Akimoto, T.; Kanekura, T.; Furue, M.; Uchi, H. Current skin symptoms of Yusho patients exposed to high levels of 2,3,4,7,8-pentachlorinated dibenzofuran and polychlorinated biphenyls in 1968. Chemosphere 2015, 137, 45–51. [Google Scholar] [CrossRef]
- Hu, T.; Pan, Z.; Yu, Q.; Mo, X.; Song, N.; Yan, M.; Zouboulis, C.C.; Xia, L.; Ju, Q. Benzo(a)pyrene induces interleukin (IL)-6 production and reduces lipid synthesis in human SZ95 sebocytes via the aryl hydrocarbon receptor signaling pathway. Environ. Toxicol. Pharmacol. 2016, 43, 54–60. [Google Scholar] [CrossRef]
- Sun, Y.V.; Boverhof, D.R.; Burgoon, L.D.; Fielden, M.R.; Zacharewski, T.R. Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucleic Acids Res. 2004, 32, 4512–4523. [Google Scholar] [CrossRef] [Green Version]
- Hidaka, T.; Ogawa, E.; Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Fujimura, T.; Aiba, S.; Nakayama, K.; Okuyama, R.; et al. The aryl hydrocarbon receptor AhR links atopic dermatitis and air pollution via induction of the neurotrophic factor artemin. Nat. Immunol. 2017, 18, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Schallreuter, K.U.; Salem, M.A.; Gibbons, N.C.; Maitland, D.J.; Marsch, E.; Elwary, S.M.; Healey, A.R. Blunted epidermal L-tryptophan metabolism in vitiligo affects immune response and ROS scavenging by Fenton chemistry, part 2: Epidermal H2O2/ONOO(-)-mediated stress in vitiligo hampers indoleamine 2,3-dioxygenase and aryl hydrocarbon receptor-mediated immune response signaling. FASEB J. 2012, 26, 2471–2485. [Google Scholar] [CrossRef] [PubMed]
- van den Bogaard, E.H.; Bergboer, J.G.; Vonk-Bergers, M.; van Vlijmen-Willems, I.M.; Hato, S.V.; van der Valk, P.G.; Schroder, J.M.; Joosten, I.; Zeeuwen, P.L.; Schalkwijk, J. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J. Clin. Investig. 2013, 123, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meglio, P.; Duarte, J.H.; Ahlfors, H.; Owens, N.D.; Li, Y.; Villanova, F.; Tosi, I.; Hirota, K.; Nestle, F.O.; Mrowietz, U.; et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity 2014, 40, 989–1001. [Google Scholar] [CrossRef] [Green Version]
- Di Meglio, P.; Perera, G.K.; Nestle, F.O. The multitasking organ: Recent insights into skin immune function. Immunity 2011, 35, 857–869. [Google Scholar] [CrossRef] [Green Version]
- Kostyuk, V.A.; Potapovich, A.I.; Lulli, D.; Stancato, A.; De Luca, C.; Pastore, S.; Korkina, L. Modulation of human keratinocyte responses to solar UV by plant polyphenols as a basis for chemoprevention of non-melanoma skin cancers. Curr. Med. Chem. 2013, 20, 869–879. [Google Scholar]
- Sheipouri, D.; Braidy, N.; Guillemin, G.J. Kynurenine Pathway in Skin Cells: Implications for UV-Induced Skin Damage. Int. J. Tryptophan Res. 2012, 5, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Podkowinska, A.; Formanowicz, D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants 2020, 9, 752. [Google Scholar] [CrossRef]
- Mo, Y.; Lu, Z.; Wang, L.; Ji, C.; Zou, C.; Liu, X. The Aryl Hydrocarbon Receptor in Chronic Kidney Disease: Friend or Foe? Front. Cell Dev. Biol. 2020, 8, 589752. [Google Scholar] [CrossRef]
- Liu, J.R.; Miao, H.; Deng, D.Q.; Vaziri, N.D.; Li, P.; Zhao, Y.Y. Gut microbiota-derived tryptophan metabolism mediates renal fibrosis by aryl hydrocarbon receptor signaling activation. Cell. Mol. Life Sci. 2021, 78, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Alkhalaf, L.M.; Ryan, K.S. Biosynthetic manipulation of tryptophan in bacteria: Pathways and mechanisms. Chem. Biol. 2015, 22, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, S.A.; Fu, J.; Chang, P.V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 19376–19387. [Google Scholar] [CrossRef] [PubMed]
- Huc, T.; Nowinski, A.; Drapala, A.; Konopelski, P.; Ufnal, M. Indole and indoxyl sulfate, gut bacteria metabolites of tryptophan, change arterial blood pressure via peripheral and central mechanisms in rats. Pharmacol. Res. 2018, 130, 172–179. [Google Scholar] [CrossRef]
- Lu, H.; Lei, X.; Klaassen, C. Gender differences in renal nuclear receptors and aryl hydrocarbon receptor in 5/6 nephrectomized rats. Kidney Int. 2006, 70, 1920–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, L.; Poitevin, S.; Sallee, M.; Addi, T.; Gondouin, B.; McKay, N.; Denison, M.S.; Jourde-Chiche, N.; Duval-Sabatier, A.; Cerini, C.; et al. Aryl hydrocarbon receptor is activated in patients and mice with chronic kidney disease. Kidney Int. 2018, 93, 986–999. [Google Scholar] [CrossRef]
- Kim, J.T.; Kim, S.H.; Min, H.K.; Jeon, S.J.; Sung, S.A.; Park, W.H.; Lee, H.K.; Choi, H.S.; Pak, Y.K.; Lee, S.Y. Effect of Dialysis on Aryl Hydrocarbon Receptor Transactivating Activity in Patients with Chronic Kidney Disease. Yonsei Med. J. 2020, 61, 56–63. [Google Scholar] [CrossRef]
- Walker, J.A.; Richards, S.; Belghasem, M.E.; Arinze, N.; Yoo, S.B.; Tashjian, J.Y.; Whelan, S.A.; Lee, N.; Kolachalama, V.B.; Francis, J.; et al. Temporal and tissue-specific activation of aryl hydrocarbon receptor in discrete mouse models of kidney disease. Kidney Int. 2020, 97, 538–550. [Google Scholar] [CrossRef]
- Ichii, O.; Otsuka-Kanazawa, S.; Nakamura, T.; Ueno, M.; Kon, Y.; Chen, W.; Rosenberg, A.Z.; Kopp, J.B. Podocyte injury caused by indoxyl sulfate, a uremic toxin and aryl-hydrocarbon receptor ligand. PLoS ONE 2014, 9, e108448. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.Y.; Yisireyili, M.; Saito, S.; Lee, C.T.; Adelibieke, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate downregulates expression of Mas receptor via OAT3/AhR/Stat3 pathway in proximal tubular cells. PLoS ONE 2014, 9, e91517. [Google Scholar] [CrossRef]
- Mutsaers, H.A.; Stribos, E.G.; Glorieux, G.; Vanholder, R.; Olinga, P. Chronic Kidney Disease and Fibrosis: The Role of Uremic Retention Solutes. Front. Med. 2015, 2, 60. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.T.; Kim, S.S.; Jun, D.W.; Hwang, Y.H.; Park, W.H.; Pak, Y.K.; Lee, H.K. Serum arylhydrocarbon receptor transactivating activity is elevated in type 2 diabetic patients with diabetic nephropathy. J. Diabetes Investig 2013, 4, 483–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.J.; Liu, S.H.; Chiang, C.K.; Lin, S.Y.; Liang, K.W.; Chen, C.H.; Tien, H.R.; Chen, P.H.; Wu, J.P.; Tsai, Y.C.; et al. Aryl Hydrocarbon Receptor Deficiency Attenuates Oxidative Stress-Related Mesangial Cell Activation and Macrophage Infiltration and Extracellular Matrix Accumulation in Diabetic Nephropathy. Antioxid. Redox Signal. 2016, 24, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Bahorun, T.; Soobrattee, M.A.; Luximon-Ramma, V.; Aruoma, O.I. Free Radicals and Antioxidants in Cardiovascular Health and Disease. Internet J. Med. Update. 2006, 1, 25–41. [Google Scholar] [CrossRef] [Green Version]
- Willcox, J.K.; Ash, S.L.; Catignani, G.L. Antioxidants and prevention of chronic disease. Crit. Rev. Food Sci. Nutr. 2004, 44, 275–295. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Saluja, R.; Kanneganti, S.; Chinta, S.; Dikshit, M. Biochemical and molecular evaluation of neutrophil NOS in spontaneously hypertensive rats. Cell. Mol. Biol. 2007, 53, 84–93. [Google Scholar]
- Ceriello, A. Possible role of oxidative stress in the pathogenesis of hypertension. Diabetes Care 2008, 31 (Suppl. 2), S181–S184. [Google Scholar] [CrossRef] [Green Version]
- Carreira, V.S.; Fan, Y.; Wang, Q.; Zhang, X.; Kurita, H.; Ko, C.I.; Naticchioni, M.; Jiang, M.; Koch, S.; Medvedovic, M.; et al. Ah Receptor Signaling Controls the Expression of Cardiac Development and Homeostasis Genes. Toxicol. Sci. 2015, 147, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N. The role of endogenous aryl hydrocarbon receptor signaling in cardiovascular physiology. J. Cardiovasc. Dis. Res. 2011, 2, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Vanholder, R.; Fouque, D.; Glorieux, G.; Heine, G.H.; Kanbay, M.; Mallamaci, F.; Massy, Z.A.; Ortiz, A.; Rossignol, P.; Wiecek, A.; et al. Clinical management of the uraemic syndrome in chronic kidney disease. Lancet Diabetes Endocrinol. 2016, 4, 360–373. [Google Scholar] [CrossRef]
- Sallee, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef] [PubMed]
- Humblet, O.; Birnbaum, L.; Rimm, E.; Mittleman, M.A.; Hauser, R. Dioxins and cardiovascular disease mortality. Environ. Health Perspect. 2008, 116, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, J. Agent Orange and heart disease: Is there a connection? FASEB J. 2014, 28, 1531–1533. [Google Scholar] [CrossRef] [PubMed]
- Oesterling, E.; Toborek, M.; Hennig, B. Benzo[a]pyrene induces intercellular adhesion molecule-1 through a caveolae and aryl hydrocarbon receptor mediated pathway. Toxicol. Appl. Pharmacol. 2008, 232, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, L.M.; Wan, M.; Ding, J.; Taylor, J.R.; Lohman, K.; Su, D.; Bennett, B.D.; Porter, D.K.; Gimple, R.; Pittman, G.S.; et al. DNA Methylation of the Aryl Hydrocarbon Receptor Repressor Associations With Cigarette Smoking and Subclinical Atherosclerosis. Circ. Cardiovasc. Genet. 2015, 8, 707–716. [Google Scholar] [CrossRef]
- Tsirpanlis, G. Cellular senescence, cardiovascular risk, and CKD: A review of established and hypothetical interconnections. Am. J. Kidney Dis. 2008, 51, 131–144. [Google Scholar] [CrossRef]
- Vogel, C.F.; Sciullo, E.; Li, W.; Wong, P.; Lazennec, G.; Matsumura, F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 2007, 21, 2941–2955. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.N.; Che, Y.; Yuan, Z.Y.; Lu, Z.L.; Li, Y.; Zhang, Z.R.; Li, N.; Li, R.D.; Wan, J.; Sun, H.D.; et al. Acetyl-macrocalin B suppresses tumor growth in esophageal squamous cell carcinoma and exhibits synergistic anti-cancer effects with the Chk1/2 inhibitor AZD7762. Toxicol. Appl. Pharmacol. 2019, 365, 71–83. [Google Scholar] [CrossRef]
- Vogel, C.F.; Sciullo, E.; Wong, P.; Kuzmicky, P.; Kado, N.; Matsumura, F. Induction of proinflammatory cytokines and C-reactive protein in human macrophage cell line U937 exposed to air pollution particulates. Environ. Health Perspect. 2005, 113, 1536–1541. [Google Scholar] [CrossRef]
- Hennig, B.; Meerarani, P.; Slim, R.; Toborek, M.; Daugherty, A.; Silverstone, A.E.; Robertson, L.W. Proinflammatory properties of coplanar PCBs: In vitro and in vivo evidence. Toxicol. Appl. Pharmacol. 2002, 181, 174–183. [Google Scholar] [CrossRef]
- Yisireyili, M.; Saito, S.; Abudureyimu, S.; Adelibieke, Y.; Ng, H.Y.; Nishijima, F.; Takeshita, K.; Murohara, T.; Niwa, T. Indoxyl sulfate-induced activation of (pro)renin receptor promotes cell proliferation and tissue factor expression in vascular smooth muscle cells. PLoS ONE 2014, 9, e109268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, K.; Meng, Q.; Zhang, Z.; Yi, T.; He, Y.; Zheng, J.; Lei, W. Aryl hydrocarbon receptor pathway: Role, regulation and intervention in atherosclerosis therapy (Review). Mol. Med. Rep. 2019, 20, 4763–4773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Hu, J.Y.; Wang, S.Q. The role of antioxidants in photoprotection: A critical review. J. Am. Acad. Dermatol. 2012, 67, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, E. Mitochondrial free radical production and cell signaling. Mol. Asp. Med. 2004, 25, 17–26. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Dandekar, A.; Mendez, R.; Zhang, K. Cross talk between ER stress, oxidative stress, and inflammation in health and disease. Methods Mol. Biol. 2015, 1292, 205–214. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Haghi Aminjan, H.; Abtahi, S.R.; Hazrati, E.; Chamanara, M.; Jalili, M.; Paknejad, B. Targeting of oxidative stress and inflammation through ROS/NF-kappaB pathway in phosphine-induced hepatotoxicity mitigation. Life Sci. 2019, 232, 116607. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Uchi, H.; Mitoma, C.; Hashimoto-Hachiya, A.; Chiba, T.; Ito, T.; Nakahara, T.; Tsuji, G. Antioxidants for Healthy Skin: The Emerging Role of Aryl Hydrocarbon Receptors and Nuclear Factor-Erythroid 2-Related Factor-2. Nutrients 2017, 9, 223. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Redman, R. Balancing the generation and elimination of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2005, 102, 3175–3176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curi, R.; Newsholme, P.; Marzuca-Nassr, G.N.; Takahashi, H.K.; Hirabara, S.M.; Cruzat, V.; Krause, M.; de Bittencourt, P.I., Jr. Regulatory principles in metabolism-then and now. Biochem. J. 2016, 473, 1845–1857. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [Green Version]
