
New Borane-Protected Derivatives of α-Aminophosphonous Acid as Anti-Osteosarcoma Agents: ADME Analysis and Molecular Modeling, In Vitro Studies on Anti-Cancer Activities, and NEP Inhibition as a Possible Mechanism of Anti-Proliferative Activity

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. New Phosphonous Acid-Borane Derivatives Exhibited Physicochemical and Pharmacokinetic Properties Typical for Small-Molecule drugs—In Silico Analysis
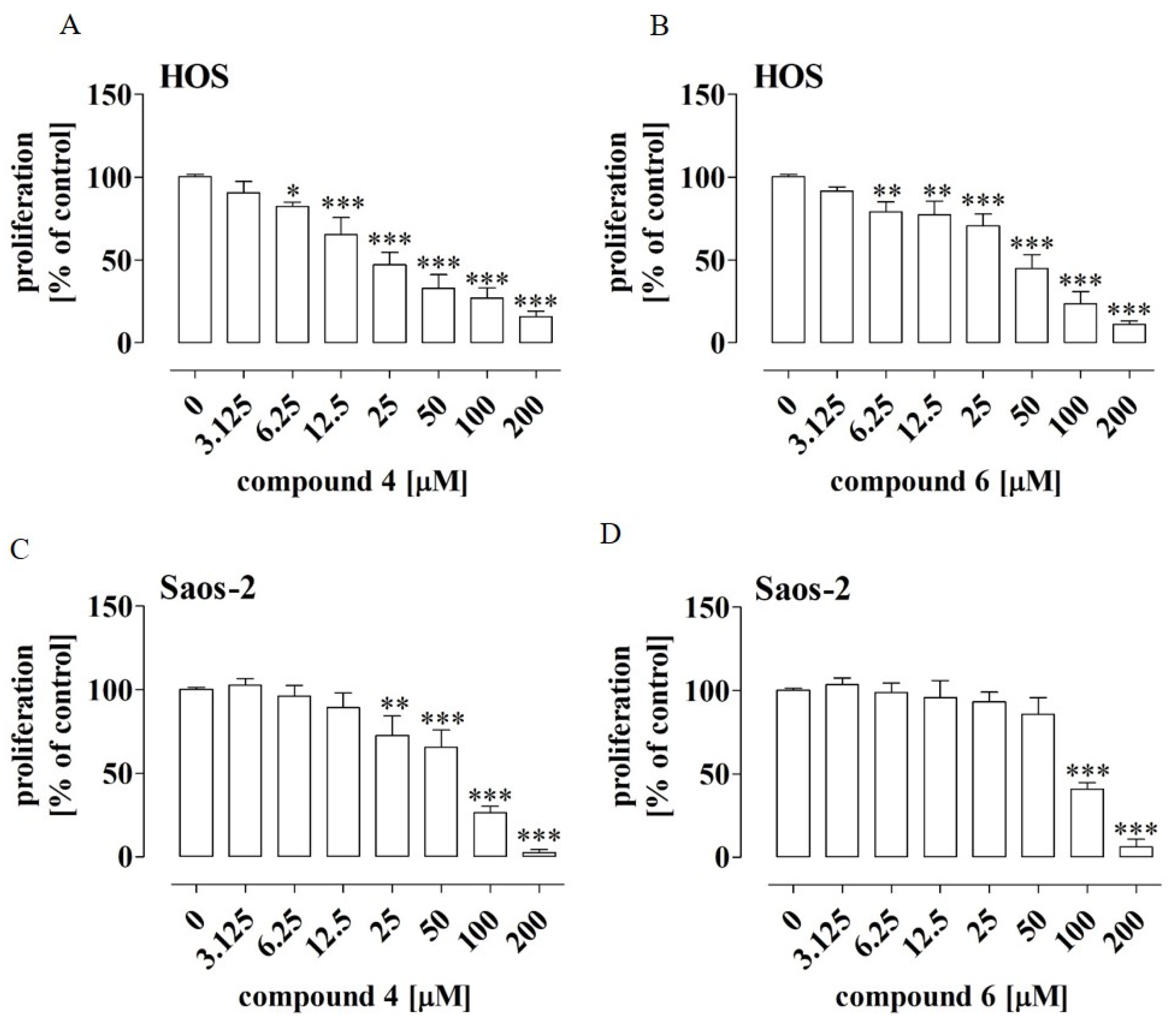
2.2. New Phosphonous Acid-Borane Derivatives Inhibited Proliferation of Osteosarcoma Cells
2.3. Compounds 4 and 6 Caused Cell Cycle Disturbances in Osteosarcoma Cells
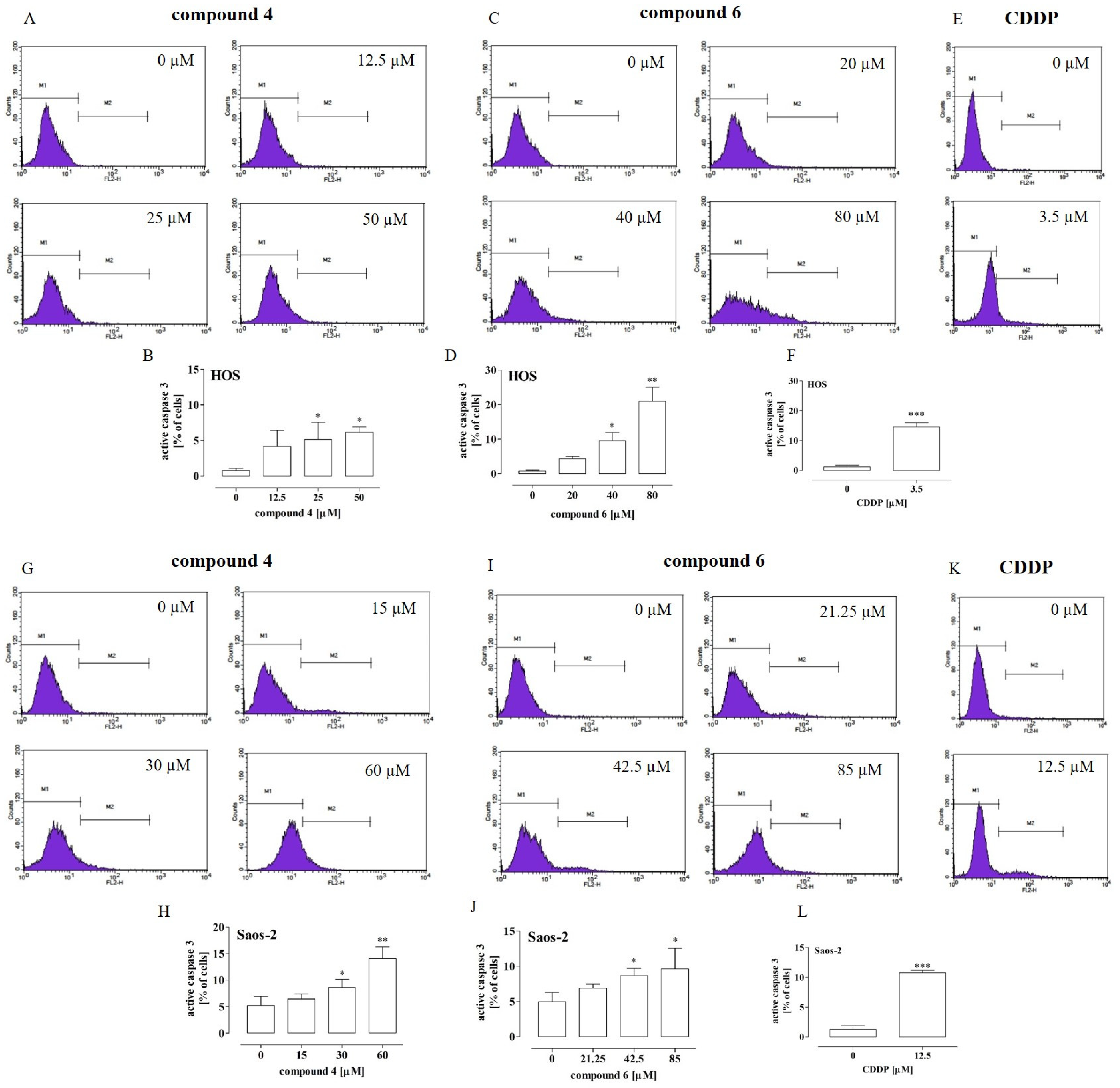
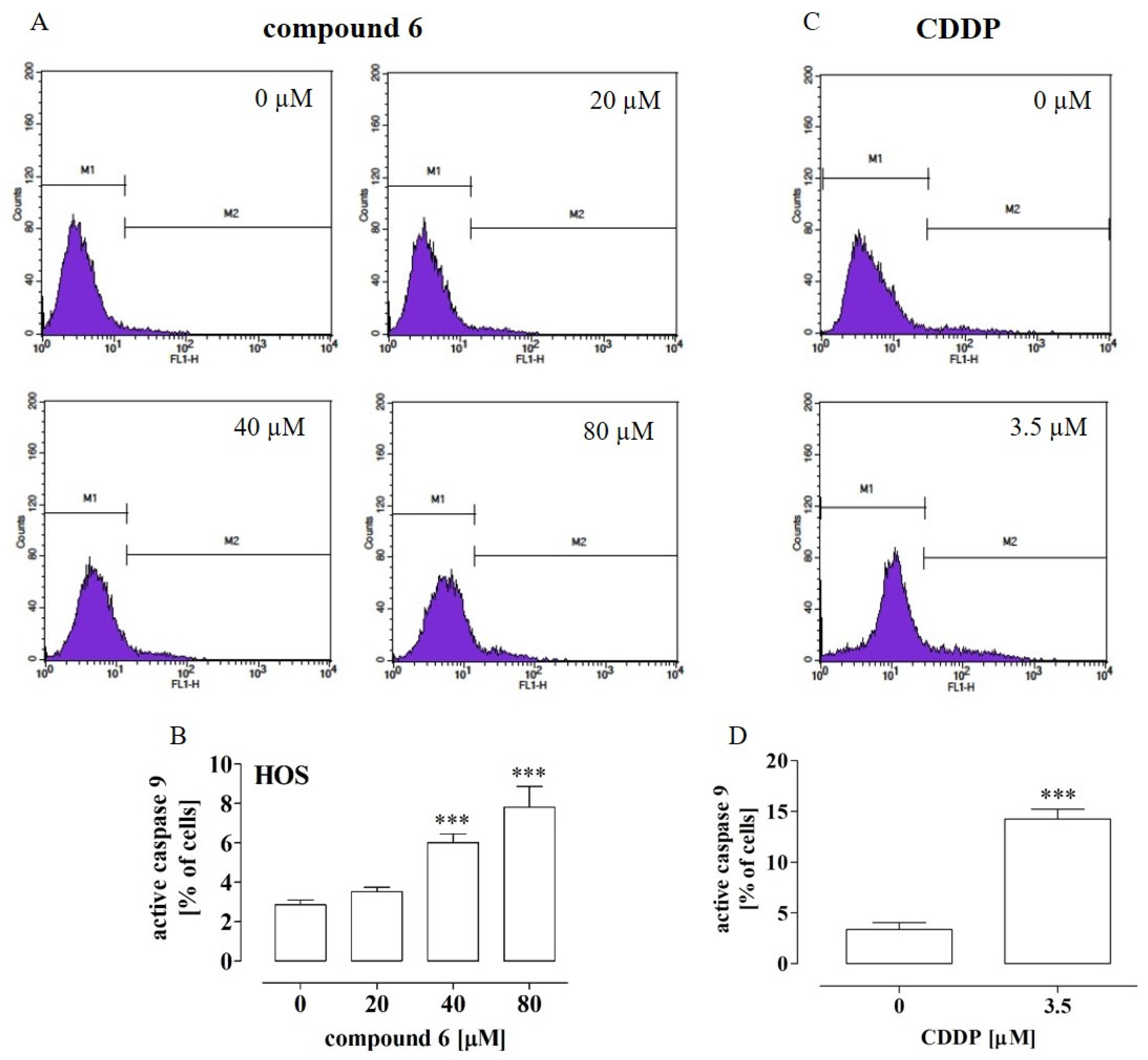
2.4. Compounds 4 and 6 Activated Caspase 3 and Compound 6 Activated Caspase 9 in Osteosarcoma Cells
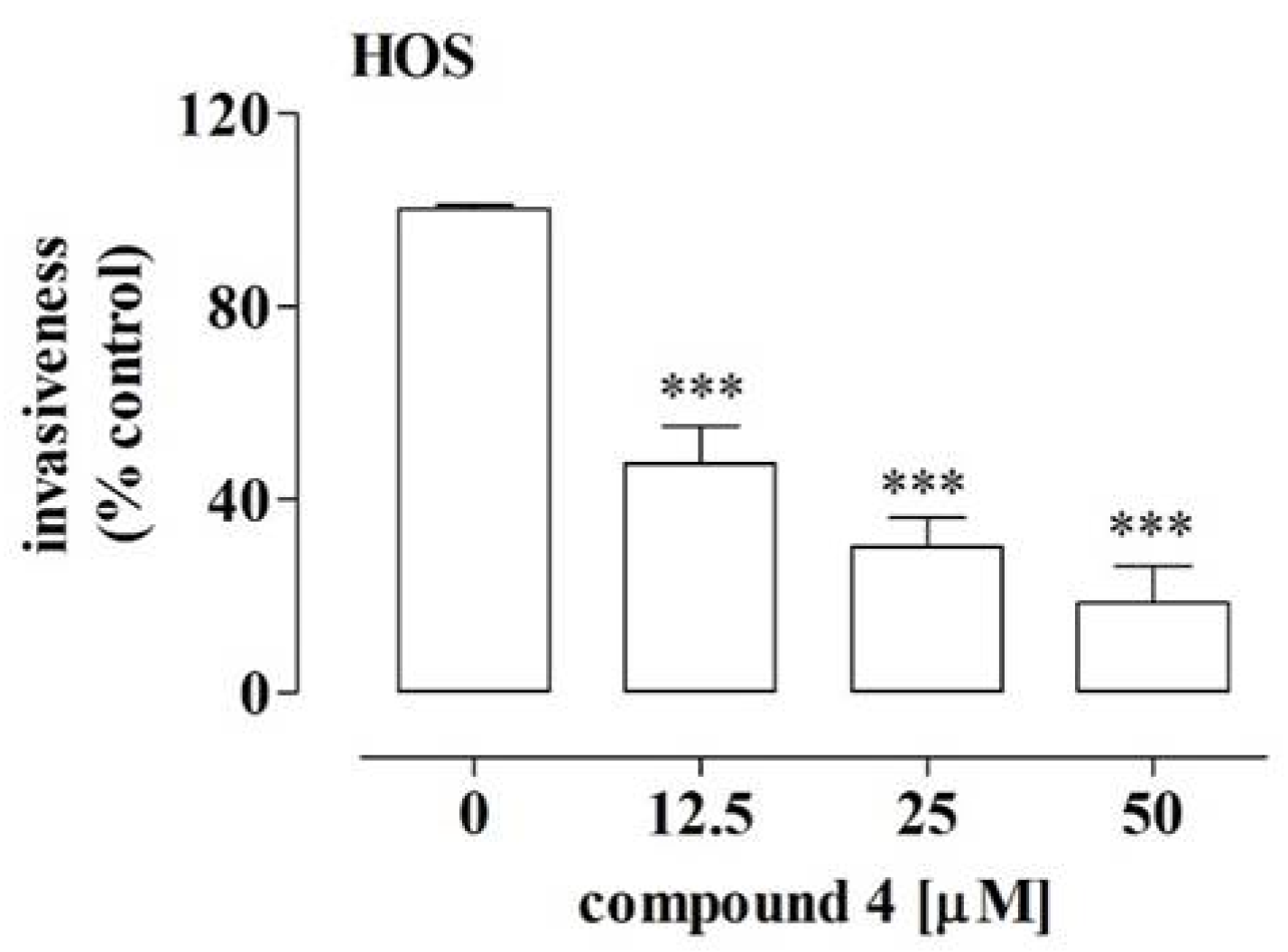
2.5. Compound 4 Inhibited the Migration and Invasiveness of Highly Aggressive HOS Osteosarcoma Cells
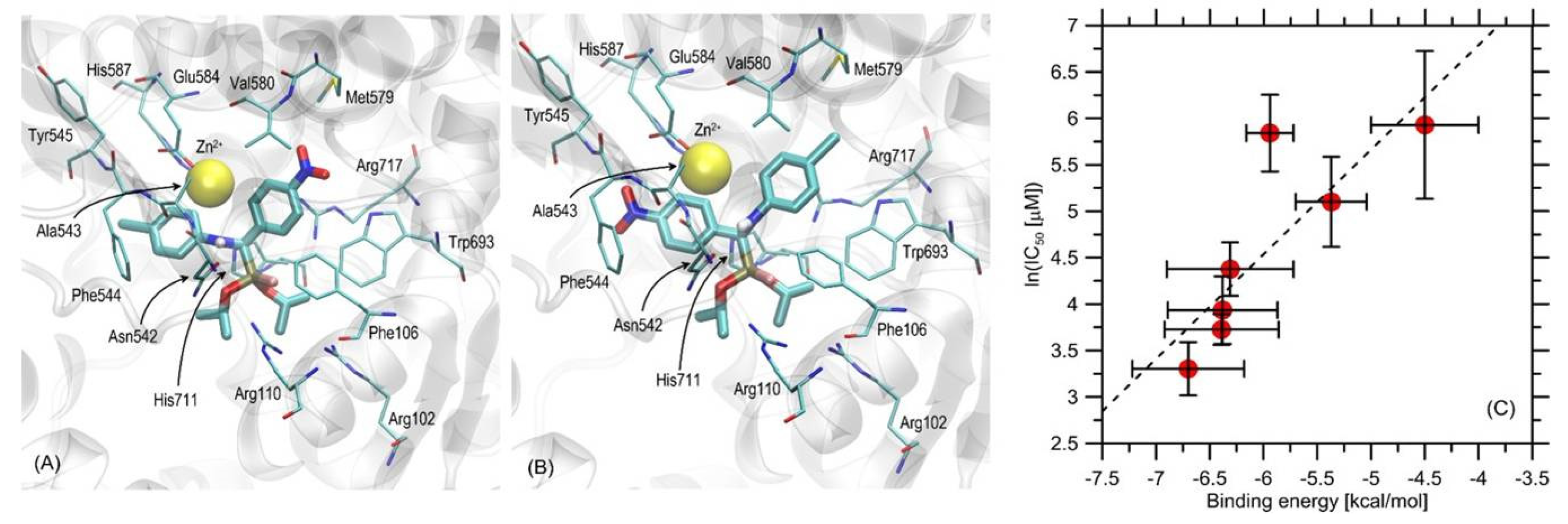
2.6. New Phosphonous Acid-Borane Derivatives Interacted with the Binding Cavity of Neutral Endopeptidase—In Silico Molecular Modeling Study
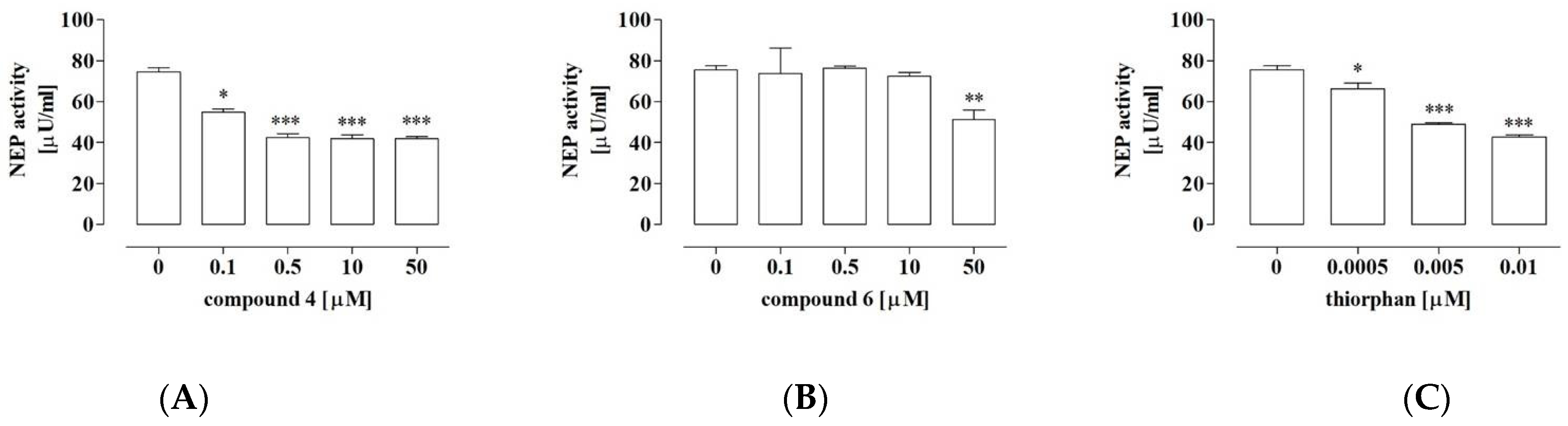
2.7. Compound 4 Exerted Significant Inhibitory Activity against NEP
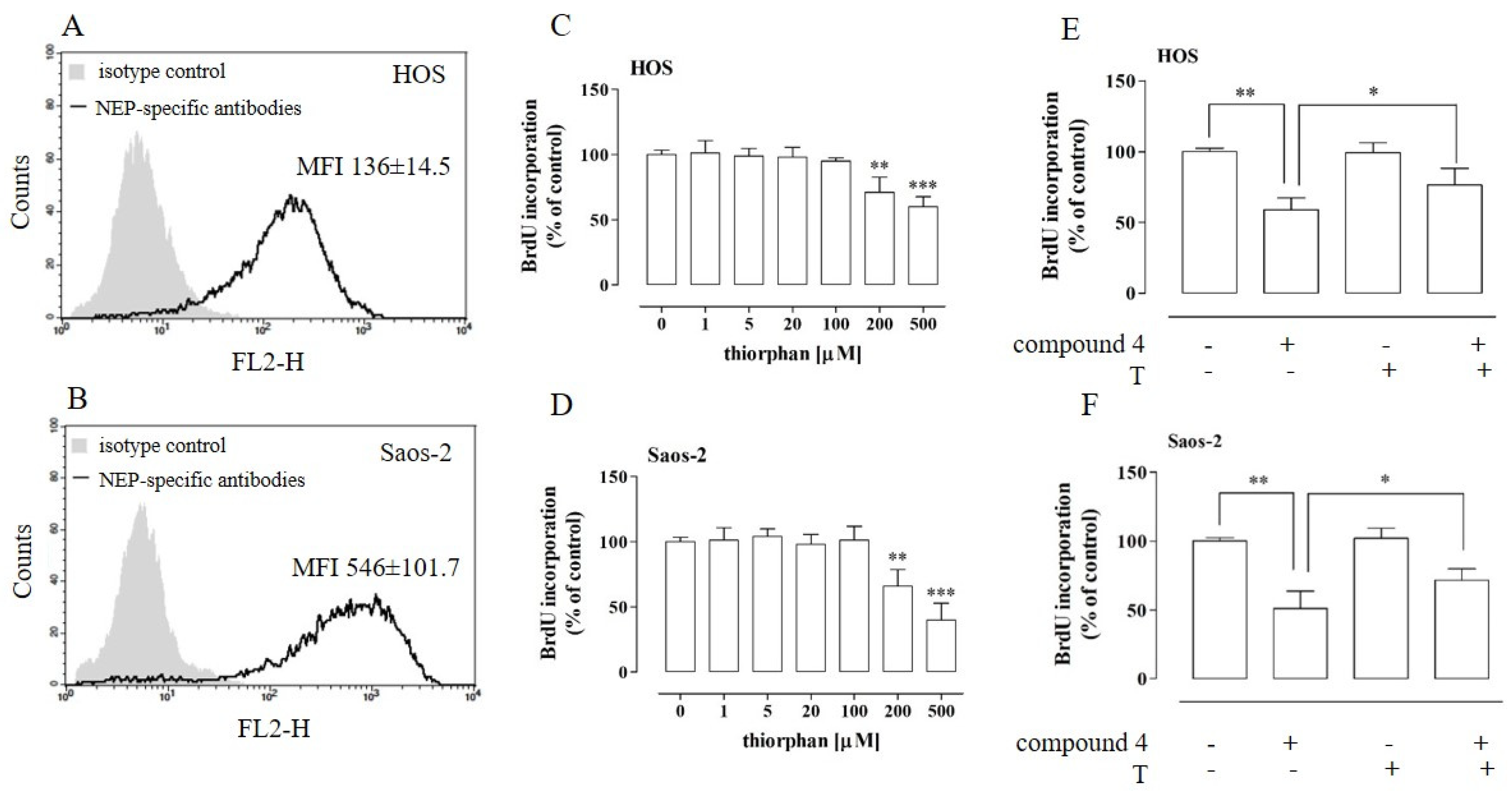
2.8. NEP Was Implicated in the Anti-Proliferative Activity of Compound 4
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Chemicals
4.2. In Silico Analysis of Drug-Likeness and Pharmacokinetic Properties
4.3. Molecular Modeling
4.4. Cell Proliferation and Viability Assays
4.5. Cytotoxicity Assay
4.6. Cell Cycle Assay
4.7. Caspase 3 and 9 Assays
4.8. Migration and Invasiveness Assays
4.9. Neutral Endopeptidase Activity Assay
4.10. Flow Cytometry Analysis of NEP Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Demkowicz, S.; Kozak, W.; Daśko, M.; Rachon, J. Phosphoroorganic Metal Complexes in Therapeutics. Mini-Reviews Med. Chem. 2016, 16, 1359–1373. [Google Scholar] [CrossRef]
- Konstantinov, S.M.; Berger, M.R. Alkylating Agents BT-Encyclopedia of Molecular Pharmacology; Offermanns, S., Rosenthal, W., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 53–57. ISBN 978-3-540-38918-7. [Google Scholar]