- To, E.E.; Vlahos, R.; Luong, R.; Halls, M.L.; Reading, P.C.; King, P.T.; Chan, C.; Drummond, G.R.; Sobey, C.G.; Broughton, B.R.S.; et al. Endosomal NOX2 oxidase exacerbates virus pathogenicity and is a target for antiviral therapy. Nat. Commun. 2017, 8, 69. [Google Scholar] [CrossRef]
- Zangar, R.C.; Davydov, D.R.; Verma, S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol. Appl. Pharmacol. 2004, 199, 316–331. [Google Scholar] [CrossRef]
- Agostinelli, E.; Tempera, G.; Viceconte, N.; Saccoccio, S.; Battaglia, V.; Grancara, S.; Toninello, A.; Stevanato, R. Potential anticancer application of polyamine oxidation products formed by amine oxidase: A new therapeutic approach. Amino Acids 2010, 38, 353–368. [Google Scholar] [CrossRef]
- Ng, M.P.; Lee, J.C.; Loke, W.M.; Yeo, L.L.; Quek, A.M.; Lim, E.C.; Halliwell, B.; Seet, R.C. Does influenza A infection increase oxidative damage? Antioxid. Redox Signal. 2014, 21, 1025–1031. [Google Scholar] [CrossRef] [Green Version]
- Masella, R.; Di Benedetto, R.; Vari, R.; Filesi, C.; Giovannini, C. Novel mechanisms of natural antioxidant compounds in biological systems: Involvement of glutathione and glutathione-related enzymes. J. Nutr. Biochem. 2005, 16, 577–586. [Google Scholar] [CrossRef]
- Gegotek, A.; Skrzydlewska, E. The role of transcription factor Nrf2 in skin cells metabolism. Arch. Dermatol. Res. 2015, 307, 385–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosh, B.E.; Hogenesch, J.B.; Bradfield, C.A. Mammalian Per-Arnt-Sim proteins in environmental adaptation. Annu. Rev. Physiol. 2010, 72, 625–645. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W. Aryl hydrocarbon receptor (AHR): “pioneer member” of the basic-helix/loop/helix per-Arnt-sim (bHLH/PAS) family of “sensors” of foreign and endogenous signals. Prog. Lipid Res. 2017, 67, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Schulte, K.W.; Green, E.; Wilz, A.; Platten, M.; Daumke, O. Structural Basis for Aryl Hydrocarbon Receptor-Mediated Gene Activation. Structure 2017, 25, 1025–1033 e1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolwick, K.M.; Schmidt, J.V.; Carver, L.A.; Swanson, H.I.; Bradfield, C.A. Cloning and expression of a human Ah receptor cDNA. Mol. Pharmacol. 1993, 44, 911–917. [Google Scholar]
- Kawajiri, K.; Fujii-Kuriyama, Y. The aryl hydrocarbon receptor: A multifunctional chemical sensor for host defense and homeostatic maintenance. Exp. Anim. 2017, 66, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, N.; Fukuda, K.; Nagata, Y.; Okada, H.; Haga, A.; Hatakeyama, S.; Yoshida, S.; Okamoto, T.; Hosaka, M.; Sekine, K.; et al. The activation mechanism of the aryl hydrocarbon receptor (AhR) by molecular chaperone HSP90. FEBS Open Bio 2014, 4, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, J.; Ihara, K.; Nakayama, H.; Hikino, S.; Satoh, K.; Kubo, N.; Iida, T.; Fujii, Y.; Hara, T. Characteristic expression of aryl hydrocarbon receptor repressor gene in human tissues: Organ-specific distribution and variable induction patterns in mononuclear cells. Life Sci. 2004, 74, 1039–1049. [Google Scholar] [CrossRef]
- Fujisawa-Sehara, A.; Sogawa, K.; Yamane, M.; Fujii-Kuriyama, Y. Characterization of xenobiotic responsive elements upstream from the drug-metabolizing cytochrome P-450c gene: A similarity to glucocorticoid regulatory elements. Nucleic Acids Res. 1987, 15, 4179–4191. [Google Scholar] [CrossRef] [Green Version]
- Fujii-Kuriyama, Y.; Kawajiri, K. Molecular mechanisms of the physiological functions of the aryl hydrocarbon (dioxin) receptor, a multifunctional regulator that senses and responds to environmental stimuli. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Gargaro, M.; Scalisi, G.; Manni, G.; Mondanelli, G.; Grohmann, U.; Fallarino, F. The Landscape of AhR Regulators and Coregulators to Fine-Tune AhR Functions. Int. J. Mol. Sci. 2021, 22, 757. [Google Scholar] [CrossRef] [PubMed]
- Abel, J.; Haarmann-Stemmann, T. An introduction to the molecular basics of aryl hydrocarbon receptor biology. Biol. Chem. 2010, 391, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.; Whitelaw, M.L. The emerging roles of AhR in physiology and immunity. Biochem. Pharmacol. 2013, 86, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Poland, A.; Palen, D.; Glover, E. Tumour promotion by TCDD in skin of HRS/J hairless mice. Nature 1982, 300, 271–273. [Google Scholar] [CrossRef]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef]
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, E.C.; Reyes, H.; Chu, F.F.; Sander, F.; Conley, L.H.; Brooks, B.A.; Hankinson, O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science 1991, 252, 954–958. [Google Scholar] [CrossRef]
- Sherr, D.H. Another important biological function for the aryl hydrocarbon receptor. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1247–1248. [Google Scholar] [CrossRef] [Green Version]
- Zablon, H.A.; Ko, C.I.; Puga, A. Converging Roles of the Aryl Hydrocarbon Receptor in Early Embryonic Development, Maintenance of Stemness, and Tissue Repair. Toxicol. Sci. 2021, 182, 1–9. [Google Scholar] [CrossRef]
- Schneider, A.J.; Branam, A.M.; Peterson, R.E. Intersection of AHR and Wnt signaling in development, health, and disease. Int. J. Mol. Sci. 2014, 15, 17852–17885. [Google Scholar] [CrossRef] [Green Version]
- Guarnieri, T.; Abruzzo, P.M.; Bolotta, A. More than a cell biosensor: Aryl hydrocarbon receptor at the intersection of physiology and inflammation. Am. J. Physiol. Cell Physiol. 2020, 318, C1078–C1082. [Google Scholar] [CrossRef] [PubMed]
- Mulero-Navarro, S.; Fernandez-Salguero, P.M. New Trends in Aryl Hydrocarbon Receptor Biology. Front. Cell Dev. Biol. 2016, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Lamorte, S.; Shinde, R.; McGaha, T.L. Nuclear receptors, the aryl hydrocarbon receptor, and macrophage function. Mol. Asp. Med. 2021, 78, 100942. [Google Scholar] [CrossRef] [PubMed]
- Kung, T.; Murphy, K.A.; White, L.A. The aryl hydrocarbon receptor (AhR) pathway as a regulatory pathway for cell adhesion and matrix metabolism. Biochem. Pharmacol. 2009, 77, 536–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, K.W. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-mediated deregulation of myeloid and sebaceous gland stem/progenitor cell homeostasis. Arch. Toxicol. 2017, 91, 2295–2301. [Google Scholar] [CrossRef]
- Casado, F.L. The Aryl Hydrocarbon Receptor Relays Metabolic Signals to Promote Cellular Regeneration. Stem Cells Int. 2016, 2016, 4389802. [Google Scholar] [CrossRef] [Green Version]
- Zaragoza-Ojeda, M.; Apatiga-Vega, E.; Arenas-Huertero, F. Role of aryl hydrocarbon receptor in central nervous system tumors: Biological and therapeutic implications. Oncol. Lett. 2021, 21, 460. [Google Scholar] [CrossRef]
- Bock, K.W. Aryl hydrocarbon receptor (AHR): From selected human target genes and crosstalk with transcription factors to multiple AHR functions. Biochem. Pharmacol. 2019, 168, 65–70. [Google Scholar] [CrossRef]
- de Tomaso Portaz, A.C.; Caimi, G.R.; Sanchez, M.; Chiappini, F.; Randi, A.S.; Kleiman de Pisarev, D.L.; Alvarez, L. Hexachlorobenzene induces cell proliferation, and aryl hydrocarbon receptor expression (AhR) in rat liver preneoplastic foci, and in the human hepatoma cell line HepG2. AhR is a mediator of ERK1/2 signaling, and cell cycle regulation in HCB-treated HepG2 cells. Toxicology 2015, 336, 36–47. [Google Scholar] [CrossRef]
- Barhoover, M.A.; Hall, J.M.; Greenlee, W.F.; Thomas, R.S. Aryl hydrocarbon receptor regulates cell cycle progression in human breast cancer cells via a functional interaction with cyclin-dependent kinase 4. Mol. Pharmacol. 2010, 77, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Nebert, D.W.; Roe, A.L.; Dieter, M.Z.; Solis, W.A.; Yang, Y.; Dalton, T.P. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem. Pharmacol. 2000, 59, 65–85. [Google Scholar] [CrossRef]
- Hatherell, S.; Baltazar, M.T.; Reynolds, J.; Carmichael, P.L.; Dent, M.; Li, H.; Ryder, S.; White, A.; Walker, P.; Middleton, A.M. Identifying and Characterizing Stress Pathways of Concern for Consumer Safety in Next-Generation Risk Assessment. Toxicol. Sci. 2020, 176, 11–33. [Google Scholar] [CrossRef] [PubMed]
- Formosa, R.; Borg, J.; Vassallo, J. Aryl hydrocarbon receptor (AHR) is a potential tumour suppressor in pituitary adenomas. Endocr. Relat. Cancer 2017, 24, 445–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Zhou, K.; Lu, S.; Bai, Y.; Qi, R.; Zhang, S. Modulation of aryl hydrocarbon receptor inhibits esophageal squamous cell carcinoma progression by repressing COX2/PGE2/STAT3 axis. J. Cell Commun. Signal. 2020, 14, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Naseri-Nosar, P.; Nogalski, M.T.; Shenk, T. The aryl hydrocarbon receptor facilitates the human cytomegalovirus-mediated G1/S block to cell cycle progression. Proc. Natl. Acad. Sci. USA 2021, 118, e2026336118. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Seyedhosseini, F.S.; Behnampour, N.; Yazdani, Y. Indole-3-carbinol induces G1 cell cycle arrest and apoptosis through aryl hydrocarbon receptor in THP-1 monocytic cell line. J. Recept. Signal. Transduct. Res. 2017, 37, 506–514. [Google Scholar] [CrossRef]
- Sahebnasagh, A.; Hashemi, J.; Khoshi, A.; Saghafi, F.; Avan, R.; Faramarzi, F.; Azimi, S.; Habtemariam, S.; Sureda, A.; Khayatkashani, M.; et al. Aromatic hydrocarbon receptors in mitochondrial biogenesis and function. Mitochondrion 2021, 61, 85–101. [Google Scholar] [CrossRef]
- Chopra, M.; Schrenk, D. Dioxin toxicity, aryl hydrocarbon receptor signaling, and apoptosis-persistent pollutants affect programmed cell death. Crit. Rev. Toxicol. 2011, 41, 292–320. [Google Scholar] [CrossRef]
- Zhang, W.; Xie, H.Q.; Li, Y.; Zhou, M.; Zhou, Z.; Wang, R.; Hahn, M.E.; Zhao, B. The aryl hydrocarbon receptor: A predominant mediator for the toxicity of emerging dioxin-like compounds. J. Hazard. Mater. 2022, 426, 128084. [Google Scholar] [CrossRef]
- Duarte-Hospital, C.; Tete, A.; Brial, F.; Benoit, L.; Koual, M.; Tomkiewicz, C.; Kim, M.J.; Blanc, E.B.; Coumoul, X.; Bortoli, S. Mitochondrial Dysfunction as a Hallmark of Environmental Injury. Cells 2021, 11, 110. [Google Scholar] [CrossRef]
- Gearhart-Serna, L.M.; Davis, J.B.; Jolly, M.K.; Jayasundara, N.; Sauer, S.J.; Di Giulio, R.T.; Devi, G.R. A polycyclic aromatic hydrocarbon-enriched environmental chemical mixture enhances AhR, antiapoptotic signaling and a proliferative phenotype in breast cancer cells. Carcinogenesis 2020, 41, 1648–1659. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Vazquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Ebert, B.; Seidel, A.; Lampen, A. Identification of BCRP as transporter of benzo[a]pyrene conjugates metabolically formed in Caco-2 cells and its induction by Ah-receptor agonists. Carcinogenesis 2005, 26, 1754–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, T.; Wang, J.; Zhu, K.; Tang, Y.; Huang, S.; Shui, X.; Ding, Y.; Chen, C.; Lei, W. Aryl Hydrocarbon Receptor: A New Player of Pathogenesis and Therapy in Cardiovascular Diseases. Biomed. Res. Int. 2018, 2018, 6058784. [Google Scholar] [CrossRef] [PubMed]
- Poland, A.; Knutson, J.C. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: Examination of the mechanism of toxicity. Annu. Rev. Pharmacol. Toxicol. 1982, 22, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Quattrochi, L.C.; Tukey, R.H. Nuclear uptake of the Ah (dioxin) receptor in response to omeprazole: Transcriptional activation of the human CYP1A1 gene. Mol. Pharmacol. 1993, 43, 504–508. [Google Scholar]
- Ciolino, H.P.; Daschner, P.J.; Yeh, G.C. Dietary flavonols quercetin and kaempferol are ligands of the aryl hydrocarbon receptor that affect CYP1A1 transcription differentially. Biochem. J. 1999, 340 Pt 3, 715–722. [Google Scholar] [CrossRef]
- Goya-Jorge, E.; Jorge Rodriguez, M.E.; Veitia, M.S.; Giner, R.M. Plant Occurring Flavonoids as Modulators of the Aryl Hydrocarbon Receptor. Molecules 2021, 26, 2315. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [Green Version]
- Phelan, D.; Winter, G.M.; Rogers, W.J.; Lam, J.C.; Denison, M.S. Activation of the Ah receptor signal transduction pathway by bilirubin and biliverdin. Arch. Biochem. Biophys. 1998, 357, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schaldach, C.M.; Riby, J.; Bjeldanes, L.F. Lipoxin A4: A new class of ligand for the Ah receptor. Biochemistry 1999, 38, 7594–7600. [Google Scholar] [CrossRef] [PubMed]
- Stejskalova, L.; Dvorak, Z.; Pavek, P. Endogenous and exogenous ligands of aryl hydrocarbon receptor: Current state of art. Curr. Drug Metab. 2011, 12, 198–212. [Google Scholar] [CrossRef] [Green Version]
- Wirthgen, E.; Hoeflich, A.; Rebl, A.; Gunther, J. Kynurenic Acid: The Janus-Faced Role of an Immunomodulatory Tryptophan Metabolite and Its Link to Pathological Conditions. Front. Immunol. 2017, 8, 1957. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, A.; Wincent, E.; Vikstrom Bergander, L.; Alsberg, T.; Bergman, J.; Rannug, A.; Rannug, U. Evidence for New Light-Independent Pathways for Generation of the Endogenous Aryl Hydrocarbon Receptor Agonist FICZ. Chem. Res. Toxicol. 2016, 29, 75–86. [Google Scholar] [CrossRef]
- Jin, U.H.; Lee, S.O.; Sridharan, G.; Lee, K.; Davidson, L.A.; Jayaraman, A.; Chapkin, R.S.; Alaniz, R.; Safe, S. Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Mol. Pharmacol. 2014, 85, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.A., 3rd. Expression of the human aryl hydrocarbon receptor complex in yeast. Activation of transcription by indole compounds. J. Biol. Chem. 1997, 272, 32824–32829. [Google Scholar] [CrossRef] [Green Version]
- Minzaghi, D.; Pavel, P.; Dubrac, S. Xenobiotic Receptors and Their Mates in Atopic Dermatitis. Int. J. Mol. Sci. 2019, 20, 4234. [Google Scholar] [CrossRef] [Green Version]
- Guzman-Navarro, G.; Leon, M.B.; Martin-Estal, I.; Duran, R.C.; Villarreal-Alvarado, L.; Vaquera-Vazquez, A.; Cuevas-Cerda, T.; Garza-Garcia, K.; Cuervo-Perez, L.E.; Barbosa-Quintana, A.; et al. Prenatal indole-3-carbinol administration activates aryl hydrocarbon receptor-responsive genes and attenuates lung injury in a bronchopulmonary dysplasia model. Exp. Biol. Med. 2021, 246, 695–706. [Google Scholar] [CrossRef]
- Moorthy, B.; Chu, C.; Carlin, D.J. Polycyclic aromatic hydrocarbons: From metabolism to lung cancer. Toxicol. Sci. 2015, 145, 5–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.; Klapczynski, A.; Kuch, N.; Arpino, F.; Simon-Keller, K.; De La Torre, C.; Sticht, C.; van Abeelen, F.A.; Oversluizen, G.; Gretz, N. Gene expression profiling reveals aryl hydrocarbon receptor as a possible target for photobiomodulation when using blue light. Sci. Rep. 2016, 6, 33847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dere, E.; Lee, A.W.; Burgoon, L.D.; Zacharewski, T.R. Differences in TCDD-elicited gene expression profiles in human HepG2, mouse Hepa1c1c7 and rat H4IIE hepatoma cells. BMC Genom. 2011, 12, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, E.A.; McCulloch, C.; Koganti, A.; Goodwin, S.B.; Sutter, T.R.; Silkworth, J.B. Divergent transcriptomic responses to aryl hydrocarbon receptor agonists between rat and human primary hepatocytes. Toxicol. Sci. 2009, 112, 257–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmahin, R.; Crump, D.; O'Brien, J.M.; Jones, S.P.; Kennedy, S.W. Time-dependent transcriptomic and biochemical responses of 6-formylindolo[3,2-b]carbazole (FICZ) and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) are explained by AHR activation time. Biochem. Pharmacol. 2016, 115, 134–143. [Google Scholar] [CrossRef]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Walker, M.K. Crosstalk between the aryl hydrocarbon receptor and hypoxia on the constitutive expression of cytochrome P4501A1 mRNA. Cardiovasc. Toxicol. 2007, 7, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.V.; Bradfield, C.A. Ah receptor signaling pathways. Annu. Rev. Cell Dev. Biol. 1996, 12, 55–89. [Google Scholar] [CrossRef] [Green Version]
- Denison, M.S.; Fisher, J.M.; Whitlock, J.P., Jr. The DNA recognition site for the dioxin-Ah receptor complex. Nucleotide sequence and functional analysis. J. Biol. Chem. 1988, 263, 17221–17224. [Google Scholar] [CrossRef]
- Soshilov, A.A.; Motta, S.; Bonati, L.; Denison, M.S. Transitional States in Ligand-Dependent Transformation of the Aryl Hydrocarbon Receptor into Its DNA-Binding Form. Int. J. Mol. Sci. 2020, 21, 2474. [Google Scholar] [CrossRef] [Green Version]
- Puga, A.; Barnes, S.J.; Dalton, T.P.; Chang, C.; Knudsen, E.S.; Maier, M.A. Aromatic hydrocarbon receptor interaction with the retinoblastoma protein potentiates repression of E2F-dependent transcription and cell cycle arrest. J. Biol. Chem. 2000, 275, 2943–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, R.E.; Hwang, K.A.; Choi, K.C. Cytochrome P450 1 family and cancers. J. Steroid Biochem. Mol. Biol. 2015, 147, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, N.; Baba, K.; Gion, K.; Sugiyama, C.; Taniura, H.; Yoneda, Y. Xenobiotic response element binding enriched in both nuclear and microsomal fractions of rat cerebellum. J. Neurochem. 2003, 85, 264–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denison, M.S.; Faber, S.C. And Now for Something Completely Different: Diversity in Ligand-Dependent Activation of Ah Receptor Responses. Curr. Opin. Toxicol. 2017, 2, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.R.; Joshi, A.D.; Elferink, C.J. The tumor suppressor Kruppel-like factor 6 is a novel aryl hydrocarbon receptor DNA binding partner. J. Pharmacol. Exp. Ther. 2013, 345, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.; Wu, D.; Goth, S.R.; Baek, J.; Lollies, A.; Domhardt, R.; Grindel, A.; Pessah, I.N. Aryl hydrocarbon receptor signaling regulates NF-kappaB RelB activation during dendritic-cell differentiation. Immunol. Cell Biol. 2013, 91, 568–575. [Google Scholar] [CrossRef]
- Jackson, D.P.; Joshi, A.D.; Elferink, C.J. Ah Receptor Pathway Intricacies; Signaling Through Diverse Protein Partners and DNA-Motifs. Toxicol. Res. 2015, 4, 1143–1158. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, Y.; Kado, S.Y.; Hoeper, C.; Harel, S.; Vogel, C.F.A. Role of NF-kB RelB in Aryl Hydrocarbon Receptor-Mediated Ligand Specific Effects. Int. J. Mol. Sci. 2019, 20, 2652. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.W.; Gazourian, L.; Quadri, S.A.; Romieu-Mourez, R.; Sherr, D.H.; Sonenshein, G.E. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene 2000, 19, 5498–5506. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A.; Naka, T.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9721–9726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szelest, M.; Walczak, K.; Plech, T. A New Insight into the Potential Role of Tryptophan-Derived AhR Ligands in Skin Physiological and Pathological Processes. Int. J. Mol. Sci. 2021, 22, 1104. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Natividad, J.M.; Sokol, H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol. 2018, 11, 1024–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeager, R.L.; Reisman, S.A.; Aleksunes, L.M.; Klaassen, C.D. Introducing the “TCDD-inducible AhR-Nrf2 gene battery”. Toxicol. Sci. 2009, 111, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Wakabayashi, N.; Misra, V.; Biswal, S.; Lee, G.H.; Agoston, E.S.; Yamamoto, M.; Kensler, T.W. NRF2 modulates aryl hydrocarbon receptor signaling: Influence on adipogenesis. Mol. Cell. Biol. 2007, 27, 7188–7197. [Google Scholar] [CrossRef] [Green Version]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar] [CrossRef] [Green Version]
- Swedenborg, E.; Pongratz, I. AhR and ARNT modulate ER signaling. Toxicology 2010, 268, 132–138. [Google Scholar] [CrossRef]
- Watabe, Y.; Nazuka, N.; Tezuka, M.; Shimba, S. Aryl hydrocarbon receptor functions as a potent coactivator of E2F1-dependent trascription activity. Biol. Pharm. Bull. 2010, 33, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Levine-Fridman, A.; Chen, L.; Elferink, C.J. Cytochrome P4501A1 promotes G1 phase cell cycle progression by controlling aryl hydrocarbon receptor activity. Mol. Pharmacol. 2004, 65, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Ge, N.L.; Elferink, C.J. A direct interaction between the aryl hydrocarbon receptor and retinoblastoma protein. Linking dioxin signaling to the cell cycle. J. Biol. Chem. 1998, 273, 22708–22713. [Google Scholar] [CrossRef] [Green Version]
- Marlowe, J.L.; Knudsen, E.S.; Schwemberger, S.; Puga, A. The aryl hydrocarbon receptor displaces p300 from E2F-dependent promoters and represses S phase-specific gene expression. J. Biol. Chem. 2004, 279, 29013–29022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, B.P.; Vorderstrasse, B.A. New insights into the aryl hydrocarbon receptor as a modulator of host responses to infection. Semin. Immunopathol. 2013, 35, 615–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Frauenstein, K.; Tigges, J.; Soshilov, A.A.; Kado, S.; Raab, N.; Fritsche, E.; Haendeler, J.; Denison, M.S.; Vogel, C.F.; Haarmann-Stemmann, T. Activation of the aryl hydrocarbon receptor by the widely used Src family kinase inhibitor 4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrimidine (PP2). Arch. Toxicol. 2015, 89, 1329–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backlund, M.; Johansson, I.; Mkrtchian, S.; Ingelman-Sundberg, M. Signal transduction-mediated activation of the aryl hydrocarbon receptor in rat hepatoma H4IIE cells. J. Biol. Chem. 1997, 272, 31755–31763. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, F. The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochem. Pharmacol. 2009, 77, 608–626. [Google Scholar] [CrossRef]
- Canga, L.; Paroli, L.; Blanck, T.J.; Silver, R.B.; Rifkind, A.B. 2,3,7,8-tetrachlorodibenzo-p-dioxin increases cardiac myocyte intracellular calcium and progressively impairs ventricular contractile responses to isoproterenol and to calcium in chick embryo hearts. Mol. Pharmacol. 1993, 44, 1142–1151. [Google Scholar]
- Dong, H.; Bonala, R.R.; Suzuki, N.; Johnson, F.; Grollman, A.P.; Shibutani, S. Mutagenic potential of benzo[a] pyrene-derived DNA adducts positioned in codon 273 of the human P53 gene. Biochemistry 2004, 43, 15922–15928. [Google Scholar] [CrossRef]
- Landvik, N.E.; Arlt, V.M.; Nagy, E.; Solhaug, A.; Tekpli, X.; Schmeiser, H.H.; Refsnes, M.; Phillips, D.H.; Lagadic-Gossmann, D.; Holme, J.A. 3-Nitrobenzanthrone and 3-aminobenzanthrone induce DNA damage and cell signalling Hepa1c1c7 cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2010, 684, 11–23. [Google Scholar] [CrossRef]
- Phillips, T.D.; Richardson, M.; Cheng, Y.S.L.; He, L.Y.; McDonald, T.J.; Cizmas, L.H.; Safe, S.H.; Donnelly, K.C.; Wang, F.; Moorthy, B.; et al. Mechanistic relationships between hepatic genotoxicity and carcinogenicity in male B6C3F1 mice treated with polycyclic aromatic hydrocarbon mixtures. Arch. Toxicol. 2015, 89, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Rossner, P.; Strapacova, S.; Stolcpartova, J.; Schmuczerova, J.; Milcova, A.; Neca, J.; Vlkova, V.; Brzicova, T.; Machala, M.; Topinka, J. Toxic Effects of the Major Components of Diesel Exhaust in Human Alveolar Basal Epithelial Cells (A549). Int. J. Mol. Sci. 2016, 17, 1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef] [PubMed]
- Burczynski, M.E.; Lin, H.K.; Penning, T.M. Isoform-specific induction of a human aldo-keto reductase by polycyclic aromatic hydrocarbons (PAHs), electrophiles, and oxidative stress: Implications for the alternative pathway of PAH activation catalyzed by human dihydrodiol dehydrogenase. Cancer Res. 1999, 59, 607–614. [Google Scholar] [PubMed]
- Hockley, S.L.; Arlt, V.M.; Brewer, D.; Giddings, I.; Phillips, D.H. Time- and concentration-dependent changes in gene expression induced by benzo(a)pyrene in two human cell lines, MCF-7 and HepG2. BMC Genom. 2006, 7, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, N.; Kanno, Y.; Saito, N.; Terai, K.; Sanada, N.; Kizu, R.; Hiruta, N.; Park, Y.; Bujo, H.; Nemoto, K. Aryl hydrocarbon receptor counteracts pharmacological efficacy of doxorubicin via enhanced AKR1C3 expression in triple negative breast cancer cells. Biochem. Biophys. Res. Commun. 2019, 516, 693–698. [Google Scholar] [CrossRef]