- Mucha, A.; Kafarski, P.; Berlicki, Y. Remarkable Potential of the r-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem 2011, 54, 5955–5980. [Google Scholar] [CrossRef]
- Collinsova, M.; Jiracek, J. Phosphinic Acid Compounds in Biochemistry, Biology and Medicine. Curr. Med. Chem. 2000, 7, 629–647. [Google Scholar] [CrossRef]
- Mucha, A.; Drag, M.; Dalton, J.P.; Kafarski, P. Metallo-aminopeptidase inhibitors. Biochimie 2010, 92, 1509–1529. [Google Scholar] [CrossRef]
- Carl-McGrath, S.; Lendeckel, U.; Ebert, M.; Röcken, C. Ectopeptidases in tumour biology: A review. Histol. Histopathol. 2006, 21, 1339–1353. [Google Scholar]
- Georgiadis, D.; Dive, V. Phosphinic Peptides as Potent Inhibitors of Zinc-Metalloproteases BT-Phosphorus Chemistry I: Asymmetric Synthesis and Bioactive Compounds; Montchamp, J.-L., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 1–38. ISBN 978-3-319-15473-2. [Google Scholar]
- Winer, A.; Adams, S.; Mignatti, P. Matrix metalloproteinase inhibitors in cancer therapy: Turning past failures into future successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Mizerska-Kowalska, M.; Kreczko-Kurzawa, J.; Zdzisińska, B.; Czerwonka, A.; Sławińska-Brych, A.; Maćkiewicz, Z.; Nidzworski, D.; Kandefer-Szerszeń, M. Neutral endopeptidase (NEP) inhibitors–thiorphan, sialorphin, and its derivatives exert anti-proliferative activity towards colorectal cancer cells in vitro. Chem. Biol. Interact. 2019, 307, 105–115. [Google Scholar] [CrossRef]
- Zhang, H.; Lin, H.; Mo, X.; Chen, G.; Lin, L. Synergistic relationship between dipeptidyl peptidase IV and neutral endopeptidase expression and the combined prognostic significance in osteosarcoma patients. Med. Oncol. 2013, 30, 1–8. [Google Scholar] [CrossRef]
- Oba, J.; Nakahara, T.; Hashimoto-Hachiya, A.; Liu, M.; Abe, T.; Hagihara, A.; Yokomizo, T.; Furue, M. CD10-Equipped Melanoma Cells Acquire Highly Potent Tumorigenic Activity: A Plausible Explanation of Their Significance for a Poor Prognosis. PLoS ONE 2016, 11, e0149285. [Google Scholar] [CrossRef] [Green Version]
- Carl-McGrath, S.; Lendeckel, U.; Ebert, M.; Wolter, A.-B.; Roessner, A.; Röcken, C. The ectopeptidases CD10, CD13, CD26, and CD143 are upregulated in gastric cancer. Int. J. Oncol. 2004, 25, 1223–1232. [Google Scholar] [CrossRef]