- Albertolle, M.E.; Guengerich, F.P. The relationships between cytochromes P450 and H2O2: Production, reaction, and inhibition. J. Inorg. Biochem. 2018, 186, 228–234. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of Cytochrome P450s in the Generation and Metabolism of Reactive Oxygen Species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef]
- Kuthan, H.; Ullrich, V. Oxidase and Oxygenase Function of the Microsomal Cytochrome-P450 Mono-Oxygenase System. Eur. J. Biochem. 1982, 126, 583–588. [Google Scholar] [CrossRef]
- Kukielka, E.; Cederbaum, A.I. Nadph-Dependent and Nadh-Dependent Oxygen Radical Generation by Rat-Liver Nuclei in the Presence of Redox Cycling Agents and Iron. Arch. Biochem. Biophys. 1990, 283, 326–333. [Google Scholar] [CrossRef]
- Wincent, E.; Bengtsson, J.; Bardbori, A.M.; Alsberg, T.; Luecke, S.; Rannug, U.; Rannug, A. Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 4479–4484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, G.; Takahara, M.; Uchi, H.; Takeuchi, S.; Mitoma, C.; Moroi, Y.; Furue, M. An environmental contaminant, benzo(a)pyrene, induces oxidative stress-mediated interleukin-8 production in human keratinocytes via the aryl hydrocarbon receptor signaling pathway. J. Dermatol. Sci. 2011, 62, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Haarmann-Stemmann, T.; Bothe, H.; Abel, J. Growth factors, cytokines and their receptors as downstream targets of arylhydrocarbon receptor (AhR) signaling pathways. Biochem. Pharmacol. 2009, 77, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Puga, A.; Ma, C.; Marlowe, J.L. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem. Pharmacol. 2009, 77, 713–722. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Smith, J.; Neupane, R.; McAmis, W.; Singh, U.; Chatterjee, S.; Raychoudhury, S. Toxicity of polycyclic aromatic hydrocarbons involves NOX2 activation. Toxicol. Rep. 2019, 6, 1176–1181. [Google Scholar] [CrossRef]
- Pinel-Marie, M.L.; Sparfel, L.; Desmots, S.; Fardel, O. Aryl hydrocarbon receptor-dependent induction of the NADPH oxidase subunit NCF1/p47(phox) expression leading to priming of human macrophage oxidative burst. Free Radic. Biol. Med. 2009, 47, 825–834. [Google Scholar] [CrossRef]
- Lee, C.W.; Lin, Z.C.; Hu, S.C.S.; Chiang, Y.C.; Hsu, L.F.; Lin, Y.C.; Lee, I.T.; Tsai, M.H.; Fang, J.Y. Urban particulate matter down-regulates filaggrin via COX2 expression/PGE2 production leading to skin barrier dysfunction. Sci. Rep. 2016, 6, 27995. [Google Scholar] [CrossRef] [Green Version]
- Amara, N.; Bachoual, R.; Desmard, M.; Golda, S.; Guichard, C.; Lanone, S.; Aubier, M.; Ogier-Denis, E.; Boczkowski, J. Diesel exhaust particles induce matrix metalloprotease-1 in human lung epithelial cells via a NADP(H) oxidase/NOX4 redox-dependent mechanism. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2007, 293, L170–L181. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Kang, K.A.; Piao, M.J.; Ahn, M.J.; Yi, J.M.; Hyun, Y.M.; Kim, S.H.; Ko, M.K.; Park, C.O.; Hyun, J.W. Particulate matter induces inflammatory cytokine production via activation of NF kappa B by TLR5-NOX4-ROS signaling in human skin keratinocyte and mouse skin. Redox Biol. 2019, 21, 101080. [Google Scholar] [CrossRef]
- Wada, T.; Sunaga, H.; Ohkawara, R.; Shimba, S. Aryl Hydrocarbon Receptor Modulates NADPH Oxidase Activity via Direct Transcriptional Regulation of p40(phox) Expression. Mol. Pharmacol. 2013, 83, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B. Redox Signaling Across Cell Membranes. Antioxid. Redox Sign 2009, 11, 1349–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiszt, M.; Leto, T.L. The Nox family of NAD(P)H oxidases: Host defense and beyond. J. Biol. Chem. 2004, 279, 51715–51718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leavey, P.J.; Gonzalez-Aller, C.; Thurman, G.; Kleinberg, M.; Rinckel, L.; Ambruso, D.W.; Freeman, S.; Kuypers, F.A.; Ambruso, D.R. A 29-kDa protein associated with p67phox expresses both peroxiredoxin and phospholipase A2 activity and enhances superoxide anion production by a cell-free system of NADPH oxidase activity. J. Biol. Chem. 2002, 277, 45181–45187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennig, B.; Hammock, B.D.; Slim, R.; Toborek, M.; Saraswathi, V.; Robertson, L.W. PCB-induced oxidative stress in endothelial cells: Modulation by nutrients. Int. J. Hyg. Environ. Health 2002, 205, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, I.; Tatebe, J.; Namba, S.; Koizumi, M.; Yamazaki, J.; Morita, T. Activation of Aryl Hydrocarbon Receptor Mediates Indoxyl Sulfate-Induced Monocyte Chemoattractant Protein-1 Expression in Human Umbilical Vein Endothelial Cells. Circ. J. 2013, 77, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Masai, N.; Tatebe, J.; Yoshino, G.; Morita, T. Indoxyl Sulfate Stimulates Monocyte Chemoattractant Protein-1 Expression in Human Umbilical Vein Endothelial Cells by Inducing Oxidative Stress Through Activation of the NADPH Oxidase-Nuclear Factor-kappa B Pathway. Circ. J. 2010, 74, 2216–2224. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi-Bardbori, A.; Bergander, L.V.; Rannug, U.; Rannug, A. NADPH Oxidase-Dependent Mechanism Explains How Arsenic and Other Oxidants Can Activate Aryl Hydrocarbon Receptor Signaling. Chem. Res. Toxicol. 2015, 28, 2278–2286. [Google Scholar] [CrossRef]
- Anwar-Mohamed, A.; Elshenawy, O.H.; Soshilov, A.A.; Denison, M.S.; Le, X.C.; Klotz, L.O.; El-Kadi, A.O.S. Methylated pentavalent arsenic metabolites are bifunctional inducers, as they induce cytochrome P450 1A1 and NAD(P)H:quinone oxidoreductase through AhR- and Nrf2-dependent mechanisms. Free Radic. Biol. Med. 2014, 67, 171–187. [Google Scholar] [CrossRef]
- Wu, J.P.; Chang, L.W.; Yao, H.T.; Chang, H.; Tsai, H.T.; Tsai, M.H.; Yeh, T.K.; Lin, P. Involvement of Oxidative Stress and Activation of Aryl Hydrocarbon Receptor in Elevation of CYP1A1 Expression and Activity in Lung Cells and Tissues by Arsenic: An In Vitro and In Vivo Study. Toxicol. Sci. 2009, 107, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Kubli, S.P.; Bassi, C.; Roux, C.; Wakeham, A.; Gobl, C.; Zhou, W.J.; Jafari, S.M.; Snow, B.; Jones, L.; Palomero, L.; et al. AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3604–3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seibert, K.; Zhang, Y.; Leahy, K.; Hauser, S.; Masferrer, J.; Perkins, W.; Lee, L.; Isakson, P. Pharmacological and Biochemical Demonstration of the Role of Cyclooxygenase-2 in Inflammation and Pain. Proc. Natl. Acad. Sci. USA 1994, 91, 12013–12017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, T.; Gupta, S.; Mukhtar, H. Cyclooxygenase-2 and prostate carcinogenesis. Cancer Lett. 2003, 191, 125–135. [Google Scholar] [CrossRef]
- Eling, T.E.; Thompson, D.C.; Foureman, G.L.; Curtis, J.F.; Hughes, M.F. Prostaglandin-H Synthase and Xenobiotic Oxidation. Annu. Rev. Pharmacol. 1990, 30, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Freyberger, A.; Schnitzler, R.; Schiffmann, D.; Degen, G.H. Prostaglandin-H-synthase competent cells derived from ram seminal vesicles: A tool for studying cooxidation of xenobiotics. Mol. Toxicol. 1987, 1, 503–512. [Google Scholar] [PubMed]
- Vogel, C.; Schuhmacher, U.S.; Degen, G.H.; Bolt, H.M.; Pineau, T.; Abel, J. Modulation of prostaglandin H synthase-2 mRNA expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice. Arch. Biochem. Biophys. 1998, 351, 265–271. [Google Scholar] [CrossRef]
- Vogel, C. Prostaglandin H synthases and their importance in chemical toxicity. Curr. Drug Metab. 2000, 1, 391–404. [Google Scholar] [CrossRef]
- Jones, D.A.; Carlton, D.P.; Mcintyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Molecular-Cloning of Human Prostaglandin Endoperoxide Synthase Type-Ii and Demonstration of Expression in Response to Cytokines. J. Biol. Chem. 1993, 268, 9049–9054. [Google Scholar] [CrossRef]
- Seibert, K.; Masferrer, J.L. Role of Inducible Cyclooxygenase (Cox-2) in Inflammation. Receptor 1994, 4, 17–23. [Google Scholar]
- Prescott, S.M.; Fitzpatrick, F.A. Cyclooxygellase-2 and carcinogenesis. BBA-Rev. Cancer 2000, 1470, M69–M78. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Li, W.; Sciullo, E.; Newman, J.; Hammock, B.; Reader, J.R.; Tuscano, J.; Matsumura, F. Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression. Am. J. Pathol. 2007, 171, 1538–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, S.K.; Sharma, R.A.; Steward, W.P.; Mellon, J.K.; Griffiths, T.R.L.; Gescher, A.J. Oxidative stress and cyclooxygenase activity in prostate carcinogenesis: Targets for chemopreventive strategies. Eur. J. Cancer 2005, 41, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Boerboom, A.M.J.F.; Baechle, C.; El-Bahay, C.; Kahl, R.; Degen, G.H.; Abel, J. Regulation of prostaglandin endoperoxide H synthase-2 induction by dioxin in rat hepatocytes: Possible c-Src-mediated pathway. Carcinogenesis 2000, 21, 2267–2274. [Google Scholar] [CrossRef]
- Fritsche, E.; Schafer, C.; Calles, C.; Bernsmann, T.; Bernshausen, T.; Wurm, M.; Hubenthal, U.; Cline, J.E.; Hajimiragha, H.; Schroeder, P.; et al. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc. Natl. Acad. Sci. USA 2007, 104, 8851–8856. [Google Scholar] [CrossRef] [Green Version]
- Munoz, M.; Sanchez, A.; Martinez, M.P.; Benedito, S.; Lopez-Oliva, M.E.; Garcia-Sacristan, A.; Hernandez, M.; Prieto, D. COX-2 is involved in vascular oxidative stress and endothelial dysfunction of renal interlobar arteries from obese Zucker rats. Free Radic. Biol. Med. 2015, 84, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Hernanz, R.; Briones, A.M.; Alonso, M.A.J.; Vila, E.; Salaices, M. Hypertension alters role of iNOS, COX-2, and oxidative stress in bradykinin relaxation impairment after LPS in rat cerebral arteries. Am. J. Physiol.-Circ. Physiol. 2004, 287, H225–H234. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M. Human Aldo-Keto Reductases and the Metabolic Activation of Polycyclic Aromatic Hydrocarbons. Chem. Res. Toxicol. 2014, 27, 1901–1917. [Google Scholar] [CrossRef]
- Park, J.H.; Troxel, A.B.; Harvey, R.G.; Penning, T.M. Polycyclic aromatic hydrocarbon (PAH) o-quinones produced by the aldo-keto-reductases (AKRs) generate abasic sites, oxidized pyrimidines, and 8-oxo-dGuo via reactive oxygen species. Chem. Res. Toxicol. 2006, 19, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Burczynski, M.E.; Harvey, R.G.; Penning, T.M. Expression and characterization of four recombinant human dihydrodiol dehydrogenase isoforms: Oxidation of trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene to the activated o-quinone metabolite benzo[a]pyrene-7,8-dione (vol 37, pg 6781, 1998). Biochemistry 1999, 38, 10626. [Google Scholar] [CrossRef] [Green Version]
- Palackal, N.T.; Burczynski, M.E.; Harvey, R.G.; Penning, T.M. The ubiquitous aldehyde reductase (AKR1A1) oxidizes proximate carcinogen trans-dihydrodiols to o-quinones: Potential role in polycyclic aromatic hydrocarbon activation. Biochemistry 2001, 40, 10901–10910. [Google Scholar] [CrossRef]
- Gelboin, H.V. Benzo[a]Pyrene Metabolism, Activation, and Carcinogenesis—Role and Regulation of Mixed-Function Oxidases and Related Enzymes. Physiol. Rev. 1980, 60, 1107–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penning, T.M.; Ohnishi, S.T.; Ohnishi, T.; Harvey, R.G. Generation of reactive oxygen species during the enzymatic oxidation of polycyclic aromatic hydrocarbon trans-dihydrodiols catalyzed by dihydrodiol dehydrogenase. Chem. Res. Toxicol. 1996, 9, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Shou, M.; Harvey, R.G.; Penning, T.M. Reactivity of Benzo[a]Pyrene-7,8-Dione with DNA—Evidence for the Formation of Deoxyguanosine Adducts. Carcinogenesis 1993, 14, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Shultz, C.A.; Quinn, A.M.; Park, J.H.; Harvey, R.G.; Bolton, J.L.; Maser, E.; Penning, T.M. Specificity of Human Aldo-Keto Reductases, NAD(P)H:Quinone Oxidoreductase, and Carbonyl Reductases to Redox-Cycle Polycyclic Aromatic Hydrocarbon Diones and 4-Hydroxyequilenin-o-quinone. Chem. Res. Toxicol. 2011, 24, 2153–2166. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, G.; Takahara, M.; Uchi, H.; Matsuda, T.; Chiba, T.; Takeuchi, S.; Yasukawa, F.; Moroi, Y.; Furue, M. Identification of ketoconazole as an AhR-Nrf2 activator in cultured human keratinocytes: The basis of its anti-inflammatory effect. J. Investig. Dermatol. 2012, 132, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Takei, K.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Nakahara, T.; Furue, M. Cynaropicrin attenuates UVB-induced oxidative stress via the AhR-Nrf2-Nqo1 pathway. Toxicol. Lett. 2015, 234, 74–80. [Google Scholar] [CrossRef]
- Takei, K.; Mitoma, C.; Hashimoto-Hachiya, A.; Uchi, H.; Takahara, M.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T.; Furue, M. Antioxidant soybean tar Glyteer rescues T-helper-mediated downregulation of filaggrin expression via aryl hydrocarbon receptor. J. Dermatol. 2015, 42, 171–180. [Google Scholar] [CrossRef]
- Nakahara, T.; Mitoma, C.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Uchi, H.; Yan, X.; Hachisuka, J.; Chiba, T.; Esaki, H.; et al. Antioxidant Opuntia ficus-indica Extract Activates AHR-NRF2 Signaling and Upregulates Filaggrin and Loricrin Expression in Human Keratinocytes. J. Med. Food 2015, 18, 1143–1149. [Google Scholar] [CrossRef]
- Haarmann-Stemmann, T.; Abel, J.; Fritsche, E.; Krutmann, J. The AhR-Nrf2 pathway in keratinocytes: On the road to chemoprevention? J. Investig. Dermatol. 2012, 132, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef]
- Niestroy, J.; Barbara, A.; Herbst, K.; Rode, S.; van Liempt, M.; Roos, P.H. Single and concerted effects of benzo[a]pyrene and flavonoids on the AhR and Nrf2-pathway in the human colon carcinoma cell line Caco-2. Toxicol. In Vitro 2011, 25, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Han, S.G.; Han, S.S.; Toborek, M.; Hennig, B. EGCG protects endothelial cells against PCB 126-induced inflammation through inhibition of AhR and induction of Nrf2-regulated genes. Toxicol. Appl. Pharmacol. 2012, 261, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Rojo de la Vega, M.; Schmidlin, C.J.; Ooi, A.; Zhang, D.D. Kelch-like ECH-associated protein 1 (KEAP1) differentially regulates nuclear factor erythroid-2-related factors 1 and 2 (NRF1 and NRF2). J. Biol. Chem. 2018, 293, 2029–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Li, W.; Su, Z.Y.; Kong, A.N. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef]
- Blank, V. Small Maf proteins in mammalian gene control: Mere dimerization partners or dynamic transcriptional regulators? J. Mol. Biol. 2008, 376, 913–925. [Google Scholar] [CrossRef]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [CrossRef]
- Telakowski-Hopkins, C.A.; King, R.G.; Pickett, C.B. Glutathione S-transferase Ya subunit gene: Identification of regulatory elements required for basal level and inducible expression. Proc. Natl. Acad. Sci. USA 1988, 85, 1000–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Kumar, H.; Kim, I.S.; More, S.V.; Kim, B.W.; Choi, D.K. Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Nat. Prod. Rep. 2014, 31, 109–139. [Google Scholar] [CrossRef] [PubMed]
- Corenblum, M.J.; Ray, S.; Remley, Q.W.; Long, M.; Harder, B.; Zhang, D.D.; Barnes, C.A.; Madhavan, L. Reduced Nrf2 expression mediates the decline in neural stem cell function during a critical middle-age period. Aging Cell 2016, 15, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- La Rosa, P.; Russo, M.; D'Amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D.; Turchi, R.; Bertini, E.S.; Piemonte, F. Nrf2 Induction Re-establishes a Proper Neuronal Differentiation Program in Friedreich's Ataxia Neural Stem Cells. Front. Cell Neurosci. 2019, 13, 356. [Google Scholar] [CrossRef] [Green Version]
- Robledinos-Anton, N.; Rojo, A.I.; Ferreiro, E.; Nunez, A.; Krause, K.H.; Jaquet, V.; Cuadrado, A. Transcription factor NRF2 controls the fate of neural stem cells in the subgranular zone of the hippocampus. Redox Biol. 2017, 13, 393–401. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Kohle, C.; Bock, K.W. Coordinate regulation of Phase I and II xenobiotic metabolisms by the Ah receptor and Nrf2. Biochem. Pharmacol. 2007, 73, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Kinneer, K.; Bi, Y.; Chan, J.Y.; Kan, Y.W. Induction of murine NAD(P)H:quinone oxidoreductase by 2,3,7,8-tetrachlorodibenzo-p-dioxin requires the CNC (cap 'n' collar) basic leucine zipper transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2): Cross-interaction between AhR (aryl hydrocarbon receptor) and Nrf2 signal transduction. Biochem. J. 2004, 377, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta 2003, 1619, 263–268. [Google Scholar] [CrossRef]
- Amakura, Y.; Tsutsumi, T.; Nakamura, M.; Kitagawa, H.; Fujino, J.; Sasaki, K.; Yoshida, T.; Toyoda, M. Preliminary screening of the inhibitory effect of food extracts on activation of the aryl hydrocarbon receptor induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biol. Pharm. Bull. 2002, 25, 272–274. [Google Scholar] [CrossRef] [Green Version]
- Amakura, Y.; Tsutsumi, T.; Sasaki, K.; Yoshida, T.; Maitani, T. Screening of the inhibitory effect of vegetable constituents on the aryl hydrocarbon receptor-mediated activity induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biol. Pharm. Bull. 2003, 26, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amakura, Y.; Tsutsumi, T.; Sasaki, K.; Nakamura, M.; Yoshida, T.; Maitani, T. Influence of food polyphenols on aryl hydrocarbon receptor-signaling pathway estimated by in vitro bioassay. Phytochemistry 2008, 69, 3117–3130. [Google Scholar] [CrossRef]
- Furue, M.; Fuyuno, Y.; Mitoma, C.; Uchi, H.; Tsuji, G. Therapeutic Agents with AHR Inhibiting and NRF2 Activating Activity for Managing Chloracne. Antioxidants 2018, 7, 90. [Google Scholar] [CrossRef] [Green Version]
- Marchand, A.; Barouki, R.; Garlatti, M. Regulation of NAD(P)H:quinone oxidoreductase 1 gene expression by CYP1A1 activity. Mol. Pharmacol. 2004, 65, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Gomez, M.; Kwak, M.K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Dinkova-Kostova, A.T.; Talalay, P. Powerful and prolonged protection of human retinal pigment epithelial cells, keratinocytes, and mouse leukemia cells against oxidative damage: The indirect antioxidant effects of sulforaphane. Proc. Natl. Acad. Sci. USA 2001, 98, 15221–15226. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef] [PubMed]
- Favreau, L.V.; Pickett, C.B. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. Identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J. Biol. Chem. 1991, 266, 4556–4561. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxidative Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, T.; Naito, Y. What Is Oxidative Stress? J. Jpn. Med. Assoc. 2002, 45, 271–276. [Google Scholar]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar]
- Neavin, D.R.; Liu, D.; Ray, B.; Weinshilboum, R.M. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int. J. Mol. Sci. 2018, 19, 3851. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Gemma, C.; Vila, J.; Bachstetter, A.; Bickford, P.C. Oxidative Stress and the Aging Brain: From Theory to Prevention. In Brain Aging: Models, Methods, and Mechanisms; Riddle, D.R., Ed.; Frontiers in Neuroscience: Boca Raton, FL, USA, 2007. [Google Scholar]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Belanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of Mitochondrial Function Impairs Lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [Green Version]
- Lefaki, M.; Papaevgeniou, N.; Chondrogianni, N. Redox regulation of proteasome function. Redox Biol. 2017, 13, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Dalton, T.P.; Puga, A.; Shertzer, H.G. Induction of cellular oxidative stress by aryl hydrocarbon receptor activation. Chem. Biol. Interact. 2002, 141, 77–95. [Google Scholar] [CrossRef]
- Liu, H.; Shi, L.; Giesy, J.P.; Yu, H. Polychlorinated diphenyl sulfides can induce ROS and genotoxicity via the AhR-CYP1A1 pathway. Chemosphere 2019, 223, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Rannug, A.; Ahlbom, E.; Hakansson, H.; Ceccatelli, S. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the expression of cytochrome P450 1A1, the aryl hydrocarbon receptor, and the aryl hydrocarbon receptor nuclear translocator in rat brain and pituitary. Toxicol. Appl. Pharmacol. 2000, 169, 159–167. [Google Scholar] [CrossRef]
- Bansal, S.; Leu, A.N.; Gonzalez, F.J.; Guengerich, F.P.; Chowdhury, A.R.; Anandatheerthavarada, H.K.; Avadhani, N.G. Mitochondrial targeting of cytochrome P450 (CYP) 1B1 and its role in polycyclic aromatic hydrocarbon-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 9936–9951. [Google Scholar] [CrossRef] [Green Version]
- Nebert, D.W.; Karp, C.L. Endogenous functions of the aryl hydrocarbon receptor (AHR): Intersection of cytochrome P450 1 (CYP1)-metabolized eicosanoids and AHR biology. J. Biol. Chem. 2008, 283, 36061–36065. [Google Scholar] [CrossRef] [Green Version]
- Perepechaeva, M.L.; Grishanova, A.Y. The Role of Aryl Hydrocarbon Receptor (AhR) in Brain Tumors. Int. J. Mol. Sci. 2020, 21, 2863. [Google Scholar] [CrossRef] [Green Version]
- Kolluri, S.K.; Jin, U.H.; Safe, S. Erratum to: Role of the aryl hydrocarbon receptor in carcinogenesis and potential as an anti-cancer drug target. Arch. Toxicol. 2017, 91, 3209. [Google Scholar] [CrossRef]
- Park, H.; Jin, U.H.; Karki, K.; Jayaraman, A.; Allred, C.; Michelhaugh, S.K.; Mittal, S.; Chapkin, R.S.; Safe, S. Dopamine is an aryl hydrocarbon receptor agonist. Biochem. J. 2020, 477, 3899–3910. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, W.J.; Quan, W.; Qiao, C.M.; Cui, C.; Hong, H.; Shi, Y.; Niu, G.Y.; Zhao, L.P.; Shen, Y.Q. Dynamic changes of activated AHR in microglia and astrocytes in the substantia nigra-striatum system in an MPTP-induced Parkinson's disease mouse model. Brain Res. Bull. 2021, 176, 174–183. [Google Scholar] [CrossRef]
- Ramos-Garcia, N.A.; Orozco-Ibarra, M.; Estudillo, E.; Elizondo, G.; Gomez Apo, E.; Chavez Macias, L.G.; Sosa-Ortiz, A.L.; Torres-Ramos, M.A. Aryl Hydrocarbon Receptor in Post-Mortem Hippocampus and in Serum from Young, Elder, and Alzheimer's Patients. Int. J. Mol. Sci. 2020, 21, 1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutierrez-Vazquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Ojo, E.S.; Tischkau, S.A. The Role of AhR in the Hallmarks of Brain Aging: Friend and Foe. Cells 2021, 10, 2729. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, M.; Malek, G. The Aryl Hydrocarbon Receptor: A Mediator and Potential Therapeutic Target for Ocular and Non-Ocular Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6777. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef]
- Rath, S.N.; Jena, L.; Patri, M. Understanding ligands driven mechanism of wild and mutant aryl hydrocarbon receptor in presence of phytochemicals combating Parkinson's disease: An in silico and in vivo study. J. Biomol. Struct. Dyn. 2020, 38, 807–826. [Google Scholar] [CrossRef]
- Datla, K.P.; Christidou, M.; Widmer, W.W.; Rooprai, H.K.; Dexter, D.T. Tissue distribution and neuroprotective effects of citrus flavonoid tangeretin in a rat model of Parkinson's disease. Neuroreport 2001, 12, 3871–3875. [Google Scholar] [CrossRef]
- Gonzalez-Barbosa, E.; Garcia-Aguilar, R.; Vega, L.; Cabanas-Cortes, M.A.; Gonzalez, F.J.; Segovia, J.; Morales-Lazaro, S.L.; Cisneros, B.; Elizondo, G. Parkin is transcriptionally regulated by the aryl hydrocarbon receptor: Impact on alpha-synuclein protein levels. Biochem. Pharmacol. 2019, 168, 429–437. [Google Scholar] [CrossRef]
- Ogura, J.; Miyauchi, S.; Shimono, K.; Yang, S.; Gonchigar, S.; Ganapathy, V.; Bhutia, Y.D. Carbidopa is an activator of aryl hydrocarbon receptor with potential for cancer therapy. Biochem. J. 2017, 474, 3391–3402. [Google Scholar] [CrossRef]