- Velazquez, E.F.; Yancovitz, M.; Pavlick, A.; Berman, R.; Shapiro, R.; Bogunovic, D.; O’Neill, D.; Yu, Y.-L.; Spira, J.; Christos, P.J.; et al. Clinical relevance of Neutral Endopeptidase (NEP/CD10) in melanoma. J. Transl. Med. 2007, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Mutsaers, A.J.; Walkley, C.R. Cells of origin in osteosarcoma: Mesenchymal stem cells or osteoblast committed cells? Bone 2014, 62, 56–63. [Google Scholar] [CrossRef]
- Wojciechowska, U.; Didkowska, J. Nowotwory Złośliwe w Polsce w 2018. Kraj. Rejestr Nowotworów 2020, 3–4, 31–63. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Lindsey, B.A.; Markel, J.E.; Kleinerman, E.S. Osteosarcoma Overview. Rheumatol. Ther. 2017, 4, 25–43. [Google Scholar] [CrossRef] [Green Version]
- Czarnecka, A.M.; Synoradzki, K.; Firlej, W.; Bartnik, E.; Sobczuk, P.; Fiedorowicz, M.; Grieb, P.; Rutkowski, P. Molecular Biology of Osteosarcoma. Cancers 2020, 12, 2130. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, Q.; Gong, X.; Liu, J.; Ma, Y. Osteosarcoma: a review of current and future therapeutic approaches. Biomed. Eng. Online 2021, 20, 24. [Google Scholar] [CrossRef]
- Hattinger, C.M.; Patrizio, M.P.; Fantoni, L.; Casotti, C.; Riganti, C.; Serra, M. Drug Resistance in Osteosarcoma: Emerging Biomarkers, Therapeutic Targets and Treatment Strategies. Cancers 2021, 13, 2878. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Sowa, S.; Michał Pietrusiewicz, K. Chemoselective Reduction of the P=O Bond in the Presence of P-O and P-N Bonds in Phosphonate and Phosphinate Derivatives. Eur. J. Org. Chem. 2019, 5, 923–938. [Google Scholar] [CrossRef]
- Modzelewski, K.; Sowa, S. Alkylation of phosphinite/phosphonite-boranes via temporary protection of the P-H bond. Synth. 2020, 52, 2410–2426. [Google Scholar]
- SwissADME. Available online: http://www.swissadme.ch/ (accessed on 15 February 2022).
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics—Anti-metastatic and Anti-invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef] [Green Version]
- Lauvrak, S.U.; Munthe, E.; Kresse, S.H.; Stratford, E.W.; Namløs, H.M.; Meza-Zepeda, L.A.; Myklebost, O. Functional characterisation of osteosarcoma cell lines and identification of mRNAs and miRNAs associated with aggressive cancer phenotypes. Br. J. Cancer 2013, 109, 2228–2236. [Google Scholar] [CrossRef]