- Qian, C.; Yang, C.; Lu, M.; Bao, J.; Shen, H.; Deng, B.; Li, S.; Li, W.; Zhang, M.; Cao, C. Activating AhR alleviates cognitive deficits of Alzheimer's disease model mice by upregulating endogenous Abeta catabolic enzyme Neprilysin. Theranostics 2021, 11, 8797–8812. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, S.; Liang, H.; Xing, Z.; Guo, L.; Shi, L.; Du, L.; Kuang, C.; Takikawa, O.; Yang, Q. Amyloid beta neurotoxicity is IDO1-Kyn-AhR dependent and blocked by IDO1 inhibitor. Signal. Transduct. Target. Ther. 2020, 5, 96. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M. Smoking: Effects on multiple sclerosis susceptibility and disease progression. Ther. Adv. Neurol Disord. 2012, 5, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeilinger, S.; Kuhnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.A.; Stanford, E.A.; Al Abdulatif, A.; Ramirez-Cardenas, A.; Ballance, H.I.; Boudeau, S.; Jeh, A.; Murithi, J.M.; Tripodis, Y.; Murphy, G.J.; et al. Dioxins and related environmental contaminants increase TDP-43 levels. Mol. Neurodegener. 2017, 12, 35. [Google Scholar] [CrossRef]
- Rothhammer, V.; Borucki, D.M.; Kenison, J.E.; Hewson, P.; Wang, Z.; Bakshi, R.; Sherr, D.H.; Quintana, F.J. Detection of aryl hydrocarbon receptor agonists in human samples. Sci. Rep. 2018, 8, 4970. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, M.A.; Quintana, F.J. Regulation of Astrocyte Functions in Multiple Sclerosis. Cold Spring Harb Perspect. Med. 2019, 9. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef] [Green Version]
- Quintana, F.J. Regulation of central nervous system autoimmunity by the aryl hydrocarbon receptor. Semin. Immunopathol. 2013, 35, 627–635. [Google Scholar] [CrossRef] [Green Version]
- Hanieh, H. Toward understanding the role of aryl hydrocarbon receptor in the immune system: Current progress and future trends. Biomed. Res. Int. 2014, 2014, 520763. [Google Scholar] [CrossRef] [Green Version]
- Duarte, J.H.; Di Meglio, P.; Hirota, K.; Ahlfors, H.; Stockinger, B. Differential influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PLoS ONE 2013, 8, e79819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaye, J.; Piryatinsky, V.; Birnberg, T.; Hingaly, T.; Raymond, E.; Kashi, R.; Amit-Romach, E.; Caballero, I.S.; Towfic, F.; Ator, M.A.; et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2016, 113, E6145–E6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.R.; Rademakers, R. The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 2008, 21, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soshilov, A.A.; Denison, M.S. Ligand promiscuity of aryl hydrocarbon receptor agonists and antagonists revealed by site-directed mutagenesis. Mol. Cell. Biol. 2014, 34, 1707–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santosa, S.; Jones, P.J. Oxidative stress in ocular disease: Does lutein play a protective role? CMAJ 2005, 173, 861–862. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.H.; Sekundo, W. Nutritional supplementation to prevent cataract formation. Dev. Ophthalmol. 2005, 38, 103–119. [Google Scholar] [CrossRef]
- Beatty, S.; Koh, H.; Phil, M.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef] [Green Version]
- Hammond, C.L.; Roztocil, E.; Gupta, V.; Feldon, S.E.; Woeller, C.F. More than Meets the Eye: The Aryl Hydrocarbon Receptor is an Environmental Sensor, Physiological Regulator and a Therapeutic Target in Ocular Disease. Front. Toxicol. 2022, 4, 791082. [Google Scholar] [CrossRef]
- Vasiliou, V.; Gonzalez, F.J. Role of CYP1B1 in glaucoma. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 333–358. [Google Scholar] [CrossRef]
- Oki, N.O.; Edwards, S.W. An integrative data mining approach to identifying adverse outcome pathway signatures. Toxicology 2016, 350–352, 49–61. [Google Scholar] [CrossRef]
- Reis, L.M.; Tyler, R.C.; Weh, E.; Hendee, K.E.; Kariminejad, A.; Abdul-Rahman, O.; Ben-Omran, T.; Manning, M.A.; Yesilyurt, A.; McCarty, C.A.; et al. Analysis of CYP1B1 in pediatric and adult glaucoma and other ocular phenotypes. Mol. Vis. 2016, 22, 1229–1238. [Google Scholar] [PubMed]
- Volotinen, M.; Maenpaa, J.; Kankuri, E.; Oksala, O.; Pelkonen, O.; Nakajima, M.; Yokoi, T.; Hakkola, J. Expression of cytochrome P450 (CYP) enzymes in human nonpigmented ciliary epithelial cells: Induction of CYP1B1 expression by TCDD. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Yang, H.J.; Chang, Y.S.; Kim, J.W.; Brooks, M.; Chew, E.Y.; Wong, W.T.; Fariss, R.N.; Rachel, R.A.; Cogliati, T.; et al. Deletion of aryl hydrocarbon receptor AHR in mice leads to subretinal accumulation of microglia and RPE atrophy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6031–6040. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, M.; Kazmin, D.; Hu, P.; Thomas, R.S.; McDonnell, D.P.; Malek, G. Aryl hydrocarbon receptor knock-out exacerbates choroidal neovascularization via multiple pathogenic pathways. J. Pathol. 2015, 235, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, A.; Takeuchi, M.; Oikawa, K.; Sonoda, K.H.; Usui, Y.; Okunuki, Y.; Takeda, A.; Oshima, Y.; Yoshida, K.; Usui, M.; et al. Effects of dioxin on vascular endothelial growth factor (VEGF) production in the retina associated with choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3410–3416. [Google Scholar] [CrossRef]
- Perepechaeva, M.L.; Kolosova, N.G.; Stefanova, N.A.; Fursova, A.; Grishanova, A.Y. The influence of changes in expression of redox-sensitive genes on the development of retinopathy in rats. Exp. Mol. Pathol. 2016, 101, 124–132. [Google Scholar] [CrossRef]
- Perepechaeva, M.L.; Grishanova, A.Y.; Rudnitskaya, E.A.; Kolosova, N.G. The Mitochondria-Targeted Antioxidant SkQ1 Downregulates Aryl Hydrocarbon Receptor-Dependent Genes in the Retina of OXYS Rats with AMD-Like Retinopathy. J. Ophthalmol. 2014, 2014, 530943. [Google Scholar] [CrossRef] [Green Version]
- Perepechaeva, M.L.; Stefanova, N.A.; Grishanova, A.Y. Expression of genes for AhR and Nrf2 signal pathways in the retina of OXYS rats during the development of retinopathy and melatonin-induced changes in this process. Bull. Exp. Biol. Med. 2014, 157, 424–429. [Google Scholar] [CrossRef]
- Gutierrez, M.A.; Davis, S.S.; Rosko, A.; Nguyen, S.M.; Mitchell, K.P.; Mateen, S.; Neves, J.; Garcia, T.Y.; Mooney, S.; Perdew, G.H.; et al. A novel AhR ligand, 2AI, protects the retina from environmental stress. Sci. Rep. 2016, 6, 29025. [Google Scholar] [CrossRef]
- Khan, A.S.; Wolf, A.; Langmann, T. The AhR ligand 2, 2′-aminophenyl indole (2AI) regulates microglia homeostasis and reduces pro-inflammatory signaling. Biochem. Biophys. Res. Commun. 2021, 579, 15–21. [Google Scholar] [CrossRef]
- Nadeem, A.; Masood, A.; Siddiqui, N. Oxidant--antioxidant imbalance in asthma: Scientific evidence, epidemiological data and possible therapeutic options. Ther. Adv. Respir. Dis. 2008, 2, 215–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.F.; Ward, P.A. Role of oxidants in lung injury during sepsis. Antioxid. Redox Signal. 2007, 9, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Mishima, M. Redox-based therapeutics for lung diseases. Antioxid. Redox Signal. 2008, 10, 701–704. [Google Scholar] [CrossRef] [PubMed]
- MacNee, W. Oxidative stress and lung inflammation in airways disease. Eur. J. Pharmacol. 2001, 429, 195–207. [Google Scholar] [CrossRef]
- White, K.E.; Ding, Q.; Moore, B.B.; Peters-Golden, M.; Ware, L.B.; Matthay, M.A.; Olman, M.A. Prostaglandin E2 mediates IL-1beta-related fibroblast mitogenic effects in acute lung injury through differential utilization of prostanoid receptors. J. Immunol. 2008, 180, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Lora, J.M.; Zhang, D.M.; Liao, S.M.; Burwell, T.; King, A.M.; Barker, P.A.; Singh, L.; Keaveney, M.; Morgenstern, J.; Gutierrez-Ramos, J.C.; et al. Tumor necrosis factor-alpha triggers mucus production in airway epithelium through an IkappaB kinase beta-dependent mechanism. J. Biol. Chem. 2005, 280, 36510–36517. [Google Scholar] [CrossRef] [Green Version]
- Perrais, M.; Pigny, P.; Copin, M.C.; Aubert, J.P.; Van Seuningen, I. Induction of MUC2 and MUC5AC mucins by factors of the epidermal growth factor (EGF) family is mediated by EGF receptor/Ras/Raf/extracellular signal-regulated kinase cascade and Sp1. J. Biol. Chem. 2002, 277, 32258–32267. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.H.; Trigg, C.J.; Devalia, J.L.; Jordan, S.; Davies, R.J. Effect of inhaled beclomethasone dipropionate on expression of proinflammatory cytokines and activated eosinophils in the bronchial epithelium of patients with mild asthma. J. Allergy Clin. Immunol. 1994, 94, 1025–1034. [Google Scholar] [CrossRef]
- Shivanna, B.; Chu, C.; Moorthy, B. The Aryl Hydrocarbon Receptor (AHR): A Novel Therapeutic Target for Pulmonary Diseases? Int. J. Mol. Sci. 2022, 23, 1516. [Google Scholar] [CrossRef]
- Chiba, T.; Uchi, H.; Tsuji, G.; Gondo, H.; Moroi, Y.; Furue, M. Arylhydrocarbon receptor (AhR) activation in airway epithelial cells induces MUC5AC via reactive oxygen species (ROS) production. Pulm. Pharmacol. Ther. 2011, 24, 133–140. [Google Scholar] [CrossRef]
- Alexandrov, K.; Rojas, M.; Satarug, S. The critical DNA damage by benzo(a)pyrene in lung tissues of smokers and approaches to preventing its formation. Toxicol. Lett. 2010, 198, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Beamer, C.A.; Shepherd, D.M. Role of the aryl hydrocarbon receptor (AhR) in lung inflammation. Semin. Immunopathol. 2013, 35, 693–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, T.; Chihara, J.; Furue, M. Role of the Arylhydrocarbon Receptor (AhR) in the Pathology of Asthma and COPD. J. Allergy 2012, 2012, 372384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zago, M.; Sheridan, J.A.; Traboulsi, H.; Hecht, E.; Zhang, Y.; Guerrina, N.; Matthews, J.; Nair, P.; Eidelman, D.H.; Hamid, Q.; et al. Low levels of the AhR in chronic obstructive pulmonary disease (COPD)-derived lung cells increases COX-2 protein by altering mRNA stability. PLoS ONE 2017, 12, e0180881. [Google Scholar] [CrossRef] [Green Version]
- Ying, S.; Meng, Q.; Zeibecoglou, K.; Robinson, D.S.; Macfarlane, A.; Humbert, M.; Kay, A.B. Eosinophil chemotactic chemokines (eotaxin, eotaxin-2, RANTES, monocyte chemoattractant protein-3 (MCP-3), and MCP-4), and C-C chemokine receptor 3 expression in bronchial biopsies from atopic and nonatopic (Intrinsic) asthmatics. J. Immunol. 1999, 163, 6321–6329. [Google Scholar]
- Yao, H.; Rahman, I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol. Appl. Pharmacol. 2011, 254, 72–85. [Google Scholar] [CrossRef] [Green Version]
- van Eeden, S.F.; Sin, D.D. Oxidative stress in chronic obstructive pulmonary disease: A lung and systemic process. Can. Respir. J. 2013, 20, 27–29. [Google Scholar] [CrossRef] [Green Version]
- Guerrina, N.; Traboulsi, H.; Eidelman, D.H.; Baglole, C.J. The Aryl Hydrocarbon Receptor and the Maintenance of Lung Health. Int. J. Mol. Sci. 2018, 19, 3882. [Google Scholar] [CrossRef] [Green Version]
- Thorley, A.J.; Tetley, T.D. Pulmonary epithelium, cigarette smoke, and chronic obstructive pulmonary disease. Int. J. Chronic Obstruct. Pulm. Dis. 2007, 2, 409–428. [Google Scholar]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Iu, M.; Zago, M.; Rico de Souza, A.; Bouttier, M.; Pareek, S.; White, J.H.; Hamid, Q.; Eidelman, D.H.; Baglole, C.J. RelB attenuates cigarette smoke extract-induced apoptosis in association with transcriptional regulation of the aryl hydrocarbon receptor. Free Radic. Biol. Med. 2017, 108, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Sarill, M.; Zago, M.; Sheridan, J.A.; Nair, P.; Matthews, J.; Gomez, A.; Roussel, L.; Rousseau, S.; Hamid, Q.; Eidelman, D.H.; et al. The aryl hydrocarbon receptor suppresses cigarette-smoke-induced oxidative stress in association with dioxin response element (DRE)-independent regulation of sulfiredoxin 1. Free Radic. Biol. Med. 2015, 89, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.S.; Vogel, C.F.; Kokosinski, K.; Matsumura, F. Arylhydrocarbon receptor activation in NCI-H441 cells and C57BL/6 mice: Possible mechanisms for lung dysfunction. Am. J. Respir. Cell Mol. Biol. 2010, 42, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, N.B.; Kerkvliet, N.I. Dioxin and immune regulation: Emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells. Ann. N. Y. Acad. Sci. 2010, 1183, 25–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, H.; Rogers, D.F. New pharmacotherapy for airway mucus hypersecretion in asthma and COPD: Targeting intracellular signaling pathways. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 219–231. [Google Scholar] [CrossRef]