- Rougeot, C.; Messaoudi, M.; Hermitte, V.; Gaëlle Rigault, A.; Blisnick, T.; Dugave, C.; Desor, D.; Rougeon, F. Sialorphin, a natural inhibitor of rat membrane-bound neutral endopeptidase that displays analgesic activity. Proc. Natl. Acad. Sci. USA 2003, 100, 8549–8554. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, M.R.; Unwin, R.J.; Kenny, A.J. Endopeptidase-24.11 and its inhibitors: Potential therapeutic agents for edematous disorders and hypertension. Kidney Int. 1993, 43, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Noble, F.; Coric, P.; Fournie-Zaluski, M.C.; Roques, B.P. Aminophosphinic inhibitors as transition state analogues of enkephalin-degrading enzymes: a class of central analgesics. Proc. Natl. Acad. Sci. USA 1998, 95, 12028–12033. [Google Scholar] [CrossRef] [Green Version]
- Jullien, N.; Makritis, A.; Georgiadis, D.; Beau, F.; Yiotakis, A.; Dive, V. Phosphinic tripeptides as dual angiotensin-converting enzyme C-domain and endothelin-converting enzyme-1 inhibitors. J. Med. Chem. 2010, 53, 208–220. [Google Scholar] [CrossRef]
- Sahli, S.; Stump, B.; Welti, T.; Schweizer, W.B.; Diederich, F.; Blum-Kaelin, D.; Aebi, J.D.; Böhm, H.-J. A New Class of Inhibitors for the Metalloprotease Neprilysin Based on a Central Imidazole Scaffold. Helv. Chim. Acta 2005, 88, 707–730. [Google Scholar] [CrossRef]
- Díaz-Rodríguez, L.; García-Martínez, O.; Arroyo-Morales, M.; Reyes-Botella, C.; Ruiz, C. Antigenic Phenotype and Phagocytic Capacity of MG-63 Osteosarcoma Line. Ann. N. Y. Acad. Sci. 2009, 1173, E46–E54. [Google Scholar] [CrossRef] [PubMed]
- Eberlin, M.; Mück, T.; Michel, M.C. A Comprehensive Review of the Pharmacodynamics, Pharmacokinetics, and Clinical Effects of the Neutral Endopeptidase Inhibitor Racecadotril. Front. Pharmacol. 2012, 3, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, C.; Ghirlanda-Keller, C.; Gosteli-Peter, M. Ascorbic acid decreases neutral endopeptidase activity in cultured osteoblastic cells. Regul. Pept. 2005, 130, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Justin, B.A.L.; Eugenie, E.M.; Kleinerman, S. Osteosarcoma Overview. Rheumatol. Ther. 2016, 4, 25–43. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Bhakay, A.; Rahman, M.; Dave, R.; Bilgili, E. Bioavailability Enhancement of Poorly Water-Soluble Drugs via Nanocomposites: Formulation–Processing Aspects and Challenges. Pharmaceutics 2018, 10, 86. [Google Scholar] [CrossRef] [Green Version]
- Mandracchia, D.; Tripodo, G. CHAPTER 1 Micro and Nano-drug Delivery Systems. In Silk-based Drug Delivery Systems; The Royal Society of Chemistry: London, UK, 2021; pp. 1–24. ISBN 978-1-78801-772-5. [Google Scholar]
- He, C.; Sun, Z.; Hoffman, R.M.; Yang, Z.; Jiang, Y.; Wang, L.; Hao, Y. P-Glycoprotein Overexpression Is Associated With Cisplatin Resistance in Human Osteosarcoma. Anticancer. Res. 2019, 39, 1711–1718. [Google Scholar] [CrossRef] [Green Version]
- Alzahrani, A.M.; Rajendran, P. The Multifarious Link between Cytochrome P450s and Cancer. Oxid. Med. Cell. Longev. 2020, 2020, 3028387. [Google Scholar] [CrossRef]
- Iwanejko, J.; Wojaczyńska, E.; Turlej, E.; Maciejewska, M.; Wietrzyk, J. Octahydroquinoxalin-2(1H)-One-Based Aminophosphonic Acids and Their Derivatives—Biological Activity towards Cancer Cells. Materials 2020, 13, 2393. [Google Scholar] [CrossRef]
- Tiwari, S.; Sharif, N.; Gajare, R.; Vazquez, J.; Sangshetti, J.; Damale, M.; Nikalje, A. New 2-Oxoindolin Phosphonates as Novel Agents to Treat Cancer: A Green Synthesis and Molecular Modeling. Molecules 2018, 23, 1981. [Google Scholar] [CrossRef] [Green Version]
- Nikalje, G.A.; Gawhane, A.P.; Tiwari, V.S.; Sangshetti, N.J.; Damale, G.M. Ultrasound Promoted Green Synthesis, Docking Study of Indole Spliced Thiadiazole, α-amino Phosphonates as Anticancer Agents and Antityrosinase Agents. Anticancer. Agents Med. Chem. 2018, 18, 1267–1280. [Google Scholar] [CrossRef] [PubMed]
- Ewies, E.F.; El-Hussieny, M.; El-Sayed, N.F.; Fouad, M.A. Design, synthesis and biological evaluation of novel α-aminophosphonate oxadiazoles via optimized iron triflate catalyzed reaction as apoptotic inducers. Eur. J. Med. Chem. 2019, 180, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.Y.; Yao, G.Y.; Pan, Y.M.; Liao, Z.X.; Zhang, Y.; Wang, H.S. Synthesis and antitumor activities of novel α-aminophosphonate derivatives containing an alizarin moiety. Eur. J. Med. Chem. 2014, 83, 116–128. [Google Scholar] [CrossRef]