- Chiba, T.; Uchi, H.; Yasukawa, F.; Furue, M. Role of the arylhydrocarbon receptor in lung disease. Int. Arch. Allergy Immunol. 2011, 155 (Suppl. 1), 129–134. [Google Scholar] [CrossRef]
- Thatcher, T.H.; Maggirwar, S.B.; Baglole, C.J.; Lakatos, H.F.; Gasiewicz, T.A.; Phipps, R.P.; Sime, P.J. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am. J. Pathol. 2007, 170, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Moorthy, B.; Parker, K.M.; Smith, C.V.; Bend, J.R.; Welty, S.E. Potentiation of oxygen-induced lung injury in rats by the mechanism-based cytochrome P-450 inhibitor, 1-aminobenzotriazole. J. Pharmacol. Exp. Ther. 2000, 292, 553–560. [Google Scholar]
- Luebke, R.W.; Copeland, C.B.; Daniels, M.; Lambert, A.L.; Gilmour, M.I. Suppression of allergic immune responses to house dust mite (HDM) in rats exposed to 2,3,7,8-TCDD. Toxicol. Sci. 2001, 62, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Moon, D.O.; Kim, M.O.; Lee, H.J.; Choi, Y.H.; Park, Y.M.; Heo, M.S.; Kim, G.Y. Curcumin attenuates ovalbumin-induced airway inflammation by regulating nitric oxide. Biochem. Biophys. Res. Commun. 2008, 375, 275–279. [Google Scholar] [CrossRef]
- Shivanna, B.; Jiang, W.; Wang, L.; Couroucli, X.I.; Moorthy, B. Omeprazole attenuates hyperoxic lung injury in mice via aryl hydrocarbon receptor activation and is associated with increased expression of cytochrome P4501A enzymes. J. Pharmacol. Exp. Ther. 2011, 339, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.; Liu, X.; Tu, W.; Do, D.C.; Yu, H.; Yang, L.; Zhou, Y.; Xu, D.; Huang, S.K.; Yang, P.; et al. Benzo(a)pyrene facilitates dermatophagoides group 1 (Der f 1)-induced epithelial cytokine release through aryl hydrocarbon receptor in asthma. Allergy 2019, 74, 1675–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, A.; Gregorc, T.; Docherty, C.K.; Harvey, K.Y.; Nilsen, M.; Morrell, N.W.; MacLean, M.R. Role of the Aryl Hydrocarbon Receptor in Sugen 5416-induced Experimental Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2018, 58, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Walston, J.; Xue, Q.; Semba, R.D.; Ferrucci, L.; Cappola, A.R.; Ricks, M.; Guralnik, J.; Fried, L.P. Serum antioxidants, inflammation, and total mortality in older women. Am. J. Epidemiol. 2006, 163, 18–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, A.; Tandon, V.R. Antioxidants and rheumatoid arthritis. J. Indian Rheumatol. Ass. 2004, 12, 139–142. [Google Scholar]
- Talbot, J.; Peres, R.S.; Pinto, L.G.; Oliveira, R.D.R.; Lima, K.A.; Donate, P.B.; Silva, J.R.; Ryffel, B.; Cunha, T.M.; Alves-Filho, J.C.; et al. Smoking-induced aggravation of experimental arthritis is dependent of aryl hydrocarbon receptor activation in Th17 cells. Arthritis Res. Ther. 2018, 20, 119. [Google Scholar] [CrossRef] [Green Version]
- Tamaki, A.; Hayashi, H.; Nakajima, H.; Takii, T.; Katagiri, D.; Miyazawa, K.; Hirose, K.; Onozaki, K. Polycyclic aromatic hydrocarbon increases mRNA level for interleukin 1 beta in human fibroblast-like synoviocyte line via aryl hydrocarbon receptor. Biol. Pharm. Bull. 2004, 27, 407–410. [Google Scholar] [CrossRef] [Green Version]
- Xi, X.; Ye, Q.; Fan, D.; Cao, X.; Wang, Q.; Wang, X.; Zhang, M.; Xu, Y.; Xiao, C. Polycyclic Aromatic Hydrocarbons Affect Rheumatoid Arthritis Pathogenesis via Aryl Hydrocarbon Receptor. Front. Immunol. 2022, 13, 797815. [Google Scholar] [CrossRef]
- Hui, W.; Dai, Y. Therapeutic potential of aryl hydrocarbon receptor ligands derived from natural products in rheumatoid arthritis. Basic Clin. Pharmacol. Toxicol. 2020, 126, 469–474. [Google Scholar] [CrossRef]
- O'Driscoll, C.A.; Gallo, M.E.; Hoffmann, E.J.; Fechner, J.H.; Schauer, J.J.; Bradfield, C.A.; Mezrich, J.D. Polycyclic aromatic hydrocarbons (PAHs) present in ambient urban dust drive proinflammatory T cell and dendritic cell responses via the aryl hydrocarbon receptor (AHR) in vitro. PLoS ONE 2018, 13, e0209690. [Google Scholar] [CrossRef]
- Villa, M.; Gialitakis, M.; Tolaini, M.; Ahlfors, H.; Henderson, C.J.; Wolf, C.R.; Brink, R.; Stockinger, B. Aryl hydrocarbon receptor is required for optimal B-cell proliferation. EMBO J. 2017, 36, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Iwamoto, N.; Takatani, A.; Igawa, T.; Shimizu, T.; Umeda, M.; Nishino, A.; Horai, Y.; Hirai, Y.; Koga, T.; et al. M1 and M2 Monocytes in Rheumatoid Arthritis: A Contribution of Imbalance of M1/M2 Monocytes to Osteoclastogenesis. Front. Immunol. 2017, 8, 1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Raemdonck, K.; Umar, S.; Palasiewicz, K.; Volkov, S.; Volin, M.V.; Arami, S.; Chang, H.J.; Zanotti, B.; Sweiss, N.; Shahrara, S. CCL21/CCR7 signaling in macrophages promotes joint inflammation and Th17-mediated osteoclast formation in rheumatoid arthritis. Cell. Mol. Life Sci. 2020, 77, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Sun, L.; Cao, J.; Yuen, T.; Lu, P.; Bab, I.; Leu, N.A.; Srinivasan, S.; Wagage, S.; Hunter, C.A.; et al. Smoke carcinogens cause bone loss through the aryl hydrocarbon receptor and induction of Cyp1 enzymes. Proc. Natl. Acad. Sci. USA 2013, 110, 11115–11120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahama, T.; Kimura, A.; Nguyen, N.T.; Chinen, I.; Hanieh, H.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor deficiency in T cells suppresses the development of collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 14222–14227. [Google Scholar] [CrossRef] [Green Version]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Miki, Y.; Hata, S.; Ono, K.; Suzuki, T.; Ito, K.; Kumamoto, H.; Sasano, H. Roles of Aryl Hydrocarbon Receptor in Aromatase-Dependent Cell Proliferation in Human Osteoblasts. Int. J. Mol. Sci. 2017, 18, 2159. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Structure of aryl hydrocarbon receptor (AhR). The basic helix–loop–helix (bHLH) motif, common among various transcription factors, is located at the N terminus of the AhR protein and is involved in DNA binding and protein–protein interactions. Per–ARNT–Sim (PAS) domains (PAS-A and PAS-B) participate in binding to ligands and to HSP90 proteins and in dimerization with partner proteins. The transactivation domain (TAD) is located at the C terminus of the AhR protein.
Figure 1.
Structure of aryl hydrocarbon receptor (AhR). The basic helix–loop–helix (bHLH) motif, common among various transcription factors, is located at the N terminus of the AhR protein and is involved in DNA binding and protein–protein interactions. Per–ARNT–Sim (PAS) domains (PAS-A and PAS-B) participate in binding to ligands and to HSP90 proteins and in dimerization with partner proteins. The transactivation domain (TAD) is located at the C terminus of the AhR protein.

Figure 2.
An outline of canonical and non-canonical AhR signaling pathways. Under physiological conditions, AhR is localized to the cytosol and forms a complex with specific proteins, such as hepatitis B virus X-associated protein 2 (XAP-2), heat shock protein 90 (HSP90), cytosolic endoplasmic-reticulum proteins, and protein tyrosine kinase c-Src. After binding to a ligand, AhR changes its conformation and relocates to the nucleus, where it dimerizes with AhR nuclear transporter (ARNT) or other partners such as transcription factor Krüppel-like factor 6 (KLF6) or transcription factors of the nuclear factor kappa B (NF-κB) family (e.g., RelB). Dissociated c-Src interacts with epidermal growth factor receptor (EGFR). AhR signaling is connected with the activity and function of estrogen receptor and E2 promoter-binding factor 1 (E2F1), which is capable of binding to pRB. The AhR–ARNT complex binds to a xenobiotic-responsive element (XRE) and induces the transcription of AhR-controlled genes. Proteins AhR and KLF6 form a heterodimer that recognizes a novel non-consensus XRE (NC-XRE) and initiates the transcription of genes involved in cell cycle regulation. Proteins AhR and RelB (an NF-κB subunit) combine into a heterodimer that recognizes a RelB–XRE complex and induces the transcription of some chemokine genes. AhR and NF-κB form a heterodimer that lead to the inducing of the expression of cytokines and chemokines B-cell-activating factor of the tumor necrosis factor family (BAFF), B-lymphocyte chemoattractant (BLC), CC-chemokine ligand 1 (CCL1), and interferon-responsive factor (IFR3). The AhR/ARNT/NF-κB interaction decreases the expression of CYP1A1. AhR and pRb form a heterodimer that lead to a blocked cell cycle progression by suppressing the expression of S-phase genes. AhR activity is controlled by negative feedback loops, including the metabolism of ligands, the disruption of the AhR/ARNT complex by AhR repressor (AhRR), and proteosomal degradation by the ubiquitin ligase complex. AhR in complex with ER promotes the proteolysis of ER by ubiquitin ligase complex.
Figure 2.
An outline of canonical and non-canonical AhR signaling pathways. Under physiological conditions, AhR is localized to the cytosol and forms a complex with specific proteins, such as hepatitis B virus X-associated protein 2 (XAP-2), heat shock protein 90 (HSP90), cytosolic endoplasmic-reticulum proteins, and protein tyrosine kinase c-Src. After binding to a ligand, AhR changes its conformation and relocates to the nucleus, where it dimerizes with AhR nuclear transporter (ARNT) or other partners such as transcription factor Krüppel-like factor 6 (KLF6) or transcription factors of the nuclear factor kappa B (NF-κB) family (e.g., RelB). Dissociated c-Src interacts with epidermal growth factor receptor (EGFR). AhR signaling is connected with the activity and function of estrogen receptor and E2 promoter-binding factor 1 (E2F1), which is capable of binding to pRB. The AhR–ARNT complex binds to a xenobiotic-responsive element (XRE) and induces the transcription of AhR-controlled genes. Proteins AhR and KLF6 form a heterodimer that recognizes a novel non-consensus XRE (NC-XRE) and initiates the transcription of genes involved in cell cycle regulation. Proteins AhR and RelB (an NF-κB subunit) combine into a heterodimer that recognizes a RelB–XRE complex and induces the transcription of some chemokine genes. AhR and NF-κB form a heterodimer that lead to the inducing of the expression of cytokines and chemokines B-cell-activating factor of the tumor necrosis factor family (BAFF), B-lymphocyte chemoattractant (BLC), CC-chemokine ligand 1 (CCL1), and interferon-responsive factor (IFR3). The AhR/ARNT/NF-κB interaction decreases the expression of CYP1A1. AhR and pRb form a heterodimer that lead to a blocked cell cycle progression by suppressing the expression of S-phase genes. AhR activity is controlled by negative feedback loops, including the metabolism of ligands, the disruption of the AhR/ARNT complex by AhR repressor (AhRR), and proteosomal degradation by the ubiquitin ligase complex. AhR in complex with ER promotes the proteolysis of ER by ubiquitin ligase complex.

Figure 3.
The scheme of putative connections between gene batteries of AhR and Nrf2. (1) Nrf2 is a target gene of AhR; (2) indirect activation of Nrf2 by CYP1A1-generated reactive oxygen species (ROS); and (3) direct interaction of complexes AhR–XRE and Nrf2–ARE in a regulatory region of the NQO1 gene.
Figure 3.
The scheme of putative connections between gene batteries of AhR and Nrf2. (1) Nrf2 is a target gene of AhR; (2) indirect activation of Nrf2 by CYP1A1-generated reactive oxygen species (ROS); and (3) direct interaction of complexes AhR–XRE and Nrf2–ARE in a regulatory region of the NQO1 gene.

Figure 4.
Pro-oxidant and antioxidant effects of AhR results in wide range of physiological and pathological processes in cells and tissues.
Figure 4.
Pro-oxidant and antioxidant effects of AhR results in wide range of physiological and pathological processes in cells and tissues.