- Fang, Y.-L.; Wu, Z.-L.; Xiao, M.-W.; Tang, Y.-T.; Li, K.-M.; Ye, J.; Xiang, J.-N.; Hu, A.-X. One-Pot Three-Component Synthesis of Novel Diethyl((2-oxo-1,2-dihydroquinolin-3-yl)(arylamino)methyl)phosphonate as Potential Anticancer Agents. Int. J. Mol. Sci. 2016, 17, 653. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.-B.; Wang, F.-Y.; Feng, H.-W.; Luo, H.; Long, Y.; Zou, T.; Chan, A.S.C.; Liu, R.; Zou, H.; Chen, Z.-F.; et al. An aminophosphonate ester ligand-containing platinum(ii) complex induces potent immunogenic cell death in vitro and elicits effective anti-tumour immune responses in vivo. Chem. Commun. 2019, 55, 13066–13069. [Google Scholar] [CrossRef]
- Ma, J.; Li, J.; Guo, P.; Liao, X.; Cheng, H. Synthesis and antitumor activity of novel indole derivatives containing α-aminophosphonate moieties. Arab. J. Chem. 2021, 14, 103256. [Google Scholar] [CrossRef]
- Pan, S.; Fan, M.; Liu, Z.; Li, X.; Wang, H. Serine, glycine and one-carbon metabolism in cancer (Review). Int J Oncol 2021, 58, 158–170. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Zhuravin, I.A.; Turner, A.J. Neprilysin expression and functions in development, ageing and disease. Mech. Ageing Dev. 2020, 192, 111363. [Google Scholar] [CrossRef]
- Sankhe, R.; Pai, S.R.K.; Kishore, A. Tumour suppression through modulation of neprilysin signaling: A comprehensive review. Eur. J. Pharmacol. 2021, 891, 173727. [Google Scholar] [CrossRef]
- COSMIC Cell Line Gene Mutation Profiles. Available online: https://maayanlab.cloud/Harmonizome/gene_set/HOS/COSMIC+Cell+Line+Gene+Mutation+Profiles (accessed on 15 March 2022).
- Roques, B.P.; Fournié-Zaluski, M.C.; Soroca, E.; Lecomte, J.M.; Malfroy, B.; Llorens, C.; Schwartz, J.-C. The enkephalinase inhibitor thiorphan shows antinociceptive activity in mice. Nature 1980, 288, 286–288. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J Comput Chem 2010, 31, 455–461. [Google Scholar]
- Mizerska-Kowalska, M.; Sławińska-Brych, A.; Kaławaj, K.; Żurek, A.; Pawińska, B.; Rzeski, W.; Zdzisińska, B. Betulin Promotes Differentiation of Human Osteoblasts In Vitro and Exerts an Osteoinductive Effect on the hFOB 1.19 Cell Line Through Activation of JNK, ERK1/2, and mTOR Kinases. Molecules 2019, 24, 2637. [Google Scholar] [CrossRef] [Green Version]
- Kaławaj, K.; Sławińska-Brych, A.; Mizerska-Kowalska, M.; Żurek, A.; Bojarska-Junak, A.; Kandefer-Szerszeń, M.; Zdzisińska, B. Alpha Ketoglutarate Exerts In Vitro Anti-Osteosarcoma Effects through Inhibition of Cell Proliferation, Induction of Apoptosis via the JNK and Caspase 9-Dependent Mechanism, and Suppression of TGF-β and VEGF Production and Metastatic Potential of Cells. Int. J. Mol. Sci. 2020, 21, 9406. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Name | Chemical Formula | Molecular Weight | |
|---|---|---|---|---|
| 1 |  | benzylphosphonous acid-borane diisopropyl ester | C13H24BO2P | 254.11 |
| 2 |  | 1-hydroxy-1-methylethylphosphonous acid-borane diisopropyl ester | C9H24BO3P | 222.07 |
| 3 |  | 1-hydroxy-1-methylethylphosphonous acid-borane isopropyl ester N,N-diethylamide | C10H27BNO2P | 235.11 |
| 4 |  | [1-(N-p-bromophenylamino)]-1-(p-nitrophenyl)methylphosphonousacid-borane diisopropyl ester | C19H27BBrN2O4P | 469.12 |
| 5 |  | [1-N-p-hydroxyphenylamino)]-1-phenylmethylphosphonous acid-borane diisopropyl ester | C19H29BNO3P | 361.22 |
| 6 |  | [1-(N-p-tolylamino)]-1-phenylmethylphosphonous acid-borane diisopropyl ester | C20H31BNO2P | 359.25 |
| 7 |  | [1-(N-p-bromophenylamino)]-1-(p-anisyl)methylphosphonous acid-borane diisopropyl ester | C20H30BBrNO3P | 454.15 |
| Compounds | logP | HBA | HBD | MW | NRB | TPSA |
|---|---|---|---|---|---|---|
| 1 | 2.05 | 2 | 0 | 254.11 | 6 | 28.27 |
| 2 | 0.77 | 3 | 1 | 222.07 | 5 | 48.50 |
| 3 | 0.81 | 3 | 1 | 235.11 | 6 | 42.51 |
| 4 | 2.96 | 4 | 1 | 469.12 | 9 | 86.12 |
| 5 | 2.58 | 3 | 2 | 361.22 | 8 | 60.53 |
| 6 | 3.21 | 2 | 1 | 359.25 | 8 | 40.30 |
| 7 | 3.41 | 3 | 1 | 454.15 | 9 | 49.53 |
| Compounds | BBB Permeant | GI Absorption | P-gp Substrate | Inhibitor of the Cytochrome P450 Isoenzymes | ||||
|---|---|---|---|---|---|---|---|---|
| CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | ||||
| 1 | Yes | High | Yes | No | No | No | Yes | No |
| 2 | Yes | High | No | No | No | No | No | Yes |
| 3 | Yes | High | No | No | No | No | No | No |
| 4 | No | High | Yes | No | Yes | No | No | No |
| 5 | Yes | High | Yes | No | No | No | Yes | Yes |
| 6 | Yes | High | Yes | No | No | No | Yes | Yes |
| 7 | Yes | High | Yes | No | No | Yes | Yes | Yes |
| Compound | IC50 (µM ± SD) | CC50 (µM ± SD) | ||
|---|---|---|---|---|
| HOS | Saos-2 | hFOB 1.19 | CCD-18Co | |
| 1 | 164.2 ± 79.5 | 91.0 ± 19.4 | 161.3 ± 22.9 | nt |