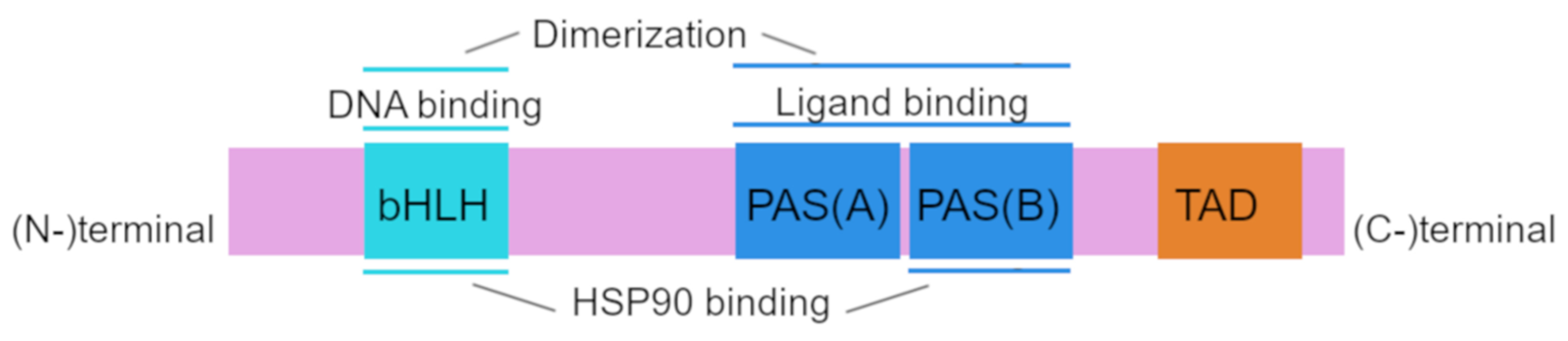{kind=link}
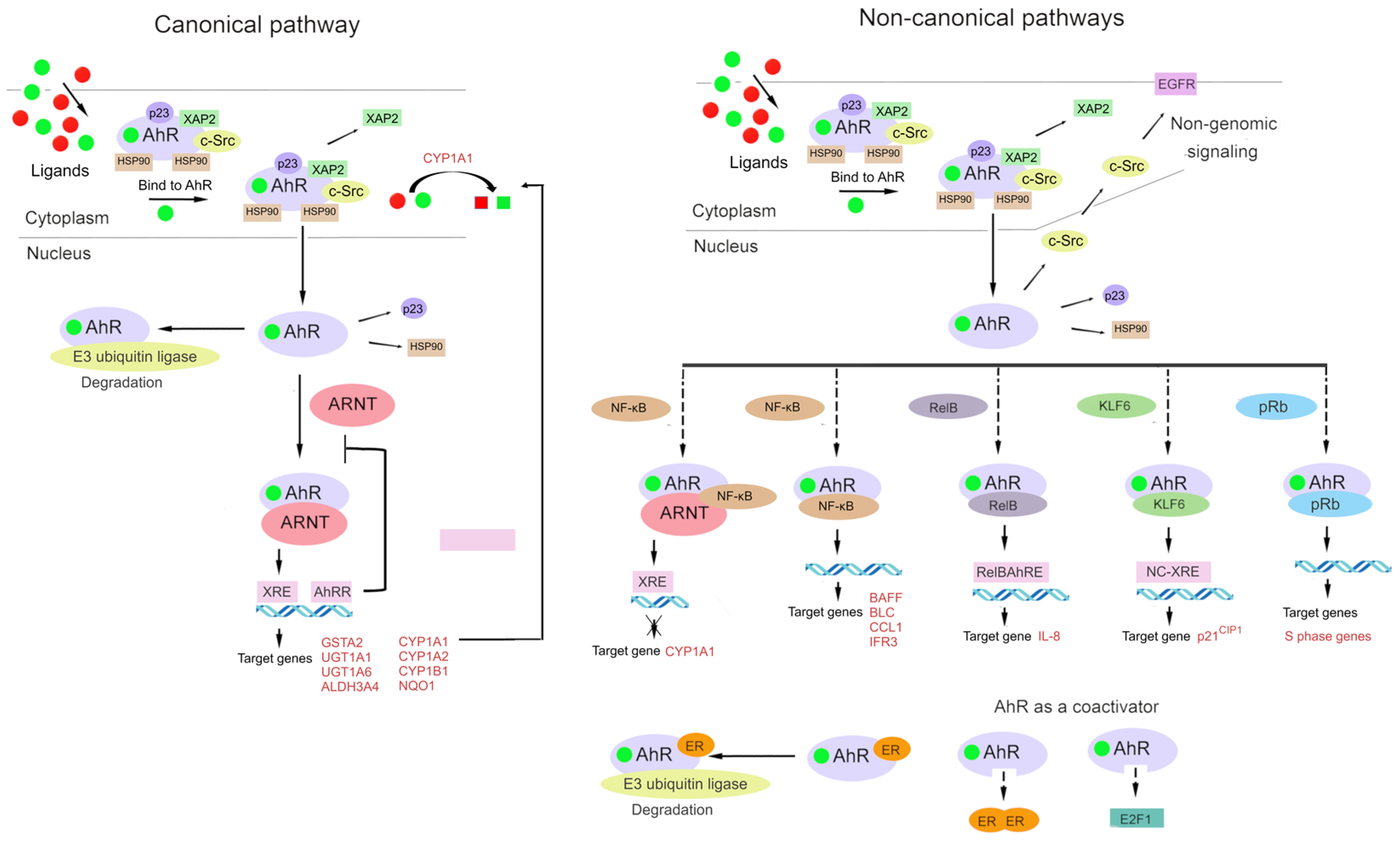{kind=link}
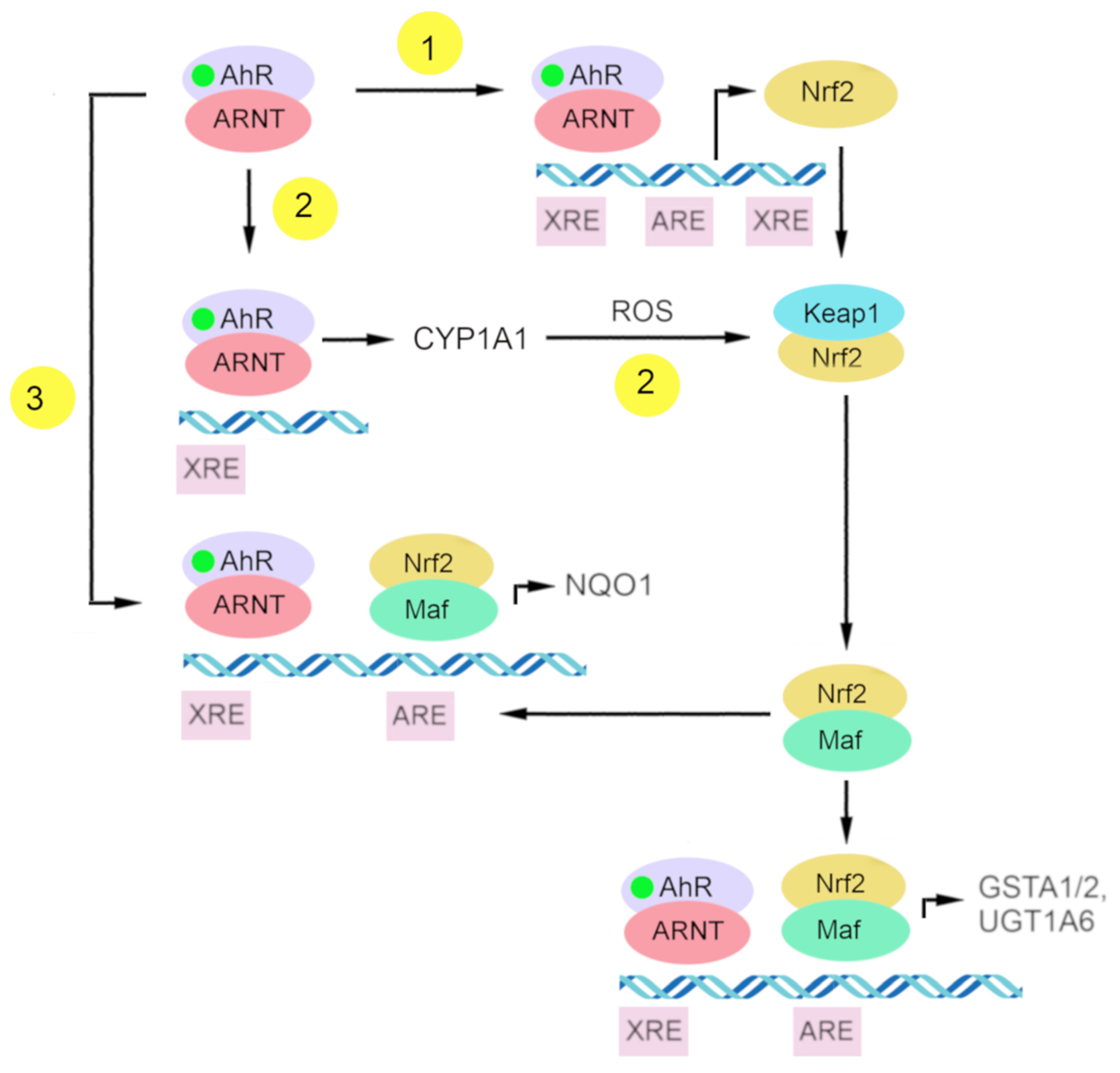{kind=link}
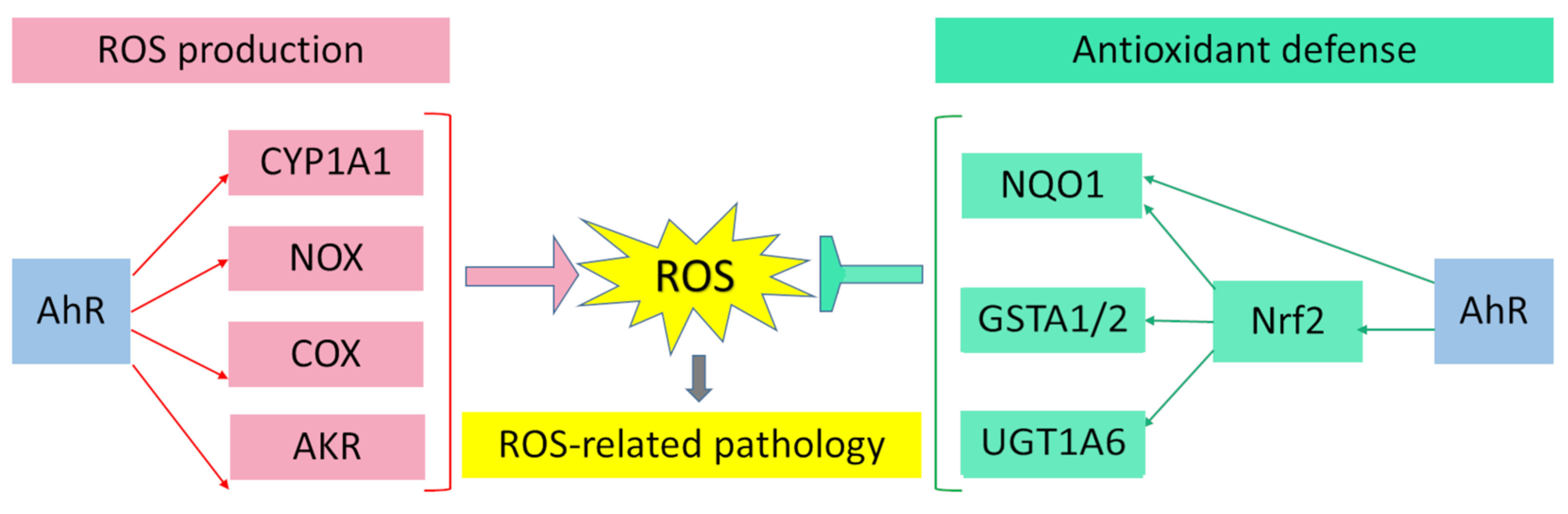{kind=link}
Table 1.
The list of AhR ligands.
| Exogenous Compounds | Endogenous Compounds | |
|---|---|---|
| Synthetic compounds | Natural Compounds | Indigoids |
| Polycyclic aromatic hydrocarbons | Polyphenols | Eicosanoids |
| Polychlorinated biphenyls | Diosmin | Tryptophan metabolites: |
| Halogeneted Dioxins and Related Compounds | Resveratrol | L-Kynurenine, |
| Other synthetic AhR Ligands: | Curcumin | Kynurenic acid, |
| Benzimidazole | Berberin | |
| Pesticides | Alcaloids | ITE—(2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester, |
| Primaquine | (Tetrandrine, | Indoxyl-3-sulfate, |
| Kinase inhibitor | Sinomenine Norisoboldin) | Indirubin, |
| Synthetic flavonoid | Dietary compounds (Indole-3-carbinol Indole-3-acetonitrile, | Tryptamine, |
| New synthetic ligands | 3,3′-Diindolylmethane Indolo(3,4)bicarbazole) | 3-Methylindole |
| (CH223191, VAF347, 4OHT, | Ultraviolet photoproducts of tryptophan: | |
| 6-MCDF) | FICZ—6-Formyl indolo (3,2-b) carbazole | |
| Heme metabolites | ||
| Bilirubin | ||
| Biliverdin | ||
| Arachidonic acid metabolites: | ||
| 12(R)-hydroxy-5(Z),8(Z),10(E) | ||
| 14(Z)-eicosatetraenoic acid | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Grishanova, A.Y.; Perepechaeva, M.L. Aryl Hydrocarbon Receptor in Oxidative Stress as a Double Agent and Its Biological and Therapeutic Significance. Int. J. Mol. Sci. 2022, 23, 6719. https://doi.org/10.3390/ijms23126719
AMA Style
Grishanova AY, Perepechaeva ML. Aryl Hydrocarbon Receptor in Oxidative Stress as a Double Agent and Its Biological and Therapeutic Significance. International Journal of Molecular Sciences. 2022; 23(12):6719. https://doi.org/10.3390/ijms23126719
Chicago/Turabian StyleGrishanova, Alevtina Y., and Maria L. Perepechaeva. 2022. "Aryl Hydrocarbon Receptor in Oxidative Stress as a Double Agent and Its Biological and Therapeutic Significance" International Journal of Molecular Sciences 23, no. 12: 6719. https://doi.org/10.3390/ijms23126719
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.