| 2 | 344 ± 142 | 83.4 ± 18.4 | 182.1 ± 28.1 | nt |
| 3 | 226.3 ± 3.56 | 79.6 ± 13.6 | 224.5 ± 95.3 | nt |
| 4 | 27.2 ± 7.8 | 59.9 ± 15 | 188.7 ± 36.4 | nt |
| 5 | 79.5 ± 22.9 | 96 ± 3.3 | 43.9 ± 3.5 | 62.51 ± 36.2 |
| 6 | 41.5 ± 6.9 | 86.8 ± 15.7 | 266.3 ± 109.7 | nt |
| 7 | 51.2 ± 18.6 | 109 ± 15.8 | 214.0 ± 39.5 | nt |
| CDDP | 3.5 ± 0.92 | 12.8 ± 3.7 | - | - |
| Compound | Partial Binding Energies ± SD [kcal/mol] | Binding Energy ± SD [kcal/mol] |
|---|---|---|
| 1 | - | −5.37 ± 0.33 |
| 2 | - | −5.94 ± 0.22 |
| 3 | - | −4.50 ± 0.50 |
| 4 | −6.76 ± 0.44 −6.64 ± 0.58 | −6.70 ± 0.52 |
| 5 | −6.66 ± 0.43 −5.97 ± 0.53 | −6.31 ± 0.59 |
| 6 | −6.67 ± 0.50 −6.10 ± 0.40 | −6.39 ± 0.53 |
| 7 | −6.46 ± 0.55 −6.30 ± 0.44 | −6.38 ± 0.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizerska-Kowalska, M.; Sowa, S.; Donarska, B.; Płaziński, W.; Sławińska-Brych, A.; Tomasik, A.; Ziarkowska, A.; Łączkowski, K.Z.; Zdzisińska, B. New Borane-Protected Derivatives of α-Aminophosphonous Acid as Anti-Osteosarcoma Agents: ADME Analysis and Molecular Modeling, In Vitro Studies on Anti-Cancer Activities, and NEP Inhibition as a Possible Mechanism of Anti-Proliferative Activity. Int. J. Mol. Sci. 2022, 23, 6716. https://doi.org/10.3390/ijms23126716
Mizerska-Kowalska M, Sowa S, Donarska B, Płaziński W, Sławińska-Brych A, Tomasik A, Ziarkowska A, Łączkowski KZ, Zdzisińska B. New Borane-Protected Derivatives of α-Aminophosphonous Acid as Anti-Osteosarcoma Agents: ADME Analysis and Molecular Modeling, In Vitro Studies on Anti-Cancer Activities, and NEP Inhibition as a Possible Mechanism of Anti-Proliferative Activity. International Journal of Molecular Sciences. 2022; 23(12):6716. https://doi.org/10.3390/ijms23126716
Chicago/Turabian StyleMizerska-Kowalska, Magdalena, Sylwia Sowa, Beata Donarska, Wojciech Płaziński, Adrianna Sławińska-Brych, Aleksandra Tomasik, Anna Ziarkowska, Krzysztof Z. Łączkowski, and Barbara Zdzisińska. 2022. "New Borane-Protected Derivatives of α-Aminophosphonous Acid as Anti-Osteosarcoma Agents: ADME Analysis and Molecular Modeling, In Vitro Studies on Anti-Cancer Activities, and NEP Inhibition as a Possible Mechanism of Anti-Proliferative Activity" International Journal of Molecular Sciences 23, no. 12: 6716. https://doi.org/10.3390/ijms23126716